

Key Points
Question
Is treatment with the antiamyloid ACI-24 vaccine safe and tolerable, and does it engage disease indicators in adults with Down syndrome (DS) who are at risk of developing a genetic form of Alzheimer disease?
Findings
In this randomized clinical trial involving 16 adults with DS, the ACI-24 vaccine was found to be safe and well tolerated. The vaccine elicited immunogenicity in 4 of 12 participants and impacted Alzheimer disease biomarkers.
Meaning
This study’s findings demonstrate that interventional clinical trials can be successfully conducted among individuals with DS and that the ACI-24 vaccine is safe, well tolerated, and warrants further investigation.
Abstract
Importance
Individuals with Down syndrome (DS) are at high risk of developing Alzheimer disease due to an increased dose of the amyloid precursor protein gene, APP, which leads to increased levels of full-length APP and its products, including amyloid-β (Aβ). The liposome-based antiamyloid ACI-24 vaccine is intended to treat neurological disorders caused by misfolded Aβ pathological protein. However, the safety, tolerability, and immunogenicity of the ACI-24 vaccine among adults with DS have not been fully examined.
Objective
To assess the safety and tolerability of the ACI-24 vaccine among adults with DS as well as its ability to induce immunogenicity measured by anti-Aβ immunoglobulin G titers.
Design, Setting, and Participants
This multicenter double-blind placebo-controlled dose-escalation phase 1b randomized clinical trial was conducted at 3 US academic medical centers with affiliated Down syndrome clinics between March 30, 2016, and June 29, 2020. A total of 20 adults with DS were screened; of those, 16 adults were eligible to participate. Eligibility criteria included men or women aged 25 to 45 years with cytogenetic diagnosis of either trisomy 21 or complete unbalanced translocation of chromosome 21. Between April 27, 2016, and July 2, 2018, participants were randomized 3:1 into 2 dose-level cohorts (8 participants per cohort, with 6 participants receiving the ACI-24 vaccine and 2 receiving placebo) in a 96-week study. Participants received 48 weeks of treatment followed by an additional 48 weeks of safety follow-up.
Interventions
Participants were randomized to receive 7 subcutaneous injections of ACI-24, 300 μg or 1000 μg, or placebo.
Main Outcomes and Measures
Primary outcomes were measures of safety and tolerability as well as antibody titers.
Results
Among 16 enrolled participants, the mean (SD) age was 32.6 (4.4) years; 9 participants were women, and 7 were men. All participants were White, and 1 participant had Hispanic or Latino ethnicity. Treatment adherence was 100%. There were no cases of meningoencephalitis, death, or other serious adverse events (AEs) and no withdrawals as a result of AEs. Most treatment-emergent AEs were of mild intensity (110 of 132 events [83.3%]) and unrelated or unlikely to be related to the ACI-24 vaccine (113 of 132 events [85.6%]). No amyloid-related imaging abnormalities with edema or cerebral microhemorrhage and no evidence of central nervous system inflammation were observed on magnetic resonance imaging scans. Increases in anti-Aβ immunoglobulin G titers were observed in 4 of 12 participants (33.3%) receiving ACI-24 (2 receiving 300 μg and 2 receiving 1000 μg) compared with 0 participants receiving placebo. In addition, a greater increase was observed in plasma Aβ1-40 and Aβ1-42 levels among individuals receiving ACI-24.
Conclusions and Relevance
In this study, the ACI-24 vaccine was safe and well tolerated in adults with DS. Evidence of immunogenicity along with pharmacodynamic and target engagement were observed, and anti-Aβ antibody titers were not associated with any adverse findings. These results support progression to clinical trials using an optimized formulation of the ACI-24 vaccine among individuals with DS.
Trial Registration
ClinicalTrials.gov Identifier: NCT02738450
This phase 1b randomized clinical trial assesses the safety and tolerability of the antiamyloid ACI-24 vaccine and investigates the vaccine’s ability to induce immunogenicity among adults with Down syndrome at risk of developing a genetic form of Alzheimer disease.
Introduction
Down syndrome (DS) is most often caused by trisomy 21, which results from an extra copy of chromosome 21 that encodes no fewer than 350 genes,1 including APP (OMIM 104760); this gene encodes the amyloid precursor protein, APP, whose cleavage products include the amyloid-β (Aβ) peptide. This process results in generation of excess full-length APP and Aβ. In individuals with DS, diffuse Aβ deposits are visible in the cerebral cortex as early as age 12 years.2 Fibrillar amyloid plaques are observed in almost all individuals with DS after age 40 years.3,4,5 The lifetime risk of AD among individuals with DS is approximately 90%6; as a result, AD is the leading cause of death among adults with DS older than 35 years.7
Genetic support for the role of APP in the pathogenesis of AD among adults with DS is compelling. Individuals with DS who have trisomy 21 but are disomic for APP develop no amyloid plaques or neurofibrillary tangles and have no symptoms of dementia.8 In the general population, a mutation in the APP gene that prevents its cleavage from producing Aβ is protective against the development of amyloid plaques and neurofibrillary tangles as well as dementia.9 Amyloid-β therefore remains a prime target for therapies intended to treat AD among individuals with DS. When examined, the neuropathology of AD in persons with DS closely resembles that of AD in persons without DS.10,11,12,13,14
It is currently estimated that 200 000 persons in the US15 and 417 000 persons in Europe have DS.16 No disease-modifying or symptomatic treatment for AD among individuals with DS is currently available. Thus, there is an important and unmet medical need for more effective treatments for AD in this genetically predisposed population.
The ACI-24 vaccine is liposome based and intended to treat neurological disorders caused by misfolded Aβ pathological protein. By incorporating the first 15 amino acids of human Aβ into a stacked β-sheet conformation on the liposome, ACI-24 induces the production of polyclonal anti-Aβ antibodies specific for soluble and insoluble aggregated forms of Aβ, including oligomers17,18,19; Aβ oligomers are one of the toxic species that create pathogenesis in both persons with AD and DS.20 Inoculation of APP×PS-1 mice for AD17 or Ts65Dn mice for DS21 using a murine version of the ACI-24 vaccine resulted in a modest, albeit statistically insignificant, reduction of Aβ levels in the Ts65Dn brain21 and significant decreases in soluble and insoluble fractions of Aβ in the APP×PS-1 brain,17 with a concomitant improvement in cognitive function. The present study was designed to assess the safety, tolerability, and immunogenicity of the ACI-24 vaccine among human adults with DS.
