

Abstract
Morusin, a water-insoluble prenylated flavonoid is known for its numerous medicinal properties. It manifests its anticancer potential by suppression of genes involved in tumor progression. However, poor solubility of the drug results in low bioavailability and rapid degradation thus hindering its clinical utilization. In order to overcome this, we have synthesized a niosome system composed of non-ionic surfactant span 60 and cholesterol using a thin-layer evaporation technique to improve the aqueous-phase solubility of the drug. Highly cytocompatible niosomes of 479 nm average size with smooth and uniform spherical morphology were synthesized in a facile manner. Unlike free morusin, nanomorusin was found to be freely dispersible in aqueous media. Having an extremely high drug entrapment efficiency (97%), controlled and sustained release of morusin resulting in enhanced therapeutic efficacy was observed in cancer cell lines of 4 different lineages. The results demonstrate that the morusin-niosome system is a promising strategy for enhanced anti-cancer activity against multiple cancer types and could be an indispensable tool for future targeted chemotherapeutic strategies.
Highly cytocompatible morusin-loaded niosomes were synthesized showing high drug loading and encapsulation efficiencies with sustained release of the drug. Enhanced therapeutic efficacy was observed against 4 different cancer cell lines.
Introduction
Cancer remains the principal cause of death in the world, regardless of economic or financial status of the respective nations.1 Clinical cancer treatment is heavily dependent upon chemotherapy in conjunction with radiation therapy and surgery, with immunotherapy playing a supportive role. Despite major advances in these treatment modules, high instances of tumor relapse and long-term permanent side effects remain unaddressed. Generally, in order to impede the growth of proliferating tumor cells, cytotoxic drugs are given which deter the proliferation of cancer cells, but play a significant role in non-target toxicity resulting in unwanted and severe side effects.
Therefore, extensive research is being carried out on using substances derived from natural origins as potent inhibitors of cancer, which have roots in numerous traditional medicinal regimes. Natural substances like curcumin,2,3 resveratrol,4,5 quercetin,6,7 and green tea catechins8,9 have displayed considerable toxicity against various cancer types with minimal non-target toxicity. However, with the rise of therapies that utilize high throughput screening methods for production of pharmaceutical molecules and drugs, the interest in natural products has declined rapidly.10
Despite showing tremendous results against few cancer types, these therapies remained unsatisfactory for many solid cancers due to undesired side-effects, which redirected the attention on natural products. Rapamycin, produced by the soil bacterium Streptomyces hygroscopicus was the first anticancer natural product to be approved in 2007 followed by other natural product derivatives.10,11 Being chemoprotective in nature, these products are classified either as potent antioxidants, phenolic or having reactive groups, which impart protective properties.12
One such natural product is morusin, a prenylated flavonoid derived from the root bark of Morus alba which has been stated to possess anti-inflammatory, anti-oxidant and anti-bacterial activities.13 Effectiveness of morusin as a potent anti-tumor agent has been elucidated by a number of studies. Various biological activities displayed by morusin include protection against nitric oxide (NO)-induced neuroblastoma cell death,14 scavenging activity against superoxide anion radicals,15 anti-diabetic activity,16 adipocyte differentiation,17etc. Predominantly, morusin has shown cytotoxic effects against cancer cell lines including breast adenocarcinoma,18,19 colorectal adenocarcinoma,20 gastric cancer,21etc. Recent studies have reported inhibition of NF-κB and STAT-3 signaling by morusin in prostrate, pancreatic and liver cancer cells.22–24 In addition, morusin is also known to inhibit the expression of metalloproteinases (MMP-2 and MMP-9), which are elevated in the tumor microenvironment.24,25 These evidences though support morusin as an effective natural anticancer drug; its full potential has not been explored mainly due to its poor aqueous solubility and bioavailability. Hydrophobicity of therapeutic moieties is a major issue in drug formulations, which results in rapid clearance by the reticuloendothelial system (RES) and poor bioavailability, restricting their full clinical potential. Additionally, non-specific accumulation of the toxic compounds cannot be monitored or controlled, leading to side effects.
Nanotechnology offers solutions for such limitations by providing a stable reservoir for hydrophobic compounds, and also minimizing off-site interactions, apart from assisting in escaping the RES and reaching the target cells with high precision.26 Numerous examples of nano-medicine formulations exist in literature27–30 with continuous updates on a near to daily basis. Of these, micellar systems have attracted much attention due to their unique properties and enhanced biocompatibility. These are a class of colloidal nano-carriers comprising liposomes or niosomes, which are known to possess significant advantages over conventional dosage forms.31 Niosomes are novel drug delivery carriers having a bilayer that can either be unilamellar, or bi-lamellar composed of various non-ionic surfactants and cholesterol. Due to the availability of a variety of non-ionic surfactants, the vesicles can be designed accordingly.32 Compared to other surfactants, spans and tweens are known to have higher entrapment efficiency and better release characteristics.33,34 Niosomes offer prime advantages like ease of preparation, reduced toxicity, increased stability, protection against degradation of active drug molecules, and enhanced bioavailability compared to liposomes, which have issues with physical and chemical stabilities.32,35,36 Due to their unique and desirable properties, niosomes have been in use as a part of the cosmetic industry since 1970.37 The drugs can either be entrapped in aqueous core (hydrophilic) or bilayer (hydrophobic) due to presence of two distinct types of microenvironments.36 Drug delivery using niosomes has been achieved using different modes of administration like intramuscular,38 intravenous,39 transdermal,40etc. and niosomes have been employed for delivery of various chemotherapeutic drugs like curcumin,36 cisplatin,41 doxorubicin,42 paclitaxel,43etc. It has been specified that entrapment of drugs inside a niosomal nano carrier can prevent the fluctuations in drug plasma concentration thus achieving controlled drug delivery and reduced systemic toxicity of anticancer drugs.31
As far to our knowledge, there is no study reporting morusin as a nano formulation for curbing cancer growth. The present study was formulated to obtain a robust drug delivery system for encapsulating morusin in nanoparticles and accordingly, a facile synthesis route was employed for producing morusin niosomes and their cytotoxic and cellular localization studies performed to assess the feasibility of this nanoformulation as a potential anti-cancer candidate. We also performed docking studies to observe binding of morusin to matrix metalloproteinases (MMP-2) and (MMP-9) which are up regulated in the tumor milieu in pancreatic cancer,44 colorectal cancer,45 ovarian cancer46,47 and breast cancer,48,49etc. thus contributing to tumor progression and metastasis.
Experimental section
Materials
Span 60 (sorbitan monostearate) and cholesterol were procured from Tokyo chemical industry Co. Ltd. Morusin was purchased from Chemfaces. Chloroform was purchased from Kanto chemicals, Japan. Human triple negative breast cancer (MDA-MB-453), colorectal adenocarcinoma (HT-29), ovarian cancer (SKOV-3), pancreatic cancer (PANC-1) cell lines and Mc-coy medium were purchased from American Type Culture Collection (ATCC), USA. Mouse fibroblast cell lines (L929) was purchased from Riken BioResource Center, Japan. Dulbecco's modified Eagle's medium (DMEM), trypsin (0.25%), fetal bovine serum (FBS), penicillin (5000 μg ml−1)/streptomycin (5000 μg ml−1) and phosphate buffer saline (PBS) were procured from Gibco (Life Technologies). Alamar blue cell viability kit reagent was purchased from Invitrogen, USA. Live/dead viability/cytotoxicity kit, Leibovitz media, Lysotracker deep red and Nuc blue live ready probes reagent were acquired form Thermo Fisher Scientific. All other chemicals and solvents were of analytical grade.
