

Abstract
Coronavirus disease 2019 (COVID‐19) is a continued leading cause of hospitalization and death. Safe, efficacious COVID‐19 antivirals are needed urgently. Nirmatrelvir (PF‐07321332), the first orally bioavailable, severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) Mpro inhibitor against the coronaviridae family, has demonstrated potent preclinical antiviral activity and benign safety profile. We report safety, tolerability, and pharmacokinetic data of nirmatrelvir with and without ritonavir as a pharmacokinetic enhancer, from an accelerated randomized, double‐blind, placebo‐controlled, phase I study. Two interleaving single‐ascending dose (SAD) cohorts were evaluated in a three‐period crossover. Multiple‐ascending dose (MAD) with nirmatrelvir/ritonavir twice daily (b.i.d.) dosing was evaluated over 10 days in five parallel cohorts. Safety was assessed, including in a supratherapeutic exposure cohort. Dose and dosing regimen for clinical efficacy evaluation in phase II/III clinical trials were supported by integrating modeling and simulations of SAD/MAD data with nonclinical data and a quantitative systems pharmacology model (QSP). In SAD, MAD, and supratherapeutic exposure cohorts, nirmatrelvir/ritonavir was safe and well‐tolerated. Nirmatrelvir exposure and half‐life were considerably increased by ritonavir, enabling selection of nirmatrelvir/ritonavir dose and regimen for phase II/III trials (300/100 mg b.i.d.), to achieve concentrations continuously above those required for 90% inhibition of viral replication in vitro. The QSP model suggested that a 5‐day regimen would significantly decrease viral load in SARS‐CoV‐2‐infected patients which may prevent development of severe disease, hospitalization, and death. In conclusion, an innovative and seamless trial design expedited establishment of phase I safety and pharmacokinetics of nirmatrelvir/ritonavir, enabling high confidence in phase II/III dose selection and accelerated pivotal trials’ initiation (NCT04756531).
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Nirmatrelvir is a potent and specific inhibitor of Mpro enzyme activity and antiviral activity across a diverse spectrum of coronaviruses. Administration in a murine severe acute respiratory syndrome‐coronavirus 2 model demonstrated dose‐dependent reduction of pulmonary viral titers and reduced tissue pathology.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This first‐in‐human study in healthy adults used an innovative and seamless operational and automated data analysis approach to evaluate safety, tolerability, and pharmacokinetics of nirmatrelvir and selected dose regimen, and duration for phase II/III studies.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The safety, tolerability, and pharmacokinetics of nirmatrelvir in healthy adults and the dose selection is explained. This study shows an innovative approach to inform and expedite the development of nirmatrelvir.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This provides an example of a seamless phase 1 program with model‐informed drug development‐based decision making to support expedited start and conduct of phase II/III study with at least 10‐fold reduction in timeline compared with industry standards.
Coronavirus disease 2019 (COVID‐19) is a continued threat to public health worldwide more than 2 years after its emergence. Despite availability of effective vaccines, 1 , 2 infection rates remain high, and COVID‐19 continues to be a leading cause of hospitalization and death. 3 Intravenously administered monoclonal antibodies (casirivimab, imdevimab, bamlanivimab, etesevimab, and sotrovimab) were the first outpatient therapies to be made available under emergency use authorization (EUA) to patients at high risk of severe COVID‐19. 4 , 5 , 6 , 7 However, logistical constraints could affect administration, and reduced effectiveness has been observed for some monoclonal antibodies, leading the US Government to recommend not using them for some severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) variants. 4 , 8 More recently, a 3‐day course of the intravenous RNA polymerase inhibitor remdesivir also received EUA for non‐hospitalized patients at risk of severe disease. 9 Nirmatrelvir (PAXLOVIDTM) is the first oral antiviral treatment authorized for the treatment of COVID‐19 under EUA. 10 The nucleoside inhibitor molnupiravir received EUA shortly after nirmatrelvir in adults at risk of severe COVID‐19 for whom other treatments are unavailable or inappropriate. 11 At the time of this study, no therapy that could be self‐administered at home was available to treat COVID‐19; there was therefore an urgent need for safe and efficacious orally administered therapies, particularly for individuals at high risk of severe illness.
The coronaviridae family, including SARS‐CoV‐2, encodes two proteases from which functional proteins are generated through proteolysis, one of which is the main protease (Mpro). 12 , 13 Mpro is a promising target for viral inhibitors because it is crucial for processing viral polyproteins into functional units, 14 is highly conserved across SARS‐CoV‐2 and other coronaviruses, 15 , 16 and has limited potential for off‐target activity with no identified human analogs. 17
Nirmatrelvir, designed to be orally administered, demonstrated potent and specific inhibition of Mpro enzyme activity and anti‐viral activity across a diverse spectrum of coronaviruses in preclinical assays. 18 Oral administration in a murine SARS‐CoV‐2 model demonstrated dose‐dependent reduction of pulmonary viral titers and reduced tissue pathology. 18 At the time of study initiation, it was not clear if nirmatrelvir concentrations several fold over the in vitro 90% effective concentration (EC90) could be maintained in humans; however because nirmatrelvir is primarily metabolized by cytochrome P450 3A4 (CYP3A4)19, dosing with a pharmacokinetic enhancing agent was included in the protocol to ensure that efficacious concentrations could be achieved. 18
In response to the public health crisis resulting in an urgent need for COVID‐19 therapeutics, we used an innovative and seamless operational approach incorporating a flexibly written protocol that allowed study conduct to adapt to emerging data within the study without the need for a protocol amendment. We simultaneously utilized model‐informed drug development to expedite dose selection and inform the design of phase II/III clinical studies. This study in healthy adults examined safety, tolerability, and pharmacokinetics of nirmatrelvir, with and without ritonavir as a pharmacokinetic enhancer to prevent rapid metabolism of nirmatrelvir by CYP3A4, thus ensuring that free concentrations well above in vitro EC90 are maintained in > 90% of the population to achieve the optimal therapeutic effect.
METHODS
Objectives, participants, and oversight
This 5‐part study (NCT04756531) assessed nirmatrelvir safety, tolerability, and pharmacokinetics following single‐ascending doses (SADs) including food effect, multiple‐ascending doses (MADs), and at supratherapeutic exposure in healthy 18–60 year old patients. MAD evaluation also included a Japanese cohort. Relative bioavailability and food effect of an oral tablet formulation without pharmacokinetic enhancer, and nirmatrelvir metabolism and excretion, were evaluated in two additional open‐label cohorts (to be reported separately). SAD, MAD, and supratherapeutic exposure cohorts were randomized and double‐blinded (to participants and investigators; open to a subset of the blinded study team for efficient decision making). Key inclusion/exclusion criteria, ethical study conduct, study responsibilities, operational conduct, and methods to ensure blinding are summarized in the Supplementary Information S1 . CONSORT 2010 checklist is included as Appendix S1.