Methods
Study Design and Participants
In this multicenter double-blind placebo-controlled dose-escalation phase 1b randomized clinical trial, participants with DS were enrolled at 3 medical centers with affiliated Down syndrome clinics in the US (Massachusetts General Hospital, Boston, Massachusetts; University of California, San Diego; and Barrow Neurological Institute, Phoenix, Arizona). The study was conducted between March 30, 2016, and June 29, 2020, and received institutional review board approval at each study site before initiation. Written informed consent was obtained from all participants and their study partners or legal representatives before any clinical trial–related activities were performed. The investigator (W.C.M., B.G.S., or A.D.B.) or a designated staff member met with the participant and the participant’s study partner or legal representative and explained the study in sufficient detail to permit an informed decision to participate. Participants provided written informed consent after demonstrating that they understood the study procedures. In cases in which participants could not describe the study procedures, the study partner or legal guardian provided written consent. The study adhered to all applicable local regulations, was conducted in accordance with the Guideline for Good Clinical Practice,22 and complied with ethical standards described in the Declaration of Helsinki.23 This study followed the Consolidated Standards of Reporting Trials (CONSORT) reporting guideline for randomized clinical trials.
Eligible participants were men or women with a cytogenetic diagnosis of either trisomy 21 or complete unbalanced translocation of chromosome 21 who were aged 25 to 45 years. This age range was selected to minimize the presence of substantial cognitive impairment due to dementia, which would have impaired participants’ ability to complete study procedures. All participants were required to have a study partner or legal representative with whom they had direct contact for a minimum of 10 hours per week and who could be asked questions about the participant. Exclusion criteria included weight less than 40 kg, IQ composite score lower than 40 (as measured by the Kaufman Brief Intelligence Test, Second Edition24), any current psychiatric or neurological illness other than DS, drug or alcohol misuse within the past 5 years, or the presence of a medical condition that could impede evaluation of the study drug. Additional inclusion and exclusion criteria are listed in the trial protocol (Supplement 1). This small study involving a unique population did not enroll a large number of members of underrepresented racial and ethnic minority groups, perhaps as a result of an unawareness of the opportunity to participate in clinical trials among these groups.
Participants were randomized between April 27, 2016, and July 2, 2018. The study consisted of 2 cohorts of 8 participants each. In cohort 1, 6 participants were randomized to receive ACI-24, 300 μg, and 2 participants were randomized to receive placebo; in cohort 2, 6 participants were randomized to receive ACI-24, 1000 μg, and 2 participants were randomized to receive placebo. A total of 7 subcutaneous injections of either ACI-24 or placebo were administered at weeks 0, 4, 8, 12, 24, 36, and 48 followed by an additional 48-week treatment-free safety follow-up period, with the last study visit conducted at week 96. The cohorts were studied sequentially in ascending dose order. Cohort 2 initiated treatment when safety and tolerability data through week 14 of the treatment period of the last participant in cohort 1 had been reviewed by the data safety monitoring board.
Randomization and Blinding
Investigators used a secure web-based system to randomize eligible participants to study groups. A central dynamic randomization model enabling a balance of treatment allocation across study groups (3:1 active drug to placebo ratio) was used at the overall study level due to the limited study population. This system was programmed to ensure that the randomization of the first 4 participants (3 to the active drug group and 1 to the placebo group) to each dose level was separated by an interval of 7 or more days to enable a sufficient safety monitoring period between each participant. The ACI-24 vials were labeled by the manufacturer (Polymun Scientific Immunbiologische Forschung). The study sponsor (AC Immune), investigators, and participants were unaware of assigned treatment groups during the active treatment and follow-up periods. Blinded clinical assessments were conducted by the study investigators (W.C.M., B.G.S., and A.D.B.). Reviews of blinded and unblinded safety data were performed on a quarterly basis by the study sponsor and the data safety monitoring board, respectively. Blinded analysis of magnetic resonance imaging (MRI) scans was conducted by an external clinical research company (BioClinica).
Procedures
The study drug or placebo was administered at weeks 0, 4, 8, 12, 24, 36, and 48. The dosing time of day and relation of dosing to meals were not controlled. Because the appearance of the liquid in the vial differed between placebo and ACI-24, a qualified unblinded person at the study site obtained the study product from the pharmacy and administered the product. The investigator assigned the responsibility of an unblinded dispenser or administrator to a person or persons who did not participate in any subjective evaluation of any participant. Assessment of adverse events (AEs), causality and cognitive and behavioral test results were subjective evaluations of study participants and were therefore not permitted to be conducted by the unblinded dispenser or administrator. Cognitive and behavioral tests included the Cambridge Neuropsychological Test Automated Battery (which measures brain function across 3 cognitive domains: visual memory, attention, and functional components of executive function),25 the Brief Praxis Test (which screens for early features of dementia in adults with intellectual disability),26 the Vineland II Adaptive Behavior Scales (which measure adaptive behaviors, including ability to cope with environmental changes, learn new skills, and demonstrate independence),27 the Neuropsychiatric Inventory (which measures neuropsychiatric symptoms),28 the Clinician Global Impression of Change (which measures symptom severity, treatment response, and treatment efficacy),29 a global assessment of tolerability (performed by W.C.M., B.G.S., and A.D.B.), and the Columbia–Suicide Severity Rating Scale (which measures suicidal ideation and behavior).30
The investigator, study coordinator, and any study participants other than the unblinded dispenser or administrator were not permitted to know the study product assigned to any participant. Site monitoring of the clinical trial was undertaken by a blinded monitor for all study data (with the exception of study drug accountability data), which were reviewed by an unblinded monitor along with the unblinded study site personnel.
The treatment was administered by subcutaneous injection. For the cohort receiving a low dose (ACI-24, 300 μg, or placebo), the preferred injection site was the arm, alternating between the left and right arm for each administration. For the cohort receiving a high dose (ACI-24, 1000 μg, or placebo), the preferred injection site was the leg, with the study drug administered as 2 concomitant subcutaneous injections because of the higher drug volume. The injection sites were alternated from right leg to left leg between visits. Free anti–Aβ1-42 immunoglobulin G (IgG) titers were determined using an assay (Meso Scale Discovery Free Assay) developed in house, then transferred to an external contract research organization (BioAgilytix Labs) for validation of good clinical laboratory practice.
Outcomes
The primary objective of this study was to assess the safety, tolerability and immunogenicity of the ACI-24 vaccine. Primary end points included AEs, global assessment of tolerability, physical and neurological examination results, vital signs, suicidal ideation or behavior, MRI and electrocardiographic findings, hematological and biochemical findings from blood and urine, inflammatory markers in blood and cerebrospinal fluid (lumbar puncture was optional), and serum anti-Aβ antibody titers.
