

Abstract
A novel vinylogous aldol addition of alkylidene oxindole with 1-trifluoromethyl-3-alkylidene-propan-2-ones is presented. The reaction, catalyzed by a bifunctional tertiary amine, provides an efficient application of the vinylogous reactivity of oxindoles for the preparation of enantioenriched trifluoromethylated allylic alcohols.
The vinylogous aldol addition of alkylidene oxindole with 1-trifluoromethyl-3-alkylidene-propan-2-ones was developed. The reaction, provides straightforward access to enantioenriched trifluoromethylated allylic alcohols.
Introduction
Vinylogy has been successfully applied to the remote functionalization of organic molecules.1 The intrinsic difficulty to introduce a functional group or an alkyl chain far away from the binding site of the catalyst, is a challenging task that makes the vinylogous reactions highly attractive.2 Alkylidene oxindoles are the central core of many biologically active compounds and intermediates for the synthesis of drugs3 (Fig. 1a).
Fig. 1. (a) Biologically active alkylidene oxindole. (b) Typical reactivity of alkylidene oxindoles.

For these reasons and due to their role in biological and pharmacological applications, the realization of novel, alkylidene oxindoles is highly desirable. Their reactivity as Michael acceptors is well known, but they are also suitable substrates for enantioselective vinylogous processes for which they have been employed with different electrophiles (Fig. 1b).4 Organofluorine compounds are important molecules employed in agro- and medicinal chemistry5 and chiral compounds containing the trifluoromethyl group (CF3) bonded to a stereogenic centre are active against various diseases. In particular, tertiary trifluoromethyl alcohols are important building blocks for the synthesis of biologically active compounds and drugs (Fig. 2).6
Fig. 2. Biologically active trifluoromethyl compounds.

For these reasons the search of new fluorinated compounds is an attractive field and many research groups developed powerful fluorination and trifluoromethylation reactions.7 The Ruppert–Prakash (R–P) reaction, which exploits the direct nucleophilic trifluoromethylation of a carbonyl group, is the most common and practical method for the synthesis of trifluoromethyl alcohols in an enantioselective fashion.7c–e,8 Recently, organocatalytic cross-aldol and Henry reactions of trifluoromethyl aryl ketones emerged as a valid alternative to the classical R–P method.9 Conversely, trifluoromethyl ketones have never been used as acceptors in vinylogous-aldol reaction.1f This observation prompted us to study the enantioselective vinylogous-aldol reaction of alkylidene oxindoles with α,β-unsaturated trifluoromethyl ketones as a powerful method for the synthesis of γ-functionalized alkylidene oxindole with tertiary allylic alcohols bearing a trifluoromethyl substituent on the chiral centre (Scheme 1).
Scheme 1. Strategic plan for the vinylogous aldol addition.

The use of 1-trifluoromethyl vinyl ketones and alkylidene oxindoles pose some challenges of chemo- and regioselectivity. In fact, because of the high reactivity imparted by the CF3-group, both the 1,2- and 1,4-addition products can be formed10 and furthermore the two γ- and γ′-positions of the oxindole can compete for the generation of the vinylogous nucleophile. The result is that a possible mixture of either E- and Z-alkenes derived from Michael and aldol adducts can be obtained. In this scenario the search of the proper catalytic machinery able to selectively drive the reaction through the desired 1,2-addition pathway is fundamental. Our idea is to concentrate the attention on a bifunctional catalyst bearing a chiral tertiary amine and a hydrogen bond donor functionality which can activate the nucleophilic γ-position of the oxindole and the electrophilic carbonyl group of the ketone. The resulting reaction would furnish a novel method of synthesis of enantioenriched trifluoromethylated allylic alcohols, a class of compounds which represent versatile intermediates for the synthesis of fine chemicals and biologically active compounds (Fig. 3).
Fig. 3. Important biologically active allylic alcohols.

Many methods have been developed for their synthesis.11 However, if we exclude rare organocatalytic examples,9e,f the preparation of chiral allylic alcohols is rare. Consequently, the development of novel reactions which realize the enantioselective preparation of trifluoromethylated allylic alcohols is highly desired.
Results and discussion
We started our investigation by exploring the reactivity of diverse cinchona alkaloids organocatalysts.12 We prepared different thiourea and squaramide derivatives of 9-NH2-9-(epi)quinine and quinidine (I–V). Using catalyst I we obtained a 9 : 1 mixture of the aldol adduct 3aa together with the 5,6-dihydropyranone 4aa13 (Table 1). Interestingly the desired trifluoromethylated allylic alcohol was isolated in a 21% yield and 87% ee. From the analysis of the crude mixture during time, it was observed that 4aa was the result of a consecutive reaction of 3aa that starts to be highly competitive after 24 hours. Unfortunately, compound 4aa was unstable under the reaction conditions for a prolonged reaction time. For this reason, we decided to stop the process after 24 hours to maximize the conversion of 3aa. As it is possible to observe from Table 1, using catalyst II, the pseudoenantiomer of I, the enantioselectivity of the reaction diminished sensibly (entry 2). After a rapid and unsuccessful survey of squaramide derivatives III and IV (entries 3 and 4), we found that the 9-epi-HQAT V gave the best enantiocontrol, albeit with low selectivity and yield (entry 5). We tested different solvents and we observed that they highly influenced the distribution of the two products and the enantiomeric excess. Contrasting results were observed with other halogenated solvents such as chloroform, dichloroethane (DCE) and trifluorotoluene (entries 6–8).
Screening of the reaction conditionsa.
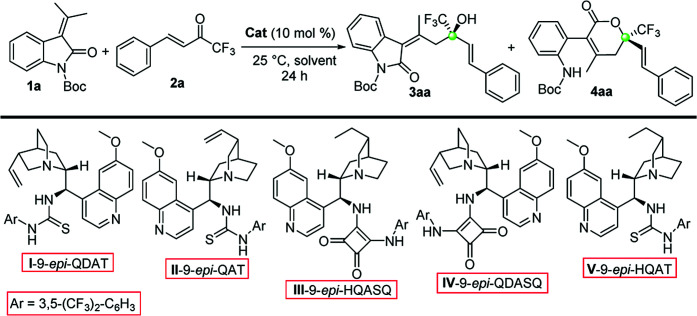
| |||||
|---|---|---|---|---|---|
| Entry | Cat | Solvent | 3aa/4aab | Yieldc [%] 3aa | eed [%] 3aa |
| 1 | I | DCM | 9.5 : 1 | 21 | 87 |
| 2 | II | DCM | 1.4 : 1 | 21 | 63 |
| 3 | III | DCM | 1 : 1.8 | 12 | 65 |
| 4 | IV | DCM | 3.5 : 1 | 42 | 75 |
| 5 | V | DCM | 3.0 : 1 | 32 | 91 |
| 6 | V | CHCl3 | 3.3 : 1 | 18 | 85 |
| 7 | V | DCE | 1 : 2.2 | 6 | 66 |
| 8 | V | PhCF3 | 1 : 3 | 10 | 61 |
| 9 | V | MeCN | 1 : 9 | 3 | 56 |
| 10 | V | Toluene | 1 : 2 | 15 | 69 |
| 11 | V | MTBE | 3 : 1 | 38 | 89 |
| 12 | V | DIPE | 1.6 : 1 | 16 | 75 |
| 13 | V | Dry THF | 13.5 : 1 | 38 | 92 |
| 14e,f | V | Dry THF | >19 : 1 | 61g | 93 |
The reactions were performed on a 0.2 mmol scale using a 1 : 1 ratio of 1a and 2a and 1 mL of solvent.
