

Voltage activation, but not channel opening, is required for RY785 to access the central cavity of Kv2 channels, where it promotes voltage sensor deactivation to trap itself in place.
Abstract
Voltage activation, but not channel opening, is required for RY785 to access the central cavity of Kv2 channels, where it promotes voltage sensor deactivation to trap itself in place.
The Kv2 family of voltage-gated K+ channels are expressed in a wide variety of cell types, including neurons, where they modulate repetitive action potential firing (1), and pancreatic β cells, where they suppress insulin secretion (2). In this issue of JGP, Marquis and Sack investigate how a drug called RY785 inhibits Kv2 channels, providing potential new insights into the mechanism of Kv2 channel gating (3).
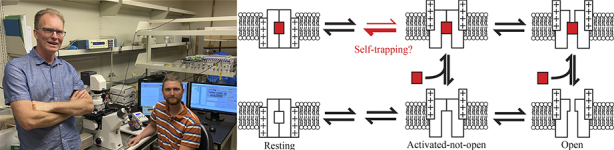
Matthew Marquis (seated) and Jon Sack (standing) determine the mechanism of use-dependence of RY785, an inhibitor of Kv2 channels. Their proposed model (right) suggests that the drug (red rectangle) can access the central cavity of Kv2 channels when they are in an activated-not-open conformation. The drug then promotes channel deactivation, trapping itself in place.
Ion channel inhibitors are useful tools for elucidating channel function, but understanding the precise mechanism of an inhibitor’s action is crucial for interpreting its physiological effects. The small molecule RY785 is a potent and selective inhibitor of Kv2 channels that shows a use-dependent onset of inhibition, i.e., voltage activation of the channel is required for the drug to begin inhibiting the K+ current (4). “We wanted to figure out how this drug works,” says Jon Sack, an associate professor at the University of California, Davis.
Sack, along with graduate student Matthew Marquis, expressed rat Kv2.1 channels in CHO-K1 cells and found that 1 μM RY785 completely inhibited channel conductance at all physiologically relevant voltages (3). As expected, voltage activation of rat Kv2.1 channels was required for the onset of RY785 inhibition, but Marquis and Sack found that recovery from inhibition is also voltage dependent: voltage activation of Kv2.1 channels speeds the restoration of K+ currents after RY785 washout, whereas channels held at negative membrane potentials barely recover any conductance at all.
The simplest explanation for these observations is that voltage activation induces a conformational change in Kv2 channels that allows RY785 to access its binding site within the channel. The drug may then become trapped at this binding site when the channel returns to its resting, deactivated conformation, and can only be released if the channel is voltage activated once more.
To learn more about the conformational changes associated with RY785 binding, Marquis and Sack analyzed Kv2.1 gating currents in the presence and absence of the drug. Remarkably, they found that RY785 accelerates the movements associated with channel deactivation. “So, it looks like a voltage-activated gate has to open for RY785 to access the channel, but then the drug pulls the gate closed behind it,” Sack says. This “self-trapping” mechanism would explain RY785’s high affinity for and slow off rate from Kv2 channels.
But where in the channel does RY785 bind? Marquis and Sack found that RY785 competes with the classic open-channel blocker tetraethylammonium, which binds to a site in the central cavity of voltage-gated K+ channels. Surprisingly, however, the researchers also determined that channel opening is not required for the onset of RY785 inhibition. Thus, Kv2 channels adopt an “activated-not-open” conformation, in which, they speculate, the central cavity is accessible from the cytosol (allowing drugs such as RY785 to enter and bind), but the channel is not open to the passage of K+ ions.
This suggests that, unlike Kv1 channels in which opening of an intracellular gate seems to be the final activation step (5), Kv2 channel opening also requires opening of a second gate. This might explain why Kv2 channels open more slowly than other voltage-gated K+ channels and are more involved in the modulation of repetitive firing than of isolated action potentials.
“The use of two activation gates could be important for the physiological role of Kv2 channels,” Sack suggests. “Now we want to know what these two gates really are and how they work together.”
References
- 1.Liu, P.W., and Bean B.P.. 2014. J. Neurosci. 10.1523/JNEUROSCI.1925-13.2014 [DOI] [Google Scholar]
- 2.Jacobson, D.A., et al. 2007. Cell Metab. 10.1016/j.cmet.2007.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marquis, M.J., and Sack J.T.. 2022. J. Gen. Physiol. 10.1085/jgp.202112981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herrington, J., et al. 2011. Mol. Pharmacol.. 10.1124/mol.111.074831 [DOI] [PubMed] [Google Scholar]
- 5.del Camino, D., et al. 2005. J. Gen. Physiol.. 10.1085/jgp.200509385 [DOI] [PMC free article] [PubMed] [Google Scholar]