The secondary objectives were to assess the study drug for clinical efficacy and effects on cognition, behavior, brain structure volumes, and blood and/or cerebrospinal fluid biomarkers. The secondary clinical assessment end points included change in cognition from baseline, as measured by the Clinician Global Impression of Change and cognitive assessments; change in motor control, reaction time, and paired associative learning from baseline, as measured by the Cambridge Neuropsychological Test Automated Battery; performance on the Brief Praxis Test; and change in behavior from baseline, as measured by the Vineland II Adaptive Behavior Scales and the Neuropsychiatric Inventory. The secondary biological end points were whole brain, ventricle, and hippocampal volumes assessed by MRI; T-cell activation; inflammatory cytokines; and AD-related biomarkers, including levels of Aβ, total tau protein, phosphorylated tau protein, neurofilament light, neurogranin, and other exploratory biomarkers, such as angiogenic proteins and vascular injury markers.
Abnormal laboratory or imaging findings and other abnormal clinical assessments that the study investigator (W.C.M., B.G.S., or A.D.B.) determined to be clinically meaningful (including severity and causal relationship to the study drug) were recorded if they met the definition of an AE or serious AE. The MRI scans were conducted at the screening visit (weeks 0-4) and at weeks 14, 26, 50, and 96; MRI scans were performed within a 14-day period after the screening visit date (start of visit plus 14 days) and within a 7-day period before or after the subsequent visit date (start of visit plus or minus 7 days).
Statistical Analysis
Analyses were 2-tailed and performed as specified in the statistical analysis plan (Supplement 1), which was finalized before unblinding of treatment assignments. Evaluations of the statistical analysis plan were performed by an external clinical research organization (Synteract). The sample size was established as 8 participants (6 receiving ACI-24 and 2 receiving placebo) per cohort. It was expected that the sample size would be sufficient to achieve the main goals of detecting common AEs and providing information about the immunogenicity of ACI-24 in this population. No statistical tests were performed because the sample size did not allow for meaningful statistical inference.
All analyses and tabulations were performed using SAS software, version 9.4 or higher (SAS Institute Inc). Four data sets were used for analysis: (1) all participants, which comprised all individuals who received initial screening; (2) safety analysis population, which comprised all randomized participants who received at least 1 dose of the study drug or placebo and at least 1 postdosing safety assessment; (3) modified intention-to-treat population, which comprised all randomized participants who received at least 1 dose of the study drug or placebo and at least 1 biological or efficacy assessment at any time during the study after the first dose administration; and (4) per protocol population, which comprised all participants included in the modified intention-to-treat population who completed the treatment period and received all injections per protocol and for whom no major protocol deviation was observed. Additional details about the statistical analysis plan are available in Supplement 1.
Results
Twenty participants were screened for eligibility; of those, 4 participants were not eligible for enrollment, and 1 participant was rescreened and deemed eligible to participate (Figure 1). Among 16 enrolled participants, the mean (SD) age was 32.6 (4.4) years; 9 participants (56.3%) were women, and 7 (43.7%) were men. All participants were White, and 1 participant in the ACI-24, 1000 μg, group had Hispanic or Latino ethnicity. The educational level was identical across all study groups (mean [SD], 12.0 [0.0] years).
Figure 1. Study Flowchart.

aOne participant was rescreened.
bTwo participants discontinued participation because they were unwilling to attend the final study visit (week 96) due to the risk of COVID-19.
A total of 6 participants were randomized to receive ACI-24, 300 μg, 6 participants were randomized to receive ACI-24, 1000 μg, and 4 participants were randomized to receive placebo. Treatment adherence was 100%; all participants completed the treatment period (through week 48) and received all study product administrations. All participants in cohort 1 who were randomized to receive ACI-24, 300 μg, or placebo completed the study through week 96 (end of safety follow-up period); however, only 4 of 6 participants (66.7%) in cohort 2 who were randomized to receive ACI-24, 1000 μg, completed week 96. These 2 participants (33.3%) discontinued the study before the end of the safety follow-up period because they were unwilling to attend the final site visit as a result of the risk of contracting SARS-CoV-2. No major protocol deviations occurred during the study; consequently, all participants from the modified intention-to-treat population were included in the per protocol population, which had the same composition as the population included in the safety analysis. Most results reported in this article refer to the modified intention-to-treat population (with the exception of data shown in Table 1, which includes baseline demographic information about participants who were deemed ineligible to participate based on initial screening results).
Table 1. Baseline Demographic Characteristics.
| Characteristic | No. (%) | ||||
|---|---|---|---|---|---|
| Total | Ineligible for study participationa | Treatment cohort 1 (ACI-24, 300 μg) | Treatment cohort 2 (ACI-24, 1000 μg) | Placebo cohorts 1 and 2 | |
| Total participants, No. | 20 | 4 | 6 | 6 | 4 |
| Age at signing of informed consent, y | |||||
| Mean (SD) | 32.9 (4.1) | 34.0 (2.6) | 33.5 (4.6) | 31.5 (4.9) | 33.0 (4.2) |
| Median (range) | 33.5 (25.0-41.0) | 34.0 (31.0-37.0) | 32.5 (28.0-41.0) | 33.0 (25.0-36.0) | 33.0 (28.0-38.0) |
| Sex | |||||
| Female | 11 (55.0) | 2 (50.0) | 2 (33.3) | 4 (66.7) | 3 (75.0) |
| Male | 9 (45.0) | 2 (50.0) | 4 (66.7) | 2 (33.3) | 1 (25.0) |
| Race | |||||
| Racial minority groupsb | 0 | 0 | 0 | 0 | 0 |
| White | 20 (100) | 4 (100) | 6 (100) | 6 (100) | 4 (100) |
| Ethnicity | |||||
| Hispanic or Latino | 1 (5.0) | 0 | 0 | 1 (16.7) | 0 |
| Non-Hispanic or non-Latino | 19 (95.0) | 4 (100) | 6 (100) | 5 (83.3) | 4 (100) |
| APOE genotype | |||||
| ε3/ε3 | 8 (40.0) | 0 | 3 (50.0) | 3 (50.0) | 2 (50.0) |
| ε2/ε3 | 2 (10.0) | 0 | 1 (16.7) | 1 (16.7) | 0 |
| ε3/ε4 | 2 (10.0) | 0 | 1 (16.7) | 0 | 1 (25.0) |
| Not reported | 8 (40.0) | 4 (100) | 1 (16.7) | 2 (33.3) | 1 (25.0) |
| BMI at screening | |||||
| Mean (SD) | 40.2 (10.1) | 42.8 (11.6) | 37.0 (9.9) | 39.9 (9.4) | 43.1 (12.7) |
| Median (range) | 39.4 (24.3-58.3) | 41.2 (30.5-58.3) | 37.3 (24.3-52.0) | 35.1 (31.9-53.4) | 47.4 (25.1-52.4) |
| KBIT-2 IQ composite | |||||
| Mean (SD) | 47.8 (10.4) | 52.8 (18.9) | 44.2 (5.5) | 49.2 (9.5) | 46.0 (8.0) |
| Median (range) | 42.0 (40.0-80.0) | 45.5 (40.0-80.0) | 42.0 (40.0-54.0) | 48.5 (40.0-60.0) | 43.5 (40.0-57.0) |
Abbreviations: APOE, apolipoprotein E; BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); KBIT-2, Kaufman Brief Intelligence Test, Second Edition.