Determined via1H-NMR on the crude mixture.
Determined on the isolated product.
Determined by HPLC on chiral stationary phase.
5 mol % of catalyst V was used.
0.5 mL of solvent were used.
Determined via1H-NMR with 1,3,5-trimethoxybenzene as internal standard.
In acetonitrile and toluene a low yield of 3aa was obtained (entries 9 and 10) and ethereal solvents gave good enantiocontrol, but low selectivity (entries 11 and 12; MTBE = methyl-t-butyl ether; DIPE = diisopropyl ether). Dry THF revealed to be the best choice because its use increased the enantiomeric excess of 3aa to 92% and gave a promising 13.5 : 1 ratio between 3aa and 4aa (entry 13). Using a 5 mol % of V and a doubly concentrated reaction, 3aa was obtained as the sole product in 93% ee and 61% yield (entry 14). With the optimized conditions we reacted different oxindoles 1a–e with trifluoroketones 2a–g (Table 2). The general trend observed is that catalyst V is able to give high control on the stereochemistry regardless of the electronic nature of the substituents at the C(5) and C(6) of the oxindole core.
Substrate scope of the reactiona.
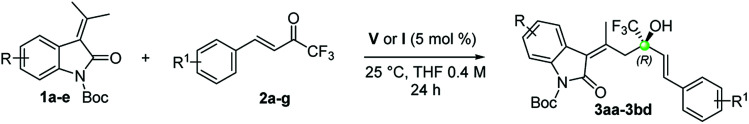
|
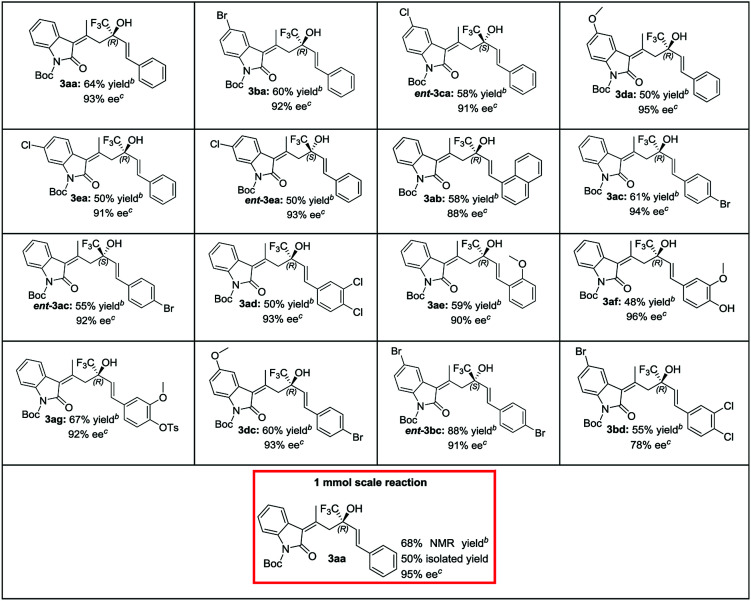
|
The reactions were performed on a 0.2 mmol scale using a 1 : 1 ratio of 1 and 2 in 0.5 mL of dry THF.
Determined via1H-NMR with 1,3,5-trimethoxybenzene as internal standard.
Determined by HPLC on chiral stationary phase.
The presence of electron-withdrawing and releasing groups is in fact well tolerated (3aa–3ea). Interestingly, the opposite enantiomers are easily accessible using catalyst I (ent-3ea and ent-3ac) and in the case of allylic alcohols ent-3ca and ent-3bc, the results are even better than using catalyst V. A good generality is observed when trifluoroketones 2b–g were studied. In all cases the corresponding allylic alcohols were obtained as sole products after 24 hours (3ab–3bd). No traces of 1,4-adducts were found as proof of the high chemoselectivity of the aldol addition. To evaluate the utility of the process we performed a 1.0 mmol scale reaction between 1a and 2a. The reaction furnished the desired compound in a 50% yield of isolated product and 95% ee.
The absolute configuration was determined to be R by means of single crystal X-ray diffraction analysis on 5aa14 which was obtained from 3aa after the removal of the Boc protecting group by reaction with pyrrolidine in DCM (Scheme 2).
Scheme 2. Synthesis and X-ray structure of compound 5aa.

A model for a plausible transition state that accounts for the bifunctional activation of catalyst V is reported in Fig. 4. The R absolute configuration can be explained as the result of a vinylogous addition of the oxindole s-cis dienolate to the prochiral Si face of the carbonyl of the trifluoroketone. Preliminary DFT calculations15 resulted in the location of seven transition states (TSs), with only three of them contributing significantly to the product enantiomer distribution. In agreement with previous studies,16 only TSs corresponding to the mode of actions A or B of catalyst V have been located. The lowest in energy (A-Si-s-cis) corresponds to the mode A addition of the oxindole to the Si-face of the ketone in the s-cis conformation and gives the (R)-enantiomer of the product. Another small fraction of the (R)-enantiomer comes from the B-Si-s-cis transition state, while the (S)-enantiomer arises from the B-Re-s-cis transition state. The results obtained account for a 96% ee in favour of the (R)-enantiomer, which is in good agreement with the observed experimental value of 93%.17
Fig. 4. Proposed transition state models derived from DFT calculations. Modes A-(Si)-s-cis and B-(Si)-s-cis contributes to the formation of the major (R)-3aa as observed. The minor enantiomer (S)-3aa is the results of the mode B-(Re)-s-cis way of action of the catalyst V.

Conclusions
In conclusion we realized the enantioselective synthesis of novel fluorinated allylic alcohols through the first vinylogous aldol addition of 3-alkylidene oxindoles and unsaturated trifluoromethyl ketones. The reaction proceeded with complete control of the chemo- and regioselectivity and represents a valuable strategy to access allylic alcohols containing a trifluoromethyl group installed on a fully substituted stereocenter.
