

Abstract
Mycobacterium tuberculosis causes a life-threatening disease known as tuberculosis (TB). In 2021, tuberculosis was the second cause of death after COVID-19 among infectious diseases. Latent life cycle and development of multidrug resistance in one hand and lack of an effective vaccine in another hand have made TB a global health issue. Here, a multi-epitope vaccine have been designed against TB using five new antigenic protein and immunoinformatic tools. To do so, immunodominant MHC-I/MHC-II binding epitopes of Rv2346, Rv2347, Rv3614, Rv3615 and Rv2031 antigenic proteins have been selected using advanced computational procedures. The vaccine was designed by linking ten epitopes from the antigenic proteins and flagellin and TpD as adjuvant. Three-dimensional (3D) structure of the vaccine was modeled, was refined and was evaluated using bioinformatics tools. The 3D structure of the vaccine was docked into the toll-like-receptors (TLR3, 4, 8) to evaluate potential interaction between the vaccine and TLRs. Evaluation of immunological and physicochemical properties of the constructed vaccine have demonstrated the vaccine construct can induce significant humoral and cellular immune responses, the vaccine is non-allergenic and can be recognized by TLR proteins. The immunoinformatic results reported in the present study demonstrates that it is worth following the designed vaccine by experimental investigations.
Keywords: Mycobacterium tuberculosis, Vaccine, Immunoinformatic, Docking, Drug resistance
Introduction
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb) and mostly affects the lungs developing symptoms including fever, night sweats and persistent cough (Natarajan et al. 2020). TB and especially drug-resistant TB are public health threats. World health organization (WHO) has estimated 9.9 million people have fell ill with TB and has estimated 1.5 million death in 2020 which makes TB the 13th cause of death and second cause of death after COVID-19 among infectious diseases. Furthermore, 5.8 million people have newly diagnosed with TB (WHO, tuberculosis fact sheet). The disease is reported from all over the world but it is widespread in developing countries including India, China, Indonesia, the Philippines, Pakistan, Nigeria, Bangladesh and South Africa (Global Tuberculosis Report 2021). Tuberculosis can be suppressed by the immune system of healthy individuals resulting a latent infection but immunocompromised patients mostly could not develop appropriate immune response resulting complicated form of TB (Hasan et al. 2018). Human immunodeficiency virus (HIV) positive patients develop 18 times more active TB than healthy individuals and TB is one of the main cause of death in the HIV positive patients. In 2020, 215,000 HIV positive patients have been died from TB (WHO, tuberculosis fact sheet).
Following transmission of Mtb to the lungs, alveolar macrophages are responsible to kill the bacteria but in 20–50% of humans infected by Mtb the bacteria circumvent the immune response of the alveolar macrophages and multiply within them (Fatima et al. 2020). Secretion of cytokines from alveolar macrophages leads to a cell-mediated response causing formation of granuloma. In the granuloma, infected macrophage is surrounded by lymphocytes, stem cells and epithelial cells reducing pathogen multiplication and leading to a clinically silent, asymptomatic form of infection (Ai et al. 2016). Cellular immunity including CD4 and CD8 positive cells is the main part of immune response against TB. Cytotoxic T-cell response is essential for eradication of TB (de Martino et al. 2019). The widespread drug resistance to isoniazid and rifampicin as first-line therapy, slow growth rate, complex pathogenesis and the dormant life cycle have complicated successful therapy of TB. Accordingly, the vaccination against TB seems promising strategy. Due to the important role of t-cytotoxic and T-helper cells in the immune response against Mtb, every proposed vaccine have to activate both classes of lymphocytes against Mtb.
The Bacillus Calmette-Guérin (BCG) vaccine is the only approved TB vaccine. It has been developed by attenuation of Mycobacterium bovis. BCG has demonstrated variable effectiveness. Although it protects children from TB, its protection against adult TB is highly variable ranging from zero to 80 percent. Efficacy of the vaccine is waned off after 10 to 20 years from time of immunization (Fatima et al. 2020). Furthermore, BCG is a live-attenuated vaccine so it cannot be administrated to immunocompromised people and there are concerns about possibility of returning to its virulent form causing disease (Fatima et al. 2020). Although many researches have focused on the development of effective and safe TB vaccine in the last decades, a better vaccine candidate is still not accessible and further investigations are encouraged (Kaufmann 2021). Sixteen TB vaccine are currently in different phases of clinical trials. Various technologies have been employed to develop these vaccines. Live attenuated form of M. tuberculosis can be found in some of these vaccine candidates including VPM1002 and MTBVAC. As mentioned for BCG, this platform has poor safety profile and its efficacy is decreased due to the pre-sensitization by environmental mycobacteria (Bibi et al. 2021). Viral vectors have also been utilized to develop TB vaccines. A recombinant vaccinia strain and an adenovirus has been employed to deliver antigens of Mycobacterium tuberculosis including 85a, 85b and Tb10.4. Pre-exposure to these viral vectors could reduce the efficacy of the vaccines (Shah et al. 2018). Subunit vaccines are consisted of pure protein or polysaccharide antigens. Various subunit TB vaccines have employed antigenic proteins/peptides including 85B, ESAT-6, TB10.4, 39A and 32A. Although there is no chance of virulence reversal in subunit vaccines but lack of appropriate immunogenicity necessitates several administrations with adjuvants (Shah et al. 2018).
The development of vaccine against TB due to its complex nature is more challenging. Furthermore, tuberculosis is much more prevalent in developing countries decreasing financial funding and investments on TB vaccine (Zhu et al. 2018). Various immunoinformatic tools have been developed in recent years to facilitate vaccine development time and cost effectively. Genetic information of pathogens can be analyzed using various software and databases to find vaccine candidates (De Groot et al. 2020). Furthermore, with the advantage of immunoinformatic, multi-epitope or chimeric vaccines can be designed by recognition of epitopes from different antigenic proteins. Combination of B-cell and T-cell epitopes in the multi-epitope vaccines can induce a broad range of immune responses. Incorporation of an adjutant in the whole sequence of the vaccine may also enhances immunogenicity and long lasting immune response. Immunoinformatic approaches have been utilized in various studies to design vaccines against pathogenic virus, bacteria and parasites (Oli et al. 2020). Multi-epitope protein vaccines have demonstrated various advantages including appropriate safety profile, lower risk of allergenic responses, low manufacturing cost and freeze deride dosage forms that do not need cold storage (Slingluff 2011, Li et al. 2014). Due to an urgent need for an effective TB vaccine, here an immunoinformatic approach was utilized to design a multi-epitope anti-TB vaccine using a few new antigenic proteins, which have not been investigated for anti-TB vaccine designing before.
Methods
Selection of Antigens and Sequence Retrieval
To select the appropriate antigenic protein for this study, literature have been searched and a few new antigenic protein, which their potential in designing multi-epitope vaccines against M. tuberculosis have not been studied, were chosen. The protein sequence of the selected antigens including Rv2346 (P9WNI7), Rv2347 (P9WNI5), Rv3614 (P9WJD5), Rv3615 (P9WJD7) and Rv2031 (P9WMK1) have been retrieved from the Universal Protein Resource (Uniprot) database (http://www.uniprot.org/)(Bairoch 2005). Sequence of flagellin of Salmonella enterica subsp. enterica serovar Dublin as adjuvant have also been retrieved with ID number of Q06971.
