

Abstract
The reaction of NH-indazoles with formaldehyde in aqueous hydrochloric acid has been experimentally studied by solution and solid-state nuclear magnetic resonance (NMR) and crystallography. The mechanism of the formation of N1-CH2OH derivatives was determined. For the first time, 2-substituted derivatives have been characterized by multinuclear NMR. Theoretically, calculations with gauge-invariant atomic orbitals (GIAOs) at the Becke three-parameter (exchange) Lee–Yang–Parr B3LYP/6-311++G(d,p) level have provided a sound basis for the experimental observations. The first X-ray structures of four (1H-indazol-1-yl)methanol derivatives are reported.
Introduction
This work was aimed at a better understanding of a characteristic reaction of N-unsubstituted azoles and their reaction with formaldehyde to afford azolylmethanols. As a model of azole, indazole was selected because it was not clear what isomer would be obtained depending on the substituents in the ring. After solving this problem for 4-, 5-, 6-, and 7-nitro derivatives, the mechanism of the reaction should be established because it is common to all azoles and that azolylmethanols are the intermediates, directly and indirectly (using hydroxymethyl as a protecting group) to other compounds relevant for their applications. The present paper reports our study of the reaction of five NH-indazoles with formaldehyde in an aqueous acid solution, Scheme 1.
Scheme 1. Reaction Studied in the Present Work.

A search in different databases shows that the chemistry of indazoles is a very active field; the numbers of items are 11 723 (Scifinder),1 5142 (ScienceDirect),2 and 4448 (Web of Science);3 and most of the papers and patents deal with biological applications.4−8 Other applications (corrosion inhibitors)9 and synthetic methods10 are less reported, and the last place is occupied by indazole reactivity.
Five indazoles 1a–1e, existing in two tautomeric forms 1H and 2H, and their protonated indazolium cations 1aH+–1eH+, covering all of the substituted nitro compounds in the six-membered ring, will be discussed (Figure 1).
Figure 1.

Five neutral 1a–1e and protonated indazoles 1aH+–1eH+.
Some nitro-1H-indazoles, bearing or not other C-substituents, are powerful inhibitors of nitric oxide synthase isoforms, nNOS, iNOS, and eNOS.11 Of the five possible C-nitro-1H-indazoles, 3-, 4-, 5-, 6-, and 7-, only 7-nitro-1H-indazoles (7-nitro, 3-bromo-7-nitro, and 3,7-dinitro) have inhibitory properties.12−14
In 1969,15 we reported that indazoles react with formaldehyde in aqueous HCl to afford (1H-indazol-1-yl)methanol derivatives. Indazole itself (1a) and 4-nitro (1b), 5-nitro (1c), and 6-nitro-1H-indazoles (1d) react, but 7-nitro-1H-indazole (1e) does not. The isolated compounds were characterized by 1H NMR in DMSO-d6, proving that they were 1-substituted indazoles. In 1986, the reaction was carried out in neutral conditions (ethanol).16 In 2004,17 the reaction of indazole 1a in acid conditions was re-examined; B3LYP/6-311++G(d,p) calculations indicated that the 1-substituted isomer (2a) was 20 kJ·mol–1 more stable than the 2-substituted isomer (3a) (Scheme 2), and the NMR data were extended to 13C and 15N nuclei together with GIAO calculations of absolute shieldings.
Scheme 2. Formal Reactions between Both Tautomers of Indazole 1a and Indazolium Cation 1aH+ with Formaldehyde Corresponding to Neutral and to Acid Conditions.