Methods
Preparation of niosomes
Drug loaded niosomes were synthesized using thin layer evaporation method with slight modifications.32 Briefly, 100 mg of span 60, 20 mg of cholesterol and 10 mg of drug (morusin) were dissolved in 20 ml of chloroform. The solution was kept for intermittent stirring with a magnetic stirrer for 1 h. After 1 h, the resultant solution was kept for evaporation in a rotary evaporator for 1 h, which results in formation of a thin film. In order to get a completely dried film the flask was kept in a vacuum desiccator for 1 h. The obtained thin film was then hydrated using 20 ml of ultrapure distilled water and 4 cycles of heating and vortexing (3 min each) were performed in order to remove any residual chloroform from the solution. The resulting solution was then centrifuged at 8000 rpm for 30 minutes for atleast 3 cycles in order to concentrate down the particles. After discarding the supernatant, the pellet was washed with ultra pure water thrice. The product mor-nio (morusin-niosomes) was freeze dried and stored at −20 °C till further use. Void nio (devoid of morusin) and C-6 nio (coumarin-6 loaded niosomes for cellular uptake studies) were also synthesized similarly.
Determination of yield, entrapment and loading efficiencies
The yield of niosomes was calculated by using the following eqn (1)50
 |
1 |
where, Wm – weight of morusin, Wspan 60 – weight of span 60, Wcholestrol – weight of cholesterol, used for niosome synthesis.
The UV-Vis absorption spectra of free morusin, void nio and mor-nio was recorded by UV-Vis spectrophotometer (DU730, Beckman Coulter) for confirming entrapment of morusin. The amount of free drug released in the supernatant was then measured quantitatively against the calibration curve of the drug and the unknown quantity was determined. Percentage of entrapment efficiency was determined with a spectrophotometer using the eqn (2)50
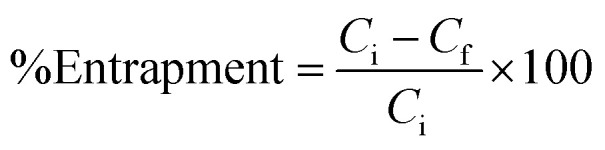 |
2 |
where, Ci is the initial drug concentration used for preparation of niosomes and Cf is the concentration of free drug in the supernatant obtained upon washing.
Loading efficiency was determined using the formula (3)51
 |
3 |
Niosome characterization
The size and morphological analysis of the synthesized nanoparticles was done using Hitachi SU6600 scanning electron microscope (SEM) and JEOL JEM-2100 transmission electron microscope (TEM). Mor-nio and void nio were dispersed in milliQ water for executing all characterization experiments. For doing SEM analysis, 50 μl of water-dispersed niosomes was dropped on a clean glass substrate and vacuum dried. The dried sample was sputter coated with platinum for 35 seconds (Hitachi E-1030, ion sputter) and observed under SEM functioning at an accelerating voltage of 10 kV. 100–125 individual particle diameters were measured and the values were averaged to find the mean diameter value of the resulting particles. For TEM, 10 μl of sample was placed on a hydrophilized Cu microgrid and air-dried with observations performed at an accelerating voltage of 100 kV. Particle size distribution and zeta-potential were measured using zetasizer (Malvern, Nano-ZS). For particle size distribution analysis, disposable polystyrene cuvettes were used and the average diameter and polydispersity index (PDI) values were acquired at 25 °C. Zeta potential measurements were carried out at 25 °C using a dip cell. All size and zeta potential measurements were carried out in triplicates.
In order to characterize the structure and explore the chemical bonding patterns between morusin and the niosome components (span 60 and cholesterol) Fourier Transform Infra-Red Spectroscopy (FTIR) (Thermo Scientific, Nicolet iS50 FT-IR) was performed. Samples were made into pellets using potassium bromide (KBr) and subjected for analysis under transmittance mode (4000–500−1 cm).
In vitro drug release
In order to study the diffusion of morusin from niosomes, drug release studies were carried out. The nanoparticles were dispersed in phosphate saline buffer (PBS) at two different pH conditions of pH 4.5 and pH 7.4. All experiments were carried out in triplicates. 15 mg of sample was dispersed in 15 ml of PBS buffer and distributed in 15 Eppendorf tubes such that concentration per tube was 1 mg ml−1. All tubes were kept at 37 °C under a continuous shaking of 130 rpm for 9 days. At predetermined time intervals, one tube was taken out and centrifuged at 15 000 rpm/30 min. Absorbance of the resulting supernatant was read by a UV-Vis spectrophotometer at fixed wavelength of the drug i.e. 269 nm and the amount of morusin released was determined. The release kinetics was calculated according to the following eqn (4)52
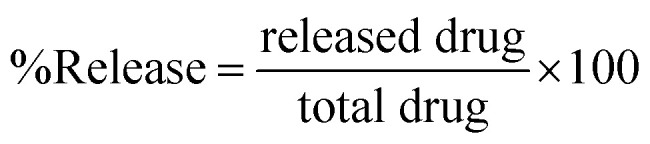 |
4 |
where ‘released drug’ is the concentration of the drug released and collected at pre-defined time intervals and ‘total drug’ is the amount of drug entrapped in the nanoparticles.
Cell culture
Human triple negative breast cancer (MDA-MB-453), Human colorectal adenocarcinoma (HT-29), Human pancreas ductal adenocarcinoma (PANC-1), Human ovarian cancer (SKOV-3) and mouse fibroblast cell lines (L929) were chosen to conduct in vitro cell studies and analyze the therapeutic efficacy of mor-nio. PANC-1 cells along with SKOV-3 and L929 were cultured in T-25 tissue culture flasks in DMEM, HT-29 in Mc-coy and MDA-MB-453 cells in Leibovitz media, supplemented with 10% FBS, 1% penicillin/streptomycin at 37 °C in a humidified atmosphere of 5% CO2 and for MDA-MB-453 without CO2 till cells were 80% confluent. The cells were sub cultured every 3–4 days or once the confluency was attained.
Cytocompatibility analysis
To assess whether the bare niosomes (void nio) exert any toxicity, cytocompatibility analysis was performed. For the assay, confluent cells were trypsinized, plated in a microtiter 96-well plate at a density of 8 × 103 cells per ml and maintained at 37 °C, 5% CO2 and for MDA-MB-453 without CO2. After incubation of 24 h, media was replenished and void niosomes were added in the concentration range of 50 μg ml−1, 100 μg ml−1, 250 μg ml−1, 500 μg ml−1, and 1000 μg ml−1. Cells without nanoparticles were considered as controls. After treatment with the desired concentration of void nio, the cells were incubated for 6, 24 and 48 hours under respective culture conditions. After defined incubation period, 10% alamar blue dye (which monitors the reducing environment of healthy cells and converts the non-fluorescent resazurin (blue color) to a highly fluorescent reduced form resorufin (red color) indicative of metabolic activity of cells) was added to the cells and plates were incubated for 4 hours following which fluorescence was recorded at 580–610 nm by a microplate reader (Power scan HT Microplate reader, Dainippon Sumitomo Pharma, Japan).