Prediction of MHC-I Binding Epitopes
MHC-I binding epitopes of every antigenic protein have been predicted using NetMHC 4 and Immune Epitope Database and Analysis Resource (IEDB) servers. Artificial neural network (ANN) algorithms have been employed to align sequences in the NetMHC 4 (http://www.cbs.dtu.dk/services/NetMHC/) server. The sequence alignment method of NetMHC 4 tolerates insertion and deletion in the alignment leading to higher performance compare to other strategies (Andreatta and Nielsen 2016). The prediction have been done with all default settings. The IEDB was also utilized to predict MHC-I binding epitopes (http://tools.iedb.org/mhci/). Various methods including ANN, stabilized matrix method (SMM), and combinatorial peptide libraries (CombLib), or NetMHCpan are employed to predict MHC-I binding epitopes in IEDB (Vita et al. 2019).
Prediction of MHC-II Binding Epitopes
MHC-II binding peptides are important for the activation of CD4 + T-cells. The prediction of MHC-II binding epitopes have been done using IEDB (http://tools.iedb.org/mhcii/) and NetMHCIIpan 4 (https://services.healthtech.dtu.dk/service.php?NetMHCIIpan-4.0) servers. The IEDB utilizes various methods including NN-align, SMM-align, CombLib, and Sturniolo. A consensus approach predicts epitopes based on the availability of predictors for the molecule or NetMHCIIpan. The performance of the predictions have been demonstrated in several studies (Vita et al. 2019). The NetMHCIIpan determines nine residues regions, which directly interact with MHC binding cleft to predict peptides with MHC-II binding potential quantitatively (Andreatta et al. 2015).
Prediction of Linear B-Cell Epitopes
BepiPred 2 (https://services.healthtech.dtu.dk/service.php?BepiPred-2.0) was utilized to predict linear B-cell epitopes. A random forest algorithm is employed by BepiPred server to predict linear B-cell epitopes (Jespersen et al. 2017).
Selection of the Epitope Segments
As above mentioned various epitope prediction tools have been employed to predict epitopes for every antigenic protein. To choose the appropriate epitopes for vaccine designing, all results have been pooled and high rank regions with overlap between various methods have been selected. Furthermore, allergenicity, immunogenicity, epitope conservancy and IFN-γ inducing potential of the epitopes have been considered. To evaluate allergenicity of the epitopes, AlgPred server (https://webs.iiitd.edu.in/raghava/algpred/submission.html) have been employed. High accuracy of allergenic peptide/protein prediction in the AlgPred server is due to the integration of various methods including blast, mast, IgEpitope and SVM to evaluate the allergenicity (Saha and Raghava 2006). Antigenicity of the epitopes have been evaluated using the VaxiJen v2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). The accuracy of the VaxiJen results varies ranging from 70 to 89% depend on the target organism. The principal chemical properties of the peptide/protein sequences instead of a sequence alignment approach is employed to assess antigenicity by VaxiJen server (Saha and Raghava 2006). Potential of IFN-γ induction could improve development of immune response by the vaccine. IFN-γ inducing potential of the epitopes have been analyzed with IFNepitope server (http://crdd.osdd.net/raghava/ifnepitope/). Various approaches, such as machine learning technique, motifs-based search, and hybrid approach are utilized in INFepitope server. The maximum accuracy of 81% have been reported for hybrid model (Dhanda et al. 2013). Epitope conservancy was also evaluated with IEDB (http://tools.iedb.org/conservancy/). Finally, for every antigenic protein one non-allergenic MHC-I binding and one non-allergenic MHC-II binding epitope have been selected. All of the selected epitopes have also demonstrated antigenicity.
Construction of the Vaccine and Evaluation of Its Properties
The selected epitopes were joined together with appropriate linkers to construct the vaccine sequence. Furthermore, flagellin as toll-like receptor (TLR) agonist and TpD as universal T-helper epitope were added to the vaccine construct to improve its efficacy. The Solpro server (http://scratch.proteomics.ics.uci.edu/) was employed to evaluate solubility of the vaccine protein in Escherichia coli. Solpro is based on a SVM approach. It has demonstrated 74% overall accuracy in an experiment using multiple runs of tenfold cross validation (Magnan et al. 2009). Different physicochemical properties of the vaccine construct including amino acid composition, molecular weight (MW), instability index, grand average of hydropathicity (GRAVY) and theoretical pI were estimated using the ProtParam tool (http://web.expasy.org/protparam/) (Gasteiger et al. 2005).
Evaluation of Antigenicity and Allergenicity of the Vaccine Construct
Allergenicity and antigenicity of the vaccine construct were also evaluated. AllergenFP v.1.0 (http://ddg-pharmfac.net/AllergenFP/) were employed to estimate allergenicity of the vaccine construct. AllergenFP specify allergens from non-allergens with accuracy of about 88%. It employs physicochemical properties of the molecules to develop a descriptor-based fingerprint approach (Dimitrov et al. 2014). To evaluate antigenicity of the vaccine construct, ANTIGENpro (http://scratch.proteomics.ics.uci.edu/) and VaxiJen v2.0 servers were employed. ANTIGENpro evaluate antigenicity of proteins by an alignment-free approach in which a final SVM classifier is combined with various machine learning algorithms (Magnan et al. 2010).
Homology Modeling
To investigate the interaction of the vaccine construct with immune system proteins such as TLRs and prediction of conformational B-cell epitopes, 3-dimential (3D) structure of the vaccine construct was modeled. 3D modeling have been done using I-Tasser software at http://zhanglab.ccmb.med.umich.edu/I-TASSER/. Four steps is followed to develop a 3D model of a protein in the I-Tasser server. First, templet proteins in protein data bank (PDB) with similar sequence to the query protein are identified with multiple alignment approaches. Second, protein structure is assembled using a modified replica-exchange Monte Carlo simulation method. Some regions including loops and tails could be modeled using ab initio approach. Third, structure decoys are clustered to select the model and fragment-guided molecular dynamics simulation (FG-MD) or ModRefiner are utilized to refine the model. Forth, the modeling is completed by a structure-based functional annotation using COACH approach. A confidence score (Cscore) is designated to the 3D models developed by I-Tasser. The higher value of Cscore demonstrates high confidence for a model. The models were retrieved from I-Tasser server in PDB format and Discovery studio 2020 was used to visualize the 3D structures and production of figures.
Refinement of the 3D Modeled Structure
GalaxyRefine and 3Drefine servers refined top two 3D models developed by I-Tasser server. GalaxyRefine (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) refines whole protein through mild and aggressive relaxation methods. Actually, repacking of the protein side-chains is followed by short molecular dynamic simulations to relax the structure (Shin et al. 2014). In the 3Drefine server, (http://sysbio.rnet.missouri.edu/3Drefine/) a two-step protocol refines the protein structure. At first, hydrogen-bonding network is iteratively optimized and then energy minimization at atomic level is performed using combination of physics and knowledge-based force fields (Bhattacharya et al. 2016).