Ineligibility determined by initial screening results.
Racial minority groups include American Indian or Alaska Native, Asian, and Black or African American.
Baseline demographic characteristics were similar across cohorts (Table 1). There were more male participants in the group receiving ACI-24, 300 μg (4 individuals [66.7%]), vs the group receiving ACI-24, 1000 μg (2 individuals [33.3%]), and the groups receiving placebo (1 individual [25.0%]). Body mass index (calculated as weight in kilograms divided by height in meters squared) was slightly lower among those receiving ACI-24, 300 μg (mean [SD], 37.0 [9.9]), vs ACI-24, 1000 μg (mean [SD], 39.9 [9.4]), and placebo (mean [SD], 43.1 [12.7]). The IQ composite score on the Kaufman Brief Intelligence Test was slightly lower among participants receiving ACI-24, 300 μg (mean [SD], 44.2 [5.5] points), vs ACI-24, 1000 μg (mean [SD], 49.2 [9.5] points), and placebo (mean [SD], 46.0 [8.0] points).
All randomized participants presented with trisomy 21. A medical history of obesity, which is a common comorbidity among individuals with DS, was recorded in one-half of participants in each study group (ie, 3 participants receiving ACI-24, 300 μg, 3 participants receiving ACI-24, 1000 μg, and 2 participants receiving placebo). However, 13 of 16 participants (81.3%) had a body mass index greater than 30, thereby meeting the medical criterion for obesity.
A total of 14 of 20 screened participants (70.0%) reported having sleep apnea (5 of 6 participants [83.3%] in each ACI-24 group, 1 of 4 participants [25.0%] in the pooled placebo group, and 3 of 4 participants [75.0%] who were ineligible to participate based on screening results). Hypothyroidism was also reported in 4 participants (66.7%) receiving ACI-24, 1000 μg, compared with 2 participants (50.0%) who were ineligible for study participation and 0 participants in the other study groups.
Safety and Tolerability Outcomes
No deaths, serious AEs, or treatment-emergent AEs (TEAEs) leading to study withdrawal were reported during the study. Overall, 15 of 16 participants (93.8%) reported TEAEs; the only participant who did not report a TEAE was in the ACI-24, 300 μg, group (Table 2). Most TEAEs were of mild intensity (110 of 132 events [83.3%]) and either unrelated or unlikely to be related to the study drug (113 of 132 events [85.6%]). Only 2 serious AEs (obstructive sleep apnea and tonsillar hypertrophy) were reported in 1 participant receiving placebo. Eight mild and self-limiting injection site reactions were reported in 3 participants (50.0%) receiving ACI-24, 1000 μg, with a mean (SD) time to onset from the last study drug administration of 32.8 (4.8) hours and a mean (SD) time to resolution of 236.9 (32.7) hours. There were no injection site reactions reported in either the ACI-24, 300 μg, group or the placebo groups.
Table 2. Treatment-Emergent Adverse Events Occurring in 50% or More of Participants in Any Study Group (Safety Analysis Population)a.
| System organ class | Total | Treatment | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cohort 1 (ACI-24, 300 μg) | Cohort 2 (ACI-24, 1000 μg) | Cohort 1 | Cohort 2 | |||||||
| Participants, No. (%) | Events, No. | Participants, No. (%) | Events, No. | Participants, No. (%) | Events, No. | Participants, No. (%) | Events, No. | Participants, No. (%) | Events, No. | |
| Preferred term, No. | 16 | NA | 6 | NA | 6 | NA | 2 | NA | 2 | NA |
| Any AE | 15 (93.8) | 132 | 5 (83.3) | 56 | 6 (100) | 56 | 2 (100) | 9 | 2 (100) | 11 |
| Infections and infestations | 9 (56.3) | 33 | 5 (83.3) | 18 | 3 (50.0) | 14 | 1 (50.0) | 1 | 0 | 0 |
| Cellulitis | 3 (18.8) | 3 | 3 (50.0) | 3 | 0 | 0 | 0 | 0 | 0 | 0 |
| Bronchitis | 2 (12.5) | 3 | 1 (16.7) | 2 | 0 | 0 | 1 (50.0) | 1 | 0 | 0 |
| Nervous system disorders | 7 (43.8) | 14 | 3 (50.0) | 5 | 3 (50.0) | 6 | 0 | 0 | 1 (50.0) | 3 |
| Headache | 4 (25.0) | 6 | 1 (16.7) | 1 | 2 (33.3) | 4 | 0 | 0 | 1 (50.0) | 1 |
| Dizziness | 2 (12.5) | 3 | 0 | 0 | 1 (16.7) | 1 | 0 | 0 | 1 (50.0) | 2 |
| Respiratory, thoracic, and mediastinal disorders | 7 (43.8) | 9 | 3 (50.0) | 4 | 3 (50.0) | 3 | 0 | 0 | 1 (50.0) | 2 |
| Sleep apnea syndrome | 2 (12.5) | 2 | 0 | 0 | 1 (16.7) | 1 | 0 | 0 | 1 (50.0) | 1 |
| Tonsillar hypertrophy | 2 (12.5) | 2 | 1 (16.7) | 1 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 |
| Gastrointestinal disorders | 6 (37.5) | 11 | 3 (50.0) | 5 | 1 (16.7) | 3 | 0 | 0 | 2 (100) | 3 |
| Diarrhea | 2 (12.5) | 3 | 1 (16.7) | 1 | 1 (16.7) | 2 | 0 | 0 | 0 | 0 |
| Vomiting | 2 (12.5) | 2 | 1 (16.7) | 1 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 |
| Abdominal hernia | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 |
| Upper abdominal pain | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 |
| General disorders and administration site conditions | 6 (37.5) | 13 | 2 (33.3) | 2 | 2 (33.3) | 9 | 1 (50.0) | 1 | 1 (50.0) | 1 |
| Fatigue | 3 (18.8) | 3 | 2 (33.3) | 2 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 |
| Peripheral swelling | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 | 0 | 0 |
| Investigations | 6 (37.5) | 8 | 2 (33.3) | 2 | 2 (33.3) | 3 | 2 (100) | 3 | 0 | 0 |
| Weight increased | 2 (12.5) | 2 | 1 (16.7) | 1 | 0 | 0 | 1 (50.0) | 1 | 0 | 0 |
| C-reactive protein increased | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 | 0 | 0 |
| Menstruation normal | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 | 0 | 0 |
| Skin and subcutaneous tissue disorders | 5 (31.3) | 13 | 1 (16.7) | 5 | 3 (50.0) | 7 | 1 (50.0) | 1 | 0 | 0 |
| Rash | 4 (25.0) | 4 | 1 (16.7) | 1 | 2 (33.3) | 2 | 1 (50.0) | 1 | 0 | 0 |
| Eye disorders | 4 (25.0) | 6 | 2 (33.3) | 4 | 1 (16.7) | 1 | 1 (50.0) | 1 | 0 | 0 |
| Lacrimation increased | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 | 0 | 0 |
| Injury, poisoning, and procedural complications | 4 (25.0) | 5 | 2 (33.3) | 2 | 1 (16.7) | 2 | 0 | 0 | 1 (50.0) | 1 |
| Lip injury | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 |
| Psychiatric disorders | 3 (18.8) | 3 | 1 (16.7) | 1 | 1 (16.7) | 1 | 1 (50.0) | 1 | 0 | 0 |
| Enuresis | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 | 0 | 0 |
| Endocrine disorders | 2 (12.5) | 2 | 1 (16.7) | 1 | 0 | 0 | 1 (50.0) | 1 | 0 | 0 |
| Hypothyroidism | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 | 0 | 0 |
| Blood and lymphatic system disorders | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 |
| Anemia | 1 (6.3) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 |
Abbreviations: AE, adverse event; NA, not applicable.