Randomization and study treatment
Within two cohorts in SAD, participants were randomly assigned using a randomization schedule to nirmatrelvir or placebo in an interleaving three‐period crossover, where dose escalations were alternated twice weekly between the two cohorts (Figure 1a ). In each period, participants received nirmatrelvir suspension or placebo, with ≥ 5 day washout between doses. In the first three dosing periods, nirmatrelvir administered fasted without ritonavir was escalated from 150 to 1,500 mg. In the next two dosing periods, nirmatrelvir administered fasted was escalated from 250 to 750 mg while co‐administered with ritonavir 100 mg. The final dosing period evaluated food effect on pharmacokinetics of nirmatrelvir 250 mg with ritonavir 100 mg. Where applicable, ritonavir was administered at −12, 0, and 12 hours relative to nirmatrelvir. For each dose escalation decision, preliminary cumulative safety, tolerability, and pharmacokinetic data from previous dosing periods were evaluated.
Figure 1.
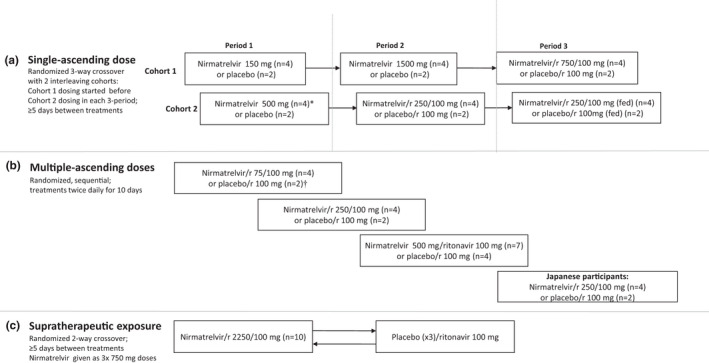
Study design, treatments and numbers of participants treated in Single‐ascending dose part (a) Multiple‐ascending dose part (b) and Supratherapeutic exposure part (c) of first‐in‐human study. *One participant in the nirmatrelvir 500 mg group during the single‐ascending dose part withdrew due to an adverse event. †One participant in the placebo/ritonavir group during the multiple‐ascending dose part discontinued due to participant withdrawal. In this study, which was conducted at the Pfizer Clinical Research Unit, nirmatrelvir was given as an oral suspension. Unless otherwise stated, nirmatrelvir was given under fasting conditions. During the single‐ascending dose and supratherapeutic exposure parts, participants remained in the clinical research unit during the washout. r = ritonavir.
In MAD, participants were randomly assigned to escalating nirmatrelvir dose levels (75, 250, and 500 mg) or placebo, all administered twice daily (b.i.d.) as a suspension for 10 days with ritonavir 100 mg, under fasting conditions (Figure 1b ). For each dose escalation decision, preliminary cumulative safety, tolerability, and pharmacokinetic data from previous treatment cohorts were evaluated. After dose escalation, a cohort of Japanese participants was randomized to receive nirmatrelvir 250 mg or placebo, with ritonavir 100 mg, under fasting conditions b.i.d. for 10 days.
To assess supratherapeutic exposure in the presence of solubility‐limited absorption, split dosing was used, where participants were randomly assigned to receive 2,250 mg of nirmatrelvir (administered as 3 × 750 mg doses at 0, 2 and 4 hours) or matching placebo in a 2‐way crossover with ≥ 5 days washout between treatments (Figure 1c ). Ritonavir was administered at −12, 0, and 12 hours relative to the first nirmatrelvir or placebo dose.
The Supplementary Information S1 provides additional details regarding randomization, including the method used to generate the random allocation sequence and the type of randomization.
Safety and tolerability
Safety and tolerability evaluations included adverse event (AE) and serious AE (SAE) assessments between signing of informed consent until 28 days after study treatment administration. AEs are presented by the Medical Dictionary for Regulatory Activities (MedDRA) version 24.0 term (MedDRA MSSO, Herndon, VA, https://www.meddra.org). Vital signs, electrocardiograms (ECGs), and laboratory tests were monitored at prespecified timepoints.
Bioanalytical methods
Human plasma samples were analyzed utilizing a validated liquid chromatography‐tandem accurate mass spectrometry assay. The assay consisted of protein precipitation in conjunction with reversed phase liquid chromatography coupled to positive ion electrospray tandem quadrupole mass spectrometry. Precipitation was conducted by aliquoting 100 µL of plasma sample to a 96‐well propylene plate then adding acetonitrile or working internal standard in acetonitrile (300 μL). The plate was mixed and centrifuged at (1421 g). Supernatant (50.0 μL) from each well was transferred to a new 96‐well propylene plate and diluted with 500 μL of 60:40 (v:v) water:acetonitrile containing 0.1% formic acid, mixed and injected on the liquid chromatography‐tandem accurate mass spectrometry system. The analytical column used was a Waters Acquity UPLC BEH C18, 1.7 μm, 2.1 × 50 mm operated at ambient temperature. Mobile phases A and B consisted of water containing 0.1% formic acid and acetonitrile containing 0.1% formic acid, respectively, with a flow rate of 0.600 mL/minute. The gradient program was initiated with a (70:30) mobile phase A:B and held for 0.1 minutes, followed by a linear gradient to (60:40) mobile phase A:B to 5.0 minutes. A column wash was applied at (15:85) mobile phase A:B to 6.0 minutes, followed by re‐equilibration at (70:30) mobile phase A:B to 7.0 minutes. A Sciex API5500 tandem quadrupole mass spectrometer equipped with a Turbo V Ion Source was operated in electrospray (TurbolonSpray) positive mode. The parent to product ion transitions 500.5/110.3 and 509.5/109.9 were utilized for nirmatrelvir and PF 07818226 (nirmatrelvir internal standard), respectively. Peak areas of the analytes and internal standards were determined by Analyst data processing software version 1.7.2 (Sciex, Framingham, MA, USA). These responses were imported into Watson LIMS system version 7.6.1 (Thermo Fisher Scientific, Waltham, MA, USA) and a calibration curve was constructed using peak area ratios of the calibration samples and applying weighted (1/X2) linear least squares regression analysis. The method was validated over the range of 10.0–50,000 ng/mL.
Pharmacokinetic evaluations
Nirmatrelvir plasma pharmacokinetic parameters following single and multiple oral doses, and urine pharmacokinetics following multiple oral doses were derived as described in Table S1 from concentration‐time data using standard noncompartmental methods.