Validation of the 3D Refined Structures
To evaluate developed 3D structures and to select the best 3D model of the vaccine construct, ProSA-web Z-score, ERRAT value, and PROCHECK Ramachandran plot were analyzed. The ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) computes Z-score and plots calculated Z-score with the Z-scores of experimentally developed 3D structures deposited in PDB. To estimate Z-score, interaction energy of each residue with the rest of protein is calculated and is compared to a certain energy criteria (Wiederstein and Sippl 2007). The Ramachandran plot have been retrieved from PROCHECH software accessible in the structural and verification analysis server (SAVE) (https://saves.mbi.ucla.edu/). In the Ramachandran plot, residues are divided to allowed and disallowed regions based on the phi-psi torsion angles (Hollingsworth and Karplus 2010). ERRAT value can also be calculated in SAVE server. Non-bonded atom–atom interactions in a database of reliable high-resolution crystallography structures is compared to the one in the query model to calculate ERRAT value (Colovos and Yeates 1993).
Prediction of Discontinuous B-Cell Epitopes
3D structure of the vaccine construct were implemented to ElliPro in IEDB database (http:// tools.immuneepitope.org/tools/ElliPro) to predict discontinuous B-cell epitopes. ElliPro identifies B-cell epitopes by combination of a residue-clustering algorithm and Thornton’s method. ElliPro have demonstrated the best performance in comparison with six other algorithms predicting epitopes (Ponomarenko et al. 2008).
Molecular Docking of the Vaccine with TLRs
Interaction of a vaccine with toll like receptors (TLR) can improve efficacy of the vaccine (Vijay 2018). Protein–protein docking have been employed to investigate interactions of the vaccine construct with TLR3, TLR4 and TLR8. Crystallographic 3D structures of TLR3 (1ZIW), TLR4 (4G8A) and TLR8 (3W3G) have been retrieved from the protein data bank (PDB; http://www.rcsb.org/pdb/) and were considered as receptor in the docking procedure. Protein–protein docking have been done using ClusPro (https://cluspro.bu.edu/login.php) (Kozakov et al. 2013, Kozakov et al. 2017, Vajda et al. 2017, Desta et al. 2020), PatchDock (https://bioinfo3d.cs.tau.ac.il/PatchDock/php.php) (Duhovny et al. 2002, Schneidman-Duhovny et al. 2005) and HawkDock (http://www.cadd.zju.edu.cn/hawkdock/) (Hou et al. 2002, Zacharias 2003, Feng et al. 2017, Weng et al. 2019) servers. Outputs of docking with PatchDock were refined with FireDock as it is recommended by the PatchDock server. To select the best docking results among the outputs of various servers, free binding energy of the docked structures have been calculated using Molecular Mechanics/Generalized Born Surface Area (MM-GBSA) at HawkDock server and the complex with the lowest free binding energy have been considered as the best docking result.
Codon Optimization and In Silico Cloning
The amino acid sequence of the vaccine construct have been reversely translated by sequence manipulation suite (https://www.bioinformatics.org/sms2/rev_trans.html) to develop a suitable gene sequence for cloning and expression. The properties of the gene sequence including Codon Adaptation Index (CAI), GC content, and Codon Frequency and Distribution (CFD) have been estimated by GenScript server (https://www.genscript.com/tools/rare-codon-analysis) (Yazdani 2020). Furthermore, to facilitate cloning of the vaccine gene into E coli, CLC Sequence viewer v.8 was employed to add NcoI and XhoI restriction sites to N and C-terminals of the sequence respectively and the sequence was inserted to pET-28a vector.
Results
Prediction and Selection of the Epitope Segments
Mtb antigenic proteins including Rv2346, Rv2347, Rv3614, Rv3615 and Rv2031 have been investigated to find antigenic epitopes using various approaches. MHC-1 (HLA-A and B) and MHC-II (DP, DQ and DR) binding epitopes have been predicted using IEDB and NetMHC 4 servers. The linear B-cell epitopes have also been predicted using BepiPred. All epitopes have been pooled and 10 epitope have been selected based on the following priorities (Table 1). From every protein, two epitopes have been selected, one MHC-I binding epitope and one MHC-II binding epitope. The epitopes with overlap in various servers were prioritized. Antigenicity, allergenicity epitope conservancy and INF-γ stimulation were also considered in epitope selection. Epitopes with antigenicity and INF-γ stimulation as well as lack of allergenicity and high degree of conservancy were preferred.
Table 1.
Selected epitopes for designing the vaccine along with their antigenicity, allergenicity, conservancy and IFN-γ inducing potential
| Protein | Epitope sequence | HLA-I (Netmhc4- IEDB) | HLA-II (RANKPEP/ IEDB) | Linear B Cell (BEPIPRED/ IEDB) | Antigenicity | IFN-γ stimulation | Allergenicity | Conservancy |
|---|---|---|---|---|---|---|---|---|
| RV2346 | QTDSAVGSSW | + | + | _ | 1.4000 | 1 | Non-applicable | 96% |
| DVLAAGDFWGGAGSVACQE | + | + | + | 0.4957 | 1 | Non-Allergen | 58% | |
| RV2347 | HAMRDMAGR | + | + | - | 0.7163 | -0.14355781 | Non-applicable | 96% |
| ARRMWASAQNISGAG | + | + | + | 0.6245 | 1 | Non-Allergen | 84% | |
| RV3614 | WTADPIIGV | + | + | + | 0.5996 | 0.35940912 | Non-applicable | 80% |
| RIDHVELSARVAWMSES | + | + | + | 0.6176 | 1 | Non-Allergen | 90% | |
| RV3615 | HTAGVDLAK | + | + | - | 1.2063 | 0.11063872 | Non-applicable | 60% |
| SSLHTAGVDLAKSLRIA | + | + | - | 0.7673 | 0.049221663 | Non-Allergen | 58% | |
| RV2031 | HPRSLFPEF | + | + | + | 0.5001 | 0.16584561 | Non-applicable | 64% |
| ELFAAFPSFAGLRPT | + | + | + | 0.5372 | 0.65327194 | Non-Allergen | 64% |
Construction of the Vaccine and Evaluation of Its Properties
To construct the multi-epitope vaccine structure, in addition to the selected epitopes, two adjuvants including flagellin of Salmonella enterica subsp. enterica serovar Dublin and TpD as 31 amino acid universal T-helper epitope have been used (Fraser et al. 2014). Adjuvants and the selected epitopes have been linked together using appropriate linkers. EAAAK was employed to connect the adjuvants to the epitopes. GPGPG and AAY linkers have connected the MHC-II and MHC-I binding epitopes respectively. The final vaccine construct have 440 amino acids and is illustrated in the Fig. 1.
Fig. 1.