A participant with more than 1 event in a specific category was only counted once. Table is sorted by descending participant count on the preferred term level. Preferred term is standard terminology for AE coding used in the Medical Dictionary for Regulatory Activities (MedDRA) software package developed by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use.
Cellulitis considered unrelated to the study drug was reported in 3 participants (50.0%) receiving ACI-24, 300 μg, and 0 participants receiving ACI-24, 1000 μg, or placebo. Rash, in all cases considered unrelated to the study drug, was reported in 2 participants (33.3%) receiving ACI-24, 1000 μg, 1 participant (16.7%) receiving ACI-24, 300 μg, and 1 participant (50.0%) in cohort 1 receiving placebo. Headache was reported by 2 participants (33.3%) in the ACI-24, 1000 μg, group and 1 participant (50.0%) in the cohort 2 placebo group.
Angiogenic proteins and vascular injury markers (ie, vascular endothelial growth factor A, C, and D; tyrosine kinase 2; C-reactive protein; vascular cell adhesion molecule 1; intracellular adhesion molecule 1, and bicinchoninic acid) were evaluated in plasma and showed no difference across treatment groups. Inflammatory cytokines (ie, interferon γ, interleukins 10 and 12, and tumor necrosis factor) evaluated in serum also showed no difference across treatment groups and no increase over time (eFigure 1 in Supplement 2). No evidence of T-cell activation was observed during the study.
No laboratory results were deemed clinically meaningful throughout the study. More weight gain was observed in participants randomized to the placebo groups vs the ACI-24 groups (gain of 5 kg vs loss of 1 kg) (eFigure 2 in Supplement 2). No corrected QT intervals (measured by electrocardiography; calculated as the time from the start of the Q wave to the end of the T wave) greater than 480 milliseconds were noted at any time in any group. No clinically meaningful abnormalities and no episodes of amyloid-related imaging abnormalities due to vasogenic edema and sulcal effusions or hemosiderin deposition or central nervous system inflammation were noted in MRI results at any time during the study. No consistent changes in brain volume were noted in participants receiving either dose of ACI-24 compared with those receiving placebo. There were no changes in results on the Columbia–Suicide Severity Rating Scale (eTable in Supplement 2).
Immunogenicity
Anti–Aβ1-42 IgG levels were measured at screening, before any injections were administered, at weeks 2 and 4 after injections were administered, and at weeks 60, 72, and 96 during the follow-up period. The mean absolute free anti–Aβ1-42 IgG titers per treatment arm are shown in Figure 2. Increased titers were observed in 4 of 12 participants (33.3%) receiving ACI-24 (2 receiving 300 μg and 2 receiving 1000 μg) compared with 0 participants receiving placebo. Individual IgG titers are shown in eFigure 3 in Supplement 2. The partial response in immunogenicity may have contributed to the good safety profile.
Figure 2. Anti–Amyloid-β (Anti-Aβ) 1-42 Immunoglobulin G Titer Values in Serum of Modified Intention-to-Treat Population by Study Visit.
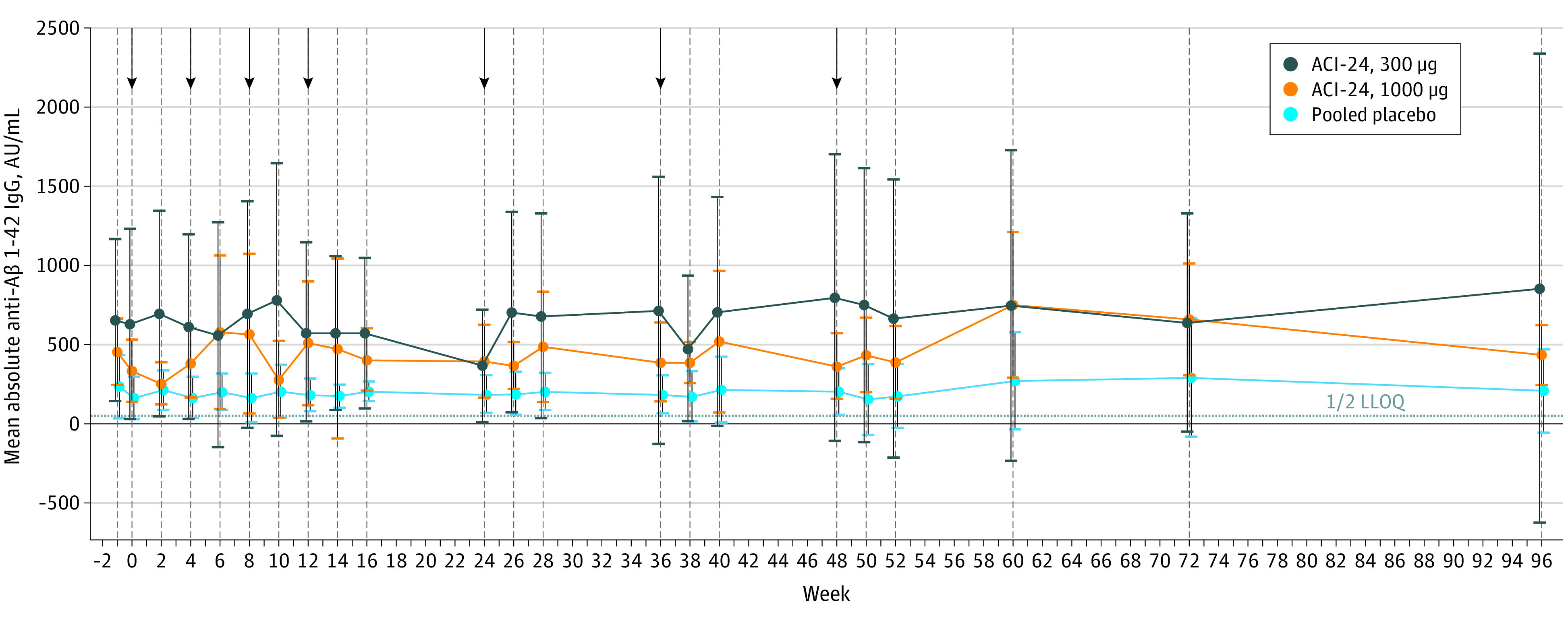
Values were obtained using the Meso Scale Discovery Free Assay. Whiskers represent 95% CIs. Arrows indicate time points of study drug administration. IgG indicates immunoglobulin G; LLOQ, lower limit of quantification.