Experimental
General methods
All the NMR spectra were recorded on Inova 300 MHz, Gemini 400 MHz or Mercury 600 MHz Varian spectrometers for 1H, 75 MHz, 100 MHz and 150 MHz for 13C and 282 MHz, 376 MHz, 564 MHz for 19F respectively. The chemical shifts (δ) for 1H, 19F and 13C are given in ppm relative to internal standard TMS (0.0 ppm) or residual signals of CHCl3 (7.26 ppm). Coupling constants are given in Hz. The following abbreviations are used to indicate the multiplicity: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; bs, broad signal. Concerning the 13C spectra of the products, we were never able to see the signal (quartet) of the fluorinated carbon regardless of the delay and the acquisition time we employed (not even a 5 day-acquisition with a 60 seconds delay at 150 MHz showed any signal). It is likely that, due to the splitting of the signal and the very high relaxation time of this particular carbon, the corresponding signal is lost in the baseline. Purification of reaction products was carried out by flash chromatography (FC) on silica gel (230–400 mesh). Organic solutions were concentrated under reduced pressure on a Büchi rotary evaporator. High Resolution Mass spectra were obtained from the CIGS facilities of the University of Modena and Reggio Emilia on a G6520AA Accurate-Mass Q-TOF LC/MS instrument. Chiral HPLC analysis was performed on an Agilent 1100-series instrumentation. HPLC traces for all compounds were compared to quasi racemic samples prepared by mixing the two product antipodes obtained performing the reactions with catalyst V and the pseudo-enantiomer I separately. Optical rotations are reported as follows:[α]25D (c in g per 100 mL, CHCl3) and the numerical values are relative to the products obtained from catalyst V. All reactions were carried out in air. Chiral catalyst I, II, III, IV and V were prepared following literature procedures.18 Alkylidene oxindoles 1a–e and α,β-unsaturated-1,1,1-trifluoroketones 2a–g were prepared according to the procedure reported in the ESI.†
General procedure for the vinylogous aldol reaction
In an ordinary vial equipped with a Teflon-coated magnetic stir bar, catalyst V or I (6 mg, 0.01 mmol, 0.05 equiv.), oxindole (0.2 mmol, 1 equiv.) and trifluoroketone (0.2 mmol, 1 equiv.) were dissolved in 0.5 mL of anhydrous THF (freshly distilled over sodium and benzophenone) at room temperature. After 24 hours of stirring, the reaction was flushed through a short silica plug with a 1 : 1 mixture of DCM : EtOAc to remove the catalyst and the crude product was concentrated to perform a 1H-NMR analysis to measure the yield (1,3,5-trimethoxybenzene was used as internal standard). At this point the product was purified with flash column chromatography and the ee% was determined through HPLC on a chiral stationary phase.
tert-Butyl (E)-(2-(4-methyl-2-oxo-6-styryl-6-(trifluoromethyl)-5,6-dihydro-2H-pyran-3-yl)phenyl)carbamate (3aa)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 64% yield for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 19 : 1) and the title compound was obtained as a yellowish oil in 93% enantiomeric excess. When the reaction was performed in 1 mmol scale the crude product showed a 68% yield based on 1H-NMR and 50% yield on the isolated pure product. The enantiomeric excess in this case was found to be 95%. The ee was determined by HPLC analysis on a Daicel Chiralpak IC column: hexane/i-PrOH 95 : 5, flow rate 1.0 mL min−1, λ = 254 nm: τV = 5.6 min, τI = 6.9 min. [α]25D + 376.5 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C26H26F3NNaO4]+ 496.1712, found 496.1721 [M + Na]+. 1H-NMR (400 MHz, CDCl3) δ 7.86–7.81 (m, 1H), 7.50 (d, J = 7.7 Hz, 1H), 7.42–7.27 (m, 6H), 7.16 (dd, J = 7.7 Hz, 1.1 Hz, 1H), 6.92 (d, J = 15.8 Hz, 1H), 6.24 (d, J = 16.3 Hz, 1H), 5.39 (s, 1H), 4.35 (d, J = 12.6 Hz, 1H), 2.83 (d, J = 12.6 Hz, 1H), 2.35 (s, 3H), 1.67 (s, 9H). 19F-NMR (376 MHz, CDCl3) δ −81.31. 13C-NMR (100 MHz, CDCl3) δ 168.7, 152.6, 148.8, 138.2, 135.9, 133.1, 128.8, 128.7, 128.2, 126.8, 124.2, 124.2, 123.7, 123.4, 114.7, 85.0, 78.0 (q, J = 28.2 Hz, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 42.0, 28.1, 27.9.
tert-Butyl (E)-(4-bromo-2-(4-methyl-2-oxo-6-styryl-6-(trifluoromethyl)-5,6-dihydro-2H-pyran-3-yl)phenyl)carbamate (3ba)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 60% yield for catalyst V and 47% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 15 : 1) and the title compound was obtained as a yellowish oil in 92% (catalyst V) and 92% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 95 : 5, flow rate 1.0 mL min−1, λ = 254 nm: τV = 5.9 min, τI = 8.1 min. [α]25D + 123.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C26H25BrF3NNaO4]+ 574.0817, found 574.0833 [M + Na]+. 1H-NMR (300 MHz, CDCl3) δ 7.80–7.73 (m, 1H), 7.61 (s, 1H), 7.47–7.24 (m, 9H), 6.91 (d, J = 15.8 Hz, 1H), 6.26 (d, J = 15.8 Hz, 1H), 5.21 (bs, 1H), 4.32 (d, J = 12.5 Hz, 1H), 2.87 (d, J = 12.5 Hz, 1H), 2.35 (s, 3H), 1.65 (s, 9H). 19F-NMR (282 MHz, CDCl3) δ −81.35. 13C-NMR (75 MHz, CDCl3) δ 167.8, 161.5, 154.7, 148.6, 137.1, 135.8, 133.2, 131.4, 128.7, 126.8, 125.9, 125.1, 117.3, 92.9, 85.3, 77.63 (m, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 55.3, 42.1, 28.2, 28.1.
tert-Butyl (Z)-5-chloro-3-((R,E)-4-hydroxy-6-phenyl-4-(trifluoromethyl)hex-5-en-2-ylidene)-2-oxoindoline-1-carboxylate (3ca)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 58% for catalyst I and 39% yield for catalyst V. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 15 : 1) and the title compound was obtained as a yellowish oil in 91% (catalyst I) and 85% (catalyst V) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 95 : 5, flow rate 1.0 mL min−1, λ = 254 nm: τV = 5.9 min, τI = 8.3 min. [α]25D + 216.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C26H25ClF3NNaO4]+ 530.1322, found 530.1329 [M + Na]+. 1H-NMR (300 MHz, CDCl3) δ 7.80–7.73 (m, 1H), 7.61 (s, 1H), 7.47–7.24 (m, 9H), 6.91 (d, J = 15.8 Hz, 1H), 6.26 (d, J = 15.8 Hz, 1H), 5.21 (bs, 1H), 4.32 (d, J = 12.5 Hz, 1H), 2.87 (d, J = 12.5 Hz, 1H), 2.35 (s, 3H), 1.65 (s, 9H). 19F-NMR (282 MHz, CDCl3) δ −81.35. 13C-NMR (75 MHz, CDCl3) δ 168.0, 154.6, 148.6, 136.6, 135.7, 133.1, 129.7, 128.7, 128.5, 128.3, 126.8, 126.0, 124.7, 124.2, 123.4, 115.8, 85.3, 78.0 (q, J = 28.2 Hz), 42.1, 28.1, 28.0.