Dose and duration selection for phase II/III
Consistent with the literature for other protease inhibitors, 20 , 21 a robust antiviral effect is predicted when unbound plasma concentrations are sustained above multiples of the EC90 (i.e., concentration at which 90% inhibition of viral replication occurs) for the entire dosing interval. The unbound target minimum plasma concentration (Cmin) to be maintained corresponded to the in vitro drug concentration at which 90% inhibition of SARS‐CoV‐2 viral replication is observed (adjusting for plasma protein binding the total plasma EC90 = 292 ng/mL). 18 A population pharmacokinetic model of nirmatrelvir with ritonavir 100 mg was built during the dose escalation based on the preliminary SAD/MAD data from healthy adults in this study to project distribution of expected nirmatrelvir concentrations at different doses and regimens. Consistent with the anticipated phase II/III dosing regimen, only the pharmacokinetic data collected from the nirmatrelvir/ritonavir treatment arms were included in the population pharmacokinetic model. This enabled selection of a phase II/III dose that would result in free Cmin above EC90 in the vast majority (≥ 90%) of future trial participants.
In the population pharmacokinetic model, logarithmically transformed plasma nirmatrelvir concentration vs. time data were analyzed with NONMEM, version 7.5.0 (ICON plc, Gaithersburg, MD, USA) and the FOCE method with interaction. A one‐ or two‐compartment model with first‐order elimination and first‐order absorption were tested as the structural model. The base model included an allometric model of baseline body weight on apparent clearances and volumes (V2 and V3) with exponentials fixed to 0.75 and 1, respectively. Interindividual variability and interoccasion variability in the pharmacokinetic parameters were assumed to be log normally distributed and modeled using multiplicative exponential random effects. Models with and without covariance for random effects were tested. Residual random effects were described with a combined proportional and additive model in the log domain.
Simulations were performed utilizing the preliminary population pharmacokinetic model with nirmatrelvir doses of 100–500 mg by 100 mg increments with ritonavir given b.i.d. for 5 days (single dose on day 5) assuming no missing doses. Interindividual variability in nirmatrelvir clearance was assumed to be 60% to account for expected increases in interindividual variability in phase II/III studies relative to this phase I study. Simulated nirmatrelvir plasma concentration profiles were used to calculate the percentage of participants achieving a trough concentration (Cmin) of at least the in vitro EC90.
To select the treatment duration, a quantitative systems pharmacology (QSP) model capable of describing viral dynamics over time was used to predict potential viral load reduction as a surrogate of efficacy. 22 , 23 The model was updated to include expected nirmatrelvir pharmacokinetic data at the proposed phase II/III dose, preclinical data from a mouse model of SARS‐CoV‐2, and publicly available viral load data from randomized controlled trials. 24 , 25 , 26 Specifically, to predict the antiviral effect and optimal dosing regimen of nirmatrelvir, the QSP model was updated to incorporate: (i) the mean simulated pharmacokinetic profile of nirmatrelvir/ritonavir 300 mg/100 mg b.i.d. 5 day and 10 day regimens from the population pharmacokinetic model described in the preceding section; (ii) preclinical data on nirmatrelvir pharmacology in a mouse model of SARS‐CoV‐2 that was used to estimate the in vivo potency of nirmatrelvir with the QSP model; and (iii) a virtual population (N = 502) that matched the placebo and treatment response of viral load and severity as reported in publicly available data. 24 , 25 , 26 , 27
A virtual population was simulated to predict the effects of different dosing durations (i.e., 5 or 10 days of dosing), 27 with a symptomatic outpatient COVID‐19 population and dosing 4 days post viral load peak/symptom onset assumed. 24 The model was used to predict the influence of the 5 and 10 day dosing regimens on the viral load time course at the estimated in vivo potency of nirmatrelvir. Moreover, given the uncertainty on the clinical potency of nirmatrelvir prior to phase II/III dose and regimen selection, sensitivity analysis was conducted to evaluate the impact of a range of potencies simulated in the model (Figure S1 ). For this sensitivity analysis, the viral load lowering effect was predicted at the start of day 7 and day 10 post‐treatment.
Statistical analysis
The study did not include statistical hypotheses. Safety end points were summarized in the safety population (Table S2 ) as counts and percentages. SAD and MAD were to include six participants per cohort (nirmatrelvir, 4 and placebo, 2). For supratherapeutic exposures, a sample size of 12 participants (with ≥ 10 completers) was chosen.
Pharmacokinetic parameters were assessed in the pharmacokinetic parameter population (Table S2 ). No formal inferential statistics were applied to pharmacokinetic data apart from comparisons of food effect in SAD (Supplementary Information S1 ).
RESULTS
Participants
Dosing for this study began on March 2, 2021, and dose selection was completed on April 30, 2021. The study randomized 13, 29, and 10 healthy adults in the SAD, MAD, and supratherapeutic parts of the study, respectively. One participant discontinued during SAD due to an AE; and one participant discontinued during MAD due to participant withdrawal. All 10 participants assessed for supratherapeutic exposure completed the study (Figure 1 ). Demographic characteristics are summarized in Table 1 .
Table 1.
Demographic and clinical characteristics
|
Single‐ascending dose (N = 13) |
Multiple‐ascending dose (N = 29) |
Supratherapeutic exposure (N = 10) |
|
|---|---|---|---|
| Age, years, n (%) | |||
| 18–44 | 8 (61.5) | 19 (65.5) | 4 (40.0) |
| 45–60 | 5 (38.5) | 10 (34.5) | 6 (60.0) |
| Mean (±SD) | 38.8 (13.03) | 39.5 (11.54) | 45.7 (13.29) |
| Median (range) | 36.0 (21, 56) | 39.0 (20, 60) | 52.0 (20, 57) |
| Male, n (%) | 11 (84.6) | 22 (75.9) | 7 (70.0) |
| Race, n (%) | |||
| White | 4 (30.8) | 6 (20.7) | 4 (40.0) |
| Black | 8 (61.5) | 16 (55.2) | 5 (50.0) |
| Other | 1 (7.7) | 0 | 0 |
| Asian | 0 | 7 (24.1) | 1 (10.0) |
| Ethnicity, n (%) | |||
| Hispanic/Latinx | 3 (23.1) | 4 (13.8) | 1 (10.0) |
| Weight, kg | |||
| Mean (±SD) | 76.3 (12.64) | 72.2 (10.07) | 77.3 (14.43) |
| Median (range) | 76.1 (60.3, 96.8) | 70.4 (58.5, 99.4) | 77.9 (58.6, 100.8) |
| Height, cm | |||
| Mean (±SD) | 172.1 (11.52) | 171.1 (11.36) | 170.9 (11.45) |
| Median (range) | 172.0 (152, 188) | 170.0 (154, 194) | 173.0 (154, 186) |
| Body mass index, kg/m2 | |||
| Mean (±SD) | 25.8 (3.4) | 24.7 (2.9) | 26.2 (2.6) |
| Median (range) | 25.3 (19.7, 30.5) | 24.4 (19.8, 30.5) | 25.90 (22.9, 29.5) |
b.i.d, twice daily; SD, standard deviation.