The arrangement of the selected epitopes and adjuvants in the vaccine. Linkers are illustrated with green arrows
Solubility and physicochemical properties of the vaccine construct have been evaluated using Solpro and ProtParam servers. The overexpressed protein of the vaccine in E coli have been predicted soluble with probability of 0.877487. ProtParam server have predicted various physicochemical properties including amino acid composition, molecular weight (MW), instability index, grand average of hydropathicity (GRAVY) and theoretical pI (Table 2). ProtParam have calculated the instability index equal to 36.90 demonstrating the vaccine is stable. Additionally, pI and MW of the vaccine have been estimated 6.26 and 46,125.53 respectively.
Table 2.
Physicochemical and immunological properties of the vaccine construct
| Physicochemical properties | Result |
|---|---|
| Predicted solubility / Solpro | Soluble / probability (0.877487) |
| Molecular weight | 46,125.53 |
| Instability index | 36.90 / stable |
| Gravy | -0.198 |
| Aliphatic index | 85.82 |
| Theoretical pI | 6.26 |
| No. Of amino acids | 440 |
| Total no. Of negatively charged residues (Asp + Glu) | 40 |
| Total no. Of positively charged residues (Arg + Lys) | 38 |
| Allergenicity | |
| AllergenFP v.1.0 | Probable Non-Allergen |
| Antigenicity | |
| ANTIGENpro | 0.926892 / Antigen |
| VaxiJen | 0.5876 / Probable antigen |
Evaluation of Antigenicity and Allergenicity of the Vaccine Construct
The protein sequence of the vaccine construct have been evaluated for antigenicity using ANTIGENpro and VaxiJen. Both server have demonstrated the vaccine as antigen. VaxiJen have estimated the antigenicity of 0.5876 for vaccine while it considers 0.5 as threshold and ANTIGENpro have estimated the antigenicity of the vaccine construct as 0.926892. AllergenFP server have shown that the vaccine is probably non-allergenic (Table 2).
Tertiary Structure Modeling, Refinement, and Validation
I-TASSER server has modeled the 3D structure of the vaccine construct. Top two models have been selected based on the Cscore and each model have been refined using GalaxyRefine and 3Drefine. Figure 2 have demonstrated the best refined model which have selected based the ProSA-web Z-score, ERRAT value, and PROCHECK Ramachandran plot. The best refined model have demonstrated Z-score of -4.21. As illustrated in the Fig. 3A the Z-score of the selected refined model is in the range of the Z-scores of experimentally developed 3D structures deposited in PDB. ERRAT values around 95 or higher demonstrates good high-resolution model. The ERRAT value of the selected model is 95.316 demonstrating appropriate 3D model for the vaccine construct (Fig. 3B). Furthermore, Ramachandran plot have demonstrated only nine residues (2.3%) in the disallowed regions (Fig. 3C). Overall, assessment of the selected refined model have demonstrated that the3D structure of the vaccine construct have the criteria of an acceptable 3D protein model and can be utilized for further investigations.
Fig. 2.
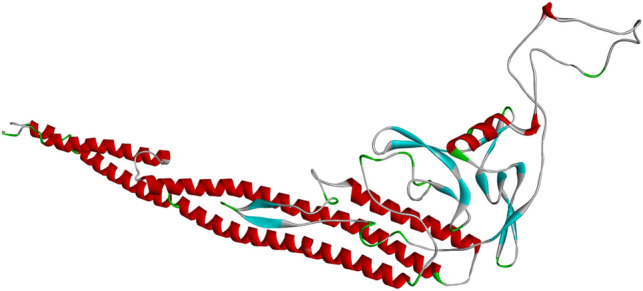
Refined 3D model of the vaccine construct. The vaccine 3D structure was modeled with I-Tasser and refined by 3Drefine
Fig. 3.

Evaluation of the refined 3D model. Part A illustrates ERRAT chart of the 3D model with ERRAT value of 95.316%. ERRAT values around 95% or higher demonstrate good quality model. Part B illustrates ProSA-web Z-score plot. The Z-score (-4.21) of the refined model is shown in a large black dote which is in the range of Z-scores of experimentally 3D structures in the PDB. Part C illustrates Ramachandran plot demonstrating nine (2.3%) amino acids in the disallowed region
Conformational B-Cell Epitope Identification
The 3D model of the vaccine construct was investigated for conformational B-Cell epitopes using ElliPro in IEDB. Table 3 demonstrates conformational B-Cell epitopes of the vaccine construct. Epitope 2 and 3 are located in the sequence of MHC-I/MHC-II binding epitopes while epitope 1 is mostly located in the flagellin sequence (Fig. 4).
Table 3.
Conformational B-Cell Epitopes in the refined 3D model of the vaccine
| No | Residues | Number of residues | Score |
|---|---|---|---|
| 1 | A:M1, A:A2, A:Q3, A:V4, A:I5, A:N6, A:T7, A:N8, A:S9, A:L10, A:S11, A:L12, A:L13, A:T14, A:Q15, A:N16, A:N17, A:L18, A:N19, A:K20, A:S21, A:Q22, A:S23, A:S24, A:L25, A:S26, A:S27, A:A28, A:I29, A:E30, A:R31, A:L32, A:S33, A:S34, A:G35, A:L36, A:R37, A:I38, A:N39, A:S40, A:A41, A:K42, A:D43, A:D44, A:A45, A:A46, A:G47, A:D402, A:A403, A:D404, A:Y405, A:A406, A:T407, A:E408, A:V409, A:S410, A:N411, A:M412, A:S413, A:K414, A:A415, A:Q416, A:I417, A:L418, A:Q419, A:Q420, A:A421, A:G422, A:T423, A:S424, A:V425, A:L426, A:A427, A:Q428, A:A429, A:N430, A:Q431, A:V432, A:P433, A:Q434, A:N435, A:V436, A:L437, A:S438, A:L439, A:L440, A:R441 | 87 | 0.77 |
| 2 | A:G186, A:P187, A:G188, A:R189, A:D190, A:V191, A:L192, A:A193, A:A194, A:G195, A:D196, A:F197, A:W198, A:G199, A:G200, A:A201, A:G202, A:S203, A:V204, A:A205, A:C206, A:Q207, A:E208, A:G209, A:P210, A:G211, A:P212, A:G213, A:S214, A:S215, A:L216, A:H217, A:T218, A:A219, A:G220, A:V221, A:D222, A:L223, A:A224, A:K225, A:S226, A:L227, A:R228, A:I229, A:A230, A:E231, A:A232, A:A233, A:A234, A:K235, A:E236, A:L237, A:A239, A:A240, A:P242, A:S243, A:F244, A:A245, A:G246, A:L247, A:R248, A:P249, A:T250, A:E251, A:A252, A:G273, A:L274, A:P275, A:Q276, A:S277, A:S282, A:L283, A:M284, A:V285, A:A286, A:Q287, A:E288, A:A289 | 78 | 0.743 |
| 3 | A:N87, A:Q90, A:R91, A:V92, A:R93, A:E94, A:L95, A:S96, A:V97, A:Q98, A:A99, A:T100, A:N101, A:G102, A:T103, A:N104, A:S105, A:D106, A:S107, A:D108, A:L109, A:S111, A:I112, A:D169, A:S174, A:A175, A:R176, A:V177, A:A178, A:W179, A:M180, A:S181, A:L257, A:M258, A:Q259, A:Y260, A:I261, A:K262, A:A263, A:N264, A:S265, A:K266, A:F267, A:I268, A:K313, A:A314, A:G347, A:S348, A:S349, A:W350, A:E351, A:A352, A:A353, A:A354, A:K355, A:A356, A:S357, A:I358, A:D359, A:S360, A:A361, A:L362, A:S363, A:D366, A:A367, A:S370 | 66 | 0.635 |
Fig. 4.