In vitro cytotoxicity studies
The therapeutic efficacy of mor-nio on different cancer cell lineages and normal cells was assessed using alamar blue. The assay procedure was similar to previous section. After incubation of cells for 24 h, media was replenished and free drug and mor-nio were added at varying concentration (50 μg ml−1, 100 μg ml−1, 250 μg ml−1, 500 μg ml−1, and 1000 μg ml−1). The culture/data acquisition conditions and parameters were similar as described in previous section. Here we used free morusin in 0.5% DMSO, mor-nio and untreated cells as control group for the experiment. The assay was performed in three autonomous experiments all in triplicates and cell viability was computed using the formula (5)51
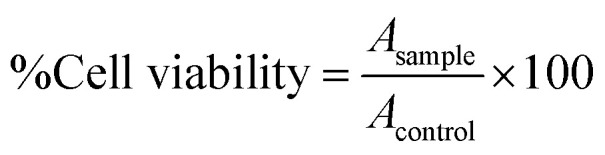 |
5 |
where Asample is the absorbance of the sample and Acontrol is the absorbance of the control.
Internalization and co-localization of niosomes
The internalization of niosomes in L929, HT-29, PANC-1, SKOV-3 and MDA-MB-453 was studied by confocal microscopy. As morusin is non-fluorescent, C-6 niosomes were utilized for the assay. The cells were grown in respective growth media/culture condition on 35 mm glass base culture dishes at a concentration of approximately 5 × 104 cells for 24 h following which 100 μg ml−1 of C-6 nio was subsequently added to the cultures and incubated for 2 hours. After incubation, the cells were washed with 1×, pH 7.4 PBS buffer to remove any excess unbound particles. Lysotracker deep red and Nuc blue were then added to the plates in order to stain the acidic compartments and nuclear region, respectively according to product instructions. The cells were examined using a high-speed confocal laser-scanning microscope (CLSM, Nikon A1 plus Tokyo, Japan) at excitation wavelengths of 402 nm (NucBlue), 488 nm (C6) and 630 nm (Lysotracker).
Live/dead assay
The qualitative determination of live and dead cells after nanoparticle treatment was carried out using CLSM. Cells at a density of 5 × 104 cells were plated in a 35 mm glass base dishes for 24 h following which 500 μg ml−1 of free morusin, void nio, and mor-nio were added and incubated for a further 24 h period. Post-incubation, cells were washed twice with PBS and stained with live/dead viability/cytotoxicity reagent as per product instructions. The plates were then incubated at RT for 1 h following which the cells were observed in CLSM at excitation and emission wavelengths of 495/515 nm for calcein and 528/617 for EthD-1. The number of dead and viable cells in the test was compared to the total number of viable cells in the control.
Docking studies
In order to check the probable binding of morusin to MMP-2 and MMP-9, molecular docking was executed using Autodock 4.0 software (USA). For performing Docking simulations, we downloaded crystal structures of proteins MMP9 (PDB id: 1GKC) and MMP2 (PDB id: 1CK 7) from protein databank (pdb). Structure of ligand (Morusin) was downloaded from PubChem (PubChem id: 5281671). Both ligand and protein structures were subjected to geometry and charge optimization using MMFF94 and Gasteiger charge calculations. Essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added with the aid of AutoDock tools. Affinity (grid) maps of 100 × 100 × 100 Å grid points and 0.375 Å spacing were produced using the Autogrid program. AutoDock parameter set-and distance-dependent dielectric functions were used in the computation of the van der Waals and the electrostatic terms, respectively. Docking simulations were accomplished using the Lamarckian genetic algorithm (LGA) and the Solis & Wets local search method. Initial position, orientation, and torsions of the ligand molecules were set randomly. All rotatable torsions were released during docking. Each docking experiment was derived from 100 different runs that were set to terminate after a maximum of 2 500 000 energy evaluations. The population size was set to 150. During the search, a translational step of 0.2 Å, and quaternion and torsion steps of 5 were applied. We selected ligand–protein complexes that offered most stable conformations based on lowest Gibbs binding energy. Short range van der Waal and electrostatic interactions, hydrogen bonding, entropy losses were included for energy-based autodock scoring function (Morris et al., 1998). The results were downloaded in pdb files and subjected to analysis using Drug Discovery Studios software.
Results and discussion
Morphological characterization of morusin loaded niosomes
Niosomes (void nio/mor-nio) were synthesized using thin-layer evaporation method in order to enhance the bioavailability, efficacy and aqueous solubility of the drug. Span 60 was used as an ionic surfactant and cholesterol as an additive for preparation of niosomes. Cholesterol imparts rigidity53 while span 60 protects the drug from proteolytic enzymes leading to higher physical stability.54,55 The synthesized niosomes were then characterized for their morphology, size distribution and surface characteristics.
As shown in SEM micrograph (Fig. 1a and d), the void nio (445 nm) and mor-nio (479 nm) have a uniform spherical morphology with smooth surface characteristics. The vesicles are formed due to self-assembly of the non-ionic surfactants upon their interaction with the aqueous medium leading to creation of a concentric bilayer structure having morphology comparable to liposomes but without phospholipids which imparts better stability.35,56 Similar observations were made in TEM as well where smooth and spherical niosomes could be visualized (Fig. 1b and e). Fig. 1c and f show Gaussian size distribution of the void and mor-loaded nanoparticles wherein majority particles in both cases lie in the size range of 400–500 nm. Detailed characterization of the particles is provided in Table 1. Further, dynamic light scattering (DLS) measurement done for void and mor-nio confirmed nanoparticles with size distribution in the range of 400–500 nm and z-average size of 445 nm and 479 nm which correlated with SEM and TEM observations. Since, the approximate size of vesicles/micelles for cellular uptake via the endocytic pathway is 100–500 nm, our synthesized mor-nio fall in the desired range for successful internalization by cancer cells.57,58
Fig. 1. (a) SEM micrograph of void nio (d) SEM micrograph of mor-nio. (b) TEM image of void nio (e) TEM image of mor-nio (c and f) Gaussian size distribution for void and mor-nio.

Physico-chemical characterization of void and mor-nio.
| Characterization parameter | Void nio | Mor-nio |
|---|---|---|
| Z average diameter (nm) | 445 | 479 |
| Poly dispersity index (PDI) | 0.3 | 0.29 |
| Zeta potential (mV) | −14.2 | −19.8 |
| Drug loading | — | 7.69 ± 0.14% |
| Entrapment efficiency | — | 97 ± 1.25% |
| % Yield | 38.46 ± 1.31% | 31.07 ± 2.43% |
The cellular interaction and uptake of nanoparticles is highly influenced by the surface charge of nanoparticles.59 Studies have reported that negatively or positively charged nanoparticles are more readily internalized as compared to their uncharged counterparts.59 Although positively charged nanoparticles have a rapid cellular uptake in comparison to negatively charged nanoparticles, they are known to cause hemolytic size-effects and cytotoxicity to normal tissues.59,60 The synthesized void and mor-nio had a zeta potential of −14.2 and −19.8, respectively, denoting the stability of the colloidal suspension of niosomes. This could be accredited to the coulombic repulsion forces occurring from their surface charge, which overcomes the van der Waals attractive forces between them.50Table 1 depicts the physico-chemical parameters of the prepared nanoformulations.