Safety
No participant had an SAE, severe AE, or the dose reduced or temporarily discontinued because of AEs. One participant in SAD discontinued from the study because of a positive COVID‐19 test. This was a protocol‐specified test occurring on day 4 and the participant was asymptomatic. All participants in MAD completed 10 days of treatment apart from one participant who withdrew after receiving placebo/ritonavir 100 mg b.i.d. (fasted) for 7 days.
In SAD, no AEs were considered treatment‐related (Table S3 ), and all AEs were mild. Five treatment‐related events of blood thyroid stimulating hormone increased occurred in MAD, and two of these events occurred in the placebo groups (Table S4 ). Three treatment‐related events of dysgeusia occurred, and all were in the treated groups. All Japanese participants in MAD reported AEs and three were considered treatment‐related (two participants with mild blood thyroid stimulating hormone increased, including one on placebo; and one participant with mild dysgeusia). At supratherapeutic exposure, one participant each had one treatment‐related AE: nausea with treatment and vomiting with placebo. Overall, no safety concerns were identified with administration of nirmatrelvir, with or without ritonavir. There was no evidence of a dose‐related increase in AEs. No clinically meaningful changes in laboratory values (≥ grade 2), vital sign measurements, or ECG measurements were observed.
Pharmacokinetics
The median plasma nirmatrelvir concentration‐time profiles for SAD, supratherapeutic exposure, and MAD cohorts are shown in Figure 2 .
Figure 2.
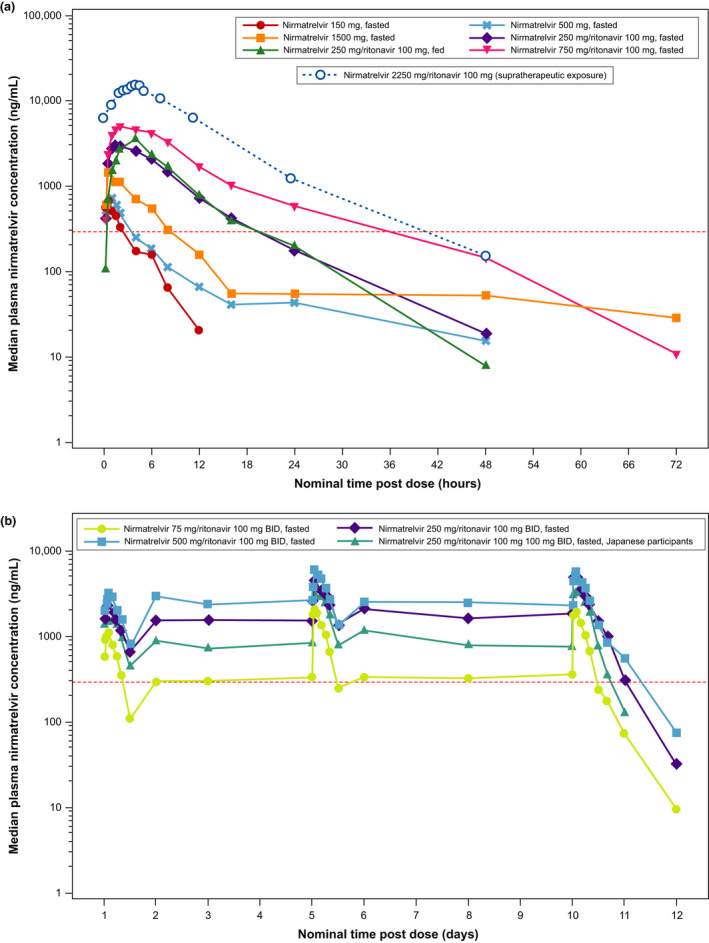
Median plasma nirmatrelvir concentration‐time profiles (semi‐log scales) for single‐ascending dose and supratherapeutic exposure cohorts (a) and multiple‐ascending dose cohort (b). For summary statistics, values below the lower limit of quantification (10 ng/mL) were set to zero. In the supratherapeutic exposure assessment, nirmatrelvir was administered as 3 × 750 mg doses at 0, 2, and 4 hours. In the single‐ascending dose assessments where applicable and supratherapeutic exposure assessments, ritonavir 100 mg was dosed at −12 hours, 0 hours, and 12 hours after dosing. In the multiple‐ascending dose assessment, ritonavir 100 mg was dosed twice daily. The red dotted line is EC90 of 292 ng/mL (accounted for plasma protein binding). b.i.d., twice daily; EC90, concentration at which 90% inhibition of viral replication is observed.
Pharmacokinetic parameters for nirmatrelvir calculated in SAD are summarized in Table 2 , and Table S5 . Following single‐dose administration of nirmatrelvir as an oral suspension at doses of 150, 500, and 1,500 mg without ritonavir under fasted conditions, less than dose proportional increases in nirmatrelvir exposure were observed. Ritonavir considerably increased nirmatrelvir exposure. Following administration of nirmatrelvir 250 or 750 mg enhanced with 100 mg ritonavir, a dose‐related increase in nirmatrelvir was observed albeit in a less than dose‐proportional manner. Following administration of a 250 mg oral suspension of nirmatrelvir with ritonavir 100 mg under fed and fasted conditions, no meaningful change in exposure was observed (~ 1.5% increase in area under the curve (AUC) and ~ 15% increase in Cmax).
Table 2.