Amyloid-Related Biomarkers
Plasma Amyloid-β 1-40 and Amyloid-β 1-42
Similar to immunogenicity, variability in plasma Aβ levels were observed at baseline. A greater increase in plasma Aβ1-40 and Aβ1-42 levels was found in participants who received either dose of ACI-24 compared with those who received placebo, with higher levels toward the end of the safety observation period (mean [SD] changes in plasma Aβ1-40 level: increase of 11.0 [41.5] ng/L in the placebo group; increase of 108.2 [49.2] ng/L in the ACI-24, 300 μg, group; and increase of 112.3 [26.9] ng/L in the ACI-24, 1000 μg, group; mean [SD] changes in plasma Aβ1-42 level: decrease of 6.5 [9.8] ng/L in the placebo groups; increase of 11.7 [5.0] ng/L in the ACI-24, 300 μg, group; and increase of 13.8 [7.4] ng/L in the ACI-24, 1000 μg, group) (Figure 3).
Figure 3. Amyloid-β (Aβ) 1-40 and Aβ 1-42 Levels in Plasma of Modified Intention-to-Treat Population by Study Visit.
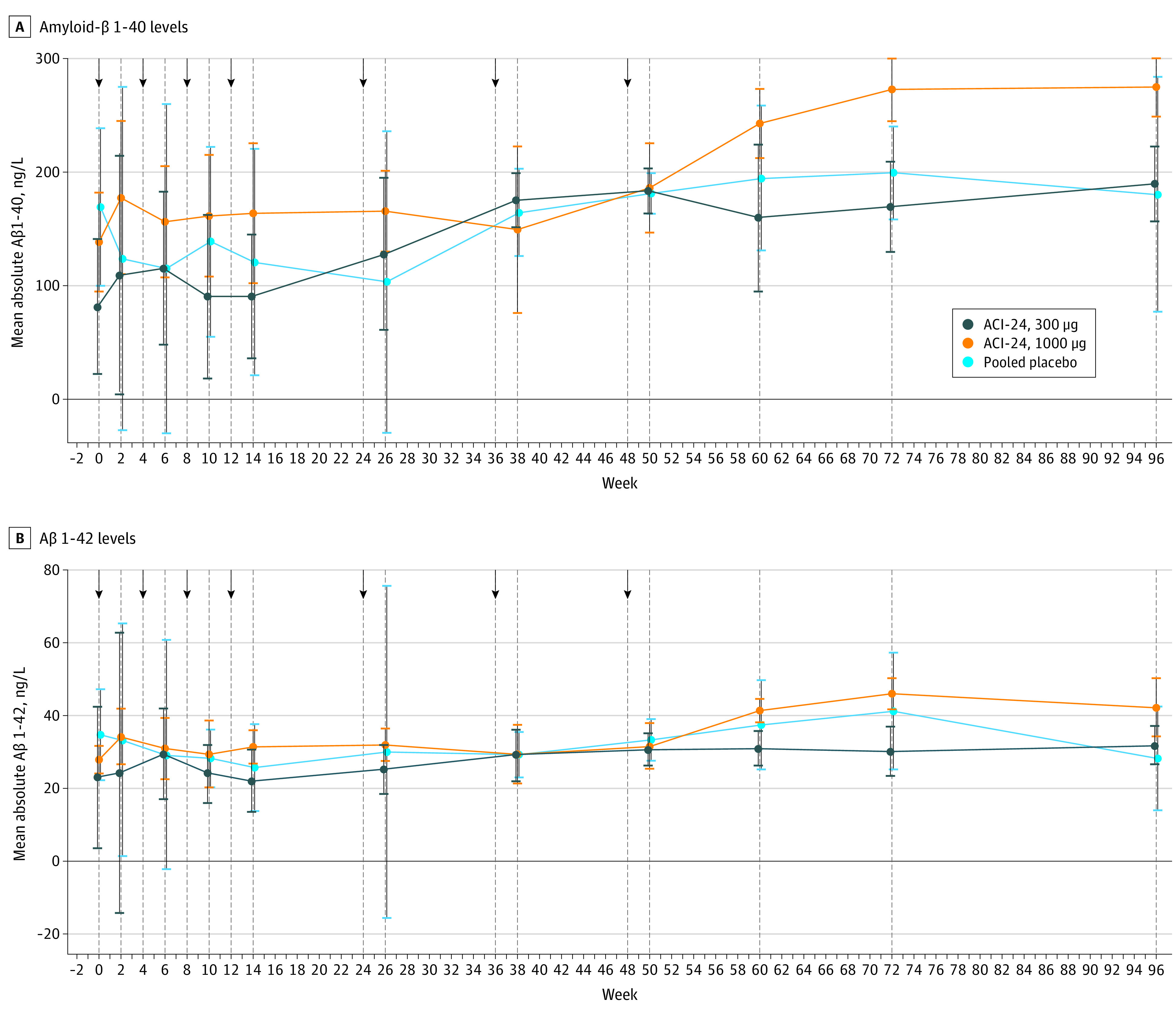
Whiskers represent 95% CIs. Arrows indicate time points of study drug administration.