Drug entrapment
Hydration of thin film of mor-nio was achieved using deionized distilled water in order to obtain drug-loaded niosomes. The hydration step is important for self-assembly of the surfactants and cholesterol as tiny polymeric vesicles entrapping morusin.61 Confirmation of entrapment efficiency was established using UV-Vis absorption spectroscopy. Fig. 2a depicts the absorbance of mor-nio in comparison with void nio and free morusin.
Fig. 2. (a) UV-absorption overlay spectra for free morusin, mor-nio and void nio. The characteristic peak for morusin is around 269 nm, which could be also observed in mor-nio. The void nio provided a flat profile (b) FT-IR spectra of mor-nio in comparison to native morusin and void nio as a control. (c) Sustained drug release profile for morusin nano formulation. An enhanced release was observed in acidic pH establishing mor-nio as an effective drug delivery system for pH dependent dug release in cancer cells.

Morusin (free drug) shows λmax at wavelength 269 nm, where a major peak was observed as shown in (Fig. 2a). Same peak was observed in the case of mor-nio as well, depicting morusin entrapment in niosomes. However the peak was not as prominent as seen in the case of free drug which maybe due to the small amount of drug entrapment inside the niosomal nano carrier. The minor peak shifts observed could be due to the interaction of morusin with niosome constituents during synthesis and encapsulation processes. In case of void nio, no peak was observed. The entrapment efficiency of the particles was around 97 ± 1.256% for every batch of particles synthesized with 70.55 μg of the drug getting encapsulated (loading efficiency − 7.69 ± 0.14%) as seen in Table 1. It has been widely reported that length of the alkyl chain of surfactants determines the diameter of vesicles. Vesicles obtained from stearyl (C18) chain are known to yield larger particles with higher entrapment efficiencies,32 which can be seen from our results by the use of span 60 as the surfactant. For every batch of nanoparticles synthesized, the encapsulation, yield and loading efficiencies had consistent values. The prepared mor-nio was totally water-dispersible as compared to free morusin, which is insoluble in water.
Surface characterization of niosomes
In order to study the chemical nature of niosomes, we performed FTIR spectroscopy studies. In the study, we used void nio and free drug as controls for the comparative analysis of chemical bonding properties and peak shifts in our drug loaded nano formulation. FTIR measures the selective absorption of light by the vibration modes of specific chemical bonds in the sample. Vibrations arising due to the functional groups are involved in such interactions, which may alter frequency and intensity.51,61Fig. 2b shows overlay of spectra from morusin, void nio and mor-nio. The small band around 2978 cm−1 is attributed to the C–H stretching, which may be caused due to the –CH, –CH2 and –CH3 stretching vibrations characteristic of morusin.62 However in case of void nio and mor-nio the band is around 2913 cm−1. Moreover the signals around 1645 cm−1 and 1585 cm −1 can be observed in both drug and drug loaded niosomes while it's completely absent in void nio. Additionally, we could also see significant overlap in the spectra of void and mor-nio, which suggests that the chemical nature of the niosome remains unaltered. The FTIR analysis implies that there was no chemical interaction between the drug and the nanoparticle that could modify its chemical structure during the study and morusin was only physically encapsulated in the niosomes.
In vitro drug release
The drug release kinetics of mor-nio was studied using UV-Vis spectrophotometer for a period of 9 days under controlled conditions of 37 °C with constant stirring at 130 rpm throughout the experiment. The most important prerequisite for a drug delivery vehicle is the slow and sustained release of drug. Percentage of morusin released from mor-nio at pH 4.5 and pH 7.4 under in vitro conditions can be observed in Fig. 2c. The drug release was biphasic with an initial burst release happening at the start, around 1st h (8.5% – pH 7.4 and 11.4% – pH 4.5) which can be attributed to the diffuse mechanism from outer layers of niosomes as well as minute surface-bound drug molecules.63 Morusin release from niosomes was perceived to be pH dependent, resulting in a slow drug release at physiological pH 7.4 and a higher release at acidic pH 4.5 (pH; depicting the acidic endosomal compartment after endocytosis), which is a prerequisite for transport of anticancer drugs in the acidic environment of cancer cells. Followed by burst release, a gradual release curve was observed after 24 h with 30.2% (21.32 μg) of drug being released till this point in acidic pH. After 48 h, the drug release increased to 43.6% (30.80 μg). At 120 h, approximately 58.1% of morusin was released at acidic condition however at physiological pH, only 43.3% was released following which there was a stationary phase. The niosome membrane ensures that the encapsulated drug crosses the membrane, which causes slower and delayed release. Also the amount of cholesterol included in the formulation affects the release kinetics, with high CH content causing slow release of the drug from the vesicle due to decreased permeability of the niosomal membrane but higher encapsulation efficiency that can be seen in case of our mor-loaded niosomes where the encapsulation efficiency was found to be around 97%.63
Cytocompatibility analysis
In order to determine whether the synthesized niosomes were non-toxic or could affect the growth of cells, we performed compatibility analysis in normal and cancer cells. Studies have proven niosomes to be safe without any cytotoxic effects.34,64,65 As could be seen from (Fig. 3), the cellular viability was above 80% in all the cell lines even at high concentration of particles (1000 μg ml−1). Thus, we confirm the particles synthesized were highly compatible to cells under study and could be utilized for further bio-applications without any modifications.
Fig. 3. Cytocompatibility of void niosomes tested against cancer (a) HT-29, (b) PANC-1, (c) MDA-MB-453 (d) SKOV-3, cells and normal cell line (e) L929. The particles were highly cytocompatible as depicted by high viability in all cell lines even at highest concentration of particles used.

Cellular uptake of mor-nio
The uptake of niosomes by cancer cells was assessed using C-6 nio. Fig. 4 depicts the CLSM images of different cell lines treated with coumarin-6 loaded niosomes. The cells were incubated with 100 μg ml−1 of C-6 nio for 2 hours following which lysotracker and nuc blue were added in order to stain the acidic organelles and nucleus. The particles can be seen accumulated predominantly in the cytoplasm as evident from the green fluorescence of coumarin-6 while controls show no fluorescence, thereby negating any auto fluorescence from the cells themselves.
Fig. 4. Confocal microscopy images demonstrating cellular internalization of coumarin-6 niosomes by cancer cells. The green fluorescent intensity from cells with C-6 nio was clearly evident, signifying the enhanced uptake of niosomes by cancer cells. Nuclei were stained with NucBlue and lysosomes with lysotracker deep red to show nanoparticles co-localization. Scale bars represent 20 μm.

HT-29 (Fig. 4b) cells had higher accumulation of particles as seen by strong green fluorescence compared to other cells. SKOV-3 (Fig. 4q) is also seen to have higher accumulation, which can be seen in the nucleus also however the cells were least susceptible to mor-nio treatment as seen from cytotoxicity studies. In PANC-1 (Fig. 4g) and MDA-MB-453 (Fig. 4l) the fluorescent signals were weak signifying lower accumulation of particles. However, MDA-MB-453 was fairly inhibited by mor-nio treatment as shown in cytotoxicity study. These observations indicate that lower particle accumulation was enough to exert significant toxicity to the cells.