Single‐ascending dose assessment of plasma nirmatrelvir pharmacokinetic parameters: descriptive summary (A) and statistical summary of food effect (B)
| A. Descriptive summary | ||||||
|---|---|---|---|---|---|---|
| Parameter (unit)a |
Nirmatrelvir 150 mg Fasted (N = 4) |
Nirmatrelvir 500 mg Fasted (N = 4) |
Nirmatrelvir 1,500 mg Fasted (N = 4) |
Nirmatrelvir/r 250/100 mg Fasted (N = 4) |
Nirmatrelvir/r 250/100 mg Fed (N = 4) |
Nirmatrelvir/r 750/100 mg Fasted (N = 4) |
| N1, N2a | 4, 3 | 4, 2 | 4, 0 | 4, 4 | 4, 4 | 4, 4 |
| AUCinf, ng hour/mL | 2,247 (42) | 5,480, 5,450b | NR | 28,220 (14) | 28,640 (17) | 66,760 (45) |
| Cmax, ng/mL | 667.7 (28) | 674.4 (38) | 1,538 (32) | 2,882 (25) | 3,323 (13) | 5,086 (25) |
| t ½, hours | 2.023 ± 0.54556 | 18.5, 25.6b | NR | 6.935 ± 1.0794 | 6.005 ± 1.6502 | 12.86 ± 8.4196 |
| Tmax, hours | 0.634 (0.550–1.50) | 1.00 (0.517–1.00) | 1.00 (0.533–2.00) | 2.75 (1.50–4.00) | 4.00 (4.00–4.00) | 2.00 (1.50–4.00) |
| B. Statistical summary—food effect | ||||
|---|---|---|---|---|
| Parameter (unit)a |
Nirmatrelvir/r 250/100 mg fed (test) |
Nirmatrelvir/r 250/100 mg fasted (test) |
Ratio (%) (test/reference) of adjusted geometric meansa |
90% CI (%) for ratioc |
| AUCinf, ng hour/mL | 28,640 | 28,220 | 101.5 | (89.57, 115.07) |
| Cmax, ng/mL | 3,323 | 2,882 | 115.3 | (99.36, 133.79) |
Results are for pharmacokinetic parameter set (defined in Table S2 ) and show the geometric mean (geometric percent coefficient of variation (%CV)) for all except for Tmax, which is the median (range) and t 1/2 which is the arithmetic mean ± SD. For the parameters analyzed on the log scale, zero values were substituted with 0.0001 prior to log transformation. The parameters are defined in Table S1 . Additional results are in Table S5 . The statistical summary of food effect used mixed effect models with treatment as a fixed effect and participant as a random effect, which were applied to the natural log transformation of AUCinf and Cmax separately using participants with data from both periods only.
AUCinf, area under the curve to infinity; CI, confidence interval; Cmax, maximum plasma concentration; NR, not reported; r, ritonavir; t 1/2, terminal half‐life; Tmax, time to maximum plasma concentration.
aN1 is the number of participants contributing to the summary statistics and N2 is the number of participants where t 1/2 and AUCinf were determined. bWhere fewer than three participants had evaluable measurements, individual values are listed. cRatios (and 90% CIs) are expressed as percentages.
Pharmacokinetic parameters for nirmatrelvir calculated in MAD are summarized in Table 3 , and Table S6 . Following multiple‐dose administration of nirmatrelvir/ritonavir at 75/100 mg, 250/100 mg, and 500/100 mg b.i.d. doses under fasted conditions, nirmatrelvir exposure on days 1, 5, and 10 increased in a less than dose‐proportional manner across the doses studied. Plasma nirmatrelvir levels increased approximately 2 fold after the first dose, with steady‐state concentrations achieved by day 2 for all doses and treatments and maintained on days 5 and 10. Urinary recovery of unchanged nirmatrelvir was substantial, indicating the kidneys as the major organs in nirmatrelvir elimination in the presence of ritonavir. Nirmatrelvir renal clearance was similar across all doses. Nirmatrelvir exposure, although numerically lower, was not meaningfully different in Japanese participants across all days.
Table 3.
Multiple‐ascending dose assessment: descriptive summary of plasma and urine nirmatrelvir pharmacokinetic parameters
| Parameter (unit)a |
Nirmatrelvir/r 75/100 mg b.i.d. Fasted (N = 4) |
Nirmatrelvir/r 250/100 mg b.i.d. Fasted (N = 4) |
Nirmatrelvir/r 500/100 mg b.i.d. Fasted (N = 7) |
Nirmatrelvir/r 250/100 mg b.i.d. Fasted, Japanese (N = 4) |
|---|---|---|---|---|
| Day 1 | ||||
| N1a | 4 | 4 | 7 | 4 |
| AUCtau, ng hour/mL | 6,017 (33) | 18,700 (43) | 22,610 (37) | 13,130 (26) |
| Cmax, ng/mL | 1,042 (28) | 2,435 (36) | 3,051 (32) | 1,925 (25) |
| Tmax, hours | 1.75 (1.00–2.00) | 1.50 (1.00–4.00) | 2.00 (1.50–2.17) | 2.75 (1.00–4.02) |
| Day 5 | ||||
| N1a | 4 | 4 | 7 | 4 |
| AUCtau, ng hour/mL | 12,570 (17) | 35,560 (26) | 38,150 (23) | 25,480 (26) |
| Cmax, ng/mL | 2,224 (27) | 4,774 (21) | 5,296 (21) | 3,674 (28) |
| Rac | 2.091 (24) | 1.901 (22) | 1.685 (29) | 1.937 (18) |
| Tmax, hours | 1.00 (1.00–1.50) | 0.750 (0.500–1.50) | 1.50 (1.00–2.02) | 1.26 (1.00–2.02) |
| Day 10 | ||||
| N1, N2a | 4, 4 | 4, 4 | 7, 7 | 4, 4 |
| AUCtau, ng hour/mL | 12,650 (16) | 37,780 (27) | 39,780 (20) | 26,930 (15) |
| Cmax, ng/mL | 2,055 (14) | 5,123 (24) | 5,607 (17) | 3,772 (21) |
| Rac | 2.104 (30) | 2.022 (16) | 1.757 (26) | 2.047 (16) |
| t ½, hours | 7.955 ± 2.0401 | 6.795 ± 1.7072 | 8.047 ± 1.7871 | 5.163 ± 2.0915 |
| Tmax, hours | 1.00 (1.00–2.00) | 1.00 (1.00–2.00) | 1.50 (1.00–2.00) | 1.50 (0.500–2.02) |
| Aetau % | 63.79 (12) | 51.81 (4) | 23.35 (121) | 54.20 (5) |
| CLr, L/hour | 3.782 (20) | 3.433 (23) | 2.934 (128) | 5.028 (11) |
Results are for pharmacokinetic parameter set (defined in Table S2 ) and show the geometric mean (geometric %CV) for all except for Tmax, which is the median (range) and t 1/2 which is the arithmetic mean ± SD. For the parameters analyzed on the log scale, zero values were substituted with 0.0001 prior to log transformation. The parameters are defined in Table S1 . Additional results are in Table S6 .
%CV, percent coefficient of variation; AUCtau, area under the concentration curve for a dosing interval; b.i.d., twice daily; CLr, renal clearance; Cmax, maximum plasma concentration; r, ritonavir; t 1/2, terminal half‐life; Tmax, time to maximum plasma concentration.
aN1 is the number of participants contributing to the summary statistics and N2 is the number of participants where t 1/2 was determined.