Discontinuous B-cell epitopes. ElliPro server have been employed to predict discontinuous B-cell epitopes. Yellow balls are illustrating amino acids in three discontinuous B-cell epitopes in the 3D structure of the vaccine
Protein–Protein Docking Investigations
Protein–protein docking have been utilized to evaluate interactions of the vaccine with TLR proteins. ClusPro, PatchDock and HawkDock servers were used to dock vaccine 3D model as ligand into the TLR3 (1ZIW), TLR4 (4G8A) and TLR8 (3W3G) as receptor. The best docking results have been selected based on the free binding energy of the ligand-receptor complexes. ClusPro docking outputs have demonstrated better free binding energy (Table 4). UCSF Chimera software have been utilized to analyze interactions between the vaccine construct and TLR proteins (Pettersen et al. 2004). Chimera H-bond analysis have found 21, 19 and 18 hydrogen bond interactions between the vaccine construct and TLR8, TLR4 and TLR 3 respectively. Furthermore, many other interactions including polar and nonpolar ones have also been formed between the vaccine construct and TLR proteins demonstrating possibility of the activation of TLR-dependent immune response by the designed vaccine. Figures 5, 6 and 7 are illustrating docking results of the vaccine 3D model with TLR 8, 4 and 3 respectively. In the figures, section A represents surface interaction between the vaccine and TLR protein, section B illustrates H-bond formation between two protein and section C illustrates all interactions including polar and non-polar in the complex.
Table 4.
Free binding energy of complexes of the vaccine 3D model and TLR proteins
| TLR (PDB ID) | ClusPro | PatchDock | HawkDock |
|---|---|---|---|
| TLR3 (1ZIW) | -58.39 | 25.82 | -8.32 |
| TLR8 (3W3G) | -114.77 | -23.21 | -3.97 |
| TLR4 (4G8A) | -123.03 | 970.62 | 10.6 |
Fig. 5.

Docking results of the vaccine with TLR8. Part A illustrates surface representation of the vaccine (Maya blue) and TLR8 (chocolate). Part B and C are illustrating potential H-bonds (red sticks) and all interactions including non-polar bonds (yellow sticks) between the vaccine and TLR8 respectively. Docking have been done in the ClusPro server and figures have been made by Chimera software
Fig. 6.

Docking results of the vaccine with TLR4. Part A illustrates surface representation of the vaccine (Maya blue) and TLR4 (chocolate). Part B and C are illustrating potential H-bonds (red sticks) and all interactions including non-polar bonds (yellow sticks) between the vaccine and TLR8 respectively. Docking have been done in the ClusPro server and figures have been made by Chimera software
Fig. 7.

Docking results of the vaccine with TLR3. Part A illustrates surface representation of the vaccine (Maya blue) and TLR3 (chocolate). Part B and C are illustrating potential H-bonds (red sticks) and all interactions including non-polar bonds (yellow sticks) between the vaccine and TLR8 respectively. Docking have been done in the ClusPro server and figures have been made by Chimera software
Codon Optimization and In Silico Cloning
Sequence manipulation suite have revers translated the amino acid into a nucleotide sequence and GenScript server have estimated Codon Adaptation Index (CAI), GC content, and Codon Frequency and Distribution (CFD) to evaluate the sequence. The CAI of 1 have been estimated which is ideal for the expression of the protein in the host (Fig. 8A). The GC content of the vaccine nucleotide sequence is 62.42%, which is in the appropriate range of 30–60%, and CFD have been calculated 100% indicating appropriate distribution and selection of the codons for every amino acid resulting effective expression in E. coli as host (Fig. 8B, C). Finally, to facilitate cloning of the vaccine gene into E coli, NcoI and XhoI restriction sites were added to the N and C terminal of the sequence respectively and the sequence was inserted to pET-28a Vector (Fig. 8D).
Fig. 8.

Evaluation of important parameters for cloning in E. coli. CAI of codon optimized sequence of the vaccine is 1 (A). CAI more than 0.8 is considered appropriate. GC content of the vaccine sequence is 62.42%, which is in the range of 30–60% as appropriate GC content (B). Codon frequency distribution of the vaccine sequence is 100 supporting maximum expression of the vaccine in E. coli (C). Vector map of the vaccine sequence in the pET-28a vector is also illustrated (D)
Discussion
Tuberculosis (TB) is a global health issue causing considerable number of death per year. Latent form of the disease, complicated pathogenesis and multidrug resistance hamper the successful eradication of TB. Various immunoinformatic approaches have been developed to design vaccines time and cost effectively. These approaches can be used to circumvent limitations like genetic variations, antigenic drift and antigenic shift. Immunoinformatic approach have attracted considerable attention for development of vaccines against infectious diseases with complicated pathogenesis and life style in the recent years (Tahir Ul Qamar et al. 2019; Akhtar et al. 2021; Fadaka et al. 2021; Joshi et al. 2021). Since the only approved TB vaccine, BCG, have demonstrated variable efficacy especially in adults and as live attenuated vaccine has low safety profile, lots of investigations have been focused on the development of anti-TB vaccines. There are also some investigations using immunoinformatic approach to design multi-epitope vaccines against TB (Bibi et al. 2021; Mahmood et al. 2021; Sharma et al. 2021). In these studies, mostly known antigenic proteins or proteins in the exosome vesicles have been employed to find antigenic epitopes while in the present study a few new proteins with antigenic properties have been selected to find epitopes and to design the vaccine. To do so, potential MHC-I and MHC-II binding epitopes of the proteins have been predicted. The multi-epitope vaccine was constructed by joining the predicted epitopes and two adjuvant sequences including a universal T-helper epitope TpD and flagellin of Salmonella enterica. The physicochemical and immunological properties of the selected epitopes and the constructed vaccine have been evaluated by various tools summarized in the method section. The vaccine construct as well as selected epitopes have demonstrated antigenicity and IFN-γ stimulation potential while have classified as non-allergenic peptide or protein. Conservation analysis of the selected epitopes have demonstrated at least 80% of conservancy for 5 epitopes demonstrating the vaccine can induce immune system response against various strains of Mtb. The protein of the vaccine was estimated soluble (probability of 0.877487) and stable (instability index of 36.90) with the pI of 6.26 demonstrating appropriate physicochemical properties. Evaluation of CAI, CFD, and GC content of the vaccine construct gene have revealed effective potential of the vaccine to be transcribed and translated in E. coli. Furthermore, 3D structure of the vaccine construct have been modeled and the potential of the vaccine in stimulation of TLR-dependent immune response have been investigated by protein–protein docking approach. Evaluation of the 3D model have indicated ERRAT value of 95.316, Z-score of − 4.21 and only nine amino acid in the disallowed regions of Ramachandran plot demonstrating acceptable criteria for the 3D model. Formation of numerous H-bonds and non-polar interactions between the vaccine construct and TLR proteins have demonstrated that the vaccine properly interacts with TLR 3, 4 and 8 (Figs. 5, 6, 7). ElliPro in IEDB have also identified three conformational B-cell epitopes in the 3D model of the vaccine playing important role in the humoral immunity.