Also, the images confirmed the endosome-mediated entry of particles as they can be readily seen accumulated in lysosomes where overlapping signals of nanoparticles and lysosomes could be visualized.
Therapeutic efficacy of mor-nio
The sustained toxic effect of the drug is stalled due to its poor bioavailability, a desired property for successful anticancer therapy. Therefore, to evaluate the therapeutic potential of mor-nio in comparison to free drug; we carried out dose and time-dependent in vitro cell viability studies using alamar blue assay in HT-29, PANC-1, MDA-MB-453, SKOV-3 cells and L929 cells. Cells were subjected to five different concentrations (50 μg ml−1, 100 μg ml−1, 250 μg ml−1, 500 μg ml−1, 1000 μg ml−1) of mor-nio and free morusin for 6, 24 and 48 hours. In cancer cell lines, we could perceive instant cell death and reduced cell viability within 24 hours when treated with mor-nio (Fig. 5). However, at the lowest dose (50–100 μg ml−1), the effect was comparatively less pronounced while maximum cytotoxic effect was observed at 1000 μg ml−1 concentration after 48 h. This could be due to the fact that mor-nio were able to reach a therapeutic concentration after 48 hours to impart cytotoxic effects to cancer cells. Toxicity of free drug was performed keeping in mind the amount of drug encapsulated in 1000 μg ml−1 of mor-nio i.e. 70.55 μg, which was calculated using eqn (2). Compared to mor-nio, the free drug displayed less cytotoxicity towards cancer cells which maybe due to its hydrophobicity indicating the advantage of encapsulating the drug inside niosomal carrier. In case of free morusin, the cell viability was - HT29 (48.4%). PANC-1 (62.77%), MDA-MB-453 (53.02%), SKOV-3 (62.84%) after 24 h at 70.55 μg ml−1 concentration (which is the amount of drug entrapped in 1000 μg ml−1 of mor-nio) however a decline in viability was seen after 48 h. On the other hand, mor-nio decreased the cell viability to a significant level after 24 h at 1000 μg ml−1 compared to the free drug -HT29 (33.05%), PANC-1 (46.13%), MDA-MB-453 (39.93%), SKOV-3 (59.17%). As seen in drug release (Fig. 2c) at 24 h, 30.23% (21.32 μg) was released in acidic environment while it increased to 43.66% (30.80 μg) after 48 h, thus correlating with the cytotoxicity data with an increase in cell death at 48 h due to augmented release of the drug. The viability after 48 h in case of morusin niosomes was – HT29 (26.32%). PANC-1 (35.88%), MDA-MB-453 (21.53%), SKOV-3 (46.3%) whereas in case of free drug it was – HT29 (35.21%), PANC-1 (55.68%), MDA-MB-453 (43.77%), SKOV-3 (53.55%). Though, similar particle accumulation was observed in case of L929 cells as well (Fig. 4v), viability remained at approximately 80% (Fig. 5i and j). The reason for this could be the high mitotic index (cell doubling rate) of L929 compared to other cell lines which could have limited the effect of the drug when compared to rapidly dividing cells.
Fig. 5. Therapeutic efficacy of morusin-loaded niosomes and free morusin towards (a and b) HT-29 (c and d) PANC-1 (e and f) MDA-MB-453 (g and h) SKOV-3 and (i and j) L929 cell line. Compared to the free drug, more inhibition was observed in the case of morusin-loaded niosomes in all cell lines.

HT-29 was seen to be the most susceptible cell line to mor-nio treatment. NF-κB activation is known to promote tumor progression in colorectal cancers. Lee et al.20 reported that morusin was able to significantly suppress constitutive activity of NF-κB via classical pathway. Dephosphorylation of Akt, PDK1 and P13 was also observed along with activation of caspase 9 due to downregulation of XIAP expression, caspase-8, and -3 due to morusin treatment in colorectal cancer cells. Thus, taking into account all the above factors, multiple pathways are known to be involved in inducing apoptosis in HT-29 cells by morusin which may be the reason of enhanced inhibition observed by mor-nio treatment. In case of pancreatic cancer, constitutive activation of STAT 3 has been reported causing tumor cell survival, proliferation and metastasis. Inhibition of STAT-3 and downregulation of its regulated gene products-IAP1, IAP2, Bcl-2, survivin, cyclin, COX-2, VEGF, MMP-9 was seen in pancreatic tumor cells in parallel with inhibition of JAK1, JAK2 upon morusin treatment leading to apoptosis.22
Triple negative breast cancers (TNBCs) are cancers that do not express progesterone receptor (PR), estrogen receptor (ER), and the HER2 isoform of epidermal growth factor receptor (EGFR) and hence due to lack of a specific therapeutic target triple negative breast cancers have limited treatment options.66 Mor-nio was seen to significantly reduce the cell viability of MDA-MB-453, a triple negative breast cancer line upon treatment. Morusin has been shown to increase expression of pro-apoptotic marker protein Bax, reduce anti-apoptotic protein survivin as well as cause cleavage of caspase 9, -3 and PARP in a dose and time dependent manner in triple negative breast cancer cell lines indicating its ability at targeting anti and pro-apoptotic signals that may increase anti-cancer activity.19 The mechanism of action however remains elusive and its known that STAT3 inhibition affects expression of downstream targets-Bax and survivin, so maybe morusin works by inhibiting STAT 3 also. Despite inducing apoptosis in breast cancer cells, apoptotic rate was low compared with cytotoxicity rate for same concentration of morusin which means morusin may induce other types of cell death like necrosis and autophagy in breast cancer cells.19 Thus, our synthesized mor-nio were able to deliver the drug into cancer cells by enhanced permeation and retention (EPR) effect for maximizing the therapeutic effects of morusin even in a drug resistant cell line and cause its inhibition while reducing systemic toxicity. A high level of receptor expression-EGFR, insulin growth factor receptor-1 (IGF-1R), folate, CXCR4 has been reported in TNBC tissues66 which may also be the mode of action by which morusin mediated toxicity in MDA-MB-453, however this remains to be confirmed by further studies.
Epithelial ovarian cancer (EOC), remains the leading cause of death among gynecological malignant tumors.67 Chemoresistance and treatment failure often occurs due to cellular resistance to apoptosis, thereby compounds, which may treat the tumor other than by inducing apoptosis, may provide a better insight in treating EOC. Recently, morusin was shown to kill ovarian cells by paraptosis rather than inducing apoptosis.67 It was also shown to inhibit colony formation in EOC cells occurring due to the dual effect of cell death and cell proliferation inhibition. However, caspase -3 nor -9 were found to be inactivated by morusin in EOC ruling out apoptosis mediated death. In our study, though significant toxicity was observed, SKOV-3 cells were the most resistant of the cell lines under study. Thus, it is presumed that morusin can activate diverse cell death pathways depending on different cancer cell types, which explains our cytotoxicity data, with sensitivity of different cancer types to morusin inhibition.