Exploratory Biomarkers
Exploratory analyses included measurement of serum neurofilament light levels in all participants and cerebrospinal fluid tau, phosphorylated tau threonine 181, and neurogranin levels in the only 3 participants (1 in the ACI-24, 300 μg, group and 2 in the ACI-24, 1000 μg, group) who underwent cerebrospinal fluid sampling. In addition, whole brain volumetric assessments were performed. No consistent changes in these biomarkers were observed in this small sample (eFigure 4 in Supplement 2).
Clinical and Cognitive Outcomes
Similar to the other exploratory biomarkers, no consistent changes from baseline were noted in scores on the Brief Praxis Test; scores on the motor control, reaction time, or paired associative learning scales on the Cambridge Neuropsychological Test Automated Battery; or scores on the Vineland II Adaptive Behavior Scales (eFigure 5 in Supplement 2). No differences in clinical or cognitive outcomes were observed; however, the study was underpowered to detect any changes.
Discussion
In this randomized clinical trial, the ACI-24 vaccine was found to be safe and well tolerated at the 2 subcutaneous doses (300 μg and 1000 μg) assessed. No deaths, serious AEs, or TEAEs leading to study withdrawal were reported during the study. There were no episodes of central nervous system inflammation or amyloid-related imaging abnormalities due to vasogenic edema and sulcal effusions or hemosiderin deposition. Injection site reactions, which were mild and self-limiting in nature, were reported in 3 of 6 participants (50.0%) in the ACI-24, 1000 μg, group, and no injection site reactions were observed in the ACI-24, 300 μg, group or the placebo groups.
Four of 12 individuals who received ACI-24 (2 in each dose cohort) showed anti-Aβ IgG response compared with 0 individuals in the placebo group. The interpretation of this finding is complicated by the existing baseline levels of anti–Aβ1-42 IgG in this group, which is thought to reflect a response to the Aβ protein in individuals with DS.31 Notably, there was evidence for peripheral target engagement with time in the actively treated groups, as assessed by an increase in plasma Aβ1-40 and Aβ1-42 levels (Figure 3).
The lack of performance decline on the cognitive scales among participants randomized to the placebo groups could be explained by the relatively young age of the participants (ie, younger than the onset of the clinical stage of AD, which is expected only after age 40 years32). This finding confirms the importance of including older participants in future studies examining effects on cognitive decline. In addition, the duration of this phase 1b study (ie, 96 weeks) may not have been sufficient to detect clinical or cognitive changes among individuals in this age range. The scales proved feasible for use among the study participants, despite their intellectual impairment. It should be noted that an original plan to include the Repeatable Battery for the Assessment of Neuropsychological Status33 was abandoned early in the study when it became clear that many participants could not complete the assessment because of the amount of time required.
Evidence of immunogenicity was observed in 4 of 12 participants who received treatment with this T-cell independent vaccine, as were pharmacodynamic and target engagement effects, which were reflected by a greater increase in plasma Aβ1-40 and Aβ1-42 levels in treatment groups compared with placebo groups. Notably, the anti-Aβ antibody titers observed were not associated with any adverse findings, which supports the performance of future clinical trials to assess the effect of an optimized vaccine formulation among individuals with DS.
Strengths and Limitations
This study has strengths. To our knowledge, the study is the first randomized clinical trial of an antiamyloid vaccine among adults with DS. The study also demonstrated that it is possible to successfully conduct multicenter clinical trials of antiamyloid immunotherapies among adults with DS. Furthermore, evidence of pharmacodynamic and peripheral target engagement suggests the generation of a polyclonal antibody response to the vaccine target.
The study also has limitations. First, currently available assays are limited in their ability to detect this polyclonal response. Second, amyloid positron emission tomographic imaging was not incorporated into the design of this small study given the expected lack of power to discern difference in treatment arms. However, it is notable that, despite clear signs of vaccine-induced immune response, none of the participants showed any adverse safety findings.
Conclusions
In this randomized clinical trial of adults with DS, the T-cell independent antiamyloid ACI-24 vaccine was safe, well tolerated, and able to induce an anti-Aβ IgG response in 4 of 12 adults with DS at the 2 doses of study drug tested. These findings support the further development of an optimized formulation of ACI-24 for the treatment of adults with DS, with the goal of improving immunogenicity and consistency of response as a potential avenue for the prevention of AD in individuals with DS.
Trial Protocol and Statistical Analysis Plan
eTable. Columbia–Suicide Severity Rating Scale (C-SSRS)
eFigure 1. Angiogenic Markers
eFigure 2. Body Mass Index
eFigure 3. Anti–Amyloid-β 1-42 Immunoglobulin G Titer in Serum Using the Meso Scale Discovery Free Assay: Change From Baseline in Modified Intention-to-Treat Population per Individual Participant
eFigure 4. Alzheimer Disease Biomarkers
eFigure 5. Cognitive Measures
Data Sharing Statement
References
- 1.Antonarakis SE, Skotko BG, Rafii MS, et al. Down syndrome. Nat Rev Dis Primers. 2020;6(1):9. doi: 10.1038/s41572-019-0143-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis. 1996;3(1):16-32. doi: 10.1006/nbdi.1996.0003 [DOI] [PubMed] [Google Scholar]
- 3.Mann DM, Yates PO, Marcyniuk B. Alzheimer’s presenile dementia, senile dementia of Alzheimer type and Down’s syndrome in middle age form an age related continuum of pathological changes. Neuropathol Appl Neurobiol. 1984;10(3):185-207. doi: 10.1111/j.1365-2990.1984.tb00351.x [DOI] [PubMed] [Google Scholar]
- 4.Mann DM, Yates PO, Marcyniuk B, Ravindra CR. The topography of plaques and tangles in Down’s syndrome patients of different ages. Neuropathol Appl Neurobiol. 1986;12(5):447-457. doi: 10.1111/j.1365-2990.1986.tb00053.x [DOI] [PubMed] [Google Scholar]
- 5.Annus T, Wilson LR, Hong YT, et al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimers Dement. 2016;12(5):538-545. doi: 10.1016/j.jalz.2015.07.490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCarron M, McCallion P, Reilly E, Dunne P, Carroll R, Mulryan N. A prospective 20-year longitudinal follow-up of dementia in persons with Down syndrome. J Intellect Disabil Res. 2017;61(9):843-852. doi: 10.1111/jir.12390 [DOI] [PubMed] [Google Scholar]