This reaction is common to all azoles (pyrazole in acid15 and neutral conditions,16,18,19 imidazole,20,21 triazoles,22−24 tetrazole,25 benzimidazole,16,26 and benzotriazole).27−29 In the case of indazole, previous to our works,15−17 Pozharskii et al. carried the reaction in 1964 in acid media.30 Some azoles have two different tautomers; this is the case for 1,2,3-triazole, 1,2,4-triazole, tetrazole, indazole and benzotriazole; for these azoles, tautomer and isomer structures (position of the NH/NR) often differ according to the Curtin–Hammett principle and the Winstein–Holnes equation.31 In the case of indazole 1a, MP2/6-31G** calculations indicate that the 1H-tautomer is 15 kJ·mol–1 more stable than the 2H tautomer.32 Similar values were obtained by other authors in the gas phase (14.5 kJ·mol–1) and in water (15.9 kJ·mol–1).33
In summary, the theoretical results we have reported above concern exclusively thermodynamic aspects, differences in energy between tautomers and isomers, and NMR chemical shifts. Note that there were no theoretical studies on the reaction mechanism.
Although the reaction can occur in neutral conditions, we have carried out our calculations on indazolium cations because our experimental procedure always includes hydrochloric acid.
There are two ways to prepare compounds 2a and 3a, from neutral indazoles 1a-1H and 1a-2H, reacting with neutral formaldehyde (Scheme 2, a and d reactions) or with protonated formaldehyde (Scheme 2, b and e reactions), or from protonated indazole 1aH+ (Scheme 2, c reaction). Obviously, the mechanism should involve protonated formaldehyde because it is a much weaker base (pKa = −4.2)34 than indazoles (1a, 1.04; 1b, 0.24; 1c, −0.96; 1d, −0.97; 1e, −0.99).35 Therefore, it is impossible to have a direct reaction between the indazolium cation and neutral or protonated formaldehyde (Scheme 2c reaction). We will see afterward how the reaction could involve indazolium cations with a relayed catalysis by a water molecule. In neutral conditions, zwitterions, zw, are intermediates to 3a and 2a.
The addition of azoles to carbonyl compounds is a reversible reaction that is more complete with aldehydes (for formaldehyde, see the Introduction section; for other aldehydes, see ref (36)) than with ketones like acetone.37,38 The reverse reaction (elimination) is very fast in the ketone adduct and rather slow in the aldehyde adduct; the combination of these two reaction rates (addition and elimination) accounts for the position of the equilibrium to the point that has incorrectly been named irreversible for formaldehyde adducts. It depends also on the azole where electron-withdrawing groups like nitro substituents increase the sensitivity to hydrolysis, i.e., to an increase of the reverse reaction rate due to the increased leaving group character for nitro derivatives. The pure samples prepared in 196815 contain in 2021 about 50% of free NH-indazole, that is, t1/2 ∼50 to 55 years in the solid state in a sealed tube (possibly formaldehyde polymerize into trioxymethylene or into paraformaldehyde). Starting from a pure adduct, crystallization in boiling water also leads to its partial decomposition.
Compounds 2aH+ and 3aH+ are in a formal way hemiaminals39−41 where the usual loss of water would lead to 1-methylene-1H-indazol-1-iums, a class of unknown nonaromatic cations. In most cases, the synthetic procedure we have used affords a pure compound (1H NMR of the crude), but crystallization in boiling water reverts the reaction and mixtures of the adduct and free indazole are obtained in proportions close to 50:50.
Results and Discussion
After reporting the synthetic schemes, we will establish the structures of the different hydroxymethyl-indazoles we have identified in this work. Since some of them are formed in small quantities or are unstable, we have followed a logical chain (1) to determine by X-ray crystallography the structure of all possible compounds, i.e., obtain crystals of all abundant and stable compounds; (2) to carry out GIAO/DFT calculations to confirm the assignment of the NMR spectra; (3) to record the solid-state NMR spectra (CPMAS) of the compounds whose X-ray structures have been determined; and (4) to register solution NMR spectra of all of the compounds and compare the NMR chemical shifts determined in solution with GIAO/DFT-calculated values to identify the structures that cannot be isolated.
Synthesis
The synthetic procedure reported in ref (15) (Scheme 3) was used with some differences. In the present work, we employ longer times and more water, particularly in the case of 7-nitro-1H-indazole (1e) that according to a previous report does not react with formaldehyde.15 In this last case, the effect of much longer times and microwave irradiation was also explored.
Scheme 3. Reaction of Indazoles with Formaldehyde; Given Also the Relative Ratios and the Isomerization Case.

Crystallization in boiling water affords pure 2a; however, in the case of 2b, 2c, and 2d, it results in the partial hydrolysis of the N-substituent with formation of 1b, 1c, and 1d (between 33 and 50% determined by integration of the 1H NMR spectrum). In Experimental Section, a detailed procedure on how to obtain suitable crystals for X-ray crystallography by avoiding decomposition is described.
In summary, according to the NMR results reported next, the reactions in HCl (aq) correspond to 1a → 2a, 1b → 2b (95%) + 3b (5%), 1c → 2c, 1d → 2d, and 1e → 3e; neutral 3e decomposes into 1e plus a small isomerization into 2e. Although the reactions in HCl (aq) should afford the indazolium salts, 2aH+, 2bH+, 3bH+, 2cH+, 2dH+, and 3eH+, the insolubility in water of the neutral indazoles makes that they precipitate by the addition of water. It is important to note that 3b is formed in an acid medium, while 2e is formed in a neutral solution.
X-ray Crystallography
No X-ray structures of N-methanol derivatives of indazoles are known, but those of benzimidazole and benzotriazole analogues are reported in the CSD;42 they correspond to the refcodes LANPOH43 and AJOQUL,17 respectively (Figure 2).
Figure 2.