tert-Butyl (Z)-3-((R,E)-4-hydroxy-6-phenyl-4-(trifluoromethyl)hex-5-en-2-ylidene)-5-methoxy-2-oxoindoline-1-carboxylate (3da)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 50% yield for catalyst V and 38% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 9 : 1) and the title compound was obtained as a yellowish oil in 95% (catalyst V) and 92% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 95 : 5, flow rate 1.0 mL min−1, λ = 254 nm: τV = 11.5 min, τI = 7.4 min. [α]25D + 280.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C27H28F3NNaO5]+ 526.1817, found 526.1803 [M + Na]+. 1H-NMR (300 MHz, CDCl3) δ 7.76 (d, J = 9.0 Hz, 1H), 7.42–7.28 (m, 5H), 7.06 (d, J = 2.6 Hz, 1H), 6.92 (d, J = 15.8 Hz, 1H), 6.85 (dd, J1 = 9.0 Hz, J2 = 2.6 Hz, 1H), 6.26 (d, J = 15.8 Hz, 1H), 4.34 (d, J = 12.5 Hz, 1H), 3.79 (s, 3H), 2.82 (d, J = 12.5 Hz, 1H), 2.33 (s, 3H), 1.66 (s, 9H). 19F-NMR (282 MHz, CDCl3) δ −81.30. 13C-NMR (75 MHz, CDCl3) δ 168.8, 156.4, 152.9, 148.9, 135.9, 133.0, 131.9, 128.7, 128.2, 127.0, 126.8, 124.4, 123.7, 115.4, 112.8, 111.4, 84.7, 77.8 (m, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 55.7, 42.0, 28.1, 27.8.
tert-Butyl (Z)-6-chloro-3-((R,E)-4-hydroxy-6-phenyl-4-(trifluoromethyl)hex-5-en-2-ylidene)-2-oxoindoline-1-carboxylate (3ea)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 50% yield for both catalyst V and I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 9 : 1) and the title compound was obtained as a yellowish oil in 91% (catalyst V) and 93% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 95 : 5, flow rate 1.0 mL min−1, λ = 254 nm: τV = 9.4 min, τI = 6.9 min. [α]25D + 238.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C26H25ClF3NNaO4]+ 530.1322, found 530.1329 [M + Na]+. 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 2.0 Hz, 1H), 7.43–7.38 (m, 3H), 7.35–7.27 (m, 3H), 7.13 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H), 6.90 (d, J = 15.8 Hz, 1H), 6.26 (d, J = 15.8 Hz, 1H), 5.18 (s, 1H), 4.30 (d, J = 12.6 Hz, 1H), 2.86 (d, J = 12.6 Hz, 1H), 2.32 (s, 3H), 1.66 (s, 9H). 19F-NMR (376 MHz, CDCl3) δ −81.34. 13C-NMR (100 MHz, CDCl3) δ 168.2, 153.2, 148.6, 139.0, 135.8, 134.6, 133.1, 128.7, 128.3, 126.8, 126.0, 124.8, 124.2, 123.5, 121.8, 115.4, 85.5, 77.8 (m, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 41.9, 28.1, 27.9.
tert-Butyl (Z)-3-((R,E)-4-hydroxy-6-(naphthalen-1-yl)-4-(trifluoromethyl)hex-5-en-2-ylidene)-2-oxoindoline-1-carboxylate (3ab)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 58% yield for catalyst V and 33% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 95 : 5) and the title compound was obtained as amorphous yellow solid in 88% (catalyst V) and 88% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 90 : 10, flow rate 0.7 mL min−1, λ = 254 nm: τV = 6.5 min, τI = 13.2 min. [α]rtD + 14.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C30H28F3NNaO4]+ 546.1868, found 546.1860 [M + Na]+. 1H-NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.4 Hz, 1H), 7.89–7.78 (m, 5H), 7.68 (d, J = 15.6 Hz, 1H), 7.59 (d, J = 7.0 Hz, 1H), 7.54–7.50 (m, 2H), 7.47 (d, J = 7.6 Hz, 2H), 7.40–7.32 (m, 1H), 7.17 (td, J1 = 7.7 Hz, J2 = 1.1 Hz, 1H), 6.28 (d, J = 15.5 Hz, 1H), 5.50 (bs, 1H), 4.41 (d, J = 12.7 Hz, 1H), 2.87 (d, J = 12.6 Hz, 1H), 2.36 (s, 3H), 1.68 (s, 9H). 19F-NMR (376 MHz, CDCl3) δ −81.32. 13C-NMR (150 MHz, CDCl3) δ 168.7, 152.5, 148.8, 138.3, 134.1, 133.5, 131.1, 130.8, 128.8, 128.4, 127.0–126.9 (m), 126.9, 126.2, 125.9, 125.5, 124.3, 124.1, 123.9, 123.8, 123.3, 85.0, 42.0, 28.2, 28.1.
tert-Butyl (Z)-3-((R,E)-6-(4-bromophenyl)-4-hydroxy-4-(trifluoromethyl)hex-5-en-2-ylidene)-2-oxoindoline-1-carboxylate (3ac)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 61% yield for catalyst V and 55% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 15 : 1) and the title compound was obtained as a yellowish oil in 94% (catalyst V) and 92% (catalyst I) enantiomeric eccess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 95 : 5, flow rate 1.0 mL min−1, λ = 254 nm: τV = 7.1 min, τI = 9.1 min. [α]25D + 92.0 (c 0.5, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C26H25BrF3NNaO4]+ 574.0817, found 574.0831 [M + Na]+. 1H-NMR (300 MHz, CDCl3) δ 7.84 (d, J = 8.2 Hz, 1H), 7.50 (d, J = 7.6 Hz, 1H), 7.37–7.27 (m, 4H), 7.19–7.12 (m, 2H), 6.87 (d, J = 15.8 Hz, 1H), 6.25 (d, J = 15.7 Hz, 1H), 5.42 (s, 1H), 4.32 (d, J = 12.6 Hz, 1H), 2.84 (d, J = 12.6 Hz, 1H), 2.33 (s, 3H), 1.66 (s, 9H). 19F-NMR (282 MHz, CDCl3) δ −81.24. 13C-NMR (75 MHz, CDCl3) δ 167.8, 161.5, 154.7, 148.6, 138.2, 137.1, 135.7, 133.2, 131.4, 128.7, 126.8, 125.9, 125.1, 124.7, 117.3, 116.2, 92.9, 85.3, 77.6 (m, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 55.3, 42.1, 28.2, 27.1.
tert-Butyl (Z)-3-((R,E)-6-(3,4-dichlorophenyl)-4-hydroxy-4-(trifluoromethyl)hex-5-en-2-ylidene)-2-oxoindoline-1-carboxylate (3ad)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 50% yield for catalyst V and 50% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 9 : 1) and the title compound was obtained as a yellowish oil in 93% (catalyst V) and 87% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 90 : 10, flow rate 1.0 mL min−1, λ = 254 nm: τV = 5.7 min, τI = 5.3 min. [α]25D + 497.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C26H24Cl2F3NNaO4]+ 564.0932, found 564.0915 [M + Na]+. 1H-NMR (300 MHz, CDCl3) δ 7.86–7.82 (m, 1H), 7.53–7.46 (m, 2H), 7.39 (d, J = 8.3 Hz, 1H), 7.36–7.29 (m, 1H), 7.22–7.13 (m, 2H), 6.84 (d, J = 15.7 Hz, 1H), 6.27 (d, J = 15.4 Hz, 1H), 5.47 (s, 1H), 4.31 (d, J = 12.7 Hz, 1H), 2.84 (d, J = 12.6 Hz, 1H), 2.32 (s, 3H), 1.66 (s, 9H). 19F-NMR (282 MHz, CDCl3) δ −81.13. 13C-NMR (75 MHz, CDCl3) δ 168.7, 151.9, 148.7, 138.2, 135.9, 132.9, 132.0, 130.9, 130.6, 128.9, 128.3, 127.8, 127.0, 126.1, 125.8, 124.2, 123.2, 114.7, 85.1, 78.1 (m, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 41.9, 28.1, 27.8.