RV2031 known as α-crystalline was one of the antigenic proteins in the present study. RV2031 have been overexpressed at reduced oxygen tension. It has also demonstrated role in non-replicating phase of mycobacteria (Cunningham and Spreadbury 1998, Rosenkrands et al. 2002). Depletion of RV2031 have decreased tolerance of Mtb in anaerobic conditions demonstrating role of RV2031 in the survival of Mtb in hypoxia and latency. It has also involved in Mtb pathogenesis by inhibition of differentiation of monocytes to dendritic cells (DCs) (Siddiqui et al. 2014). RV2031 is a member of an operon known as Rv2028-Rv2031. A bioinformatics study has been reported that this operon is involved in response of Mtb to stress and its dormant state. Proteins of this operon have been overexpressed during latency and have also demonstrated positive effects on the expression of other latency associated genes. The antigenic properties of RV2031 have been reported in various studies (Chegou et al. 2012; Hozumi et al. 2013; Serra-Vidal et al. 2014; Belay et al. 2015). It have also demonstrated INF-gamma and TNF-α inducing potential (Leyten et al. 2006; Mushtaq et al. 2015; Meier et al. 2021a). Overall demonstrated role of Rv2031 in the latency and pathogenesis of Mtb as well as its antigenic and IFN-γ inducing properties make it an attractive candidate for vaccine design.
Rv2346/47 and Rv3614/15 were also employed in vaccine design in the present study. These are all associated with the 6-kDa early secretory antigenic target of Mycobacterium tuberculosis (ESAT-6) secretion system demonstrating their role in the pathogenesis of Mtb. Rv2346/47 have induced TNF-α and Rv3514/15 have induced IFN-γ in the peripheral blood mononuclear cells of the HIV positive individuals developing TB disease, demonstrating antigenic potential of these proteins (Meier et al. 2021a). Antigenic potential of Rv2346/47 have also demonstrated when recombinant form of Rv2346/47 have developed positive delayed-type hypersensitivity (DTH) in guinea pigs which were treated with heat-killed Mtb (Mustafa 2012). Furthermore, absence of these genes in the BCG strain decreases concerns about vaccine efficacy in individuals pre-exposured to BCG (Mahairas et al. 1996). Rv2346 induces genomic instability in the infected macrophages through induction of oxidative stress modulating immune function of macrophages. It also supports intracellular bacillary persistence (Mohanty et al. 2016). Infection of mice with a Rv2346 knockout strain of Mtb have led to mortality reduction, long inflammation markers decrement and lower CFU count in vitro demonstrating pivotal role of Rv2346 in the pathogenesis of Mtb (Chen et al. 2018).
Rv3614/15 are involved in the ESX-1 secretion system playing role in the Mtb pathogenesis (Champion et al. 2009, Chen et al. 2012). Rv3614/15 have induced secretion of various cytokines including IFN-γ, and TNF-α in the blood sampled from latently Mtb-infected individuals demonstrating their antigenic potential (Coppola et al. 2016). ESAT6 and the 10-kDa culture filtrate antigen (CFP-10) have been considered as the most immunodominant and highly Mtb-specific antigens (Millington et al. 2011). Immunodominance effects of Rv3615 have been reported as equal as ESAT-6 and CFP-10 in the active and latent TB infection. Furthermore, Rv3615 has demonstrated T-cell responses as specific as ESAT-6 and CFP-10 for Mtb infection. The high immunodominance and specificity of Rv3615 propose it as appropriate candidate in vaccine designing and immunodiagnostic (Millington et al. 2011). In addition to all above-mentioned studies, Rv2346/47, Rv3614/15c and Rv2031 have demonstrated appropriate potential in immunodiagnosis of Mtb in children. In this study, whole blood samples from 80 TB infected or healthy children were stimulated with 10 antigenic Mtb protein and cytokine secretion was measured. Machine-learning algorithms were employed to analyze the results and identification of the best antigenic protein (Meier et al. 2021b).
To connect various epitopes and adjuvants in the vaccine construct, appropriate linkers were employed. EAAAK have been used to connect the adjuvants to the epitopes. EAAAK is a rigid linker facilitating development of 3D structure of adjuvants and preventing epitopes to interfere with the interaction of adjuvants with their targets. MHC-I and MHC-II binding epitopes have been linked with AAY and GPGPG linkers respectively (Bibi et al. 2021). The sequence of two adjuvants have been utilized in the vaccine construct to improve the vaccine immunogenicity. Flagellin can enhance systemic and mucosal adaptive immune response when added to the vaccine antigen. Cui et al. (2018) have reviewed various investigations demonstrating adjuvant activity of flagellin in human and animal. Agonistic effects on TLR5, induction of cytokines and nitric oxide, activation of dendritic cells and neutrophils, activation of adaptive immune responses manly production of IgA and Th2-type have been proposed as mechanisms of flagellin (Cui et al. 2018). The second adjuvant was known as TpD with the sequence of "ILMQYIKANSKFIGIPMGLPQSIALSSMVAQ". It is a universal memory T-cell helper peptide designed to be active in human, mice and non-human primates (Fraser et al. 2014).
Conclusion
In this study five new antigenic protein from Mtb have been selected to design a multi-epitope vaccine against TB. Extensive immunoinformatic tools have been utilized to find immunodominant and non-allergenic peptides from the antigenic proteins. Flagellin and TpD were added to the vaccine construct to improve immunization potency of the vaccine. According to the evaluation of various immunological and physicochemical properties of the vaccine construct, it can be expected that the vaccine develops appropriate immune responses against Mtb. Obviously, the immunoinformatic results reported in the present study should be followed by experimental investigations but with the advantage of immunoinformatic tools development of new generations of vaccines against various infectious and cancerous diseases is now accessible time and cost effectively. For the diseases with complicated pathogenesis and life cycle which no effective vaccine have already been developed including TB, this can be a great opportunity.
Author contributions
MG designed and performed the study and wrote the manuscript.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Global Tuberculosis Report (2021) Geneva: World Health Organization; 2021. Licence: CC BY-NC-SA 3.0 IGO
- https://www.who.int/news-room/fact-sheets/detail/tuberculosis.