1-Methanol derivatives of the three parent benzazoles (top) and 1-methanol derivatives of three C-nitro indazoles (bottom). The structure of 2c contains a molecule of dioxane (not represented).
We have succeeded in obtaining crystals good enough to solve the structures of parent compound 2a and those of the three nitro derivatives in Figure 2.
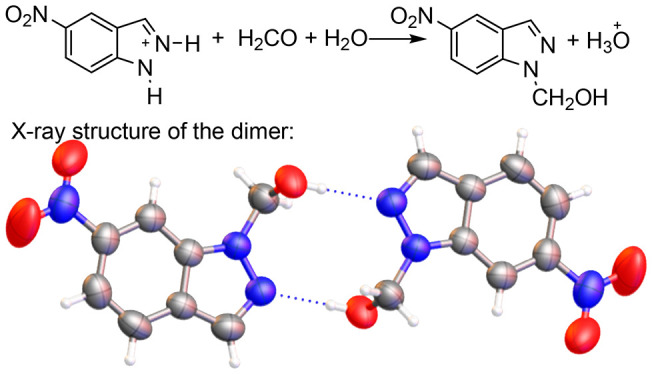
The four structures very similarly form dimers through intermolecular O–H···N hydrogen bonds (HBs) (Figure 3). For compound 2c, the crystallization molecule of dioxane is not represented.
Figure 3.

Four X-ray structures. The thermal ellipsoids are set at a 50% probability level.
The torsion angles of the 1-methanol substituent (N2–N1–C–O, Figure 4) and (N1–C–O–H) are 75.4/105.5°, 85.6/98.8°, 85.0/100.7°, and 86.4/101.3° for 2a, 2b, 2c, and 2d, respectively. The three nitro derivatives have average values of 85.7/100.3°, which differ from the unsubstituted derivative, 75.4/105.5°.
Figure 4.