- 7.Hithersay R, Startin CM, Hamburg S, et al. Association of dementia with mortality among adults with Down syndrome older than 35 years. JAMA Neurol. 2019;76(2):152-160. doi: 10.1001/jamaneurol.2018.3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doran E, Keator D, Head E, et al. Down syndrome, partial trisomy 21, and absence of Alzheimer’s disease: the role of APP. J Alzheimers Dis. 2017;56(2):459-470. doi: 10.3233/JAD-160836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488(7409):96-99. doi: 10.1038/nature11283 [DOI] [PubMed] [Google Scholar]
- 10.Lott IT, Head E. Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat Rev Neurol. 2019;15(3):135-147. doi: 10.1038/s41582-018-0132-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rafii MS, Wishnek H, Brewer JB, et al. The Down Syndrome Biomarker Initiative (DSBI) pilot: proof of concept for deep phenotyping of Alzheimer’s disease biomarkers in Down syndrome. Front Behav Neurosci. 2015;9:239. doi: 10.3389/fnbeh.2015.00239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rafii MS, Lukic AS, Andrews RD, et al. ; Down Syndrome Biomarker Initiative and the Alzheimer’s Disease Neuroimaging Initiative . PET imaging of tau pathology and relationship to amyloid, longitudinal MRI, and cognitive change in Down syndrome: results from the Down Syndrome Biomarker Initiative (DSBI). J Alzheimers Dis. 2017;60(2):439-450. doi: 10.3233/JAD-170390 [DOI] [PubMed] [Google Scholar]
- 13.Fortea J, Vilaplana E, Carmona-Iragui M, et al. Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: a cross-sectional study. Lancet. 2020;395(10242):1988-1997. doi: 10.1016/S0140-6736(20)30689-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lott IT, Head E. Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol Aging. 2005;26(3):383-389. doi: 10.1016/j.neurobiolaging.2004.08.005 [DOI] [PubMed] [Google Scholar]
- 15.de Graaf G, Buckley F, Skotko BG. Estimation of the number of people with Down syndrome in the United States. Genet Med. 2017;19(4):439-447. doi: 10.1038/gim.2016.127 [DOI] [PubMed] [Google Scholar]
- 16.de Graaf G, Buckley F, Skotko BG. Estimation of the number of people with Down syndrome in Europe. Eur J Hum Genet. 2021;29(3):402-410. doi: 10.1038/s41431-020-00748-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muhs A, Hickman DT, Pihlgren M, et al. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc Natl Acad Sci U S A. 2007;104(23):9810-9815. doi: 10.1073/pnas.0703137104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hickman DT, López-Deber MP, Ndao DM, et al. Sequence-independent control of peptide conformation in liposomal vaccines for targeting protein misfolding diseases. J Biol Chem. 2011;286(16):13966-13976. doi: 10.1074/jbc.M110.186338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pihlgren M, Silva AB, Madani R, et al. TLR4- and TRIF-dependent stimulation of B lymphocytes by peptide liposomes enables T cell–independent isotype switch in mice. Blood. 2013;121(1):85-94. doi: 10.1182/blood-2012-02-413831 [DOI] [PubMed] [Google Scholar]
- 20.Teller JK, Russo C, DeBusk LM, et al. Presence of soluble amyloid beta-peptide precedes amyloid plaque formation in Down’s syndrome. Nat Med. 1996;2(1):93-95. doi: 10.1038/nm0196-93 [DOI] [PubMed] [Google Scholar]
- 21.Belichenko PV, Madani R, Rey-Bellet L, et al. An anti–β-amyloid vaccine for treating cognitive deficits in a mouse model of Down syndrome. PLoS One. 2016;11(3):e0152471. doi: 10.1371/journal.pone.0152471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Guideline for Good Clinical Practice E6 (R1). June 10, 1996; World Health Organization. https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf [Google Scholar]
- 23.World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191-2194. doi: 10.1001/jama.2013.281053 [DOI] [PubMed] [Google Scholar]
- 24.Kaufman AS, Kaufman NL. Kaufman Brief Intelligence Test, Second Edition. Pearson; 2004. [Google Scholar]
- 25.Wild KV, Musser ED. The Cambridge Neuropsychological Test Automated Battery in the assessment of executive functioning. In: Goldstein S, Naglieri JA, eds. Handbook of Executive Functioning. Springer; 2014:171-190. [Google Scholar]
- 26.Dalton AJ. The Dyspraxia Scale for adults with Down syndrome. In: Prasher V, ed. Neuropsychological Assessments of Dementia in Down Syndrome and Intellectual Disabilities. Springer; 2009:67-89. [Google Scholar]
- 27.Sparrow SS, Balla DA, Cicchetti DV, Doll EA. Vineland-II, Vineland Adaptive Behavior Scales: Survey Forms Manual. 2nd ed. AGS Publishing; 2005. [Google Scholar]
- 28.Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308-2314. doi: 10.1212/WNL.44.12.2308 [DOI] [PubMed] [Google Scholar]
- 29.Staunton H, Trennery C, Arbuckle R, et al. Development of a Clinical Global Impression of Change (CGI-C) and a Caregiver Global Impression of Change (CaGI-C) measure for ambulant individuals with Duchenne muscular dystrophy. Health Qual Life Outcomes. 2021;19:184. doi: 10.1186/s12955-021-01813-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Posner K, Brent D, Lucas C, et al. Columbia–Suicide Severity Rating Scale. Version 6/23/10. Research Foundation for Mental Hygiene; 2010. Accessed June 1, 2015. https://cssrs.columbia.edu/wp-content/uploads/C-SSRS_Pediatric-SLC_11.14.16.pdf
- 31.Conti E, Galimberti G, Piazza F, Raggi ME, Ferrarese C. Increased soluble APPalpha, Abeta 1-42, and anti–Abeta 1-42 antibodies in plasma from Down syndrome patients. Alzheimer Dis Assoc Disord. 2010;24(1):96-100. doi: 10.1097/WAD.0b013e3181aba63a [DOI] [PubMed] [Google Scholar]
- 32.Strydom A, Coppus A, Blesa R, et al. Alzheimer’s disease in Down syndrome: an overlooked population for prevention trials. Alzheimers Dement (N Y). 2018;4:703-713. doi: 10.1016/j.trci.2018.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Randolph C, Tierney MC, Mohr E, Chase TN. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): preliminary clinical validity. J Clin Exp Neuropsychol. 1998;20(3):310-319. doi: 10.1076/jcen.20.3.310.823 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol and Statistical Analysis Plan
eTable. Columbia–Suicide Severity Rating Scale (C-SSRS)
eFigure 1. Angiogenic Markers
eFigure 2. Body Mass Index
eFigure 3. Anti–Amyloid-β 1-42 Immunoglobulin G Titer in Serum Using the Meso Scale Discovery Free Assay: Change From Baseline in Modified Intention-to-Treat Population per Individual Participant
eFigure 4. Alzheimer Disease Biomarkers
eFigure 5. Cognitive Measures
Data Sharing Statement