The enhancement of cell death within 24 h treatment of morusin niosomes compared to free morusin may be due to the advantage of nanoparticulate mode of drug delivery. Also it has been seen that use of span 60 as a surfactant has been known to increase the anti-tumor action of the drug.68 Since nanoscale particles are easy to internalize within cells, increased drug retention could be achieved. The cytotoxicity data implies that mor-nio can competitively demonstrate excellent anticancerous effect than free morusin.
Live/dead viability assay
In order to analyze activation of apoptosis in cancer cells when treated with morusin niosomes, we performed live/dead viability assay to visualize early apoptotic and necrotic cells. The viability of cells was analyzed post 24 h using live dead viability staining. EthD-1 enters dead cells via the damaged membrane, binds to their nuclei, and releases red fluorescence while live cells enzymatically convert calcein AM in fluorescent calcein which emits green fluorescence. 500 μg ml−1 concentration was sufficient to elicit cytotoxicity to cancer cells as confirmed by cytotoxicity analysis therefore the same concentration was utilized for this assay. Significant number of dead cells could be observed with morusin loaded niosome treated groups compared to free drug at the same concentration (Fig. 6).
Fig. 6. Therapeutic efficacy of morusin loaded, free morusin and void niosomes against cancer cell lines using Live/Dead Assay. The control cells and cells treated with void niosomes were live as evident by the green fluorescence. The cells treated with morusin niosomes showed considerable toxicity with minimal number of live cells and increased amount of dead cells (red fluorescence) compared to free morusin where there were both viable and dead cell populations. Scale bars represent 500 μm.

In case of HT-29 and MDA-MB-453, when cells were treated with free morusin, a large number of viable cell population (Fig. 6b′ and j′) could be observed with some necrotic cells – (Fig. 6c′ and k′) as well. Compared to free morusin, significant amount of dead cells were observed when cells were treated with mor-nio (Fig. 6c* and k*) as established by enhanced fluorescent intensity of Etbr (red). Similar effects could be observed in PANC-1- (Fig. 6g*), however the fluorescent intensity of Etbr was comparatively less compared to HT-29 and MDA-MB-453 which can be confirmed by cytotoxicity analysis wherein at 500 μg ml−1 concentration, cell viability for mor-nio was 48.1% for HT-29 and 50.17% for MDA-MB-453 while for PANC-1 it was 61.65% after 24 h. However, in case of SKOV-3, both morusin (Fig. 6o′) and mor-nio (Fig. 6o*) had similar effect on the cells but the red fluorescence coming from the dead cells was more pronounced in the case of mor-nio as inferred from cytotoxicity data where cell viability after 24 h was 77.45% when free drug was used and 70.78% when cells were treated with mor-nio. Both control cells (Fig. 6d, h, l and p) and cells that were treated with void nio (Fig. 6d′′, h′′, l′′ and p′′) showed higher percentage of live cells as evident by high green fluorescent intensity thus indicative of the healthy state of cells and establishing that cell death was induced by the drug and not by the niosome nano-carrier. The results provide visual quantitative support to the cytotoxicity observations that mor-nio could induce cytotoxic effects in cancer cells.
Docking studies
It has been reported by a number of studies that morusin exerts its inhibitory effect on cancer cells by inhibiting one or two key molecules, which are required for cancer pathogenesis like STAT-3 and NF-κB pathways. The inhibition of the matrix metalloproteinases gelatinases (MMP-2 and MMP-9) by morusin has also been described in literature.20,24,25 Also, there is an elevation of these MMPs in colorectal cancer, pancreatic cancer, ovarian cancer and breast cancer as discussed previously. Thus, in order to check whether there was any interaction between morusin and MMPs, we performed molecular docking studies using Autodock tool. The study showed that the MMPs interaction with morusin was influenced by hydrogen bonding with active site residues. We performed blind docking study to get an unbiased estimate of the binding site for morusin on MMPs. The Gibbs free binding energy (ΔG) of the MMP-9 morusin complex was ΔG = −7.35 kJ mol−1 with inhibitory constant (Ki) value of 4.09 μM. Whereas, for MMP-2 morusin complex ΔG = −8.33 kJ mol−1 with Ki value of 786.78 nM. In case of MMP-9-morusin complex, there was H-bonding of Tyr 393 with morusin while Tyr 423 was engaged in cationic-pi interactions. Leu 187, Met 422 and His 401 provided the hydrophobic interactions. There was no interaction between morusin and Zn of MMPs (Fig. 7a and b). In case of MMP-2, active site amino acids Asn 111, Asn 55 and His 193 were engaged in polar contact with morusin, while Tyr 182 was found to have cation-pi interactions. Pro 105 and Leu 190 were the two amino acid residues responsible for the hydrophobic interactions between the MMP-2-morusin complexes (Fig. 7c and d). This is the first computational study to show the probable binding interaction site for morusin on MMPs.
Fig. 7. Molecular docking studies showing interaction between the metalloproteinases (MMP-9 and MMP-2) and morusin. (a and b) It shows the amino acid residues of MMP-9 interacting with morusin (c and d) Probable binding pocket for morusin on MMP-2 and its interacting residues, which help in attaining stable conformational state via polar and non-polar interactions.

Conclusion & future perspectives
The present research work details the development of morusin loaded niosomal nano-formulation for cancer inhibition. The therapeutic efficacy was imparted by morusin, which is a potent naturally derived chemotherapeutic agent as confirmed by the results presented in this report. The particles, with an average size of 479 nm, displayed high drug loading and encapsulation efficiencies, in addition to rendering aqueous dispersibility to the usually hydrophobic morusin. A steady and sustained drug release could be observed for morusin from the niosomes along-with increased release in acidic pH thus not only proposing its efficient release in the acidic environment of cancer cells, but additionally reducing the spontaneous drug release under physiological pH of normal cells. The toxicity of morusin niosomes was tested against 4 cancer cell lines of different lineages to assess the degree of toxicity depending on the cell lineage. According to our study, the cancer cells susceptibility to mor-nio treatment was in the following order: HT-29>MDA-MB-453>PANC-1>SKOV-3. Our findings suggest that, regardless of the cell type or origin, all the cells under investigation were highly susceptible to the nano formulation in a concentration dependent manner, predicting the utilization of this morusin-niosomal nanoformulation for multiple types of cancers. Morusin-nio was able to inhibit the survival of MDA-MB-453, which is a highly drug resistant triple negative breast cancer cell line. We also performed docking studies to analyze probable binding of morusin with MMPs. We anticipate that morusin loaded niosomes will open new scenarios for precise delivery of morusin to cancer sites as well as lay foundation for the development of novel targeted therapies in future.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Srishti Agarwal would like to acknowledge her sincere gratitude to the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan for the financial support under the Monbukagakusho fellowship during the research. S. A. would also like to express her sincere gratitude to the Inoue Enryo Research Grant (Toyo University).