tert-Butyl (Z)-3-((R,E)-4-hydroxy-6-(2-methoxyphenyl)-4-(trifluoromethyl)hex-5-en-2-ylidene)-2-oxoindoline-1-carboxylate (3ae)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 59% yield for catalyst V and 41% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 15 : 1) and the title compound was obtained as a yellowish oil in 90% (catalyst V) and 86% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 95 : 5, flow rate 1.0 mL min−1, λ = 254 nm: τV = 6.5 min, τI = 9.1 min. [α]25D + 72.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C27H28F3NNaO5]+ 526.1817, found 526.1832 [M + Na]+. 1H-NMR (400 MHz, CDCl3) δ 7.87–7.81 (m, 2H), 7.51 (dd, J1 = 8.1, J2 = 1.1 Hz, 1H), 7.41 (dd, J1 = 7.6 Hz, J2 = 1.8 Hz, 1H), 7.31 (td, J1 = 8.0, J2 = 1.2 Hz, 2H), 7.27–7.22 (m, 1H), 7.18–7.11 (m, 2H), 6.93 (td, J1 = 7.5 Hz, J2 = 1.1 Hz, 1H), 6.86 (dd, J1 = 8.3 Hz, J2 = 1.1 Hz, 1H), 5.26 (bs, 1H) 4.33 (d, J = 12.5 Hz, 1H), 3.78 (s, 3H), 2.36 (s, 3H), 1.66 (s, 9H). 19F-NMR (376 MHz, CDCl3) δ −81.38. 13C-NMR (75 MHz, CDCl3) δ 168.6, 157.2, 153.1, 148.9, 138.2, 129.1, 128.6, 128.3, 127.6, 126.6, 125.0, 124.5, 124.2, 124.1, 123.5, 120.6, 114.6, 110.9, 84.7, 55.3, 42.0, 28.1, 28.1.
tert-Butyl (Z)-3-((R,E)-4-hydroxy-6-(4-hydroxy-3-methoxyphenyl)-4-(trifluoromethyl)hex-5-en-2-ylidene)-2-oxoindoline-1-carboxylate (3af)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 48% yield for catalyst V and 45% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 4 : 1) and the title compound was obtained as a yellowish oil in 96% (catalyst V) and 92% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 90 : 10, flow rate 1.0 mL min−1, λ = 254 nm: τV = 10.4 min, τI = 12.2 min. [α]25D + 60.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C27H28F3NNaO6]+ 542.1766, found 542.1760 [M + Na]+. 1H-NMR (300 MHz, CDCl3) δ 7.84 (d, J = 8.1 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.31 (d, J = 8.1 Hz, 1H), 7.19–7.11 (m, 3H), 7.01–6.85 (m, 3H), 6.82 (d, J = 15.7 Hz, 1H), 6.10 (d, J = 15.7 Hz, 1H), 4.31 (d, J = 12.5 Hz, 1H), 3.90 (s, 3H), 2.85 (d, J = 12.5 Hz, 1H), 2.35 (s, 3H), 1.66 (s, 9H). 19F-NMR (282 MHz, CDCl3) δ −81.35. 13C-NMR (75 MHz, CDCl3) δ 167.6, 151.8, 147.8, 145.6, 144.9, 137.2, 131.7, 127.7, 127.5, 125.7, 123.2, 123.1, 122.4, 120.3, 119.2, 113.6, 113.5, 108.1, 83.9, 77.2 (m, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 54.9, 27.1, 26.9, 24.3.
tert-Butyl (Z)-3-((R,E)-4-hydroxy-6-(3-methoxy-4-(tosyloxy)phenyl)-4-(trifluoromethyl)hex-5-en-2-ylidene)-2-oxoindoline-1-carboxylate (3ag)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 67% yield for catalyst V and 50% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 9 : 1) and the title compound was obtained in as a yellowish oil 92% (catalyst V) and 92% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 90 : 10, flow rate 1.0 mL min−1, λ = 254 nm: τV = 11.1 min, τI = 14.6 min. [α]25D + 165.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C34H34F3NNaO8S]+ 696.1855, found 696.1878 [M + Na]+. 1H-NMR (300 MHz, CDCl3) δ 7.83 (d, J = 8.2 Hz, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 7.8 Hz, 1H), 7.38–7.27 (m, 4H), 7.17 (td, J1 = 7.7 Hz, J2 = 1.0 Hz, 1H), 7.10 (d, J = 8.3 Hz, 1H), 6.94 (dd, J1 = 8.4 Hz, J2 = 1.7 Hz, 1H), 6.81 (d, J = 1.8 Hz, 2H), 6.21 (d, J = 15.7 Hz, 1H), 5.43 (s, 1H), 4.29 (d, J = 12.6 Hz, 1H), 3.56 (s, 3H), 2.86 (d, J = 12.6 Hz, 1H), 2.44 (s, 3H), 2.34 (s, 3H), 1.66 (s, 9H). 19F-NMR (282 MHz, CDCl3) δ −81.16. 13C-NMR (75 MHz, CDCl3) δ 168.7, 152.2, 151.9, 148.7, 145.1, 138.2, 135.9, 133.3, 132.2, 129.4, 128.9, 128.6, 126.9, 124.9, 124.3, 124.2, 124.2, 123.3, 118.7, 114.7, 111.3, 85.1, 78.1 (m, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 31.6, 28.1, 27.9, 22.6, 14.1.
tert-Butyl (Z)-3-((R,E)-6-(4-bromophenyl)-4-hydroxy-4-(trifluoromethyl)hex-5-en-2-ylidene)-5-methoxy-2-oxoindoline-1-carboxylate (3dc)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 60% yield for catalyst V and 55% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 9 : 1) and the title compound was obtained as a yellowish oil in 93% (catalyst V) and 92% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 95 : 5, flow rate 1.0 mL min−1, λ = 254 nm: τV = 10.6 min, τI = 8.8 min. [α]25D + 292.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C27H27BrF3NNaO5]+ 604.0922, found 604.0928 [M + Na]+.1H-NMR (300 MHz, CDCl3) δ 7.76 (d, J = 8.9 Hz, 1H), 7.34–7.27 (m, 4H), 7.06 (d, J = 2.6 Hz, 1H), 6.92–6.79 (m, 2H), 6.24 (d, J = 15.7 Hz, 1H), 5.45 (s, 1H), 4.31 (d, J = 12.6 Hz, 1H), 3.79 (s, 3H), 2.83 (d, J = 12.6 Hz, 1H), 2.31 (s, 3H), 1.65 (s, 9H). 19F-NMR (282 MHz, CDCl3) δ −81.30. 13C-NMR (75 MHz, CDCl3) δ 168.8, 156.4, 152.6, 148.8, 134.4, 133.9, 131.9, 131.9, 130.3, 129.6, 128.9, 128.0, 127.1, 124.4, 124.3, 115.4, 112.8, 111.5, 84.8, 77.8 (m, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 55.7, 42.0, 28.1, 27.8.