Pharmacokinetic parameters for nirmatrelvir calculated in the supratherapeutic assessment are summarized in Table 4 and Table S7 . Following a supratherapeutic oral dose of nirmatrelvir as a 2,250‐mg suspension (dosed as 3 split doses of 750 mg administered at 0, 2, and 4 hours) enhanced with 100 mg ritonavir, the mean Cmax was ~ 5 times higher than the mean Cmax observed in the nirmatrelvir/ritonavir 500/100 mg b.i.d. cohort on day 1 in the MAD part of the study.
Table 4.
Descriptive summary of plasma nirmatrelvir pharmacokinetic parameters for supratherapeutic exposure
| Parameter (unit) |
Nirmatrelvir/r 2250a/100 mg (N = 10) |
|---|---|
| N1, N2b | 10, 10 |
| AUCinf, ng hour/mL | 188,800 (35) |
| Cmax, ng/mL | 15,940 (27) |
| Tmax, hours | 5.00 (3.02–6.03) |
Results are for pharmacokinetic parameter set (defined in Table S2 ) and show the geometric mean (geometric %CV) for all except for Tmax, which is the median (range). For the parameters analyzed on the log scale, zero values were substituted with 0.0001 before log transformation. The parameters are defined in Table S1 . Additional results are in Table S7 .
%CV, percent coefficient of variation; AUCinf, area under the concentration curve to infinity; Cmax, maximum plasma concentration; Tmax, time to maximum plasma concentration.
aGiven as three doses of 750 mg at 0, 2, and 4 hours. bN1 is the number of participants contributing to the summary statistics and N2 is the number of participants where AUCinf was determined.
Dose and duration selection in phase II/III
The pharmacokinetics of nirmatrelvir with ritonavir 100 mg following oral administration was adequately characterized by a 2‐compartment disposition model with first‐order absorption. Separate power functions were used to describe the dose effect on k a and F1. The parameter estimates for a nirmatrelvir dose of 300 mg with ritonavir were clearance 8.2 L/hour, volume of distribution 111 L, and k a 1.11 hour−1. No obvious time‐dependent change in nirmatrelvir clearance was noted.
Concentrations were simulated for a sample size commensurate with phase II/III studies for a range of nirmatrelvir doses. With the population pharmacokinetic model based on the preliminary data from this clinical study, the nirmatrelvir/ritonavir 300/100 mg b.i.d. simulation showed that > 90% of future trial participants would achieve the target free Cmin above in vitro EC90 after the first and subsequent dose and with interindividual variability in clearance inflated to 60% (Table S8 ). The projected median Cmin on day 1 and at steady‐state was 987 and 1,800 ng/mL, respectively, which are ~ 3 and 6 times higher than the in vitro EC90, and therefore projected to achieve robust antiviral effects. Simulated steady‐state (day 5) Cmin at different doses is shown in Figure 3a .
Figure 3.

Distribution of simulated Cmin at steady‐state with different doses (b.i.d.) enhanced with ritonavir (a), and simulation of a virtual population (n = 502) to predict viral load effect for nirmatrelvir/ritonavir 300/100 mg twice daily in symptomatic patients with COVID‐19 (b). In panel a, red dots indicate the means, grey lines indicate the medians, boxes are 25th and 75th percentiles, error bars show 10th and 90th percentiles, and the red dotted line is EC90 of 292 ng/mL (accounted for plasma protein binding). COVID‐19, coronavirus disease 2019; EC90, concentration at which 90% inhibition of viral replication is observed; PI, prediction interval; RNA, ribonucleic acid.
QSP model simulations of a virtual population treated with nirmatrelvir/ritonavir 300/100 mg b.i.d. (Figure 3b ) at 4 days post symptom onset predicted that 5 days of treatment is needed for robust viral reduction (based on clinical trials of monoclonal antibodies 25 , 28 ) in patients with symptomatic COVID‐19, with no meaningful additional benefit with longer dosing. Additional simulations with onset of treatment of ≤ 5 days after symptom onset supported a similar conclusion.
DISCUSSION
An urgent need exists for safe and effective COVID‐19 therapeutics in community settings that can reduce viral load and transmission, improve time to clinical recovery, and prevent progression to adverse outcomes. 29 , 30 , 31 , 32 Patient populations to benefit include those who are immunocompromised, unvaccinated, or are fully vaccinated but have waning immunity or breakthrough infections. At the time of this study, no orally administered antiviral had received EUA to treat non‐hospitalized patients with COVID‐19. On December 22, 2021, the US Food and Drug Administration (FDA) issued an EUA for nirmatrelvir/ritonavir for the treatment of mild‐to‐moderate COVID‐19 in patients 12 years and older who are at high risk of progression to severe disease, including hospitalization and death. 10 The primary data supporting EUA for nirmatrelvir/ritonavir was from the phase III, placebo‐controlled Evaluation of Protease Inhibition for COVID‐19 in High‐Risk Patients (EPIC‐HR) study, which showed nirmatrelvir/ritonavir to be 88% efficacious at reducing hospitalization or death, with no deaths recorded in the treatment group in patients who started treatment up to 5 days after symptom onset. 10 , 33
Preclinical nirmatrelvir evaluations demonstrated potent antiviral effect, oral bioavailability, and a clean toxicity profile, making a compelling case for clinical development. 18 In response to the urgent need for COVID‐19 therapeutics, we used an innovative and seamless approach to generate safety and pharmacokinetic data to support rapid progression to phase II/III studies in patients with mild‐to‐moderate COVID‐19, by designing this first‐in‐human study with multiple parts and flexibility. The interleaving design, rapid sample analysis, and implementation of automated data analysis allowed twice a week dose escalation, with the in‐clinic phase of SAD and MAD completed within 16 days and 6 weeks, respectively, compared with ~ 6 months following a normal paradigm. The design allowed optional implementation of ritonavir as a pharmacokinetic enhancer based on emerging pharmacokinetic data. A cohort to study safety and tolerability at supratherapeutic exposure was added to reduce the need for ECG monitoring in phase II/III studies and demonstrate lack of pro‐arrhythmic risk of nirmatrelvir. 34 The study conduct and rapid adaption to emerging data together with near real‐time data analysis and model‐informed drug development offered a substantial decrease in the overall development and decision timelines.
Nirmatrelvir was safe and well‐tolerated in SAD, MAD, and supratherapeutic dose cohorts. All treatment‐emergent AEs were mild in severity. No severe AEs or SAEs were reported.