References
- Prager G. W. Braga S. Bystricky B. Qvortrup C. Criscitiello C. Esin E. et al. ESMO Open. 2018;3(2):e000285. doi: 10.1136/esmoopen-2017-000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J. Fan Z. Li Y. Zhang Y. Yu F. Su G. Xie L. Hou Z. Int. J. Nanomed. 2018;13:1381–1398. doi: 10.2147/IJN.S152312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davatgaran-Taghipour Y. Masoomzadeh S. Farzaei M. H. Bahramsoltani R. Karimi-Soureh Z. Rahimi R. Abdollahi M. Int. J. Nanomed. 2017;12:2689–2702. doi: 10.2147/IJN.S131973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. K. Lillard Jr. J. W. Singh R. Cancer Lett. 2018;427:49–62. doi: 10.1016/j.canlet.2018.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal R. Chadha V. D. Dhawan D. K. Nanomedicine. 2018;14:1059–1071. doi: 10.1016/j.nano.2018.01.008. [DOI] [PubMed] [Google Scholar]
- Aghapour F. Moghadamnia A. A. Nicolini A. Kani S. N. M. Barari L. Morakabati P. Rezazadeh L. Kazemi S. Biochem. Biophys. Res. Commun. 2018;500:860–865. doi: 10.1016/j.bbrc.2018.04.174. [DOI] [PubMed] [Google Scholar]
- Patra A. Satpathy S. Shenoy A. K. Bush J. A. Kazi M. Hussain M. D. Int. J. Nanomed. 2018;13:2869–2881. doi: 10.2147/IJN.S153094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisha Tyagi R. D. Begun J. Popat A. Int. J. Pharm. 2017:518. doi: 10.1016/j.ijpharm.2016.12.030. [DOI] [PubMed] [Google Scholar]
- Liang K. Chung J. E. Gao S. J. Yongvongsoontorn N. Kurisawa M. Adv. Mater. 2018;30:e1706963. doi: 10.1002/adma.201706963. [DOI] [PubMed] [Google Scholar]
- Basmadjian C. Zhao Q. Bentouhami E. Djehal A. Nebigil C. G. Johnson R. A. Serova M. de Gramont A. Faivre S. Raymond E. Desaubry L. G. Front. Chem. 2014;2:20. doi: 10.3389/fchem.2014.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Kim S. G. Blenis J. Cell Metab. 2014;19:373–379. doi: 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy L. Odhav B. Bhoola K. D. Pharmacol. Ther. 2003;99:1–13. doi: 10.1016/S0163-7258(03)00042-1. [DOI] [PubMed] [Google Scholar]
- Geng C. Yao S. Xue D. Zuo A. Zhang X. Jiang Z. Ma Y. Chen J. Zhongguo Zhongyao Zazhi. 2010;35:1560–1565. [PubMed] [Google Scholar]
- Lee H. J. Lyu da H. Koo U. Nam K. W. Hong S. S. Kim K. O. Kim K. H. Lee D. Mar W. Arch. Pharmacal Res. 2012;35:163–170. doi: 10.1007/s12272-012-0118-7. [DOI] [PubMed] [Google Scholar]
- Ko H. H. Wang J. J. Lin H. C. Wang J. P. Lin C. N. Biochim. Biophys. Acta. 1999;1428:293–299. doi: 10.1016/S0304-4165(99)00084-7. [DOI] [PubMed] [Google Scholar]
- Abd El-Mawla A. M. Mohamed K. M. Mostafa A. M. Sci. Pharm. 2011;79:951–961. doi: 10.3797/scipharm.1101-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. G. Matsuzaki K. Takamatsu S. Kitanaka S. Molecules. 2011;16:6010–6022. doi: 10.3390/molecules16076010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Wang Q. Dong L. Liu C. Sun Z. Gao L. Wang X. J. Exp. Clin. Cancer Res. 2015;34:137. doi: 10.1186/s13046-015-0252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S. Kim E. O. Kim S. H. Lee J. H. Ahn K. S. Yun M. Lee S. G. Oncol. Lett. 2017;13:4558–4562. doi: 10.3892/ol.2017.6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. C. Won S. J. Chao C. L. Wu F. L. Liu H. S. Ling P. Lin C. N. Su C. L. Biochem. Biophys. Res. Commun. 2008;372:236–242. doi: 10.1016/j.bbrc.2008.05.023. [DOI] [PubMed] [Google Scholar]
- Wang F. Zhang D. Mao J. Ke X. X. Zhang R. Yin C. Gao N. Cui H. Oncotarget. 2017;8:57187–57200. doi: 10.18632/oncotarget.19231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C. Kim J. H. Oh E. Y. Nam D. Lee S. G. Lee J. Kim S. H. Shim B. S. Ahn K. S. Pancreas. 2016;45:409–419. doi: 10.1097/MPA.0000000000000496. [DOI] [PubMed] [Google Scholar]
- Lim S. L. Park S. Y. Kang S. Park D. Kim S. H. Um J. Y. Jang H. J. Lee J. H. Jeong C. H. Jang J. H. Ahn K. S. Lee S. G. Am. J. Cancer Res. 2015;5:289–299. [PMC free article] [PubMed] [Google Scholar]
- Lin W. L. Lai D. Y. Lee Y. J. Chen N. F. Tseng T. H. Toxicol. Lett. 2015;232:490–498. doi: 10.1016/j.toxlet.2014.11.031. [DOI] [PubMed] [Google Scholar]
- Vochyanova Z. Pokorna M. Rotrekl D. Smekal V. Fictum P. Suchy P. Gajdziok J. Smejkal K. Hosek J. PLoS One. 2017;12:e0182464. doi: 10.1371/journal.pone.0182464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parveen S. Misra R. Sahoo S. K. Nanomedicine. 2012;8:147–166. doi: 10.1016/j.nano.2011.05.016. [DOI] [PubMed] [Google Scholar]
- Orunoglu M. Kaffashi A. Pehlivan S. B. Sahin S. Soylemezoglu F. Oguz K. K. Mut M. Mater. Sci. Eng., C. 2017;78:32–38. doi: 10.1016/j.msec.2017.03.292. [DOI] [PubMed] [Google Scholar]
- Sauraj Kumar S. U. Kumar V. Priyadarshi R. Gopinath P. Negi Y. S. Carbohydr. Polym. 2018;188:252–259. doi: 10.1016/j.carbpol.2018.02.006. [DOI] [PubMed] [Google Scholar]
- Loureiro J. A. Andrade S. Duarte A. Neves A. R. Queiroz J. F. Nunes C. Sevin E. Fenart L. Gosselet F. Coelho M. A. Pereira M. C. Molecules. 2017;22:277. doi: 10.3390/molecules22020277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M. Bhatnagar P. Mishra S. Kumar P. Shukla Y. Gupta K. C. Int. J. Nanomed. 2015;10:6789–6809. doi: 10.2147/IJN.S79489. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bansal S. Aggarwal G. Chandel P. Harikumar S. L. J. Pharm. BioAllied Sci. 2013;5:318–325. doi: 10.4103/0975-7406.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranya Manosroi P. W. Manosroi J. Sakai H. Sugawara F. Yuasa M. Abe M. Colloids Surf., B. 2003;30:129–138. doi: 10.1016/S0927-7765(03)00080-8. [DOI] [Google Scholar]