tert-Butyl (Z)-5-bromo-3-((S,E)-6-(4-bromophenyl)-4-hydroxy-4-(trifluoromethyl)hex-5-en-2-ylidene)-2-oxoindoline-1-carboxylate (ent-3bc)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 57% yield for catalyst V and 88% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 9 : 1) and the title compound was obtained in as a yellowish oil 90% (catalyst V) and 91% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 95 : 5, flow rate 1.0 mL min−1, λ = 254 nm: τV = 6.7 min, τI = 7.8 min. [α]25D + 236.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C26H24Br2F3NNaO4]+ 651.9922 found 651.9916 [M + Na]+. 1H-NMR (400 MHz, CDCl3) δ 7.75 (d, J = 8.7 Hz, 1H), 7.61 (d, J = 2.0 Hz, 1H), 7.44 (dd, J1 = 8.7 Hz, J2 = 2.0 Hz, 1H), 7.32 (d, J = 2.8 Hz, 4H), 6.86 (d, J = 15.8 Hz, 1H), 6.24 (d, J = 15.8 Hz, 1H), 5.22 (bs, 1H), 4.29 (d, J = 12.5 Hz, 1H), 2.88 (d, J = 12.5 Hz, 1H), 2.33 (s, 3H), 1.65 (s, 9H). 19F-NMR (376 MHz, CDCl3) δ −81.29. 13C-NMR (100 MHz, CDCl3) δ 167.9, 165.0, 158.9, 154.4, 148.6, 137.1, 134.2, 134.1, 132.0, 131.49, 130.4, 128.9, 128.0, 126.9, 126.0, 125.0, 124.1, 117.3, 116.7, 116.2, 85.4, 77.9 (m, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 28.2, 28.1, 26.1.
tert-Butyl (Z)-5-bromo-3-((R,E)-6-(3,4-dichlorophenyl)-4-hydroxy-4-(trifluoromethyl)hex-5-en-2-ylidene)-2-oxoindoline-1-carboxylate (3bd)
The reaction was carried out following the general procedure. 1H-NMR of the crude showed a 39% yield for catalyst V and 55% for catalyst I. The crude mixture was purified by flash column chromatography (hexane : EtOAc = 9 : 1) and the title compound was obtained as a yellowish oil in 83% (catalyst V) and 78% (catalyst I) enantiomeric excess. The ee was determined by HPLC analysis on a Daicel Chiralpak AD-H column: hexane/i-PrOH 95 : 5, flow rate 1.0 mL min−1, λ = 254 nm: τV = 7.7 min, τI = 6.3 min. [α]25D + 159.0 (c 1.0, CHCl3). HRMS-ESI-ORBITRAP (+): calculated for [C26H23BrCl2F3NNaO4]+ 642.0037, found 642.0020 [M + Na]+. 1H-NMR (300 MHz, CDCl3) δ 7.76 (d, J = 8.7 Hz, 1H), 7.62 (d, J = 2.0 Hz, 1H), 7.49–7.38 (m, 3H), 7.21 (dd, J1 = 8.3 Hz, J2 = 2.1 Hz, 1H), 6.82 (d, J = 15.7 Hz, 1H), 6.27 (d, J = 15.3 Hz, 1H), 5.26 (s, 1H), 4.29 (d, J = 12.6 Hz, 1H), 2.88 (d, J = 12.6 Hz, 1H), 2.33 (s, 3H), 1.65 (s, 9H). 19F-NMR (282 MHz, CDCl3) δ −81.18. 13C-NMR (75 MHz, CDCl3) δ 167.8, 154.0, 148.5, 137.1, 135.8, 133.0, 132.2, 131.5, 131.1, 130.7, 128.4, 127.0, 126.1, 125.5, 124.9, 117.3, 116.2, 85.4, 77.8 (m, tetrasubstituted aliphatic carbon, partially overlapped with CDCl3), 41.9, 28.1, 27.9.
(Z)-3-((R,E)-4-hydroxy-6-phenyl-4-(trifluoromethyl)hex-5-en-2-ylidene)indolin-2-one (5aa)
In an ordinary vial equipped with a teflon-coated magnetic stir bar, catalyst V (6 mg, 0.01 mmol, 0.05 equiv.), oxindole 1a (0.2 mmol, 1 equiv.) and trifluoroketone 2a (0.2 mmol, 1 equiv.) were dissolved in 0.5 mL of anhydrous THF (freshly distilled over sodium and benzophenone). After 24 hours of stirring, pyrrolidine (0.2 mmol, 17 μL) was added and the resulting solution was stirred for 2 hours. The crude mixture was purified by column chromatography (hexane/AcOEt 85 : 15) to give 5aa in 60% yield (0.12 mmol, 37.3 mg). Suitable crystals of enantiopure 5aa were obtained from CDCl3 directly in the NMR tube.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are grateful to the University of Bologna for the financial support. We are grateful to the C.I.G.S. of the University of Modena and Reggio Emilia for the HRMS analyses.
Electronic supplementary information (ESI) available: Experimental procedures, products and starting materials characterization, copy of NMR and HPLC spectra. CCDC 1834665. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06615a
Notes and references
- (a) Fuson R. C. Chem. Rev. 1935;16:1. doi: 10.1021/cr60053a001. [DOI] [Google Scholar]; (b) Battisitini L. Synthesis. 2017;49:2297. doi: 10.1055/s-0036-1589487. [DOI] [Google Scholar]; (c) Jurberg I. D. Chatterjee I. Tannert R. Melchiorre P. Chem. Commun. 2013;49:4869. doi: 10.1039/C3CC41270A. [DOI] [PubMed] [Google Scholar]; (d) Casiraghi G. Battistini L. Curti C. Rassu G. Zanardi F. Chem. Rev. 2011;111:3076. doi: 10.1021/cr100304n. [DOI] [PubMed] [Google Scholar]; (e) Pansare S. V. Eldho K. P. Chem.–Eur. J. 2011;17:8770. doi: 10.1002/chem.201101269. [DOI] [PubMed] [Google Scholar]; (f) Denmark S. E. Heemstra J. R. Beutner G. L. Angew. Chem., Int. Ed. 2005;44:4682. doi: 10.1002/anie.200462338. [DOI] [PubMed] [Google Scholar]; (g) Hepburn H. B. Dell’ Amico L. Melchiorre P. Chem. Rec. 2016;16:1787. doi: 10.1002/tcr.201600030. [DOI] [PubMed] [Google Scholar]