A pharmacokinetic enhancer (ritonavir) 35 was used to maximize clinical concentrations to increase confidence in achieving a clinical antiviral effect against existing and new variants of concern through maintaining free exposures multiple times above the in vitro EC90 and provide a potential barrier against resistance development. As indicated by the urinary excretion data, in the presence of ritonavir (limiting metabolism of nirmatrelvir), renal excretion was the major pathway of elimination. Co‐administration resulted in an approximately 8 fold increase in nirmatrelvir concentrations. Simulations showed coadministration of nirmatrelvir 300 mg with ritonavir 100 mg maintained a free concentration at 12 hours postdose (Cmin) above efficacious concentrations in > 90% of potential trial participants from the very first dose, with median free Cmin > 6 times the in vitro EC90 at steady‐state. 18
The effect of food after oral administration of a nirmatrelvir suspension, when enhanced with ritonavir, was minimal. Given the intended use of co‐administration of nirmatrelvir with ritonavir, nirmatrelvir is being dosed without regard to food.
Population pharmacokinetic modeling and simulations enabled selection of the nirmatrelvir/ritonavir dose and dosing regimen for phase II/III (300/100 mg b.i.d.), predicted to achieve efficacious concentrations continuously in ≥ 90% of dosed participants. This target, which was successfully implemented for the first time for SARS‐CoV‐2 in the current study, was selected based on the literature, where exposure in multiples of EC90 for oral protease and non‐nucleoside protease inhibitors correlates with clinical potency. 20 , 21 This was also supported by efficacy studies in preclinical species infected with SARS‐CoV‐2, 18 and projected to translate to human antiviral effect and relative risk reduction benefits when applied in the QSP modeling. In fact, QSP modeling predicted that 5 days of dosing of oral nirmatrelvir/ritonavir 300/100 mg b.i.d. would demonstrate meaningful antiviral effect, supporting this duration for phase II/III trials. Dosing for > 5 days in symptomatic patients was predicted to have no additional benefit. The physiological rationale for this observation is that after 5 days of dosing (~ 9 days after symptom onset and about 2 weeks after infection 25 , 36 , 37 ) viral dynamics may be almost entirely driven by viral clearance rates, and not by viral replication.
Limitations of the study include that only a small number of participants were enrolled to provide an initial evaluation of safety and pharmacokinetics in healthy participants. This study offered an initial assessment of the safety and tolerability profile of nirmatrelvir/ritonavir and provided the basis for robust dose selection; however, larger trials across various patient populations were needed to confirm nirmatrelvir/ritonavir safety and evaluate efficacy. Such large studies in patients with COVID‐19 (EPIC‐HR, in Standard Risk Patients (EPIC‐SR), and Post‐Exposure Prophylaxis (EPIC‐PEP)) along with clinical pharmacology studies (e.g., in participants with renal or hepatic impairment), are either completed or ongoing. Study participants were also of limited ethnic diversity. However, a small number of Japanese participants were included, and no meaningful differences in safety and pharmacokinetic results were identified between Japanese and non‐Japanese participants.
In conclusion, in response to the urgent need for COVID‐19 treatments, our innovative, seamless, and efficient approach in this study enabled accelerated dose and regimen selection for use in late phase clinical studies within 2 months from the first dose in humans without compromising the quality of safety and pharmacokinetic assessment, while providing the appropriate confidence to initiate pivotal efficacy trials. Nirmatrelvir/ritonavir was safe and well‐tolerated in healthy participants. Ritonavir enhanced nirmatrelvir pharmacokinetics, hereby expected to achieve plasma concentrations and maintain free Cmin values in multiples of the in vitro EC90 when dosed b.i.d. at steady‐state. These data enable high confidence in selection of a regimen of nirmatrelvir 300 mg in combination with ritonavir 100 mg administered b.i.d. over a 5‐day period for phase II/III clinical trials in patients with COVID‐19.
FUNDING
This study was sponsored by Pfizer.
CONFLICT OF INTEREST
All authors are employees of Pfizer Inc. and may hold stock or stock options.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. R.S., S.T., F.H., R.R., R.A., L.V.E., S.P., A.A., M.B., S.M., G.N., and A.B. designed the research. R.R., R.A., L.V.E., S.P., E.K., F.C., and H.S. performed the research. R.S., S.T., F.H., P.C., L.V.E., S.P., A.A., G.N., and A.B. analyzed the data.
PROTOCOL SHARING STATEMENT
The protocol for this study is available from the corresponding author upon request.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Tricia Newell, PhD, and Sheena Hunt, PhD, of ICON (Blue Bell, PA, USA), who wrote the first draft under direction from the authors, with funding from Pfizer Inc. We would like to thank all the participants who volunteered for this study. We also thank all the study site personnel for their contributions to this study. The authors would also like to acknowledge the significant number of Pfizer colleagues who have contributed to this first‐in‐human study. In particular, we acknowledge Scott White for his contribution to the study conduct, Joanne Salageanu for NCA analysis, Claudine Fredette and Geraldine Gigou for their contribution with study conduct, Karen Bartsch for study management, NH CRU and BR CRU staffs supporting the conduct of this study, C.J. Musante for support of QSP modeling, Li Di for PBPK simulations, Amit Kalgutkar for the PDM contributions, Charlotte Allerton for strategic input, and Britton Boras for the assessment of efficacious concentrations.
Investigational sites: Brussels Clinical Research Institute, Brussels, Belgium, and New Haven Clinical Research Unit, New Haven, CT, USA.
DATA AVAILABILITY STATEMENT
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de‐identified participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.
- 1. Harder, T. et al. Effectiveness of COVID‐19 vaccines against SARS‐CoV‐2 infection with the Delta (B.1.617.2) variant: second interim results of a living systematic review and meta‐analysis, 1 January to 25 August 2021. Euro. Surveill. 26, 2100920 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zheng, C. et al. Real‐world effectiveness of COVID‐19 vaccines: a literature review and meta‐analysis. Int. J. Infect. Dis. 114, 252–260 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Centers for Disease Control and Prevention Estimated COVID‐19 Burden <https://www.cdc.gov/coronavirus/2019‐ncov/cases‐updates/burden.html> (2021). Accessed February 21, 2022.
- 4. National Institutes of Health . COVID‐19 Treatment Guidelines Panel. Coronavirus Disease 2019 (COVID‐19) Treatment Guidelines <https://www.covid19treatmentguidelines.nih.gov/> (2021). Accessed February 21, 2022. [PubMed]
- 5. US Food and Drug Administration . Emergency Use Authorization 091 (Casirivimab and Imdevimab) <https://www.fda.gov/media/145610/download> (2021). Accessed October 21, 2021.
- 6. US States Food and Drug Administration . Emergency Use Authorization 094 (Bamlanivimab and Etesevimab) <https://www.fda.gov/media/145801/download> (2021) Accessed October 21, 2021.