- Ai JW, Ruan QL, Liu QH, Zhang WH. Updates on the risk factors for latent tuberculosis reactivation and their managements. Emerg Microbes & Infect. 2016;5(2):e10. doi: 10.1038/emi.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar N, Joshi A, Singh J, Kaushik V. Design of a novel and potent multivalent epitope based human cytomegalovirus peptide vaccine: an immunoinformatics approach. J Mol Liq. 2021;335:116586. doi: 10.1016/j.molliq.2021.116586. [DOI] [Google Scholar]
- Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, et al. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics. 2015;67(11–12):641–650. doi: 10.1007/s00251-015-0873-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32(4):511–517. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, et al. The universal protein resource (UniProt) Nucleic Acids Res. 2005;33(Database Issue):D154–159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belay M, Legesse M, Mihret A, Bekele Y, Ottenhoff THM, et al. Pro- and anti-inflammatory cytokines against Rv2031 are elevated during latent tuberculosis: a study in cohorts of tuberculosis patients, household contacts and community controls in an endemic setting. PLoS ONE. 2015;10(4):e0124134–e0124134. doi: 10.1371/journal.pone.0124134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya D, Nowotny J, Cao R, Cheng J. 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016;44(W1):W406–409. doi: 10.1093/nar/gkw336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibi S, Ullah I, Zhu B, Adnan M, Liaqat R, et al. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci Rep. 2021;11(1):1249. doi: 10.1038/s41598-020-80899-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champion PAD, Champion MM, Manzanillo P, Cox JS. ESX-1 secreted virulence factors are recognized by multiple cytosolic AAA ATPases in pathogenic mycobacteria. Mol Microbiol. 2009;73(5):950–962. doi: 10.1111/j.1365-2958.2009.06821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chegou NN, Essone PN, Loxton AG, Stanley K, Black GF, et al. Potential of host markers produced by infection phase-dependent antigen-stimulated cells for the diagnosis of tuberculosis in a highly endemic area. PLoS ONE. 2012;7(6):e38501. doi: 10.1371/journal.pone.0038501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JM, Boy-Röttger S, Dhar N, Sweeney N, Buxton RS, et al. EspD is critical for the virulence-mediating ESX-1 secretion system in Mycobacterium tuberculosis. J Bacteriol. 2012;194(4):884–893. doi: 10.1128/JB.06417-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Chen S, Yao J, Shu L, Deng K et al (2018) Study on the construction and virulence observation of Rv2346 c gene knockout strains of Mycobacterium tuberculosis mediated by bacteriophage. Chin J Anim Infect Dis: 490–495.
- Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola M, van Meijgaarden KE, Franken KL, Commandeur S, Dolganov G, et al. New genome-wide algorithm identifies novel in-vivo expressed Mycobacterium tuberculosis antigens inducing human T-cell responses with classical and unconventional cytokine profiles. Sci Rep. 2016;6:37793. doi: 10.1038/srep37793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui B, Liu X, Fang Y, Zhou P, Zhang Y, et al. Flagellin as a vaccine adjuvant. Expert Rev Vaccines. 2018;17(4):335–349. doi: 10.1080/14760584.2018.1457443. [DOI] [PubMed] [Google Scholar]
- Cunningham AF, Spreadbury CL. Mycobacterial stationary phase induced by low oxygen tension: cell wall thickening and localization of the 16-kilodalton alpha-crystallin homolog. J Bacteriol. 1998;180(4):801–808. doi: 10.1128/JB.180.4.801-808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot, A. S., L. Moise, F. Terry, A. H. Gutierrez, P. Hindocha, et al. (2020) Better Epitope Discovery, Precision Immune Engineering, and Accelerated Vaccine Design Using Immunoinformatics Tools. Fron Immunol 11. [DOI] [PMC free article] [PubMed]
- de Martino, M., L. Lodi, L. Galli and E. Chiappini (2019) Immune Response to Mycobacterium tuberculosis: a narrative review. Front Pediatr 7. [DOI] [PMC free article] [PubMed]
- Desta IT, Porter KA, Xia B, Kozakov D, Vajda S. Performance and its limits in rigid body protein-protein docking. Structure. 2020;28(9):1071–1081. doi: 10.1016/j.str.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8:30. doi: 10.1186/1745-6150-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov I, Naneva L, Doytchinova I, Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30(6):846–851. doi: 10.1093/bioinformatics/btt619. [DOI] [PubMed] [Google Scholar]
- Duhovny, D., R. Nussinov and H. J. Wolfson (2002). Efficient Unbound Docking of Rigid Molecules. Algorithms in Bioinformatics, Berlin, Heidelberg, Springer Berlin Heidelberg.
- Fadaka AO, Sibuyi NRS, Martin DR, Goboza M, Klein A, et al. Immunoinformatics design of a novel epitope-based vaccine candidate against dengue virus. Sci Rep. 2021;11(1):19707. doi: 10.1038/s41598-021-99227-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatima S, Kumari A, Das G, Dwivedi VP. Tuberculosis vaccine: A journey from BCG to present. Life Sci. 2020;252:117594. doi: 10.1016/j.lfs.2020.117594. [DOI] [PubMed] [Google Scholar]
- Feng T, Chen F, Kang Y, Sun H, Liu H, et al. HawkRank: a new scoring function for protein–protein docking based on weighted energy terms. J Cheminform. 2017;9(1):66. doi: 10.1186/s13321-017-0254-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CC, Altreuter DH, Ilyinskii P, Pittet L, LaMothe RA, et al. Generation of a universal CD4 memory T cell recall peptide effective in humans, mice and non-human primates. Vaccine. 2014;32(24):2896–2903. doi: 10.1016/j.vaccine.2014.02.024. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud SE, Wilkins MR, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, et al., editors. The proteomics protocols handbook. Totowa, NJ, Humana: Press; 2005. pp. 571–607. [Google Scholar]
- Hasan T, Au E, Chen S, Tong A, Wong G. Screening and prevention for latent tuberculosis in immunosuppressed patients at risk for tuberculosis: a systematic review of clinical practice guidelines. BMJ Open. 2018;8(9):e022445–e022445. doi: 10.1136/bmjopen-2018-022445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth SA, Karplus PA. A fresh look at the Ramachandran plot and the occurrence of standard structures in proteins. Biomol Concepts. 2010;1(3–4):271–283. doi: 10.1515/bmc.2010.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou T, Qiao X, Zhang W, Xu X. Empirical Aqueous Solvation Models Based on Accessible Surface Areas with Implicit Electrostatics. J Phys Chem. 2002;106(43):11295–11304. doi: 10.1021/jp025595u. [DOI] [Google Scholar]
- Hozumi H, Tsujimura K, Yamamura Y, Seto S, Uchijima M, et al. Immunogenicity of dormancy-related antigens in individuals infected with Mycobacterium tuberculosis in Japan. Int J Tuberc Lung Dis. 2013;17(6):818–824. doi: 10.5588/ijtld.12.0695. [DOI] [PubMed] [Google Scholar]
- Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45(W1):W24–W29. doi: 10.1093/nar/gkx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Ray NM, Singh J, Upadhyay AK, Kaushik V. T-cell epitope-based vaccine designing against Orthohantavirus: a causative agent of deadly cardio-pulmonary disease. Netw Model Anal Health Inform Bioinform. 2021;11(1):2. doi: 10.1007/s13721-021-00339-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann SHE (2021) Vaccine development against tuberculosis over the last 140 years: failure as part of success. Front Microb 12. [DOI] [PMC free article] [PubMed]
- Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, et al. How good is automated protein docking? Proteins. 2013;81(12):2159–2166. doi: 10.1002/prot.24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyten EM, Lin MY, Franken KL, Friggen AH, Prins C, et al. Human T-cell responses to 25 novel antigens encoded by genes of the dormancy regulon of Mycobacterium tuberculosis. Microbes Infect. 2006;8(8):2052–2060. doi: 10.1016/j.micinf.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK. Peptide vaccine: progress and challenges. Vaccines. 2014;2(3):515–536. doi: 10.3390/vaccines2030515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan CN, Randall A, Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics. 2009;25(17):2200–2207. doi: 10.1093/bioinformatics/btp386. [DOI] [PubMed] [Google Scholar]
- Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26(23):2936–2943. doi: 10.1093/bioinformatics/btq551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J Bacteriol. 1996;178(5):1274–1282. doi: 10.1128/jb.178.5.1274-1282.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood MS, Bin-T-Abid D, Irshad S, Batool H. Analysis of putative epitope candidates of Mycobacterium tuberculosis against Pakistani human leukocyte antigen background: an immunoinformatic study for the development of future vaccine. Int J Pept Res Ther. 2021;27(1):597–614. doi: 10.1007/s10989-020-10111-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier NR, Battegay M, Ottenhoff THM, Furrer H, Nemeth J, et al. HIV-infected patients developing tuberculosis disease show early changes in the immune response to novel Mycobacterium tuberculosis antigens. Front Immunol. 2021;12:620622–620622. doi: 10.3389/fimmu.2021.620622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier NR, Sutter M, Jacobsen M, Ottenhoff THM, Vogt JE, et al. Machine learning algorithms evaluate immune response to novel Mycobacterium tuberculosis antigens for diagnosis of tuberculosis. Front Cell Infect Microbiol. 2021 doi: 10.3389/fcimb.2020.594030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millington KA, Fortune SM, Low J, Garces A, Hingley-Wilson SM, et al. Rv3615c is a highly immunodominant RD1 (Region of Difference 1)-dependent secreted antigen specific for Mycobacterium tuberculosis infection. PNAS. 2011;108(14):5730. doi: 10.1073/pnas.1015153108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty S, Dal Molin M, Ganguli G, Padhi A, Jena P, et al. Mycobacterium tuberculosis EsxO (Rv2346c) promotes bacillary survival by inducing oxidative stress mediated genomic instability in macrophages. Tuberculosis (edinb) 2016;96:44–57. doi: 10.1016/j.tube.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Mushtaq K, Sheikh JA, Amir M, Khan N, Singh B, et al. Rv2031c of Mycobacterium tuberculosis: a master regulator of Rv2028-Rv2031 (HspX) operon. Front Microbiol. 2015;6:351–351. doi: 10.3389/fmicb.2015.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa A. Recombinant proteins encoded by genes present in Mycobacterium tuberculosis-specific regions of difference induce delayed-type hypersensitivity skin responses (43.31) J Immunol Res. 2012;188(1):43.31. [Google Scholar]
- Natarajan A, Beena PM, Devnikar AV, Mali S. A systemic review on tuberculosis. Indian J Tuberc. 2020;67(3):295–311. doi: 10.1016/j.ijtb.2020.02.005. [DOI] [PubMed] [Google Scholar]
- Oli AN, Obialor WO, Ifeanyichukwu MO, Odimegwu DC, Okoyeh JN, et al. Immunoinformatics and vaccine development: an overview. Immunotargets Ther. 2020;9:13–30. doi: 10.2147/ITT.S241064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkrands I, Slayden RA, Crawford J, Aagaard C, Barry CE, 3rd, et al. Hypoxic response of Mycobacterium tuberculosis studied by metabolic labeling and proteome analysis of cellular and extracellular proteins. J Bacteriol. 2002;184(13):3485–3491. doi: 10.1128/JB.184.13.3485-3491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Raghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34(Web Server Issue):W202–209. doi: 10.1093/nar/gkl343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33(Web Server Issue):W363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Vidal MM, Latorre I, Franken KLCM, Díaz J, de Souza-Galvão ML, et al. Immunogenicity of 60 novel latency-related antigens of Mycobacterium tuberculosis. Front Microbiol. 2014;5:517–517. doi: 10.3389/fmicb.2014.00517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P, Mistry J, Reche PA, Gatherer D, Flower DR. In silico design of Mycobacterium tuberculosis epitope ensemble vaccines. Mol Immunol. 2018;97:56–62. doi: 10.1016/j.molimm.2018.03.007. [DOI] [PubMed] [Google Scholar]
- Sharma R, Rajput VS, Jamal S, Grover A, Grover S. An immunoinformatics approach to design a multi-epitope vaccine against Mycobacterium tuberculosis exploiting secreted exosome proteins. Sci Rep. 2021;11(1):13836. doi: 10.1038/s41598-021-93266-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin W-H, Lee GR, Heo L, Lee H, Seok C. Prediction of protein structure and interaction by GALAXY protein modeling programs. Bio Design. 2014;2(1):1–11. [Google Scholar]
- Siddiqui KF, Amir M, Gurram RK, Khan N, Arora A, et al. Latency-associated protein Acr1 impairs dendritic cell maturation and functionality: a possible mechanism of immune evasion by Mycobacterium tuberculosis. J Infect Dis. 2014;209(9):1436–1445. doi: 10.1093/infdis/jit595. [DOI] [PubMed] [Google Scholar]
- Slingluff CL. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination? Cancer J. 2011;17(5):343–350. doi: 10.1097/PPO.0b013e318233e5b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir Ul Qamar M, Saleem S, Ashfaq UA, Bari A, Anwar F, Alqahtani S. Epitope-based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: An immune-informatics study. J Transl Med. 2019;17(1):1–4. doi: 10.1186/s12967-019-2116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajda S, Yueh C, Beglov D, Bohnuud T, Mottarella SE, et al. New additions to the ClusPro server motivated by CAPRI. Proteins. 2017;85(3):435–444. doi: 10.1002/prot.25219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay K. Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int Immunopharmacol. 2018;59:391–412. doi: 10.1016/j.intimp.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019;47(D1):D339–d343. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng G, Wang E, Wang Z, Liu H, Zhu F, et al. HawkDock: a web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019;47(W1):W322–w330. doi: 10.1093/nar/gkz397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(Web Server Issue):407–410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani Z, Rafiei A, Yazdani M, Valadan R. Design an efficient multi-epitope peptide vaccine candidate against SARS-CoV-2: an in silico analysis. Infect Drug Resist. 2020;13:3007–3022. doi: 10.2147/IDR.S264573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias M. Protein-protein docking with a reduced protein model accounting for side-chain flexibility. Protein Sci. 2003;12(6):1271–1282. doi: 10.1110/ps.0239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Dockrell HM, Ottenhoff THM, Evans TG, Zhang Y. Tuberculosis vaccines: opportunities and challenges. Respirology. 2018;23(4):359–368. doi: 10.1111/resp.13245. [DOI] [PubMed] [Google Scholar]