- (a) Clayden J. Chem. Soc. Rev. 2009;38:817. doi: 10.1039/B801639A. [DOI] [PubMed] [Google Scholar]; (b) Jiang H. Albrecht Ł. Jørgensen K. A. Chem. Sci. 2013;4:2287. doi: 10.1039/C3SC50405K. [DOI] [Google Scholar]; . For representative examples of vinylogous reactions: ; (c) Tian X. Hofmann N. Melchiorre P. Angew. Chem., Int. Ed. 2014;53:2997. doi: 10.1002/anie.201310487. [DOI] [PubMed] [Google Scholar]; (d) Dell’ Amico L. Albrecht Ł. Naicker T. Poulsen P. H. Jørgensen K. A. J. Am. Chem. Soc. 2013;135:8063. doi: 10.1021/ja4029928. [DOI] [PubMed] [Google Scholar]; (e) Stiller J. Poulsen P. H. Cruz Cruz D. Dourado J. Davis R. L. Jørgensen K. A. Chem. Sci. 2014;5:2052. doi: 10.1039/C4SC00081A. [DOI] [Google Scholar]; (f) Jia Z.-J. Jiang H. Li J.-L. Gschwend B. Li Q.-Z. Yin X. Grouleff J. Chen Y.-C. Jørgensen K. A. J. Am. Chem. Soc. 2011;133:5053. doi: 10.1021/ja1112194. [DOI] [PubMed] [Google Scholar]; (g) Xiong X.-F. Zhou Q. Gu J. Dong L. Liu T.-Y. Chen Y.-C. Angew. Chem., Int. Ed. 2012;51:4401. doi: 10.1002/anie.201200248. [DOI] [PubMed] [Google Scholar]; (h) Silvi M. Chatterjee I. Liu Y. Melchiorre P. Angew. Chem., Int. Ed. 2013;52:10780. doi: 10.1002/anie.201305870. [DOI] [PubMed] [Google Scholar]; (i) Bencivenni G. Galzerano P. Mazzanti A. Bartoli G. Melchiorre P. Proc. Natl. Acad. Sci. U. S. A. 2010;107:20642. doi: 10.1073/pnas.1001150107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Di Iorio N. Righi P. Mazzanti A. Mancinelli M. Ciogli A. Bencivenni G. J. Am. Chem. Soc. 2014;136:10250. doi: 10.1021/ja505610k. [DOI] [PubMed] [Google Scholar]; (k) Albrecht Ł. Dickmeiss G. Cruz Acosta F. Rodríguez-Escrich C. Davis R. L. Jørgensen K. A. J. Am. Chem. Soc. 2012;134:5053. doi: 10.1021/ja211878x. [DOI] [PubMed] [Google Scholar]; (l) Qi L.-W. Yang Y. Gui Y.-Y. Zhang Y. Chen F. Tian F. Peng L. Wang L.-X. Org. Lett. 2014;16:6436. doi: 10.1021/ol503266q. [DOI] [PubMed] [Google Scholar]
- (a) Ganesh M. Rao P. M. Mirajakar J. S. Asian J. Org. Chem. 2018;7:250. doi: 10.1002/ajoc.201700410. [DOI] [Google Scholar]; (b) Millemaggi A. Taylor R. J. K. Eur. J. Org. Chem. 2010:4527. doi: 10.1002/ejoc.201000643. [DOI] [Google Scholar]
- For review on the electrophilic reactivity see: ; (a) Chris V. Galliford C. V. Scheidt K. A. Angew. Chem., Int. Ed. 2007;46:8748. doi: 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]; (b) Dalpozzo R. Bartoli G. Bencivenni G. Chem. Soc. Rev. 2012;41:7247. doi: 10.1039/C2CS35100E. [DOI] [PubMed] [Google Scholar]; (c) Dalpozzo R. Adv. Synth. Catal. 2017;359:1772. doi: 10.1002/adsc.201700361. [DOI] [Google Scholar]; (d) Cheng D.-J. Ishihara Y. Tan B. Barbas III C. F. ACS Catal. 2014;4:743. doi: 10.1021/cs401172r. [DOI] [Google Scholar]; . For vinylogous reactivity see: ; (e) Rassu G. Zambrano V. Tanca R. Sartori A. Battistini L. Zanardi F. Curti C. Casiraghi G. Eur. J. Org. Chem. 2012:466. doi: 10.1002/ejoc.201101446. [DOI] [Google Scholar]; (f) Curti C. Rassu G. Zambrano V. Pinna L. Pelosi G. Sartori A. Battistini L. Zanardi F. Casiraghi G. Angew. Chem., Int. Ed. 2012;51:6200. doi: 10.1002/anie.201202027. [DOI] [PubMed] [Google Scholar]; (g) Chen Q. Wang G. Jiang X. Xu Z. Lin L. Wang R. Org. Lett. 2014;16:1394. doi: 10.1021/ol500157b. [DOI] [PubMed] [Google Scholar]; (h) Zhong Y. Ma S. Xu Z. Changa M. Wang R. RSC Adv. 2014;4:49930. doi: 10.1039/C4RA09128K. [DOI] [Google Scholar]; (i) Feng J. Li X. Cheng J.-P. Chem. Commun. 2015;51:14342. doi: 10.1039/C5CC06182B. [DOI] [PubMed] [Google Scholar]; (j) Di Iorio N. Righi P. Ranieri S. Mazzanti A. Margutta R. G. Bencivenni G. J. Org. Chem. 2015;80:7158. doi: 10.1021/acs.joc.5b01022. [DOI] [PubMed] [Google Scholar]; (k) Xiao X. Mei H. Chen Q. Zhao L. Lin X. Liu X. Feng X. Chem. Commun. 2015;51:580. doi: 10.1039/C4CC07204A. [DOI] [PubMed] [Google Scholar]; (l) Liu Y. Yang Y. Huang Y. Xu X.-H. Qing F.-L. Synlett. 2015;26:72. [Google Scholar]; (m) Feng J. Li X. Cheng J.-P. J. Org. Chem. 2017;82:1412. doi: 10.1021/acs.joc.6b02582. [DOI] [PubMed] [Google Scholar]; (n) Jadhav A. P. Ali A. Singh R. P. Adv. Synth. Catal. 2017;359:1508. doi: 10.1002/adsc.201601265. [DOI] [Google Scholar]
- (a) Petrov V. A., Fluorinated Heterocyclic Compounds: Synthesis, Chemistry, and Applications, John Wiley & Sons, Hoboken, NJ, 2009, p. 397 [Google Scholar]; (b) O'Hagan D. J. Fluorine Chem. 2010;131:1071. doi: 10.1016/j.jfluchem.2010.03.003. [DOI] [Google Scholar]
- (a) Purser S. Moore P. R. Swallow S. Gouverneur V. Chem. Soc. Rev. 2008;37:320. doi: 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]; (b) Wang J. Sánchez-Roselló M. Aceña J. L. del Pozo C. Sorochinsky A. E. Fustero S. Soloshonok V. A. Liu H. Chem. Rev. 2014;114:2432. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- (a) Ma J.-A. Cahard D. J. Fluorine Chem. 2007;128:975. doi: 10.1016/j.jfluchem.2007.04.026. [DOI] [Google Scholar]; (b) Nagib D. A. Scott M. E. MacMillan D. W. C. J. Am. Chem. Soc. 2009;131:10875. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ruppert I. Schlich K. Volbach W. Tetrahedron Lett. 1984;25:2195. doi: 10.1016/S0040-4039(01)80208-2. [DOI] [Google Scholar]; (d) Prakash G. K. S. Krishnamurti R. Olah G. A. J. Am. Chem. Soc. 1989;111:393. doi: 10.1021/ja00183a073. [DOI] [Google Scholar]; (e) Prakash G. K. S. Yudin A. K. Chem. Rev. 1997;97:757. doi: 10.1021/cr9408991. [DOI] [PubMed] [Google Scholar]; (f) Kieltsch I. Eisenberger P. Togni A. Angew. Chem., Int. Ed. 2007;46:754. doi: 10.1002/anie.200603497. [DOI] [PubMed] [Google Scholar]; (g) Umemoto T. Ishihara S. J. Am. Chem. Soc. 1993;115:2156. doi: 10.1021/ja00059a009. [DOI] [Google Scholar]; (h) Ventre S. Petronijevic F. R. MacMillan D. W. C. J. Am. Chem. Soc. 2015;137:5654. doi: 10.1021/jacs.5b02244. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Braun M.-G. Doyle A. G. J. Am. Chem. Soc. 2013;135:12990. doi: 10.1021/ja407223g. [DOI] [PubMed] [Google Scholar]; (j) Bloom S. Pitts C. R. Miller D. C. Haselton N. Holl M. G. Urheim E. Lectka T. Angew. Chem., Int. Ed. 2012;51:10580. doi: 10.1002/anie.201203642. [DOI] [PubMed] [Google Scholar]; (k) Kwiatkowski P. Beeson T. D. Conrad J. C. MacMillan D. W. C. J. Am. Chem. Soc. 2011;133:1738. doi: 10.1021/ja111163u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Beeson T. D. MacMillan D. W. C. J. Am. Chem. Soc. 2005;127:8826. doi: 10.1021/ja051805f. [DOI] [PubMed] [Google Scholar]
- For interesting reviews see: ; (a2) Man J.-N. Cahard D. Chem. Rev. 2008;108:PR1. doi: 10.1021/cr050005k. [DOI] [PubMed] [Google Scholar]; (b) Xiaoyu Yang X. Wu T. Phipps R. J. Toste F. D. Chem. Rev. 2015;115:826. doi: 10.1021/cr500277b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For aldol reactions: ; (a) Duangdee N. Harnying W. Rulli G. Neudörfl J.-M. Gröger H. Berkessel A. J. Am. Chem. Soc. 2012;134:11196. doi: 10.1021/ja302511t. [DOI] [PubMed] [Google Scholar]; (b) Hara N. Tamura R. Funahashi Y. Nakamura S. Org. Lett. 2011;13:1662. doi: 10.1021/ol2001039. [DOI] [PubMed] [Google Scholar]; (c) Mizuta S. Shibata N. Akiti S. Fujimoto H. Nakamura S. Toru T. Org. Lett. 2007;9:3707. doi: 10.1021/ol701791r. [DOI] [PubMed] [Google Scholar]; (d) Kokotos C. G. J. Org. Chem. 2012;77:1131. doi: 10.1021/jo2020104. [DOI] [PubMed] [Google Scholar]; (e) Wang X.-J. Zhao Y. Liu J.-T. Org. Lett. 2007;9:1343. doi: 10.1021/ol070217z. [DOI] [PubMed] [Google Scholar]; (f) Zang D. Yuan C. Tetrahedron. 2008;64:2480. doi: 10.1016/j.tet.2007.12.035. [DOI] [Google Scholar]; (g) Wang P. Li H.-F. Zhao J.-Z. Du Z.-H. Da C.-S. Org. Lett. 2017;19:2634. doi: 10.1021/acs.orglett.7b00828. [DOI] [PubMed] [Google Scholar]; (h) Yang W. Cui Y.-M. Zhou W. Li L. Yang K.-F. Zheng Z.-J. Lu Y. Xu L.-W. Synlett. 2014;25:1461. doi: 10.1055/s-0033-1341281. [DOI] [Google Scholar]; . For nitro-aldol: ; (i) Bandini M. Sinisi R. Umani-Ronchi A. Chem. Commun. 2008:4360. doi: 10.1039/B807640E. [DOI] [PubMed] [Google Scholar]; (j) Palacio C. Connon S. J. Org. Lett. 2011;13:1298. doi: 10.1021/ol103089j. [DOI] [PubMed] [Google Scholar]
- For examples of 1,2-addition: ; (a) Zhang G.-W. Meng W. Ma H. Nie J. Zhang W.-Q. Ma J.-A. Angew. Chem., Int. Ed. 2011;50:3538. doi: 10.1002/anie.201007341. [DOI] [PubMed] [Google Scholar]; (b) Liu Z.-J. Liu J.-T. Chem. Commun. 2008:5233. doi: 10.1039/B810459J. [DOI] [PubMed] [Google Scholar]; . For examples of 1,4-addition: ; (c) Morril L. C. Douglas J. Lebl T. Slawin A. M. Z. Fox D. J. Smith A. D. Chem. Sci. 2013;4:4146. doi: 10.1039/C3SC51791H. [DOI] [Google Scholar]; (d) Li P. Chai Z. Zhao S.-L. Yang Y.-Q. Wang H.-F. Zheng C.-W. Cai Y.-P. Zhao G. Zhu S.-Z. Chem. Commun. 2009:7369. doi: 10.1039/B915210E. [DOI] [PubMed] [Google Scholar]
- Lumbroso A. Cooke M. L. Bernhard B. Angew. Chem., Int. Ed. 2013;52:1890. doi: 10.1002/anie.201204579. [DOI] [PubMed] [Google Scholar]
- (a) Alemán J. Parra A. Jiang H. Jørgensen K. A. Chem. - Eur. J. 2011;17:6890. doi: 10.1002/chem.201003694. [DOI] [PubMed] [Google Scholar]; (b) Song C. E., in Cinchona Alkaloids in Synthesis and Catalysis, Wiley-VCH, Weinheim, 2009 [Google Scholar]; (c) Connon S. J. Chem. Commun. 2008:2499. doi: 10.1039/B719249E. [DOI] [PubMed] [Google Scholar]; (d) Doyle A. G. Jacobsen E. N. Chem. Rev. 2007;107:5713. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- When aromatic trifluoromethyl ketones are employed the trifluoromethyl α,β-unsaturated δ-lactones, as the result of an aldol-lactonization sequence, is the only product observed: ; Crotti S. Di Iorio N. Mazzanti A. Righi P. Bencivenni G. J. Org. Chem. 2018 doi: 10.1021/acs.joc.8b01672. [DOI] [PubMed] [Google Scholar]
- CCDC-1834665 for 5aa.
- For this preliminary investigation DFT calculations were performed using Gaussian 16 at the B3LYP/6-31G(d) level in the gas phase.
- (a) Grayson M. N. J. Org. Chem. 2017;82:4396. doi: 10.1021/acs.joc.7b00521. [DOI] [PubMed] [Google Scholar]; (b) Grayson M. N. Houk K. N. J. Am. Chem. Soc. 2016;138:9041. doi: 10.1021/jacs.6b05074. [DOI] [PubMed] [Google Scholar]
- See ESI† for details.
- (a) Cassani C. Martín-Rapún R. Arceo E. Bravo F. Melchiorre P. Nat. Protoc. 2013;8:325. doi: 10.1038/nprot.2012.155. [DOI] [PubMed] [Google Scholar]; (b) Malerich J. P. Hagihara K. Rawal V. H. J. Am. Chem. Soc. 2008;130:14416. doi: 10.1021/ja805693p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vakulya B. Varga S. Csámpai A. Soós T. Org. Lett. 2005;7:1967. doi: 10.1021/ol050431s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.