- 7. US Food and Drug Administration . Emergency Use Authorization 100 (Sotrovimab). <https://www.fda.gov/media/149532/download> (2021). Accessed October 21, 2021.
- 8. US Food and Drug Administration . Bamlanivimab and Etesevimab Authorized States, Territories, and U.S. Jurisdictions <https://www.fda.gov/media/151719/download> (2021). Accessed January 18, 2022.
- 9. US Food and Drug Administration . FDA Takes Actions to Expand Use of Treatment for Outpatients with Mild‐to‐Moderate COVID‐19 <https://www.fda.gov/news‐events/press‐announcements/fda‐takes‐actions‐expand‐use‐treatment‐outpatients‐mild‐moderate‐covid‐19> (2022. January 21). Accessed February 21, 2022.
- 10. US Food and Drug Administration . Coronavirus (COVID‐19) update: FDA authorizes first oral antiviral for treatment of COVID‐19. December 22, 2021 <https://www.fda.gov/news‐events/press‐announcements/coronavirus‐covid‐19‐update‐fda‐authorizes‐first‐oral‐antiviral‐treatment‐covid‐19> (2021).
- 11. US Food and Drug Administration . Coronavirus (COVID‐19) update: FDA authorizes additional oral antiviral for treatment of COVID‐19 in certain adults. December 23, 2021 <https://www.fda.gov/news‐events/press‐announcements/coronavirus‐covid‐19‐update‐fda‐authorizes‐additional‐oral‐antiviral‐treatment‐covid‐19‐certain> (2021).
- 12. Jin, Z. et al. Structure of M(pro) from SARS‐CoV‐2 and discovery of its inhibitors. Nature 582, 289–293 (2020). [DOI] [PubMed] [Google Scholar]
- 13. Pillaiyar, T. , Manickam, M. , Namasivayam, V. , Hayashi, Y. & Jung, S.H. An overview of severe acute respiratory syndrome‐coronavirus (SARS‐CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 59, 6595–6628 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hilgenfeld, R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 281, 4085–4096 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang, H. et al. Design of wide‐spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 3, e324 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang, L. et al. Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved alpha‐ketoamide inhibitors. Science 368, 409–412 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anand, K. , Ziebuhr, J. , Wadhwani, P. , Mesters, J.R. & Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti‐SARS drugs. Science 300, 1763–1767 (2003). [DOI] [PubMed] [Google Scholar]
- 18. Owen, D.R. et al. An oral SARS‐CoV‐2 M(pro) inhibitor clinical candidate for the treatment of COVID‐19. Science 374, 1586–1593 (2021). [DOI] [PubMed] [Google Scholar]
- 19. Eng H. et al. Disposition of PF‐07321332 (Nirmatrelvir), an Orally Bioavailable Inhibitor of SARS‐CoV‐2 3CL Protease, across Animals and Humans. Drug Metabolism and Disposition (2022). 10.1124/dmd.121.000801 [DOI] [PubMed] [Google Scholar]
- 20. Reddy, M.B. et al. Pharmacokinetic/Pharmacodynamic predictors of clinical potency for hepatitis C virus nonnucleoside polymerase and protease inhibitors. Antimicrob. Agents Chemother. 56, 3144–3156 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bertz, R.J. et al. Pharmacokinetics and pharmacodynamics of atazanavir‐containing antiretroviral regimens, with or without ritonavir, in patients who are HIV‐positive and treatment‐naive. Pharmacotherapy 33, 284–294 (2013). [DOI] [PubMed] [Google Scholar]
- 22. Dai, W. et al. A prototype QSP model of the immune response to SARS‐CoV‐2 for community development. CPT Pharmacometrics Syst. Pharmacol. 10, 18–29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rao, R. , Musante, C.J. & Allen, R. A quantitative systems pharmacology model of the pathophysiology and treatment of COVID‐19 predicts optimal timing of pharmacological interventions. medRxiv preprint. 10.1101/2021.12.07.21267277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. US Food and Drug Administration . Emergency Use Authorization (EUA) for bamlanivimab 700 mg and etesevimab 1,400 IV Center for Drug Evaluation and Research (CDER). Memorandum <https://www.fda.gov/media/151973/download> (2021). Accessed August 27, 2021.
- 25. Weinreich, D.M. et al. REGN‐COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid‐19. N. Engl. J. Med. 384, 238–251 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fischer, W. et al. Molnupiravir, an oral antiviral treatment for COVID‐19. medRxiv preprint. 10.1101/2021.06.17.21258639 [DOI] [Google Scholar]
- 27. Allen, R.J. , Rieger, T.R. & Musante, C.J. Efficient generation and selection of virtual populations in quantitative systems pharmacology models. CPT Pharmacometrics Syst. Pharmacol. 5, 140–146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eli Lilly . SARS‐CoC‐2 Neutralizing Antibody Program Update <https://investor.lilly.com/static‐files/081a5ef7‐f5d6‐4acc‐b0d2‐7ae4daf9e953>. Accessed January 26, 2021.
- 29. Brown, C.M. et al. Outbreak of SARS‐CoV‐2 infections, including COVID‐19 vaccine breakthrough infections, associated with large public gatherings – Barnstable County, Massachusetts, July 2021. MMWR Morb. Mortal. Wkly. Rep. 70, 1059–1062 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thangaraj, J.W.V. et al. Predominance of delta variant among the COVID‐19 vaccinated and unvaccinated individuals, India, May 2021. J. Infect. 84, 94–118 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. The Lancet Infectious Diseases . Unmet need for COVID‐19 therapies in community settings. Lancet Infect Dis 21, 1471 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bergwerk, M. et al. Covid‐19 breakthrough infections in vaccinated health care workers. N. Engl. J. Med. 385, 1474–1484 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hammond, J. et al. Oral nirmatrelvir for high‐risk, nonhospitalized adults with Covid‐19. N. Engl. J. Med. 386, 1397‐1408 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. European Medicines Agency . Assessment report: Paxlovid <https://www.ema.europa.eu/en/documents/referral/paxlovid‐pf‐07321332‐ritonavir‐covid‐19‐article‐53‐procedure‐assessment‐report_en.pdf> (2021). Accessed January 18, 2022.
- 35. Sevrioukova, I.F. & Poulos, T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 107, 18422–18427 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dougan, M. et al. Bamlanivimab plus etesevimab in mild or moderate Covid‐19. N. Engl. J. Med. 385, 1382–1392 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lauer, S.A. et al. The incubation period of coronavirus disease 2019 (COVID‐19) from publicly reported confirmed cases: estimation and application. Ann. Intern. Med. 172, 577–582 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de‐identified participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.