- Kumar P. G. P. Acta Pharm. Sin. B. 2011;1:208–219. doi: 10.1016/j.apsb.2011.09.002. [DOI] [Google Scholar]
- Taymouri S. Varshosaz J. Adv. Biomed. Res. 2016;5:48. doi: 10.4103/2277-9175.178781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. I. Asadullah M. Saeed A. Mahmood M. A. Rehman M. Ashfaq M. Braz. J. Pharm. Sci. 2015;51:607–615. doi: 10.1590/S1984-82502015000300012. [DOI] [Google Scholar]
- Mandal S. Banerjee C. Ghosh S. Kuchlyan J. Sarkar N. J. Phys. Chem. B. 2013;117:6957–6968. doi: 10.1021/jp403724g. [DOI] [PubMed] [Google Scholar]
- Thakur Gurjeet Singh N., in Nanobiomaterials in Galenic Formulations and Cosmetics, William Andrew Applied Science Publishers, 2016, ch. 7, vol. 10, pp. 149–174 [Google Scholar]
- Arunothayanun P. Turton J. A. Uchegbu I. F. Florence A. T. J. Pharm. Sci. 1999;88:34–38. doi: 10.1021/js980286u. [DOI] [PubMed] [Google Scholar]
- Namdeo A. Jain N. K. J. Microencapsulation. 1999;16:731–740. doi: 10.1080/026520499288672. [DOI] [PubMed] [Google Scholar]
- Uchegbu I. F. Double J. A. Turton J. A. Florence A. T. Pharm. Res. 1995;12:1019–1024. doi: 10.1023/A:1016210515134. [DOI] [PubMed] [Google Scholar]
- Kanaani L. Javadi I. Ebrahimifar M. Ebrahimi Shahmabadi H. Akbarzadeh Khiyav A. Mehrdiba T. Asian Pac. J. Cancer Prev. 2017;18:365–368. doi: 10.22034/APJCP.2017.18.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V. Anandhakumar S. Sasidharan M. Mater. Sci. Eng., C. 2015;56:393–400. doi: 10.1016/j.msec.2015.06.049. [DOI] [PubMed] [Google Scholar]
- Bayindir Z. S. Yuksel N. J. Pharm. Sci. 2010;99:2049–2060. doi: 10.1002/jps.21944. [DOI] [PubMed] [Google Scholar]
- Jakubowska K. Pryczynicz A. Januszewska J. Sidorkiewicz I. Kemona A. Niewinski A. Lewczuk L. Kedra B. Guzinska-Ustymowicz K. Dis. Markers. 2016;2016:9895721. doi: 10.1155/2016/9895721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said A. H. Raufman J. P. Xie G. Cancers. 2014;6:366–375. doi: 10.3390/cancers6010366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roomi M. W. Monterrey J. C. Kalinovsky T. Rath M. Niedzwiecki A. Oncol. Rep. 2010;23:605–614. doi: 10.3892/or_00000675. [DOI] [PubMed] [Google Scholar]
- Hu X. Li D. Zhang W. Zhou J. Tang B. Li L. Arch. Gynecol. Obstet. 2012;286:1537–1543. doi: 10.1007/s00404-012-2456-6. [DOI] [PubMed] [Google Scholar]
- Li H. Qiu Z. Li F. Wang C. Oncol. Lett. 2017;14:5865–5870. doi: 10.3892/ol.2017.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan-Solis K. D. Schneider S. S. Pentecost B. T. Bentley B. A. Otis C. N. Gierthy J. F. Arcaro K. F. J. Cell. Biochem. 2013;114:1385–1394. doi: 10.1002/jcb.24480. [DOI] [PubMed] [Google Scholar]
- Ankita Borah V. P. Girija A. R. Balasubramanian S. Rochani A. K. Maekawa T. Sakthi Kumar D. NanoWorld J. 2017;3(1):1–10. [Google Scholar]
- Rochani A. K. Balasubramanian S. Ravindran Girija A. Raveendran S. Borah A. Nagaoka Y. Nakajima Y. Maekawa T. Kumar D. S. Int. J. Pharm. 2016;511:648–658. doi: 10.1016/j.ijpharm.2016.07.048. [DOI] [PubMed] [Google Scholar]
- Sivakumar B A. R. Nagaoka Y. Iwai S. Venugopal K. et al. . RSC Adv. 2013;3:20579–20598. doi: 10.1039/C3RA42645A. [DOI] [Google Scholar]
- Abdelkader H. Alani A. W. Alany R. G. Drug Delivery. 2014;21:87–100. doi: 10.3109/10717544.2013.838077. [DOI] [PubMed] [Google Scholar]
- Varshosaz J. Pardakhty A. Hajhashemi V. I. Najafabadi A. R. Drug Delivery. 2003;10:251–262. doi: 10.1080/drd_10_4_251. [DOI] [PubMed] [Google Scholar]
- Nasr M. Mansour S. Mortada N. D. Elshamy A. A. J. Microencapsulation. 2008;25:499–512. doi: 10.1080/02652040802055411. [DOI] [PubMed] [Google Scholar]
- Hood E. Gonzalez M. Plaas A. Strom J. VanAuker M. Int. J. Pharm. 2007;339:222–230. doi: 10.1016/j.ijpharm.2006.12.048. [DOI] [PubMed] [Google Scholar]
- Zhang S. Gao H. Bao G. ACS Nano. 2015;9:8655–8671. doi: 10.1021/acsnano.5b03184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L. Nienhaus K. Nienhaus G. U. J. Nanobiotechnol. 2014;12:5. doi: 10.1186/1477-3155-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich E. Int. J. Nanomed. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoor Ahmad Gatoo S. N. Arfat M. Y. Dar A. M. Qasim K. Zubair S. BioMed Res. Int. 2014:1–8. doi: 10.1155/2014/498420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed M. S. Veeranarayanan S. Baliyan A. Poulose A. C. Nagaoka Y. Minegishi H. Iwai S. Shimane Y. Yoshida Y. Maekawa T. Kumar D. S. Macromol. Biosci. 2014;14:1696–1711. doi: 10.1002/mabi.201400248. [DOI] [PubMed] [Google Scholar]
- Quality Analysis of traditional Chinese medicines, http://qatcm.nricm.edu.tw/analysis.php?id=2720, accessed June 2018
- Shaker D. S. Shaker M. A. Hanafy M. S. Int. J. Pharm. 2015;493:285–294. doi: 10.1016/j.ijpharm.2015.07.041. [DOI] [PubMed] [Google Scholar]
- Ullah S. Shah M. R. Shoaib M. Imran M. Elhissi A. M. Ahmad F. Ali I. Shah S. W. Drug Delivery. 2016;23:3480–3491. doi: 10.1080/10717544.2016.1196768. [DOI] [PubMed] [Google Scholar]
- Garcia-Salinas S. Himawan E. Mendoza G. Arruebo M. Sebastian V. ACS Appl. Mater. Interfaces. 2018;10:19197–19207. doi: 10.1021/acsami.8b02994. [DOI] [PubMed] [Google Scholar]
- Miller-Kleinhenz J. M. Bozeman E. N. Yang L. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2015;7:797–816. doi: 10.1002/wnan.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J. Li R. Zhao X. Ma C. Lv X. Liu L. Liu P. Chem.-Biol. Interact. 2018;283:59–74. doi: 10.1016/j.cbi.2018.02.003. [DOI] [PubMed] [Google Scholar]
- Naresh R. A. Udupa N. Devi P. U. Indian J. Exp. Biol. 1996;34:764–772. [PubMed] [Google Scholar]