Superposition of the four structures.
The nitro groups are almost coplanar with the benzene ring, with a mean value of 2.0° (lower and higher values of 1.75 and 2.65°, respectively). The O–H···N2 angles are 168.6, 149.3, 172.3, and 162.3° for 2a, 2b, 2c, and 2d, respectively (mean value of 163.1°). Note that an intermolecular O–H···N2 HB leading to dimers is preferred to the possible intramolecular HB of the monomer; this is due to angular strains in the HB that are much more favorable for the dimer.
GIAO/B3LYP/6-311++G(d,p) Calculations of NMR Chemical Shifts and Some Coupling Constants of the 10 Isomers, 2n to 3n, for n = a, b, c, d, e
This method has provided excellent results as long as there are no heavy atoms linked to the carbon atoms, i.e., HALA effects.44,45 Since the calculations afford absolute shieldings, σ ppm in the gas phase, it is necessary to use empirical equations to transform these data into chemical shifts, δ ppm in solution, equations that we have already established from a large set of data for 1H, 13C, and 15N NMR chemical shifts.46 The spin–spin coupling constants, SSCCs, do not need any transformation. The calculated values are reported in Table 1; the remaining coupling constants are given in the Supporting Information.
Table 1. GIAO/B3LYP/6-311++G(d,p)-Calculated 1H, 13C, and 15N NMR Chemical Shifts (δ in ppm) and Coupling Constants (J in Hz).
| 2a | 3a | 2b | 3b | 2c | 3c | 2d | 3d | 2e | 3e | |
|---|---|---|---|---|---|---|---|---|---|---|
| nuclei | H | H | 4-NO2 | 4-NO2 | 5-NO2 | 5-NO2 | 6-NO2 | 6-NO2 | 7-NO2 | 7-NO2 |
| 1H (ppm) | ||||||||||
| H3 | 7.83 | 7.82 | 8.79 | 8.70 | 8.00 | 8.05 | 7.94 | 7.88 | 7.96 | 7.97 |
| H4 | 7.65 | 7.62 | NO2 | NO2 | 8.70 | 8.78 | 7.64 | 7.61 | 7.87 | 7.92 |
| H5 | 7.16 | 7.06 | 8.26 | 8.25 | NO2 | NO2 | 8.16 | 8.05 | 7.15 | 7.08 |
| H6 | 7.37 | 7.26 | 7.35 | 7.29 | 8.40 | 8.23 | NO2 | NO2 | 8.17 | 8.49 |
| H7 | 7.48 | 7.72 | 7.77 | 8.03 | 7.40 | 7.64 | 8.55 | 8.82 | NO2 | NO2 |
| CH2 | 5.64 | 5.48 | 5.70 | 5.54 | 5.63 | 5.48 | 5.72 | 5.52 | 6.16 | 5.54 |
| OH | 1.59 | 2.15 | 1.74 | 2.29 | 1.79 | 2.33 | 1.80 | 2.28 | 1.42 | 2.59 |
| 13C (ppm) | ||||||||||
| C3 | 134.0 | 120.8 | 135.4 | 125.3 | 136.7 | 125.8 | 134.1 | 122.0 | 134.7 | 122.9 |
| C3a | 126.6 | 123.7 | 120.3 | 116.7 | 125.5 | 121.6 | 129.4 | 125.8 | 131.2 | 126.7 |
| C4 | 120.6 | 120.2 | 142.1 | 142.6 | 118.6 | 120.0 | 120.1 | 120.2 | 126.8 | 128.8 |
| C5 | 120.8 | 122.3 | 119.3 | 121.9 | 144.5 | 145.3 | 117.0 | 117.6 | 119.5 | 120.0 |
| C6 | 126.0 | 126.1 | 124.3 | 123.9 | 122.5 | 121.7 | 147.8 | 148.4 | 125.8 | 126.4 |
| C7 | 108.4 | 119.0 | 115.7 | 128.0 | 107.8 | 118.6 | 106.3 | 117.6 | 138.8 | 139.8 |
| C7a | 139.7 | 150.8 | 140.7 | 151.2 | 141.2 | 151.3 | 138.0 | 148.7 | 131.4 | 142.6 |
| CH2 | 72.4 | 76.7 | 73.0 | 77.0 | 72.8 | 77.0 | 72.7 | 77.2 | 76.5 | 77.2 |
| 15N (ppm) | ||||||||||
| N1 | –181.1 | –93.3 | –177.8 | –91.4 | –177.2 | –91.4 | –174.7 | –81.9 | –172.4 | –87.7 |
| N2 | –60.7 | –144.5 | –50.1 | –136.0 | –52.4 | –136.2 | –45.4 | –133.5 | –48.4 | –136.3 |
| NO2 | –15.8 | –16.1 | –17.3 | –16.7 | –16.8 | –16.2 | –14.1 | –17.6 | ||
| SSCC (Hz) | ||||||||||
| 3JCH2OH | –10.7 | –9.3 | –10.9 | –9.4 | –10.9 | –9.5 | –10.9 | –9.5 | –10.3 | –9.3 |
| 3JH4H5 | 7.4 | 7.8 | NO2 | NO2 | NO2 | NO2 | 8.2 | 8.6 | 7.2 | 7.6 |
| 3JH5H6 | 6.4 | 6.2 | 7.3 | 7.2 | NO2 | NO2 | NO2 | NO2 | 7.3 | 7.2 |
| 3JH6H7 | 7.7 | 8.1 | 7.6 | 7.9 | 8.6 | 8.5 | NO2 | NO2 | NO2 | NO2 |
| 4JH4H6 | 0.4 | 0.5 | 1.5 | 1.5 | 0.3 | 0.5 | ||||
| 4JH5H7 | 0.3 | 0.3 | 0.2 | 0.2 | 1.3 | 1.4 | ||||
| 5JH3H7 | 0.6 | 0.8 | 0.6 | 0.7 | 0.6 | 0.7 | 0.8 | 0.8 | NO2 | NO2 |
Obviously, the 1H and 13C chemical shifts of the aromatic indazole ring in Table 1 depend on the presence and position of the nitro group. In 1H NMR, in what concerns the methanol group, the OH proton shows some interesting variations but, since this signal is strongly dependent on the solvent, they are of little interest. Note, however, that the difference between the 2 and 3 isomers is about 0.5 ppm except in the e series where it reaches 1.2 ppm. The CH2 group appears between 5.5 and 5.6 ppm; only in compound 2e, it resonates at 6.2 ppm due to the proximity of the 7-nitro group.
The 13C chemical shifts are very different in isomers 2 and 3, a fact well known for other N-substituted indazoles.47,48 The signal of C3, a singlet or a doublet with a small coupling constant, is also a useful probe to determine the position of the CH2OH group: 135 ppm (2) and 123 ppm (3) in average. The 15N chemical shifts of N1 and N2 atoms are also very different for isomers 2 and 3.
The SSCCs of the methanol group are slightly larger in the 2 series (−10.7 Hz) than in the 3 series (−9.4 Hz). The ortho SSCCs are normal and the W coupling,49,505JHH between protons H3 and H7,15,17,47 is calculated to be 0.7–0.8 Hz. The 4JHH coupling constants are small (between 0.2 and 0.5 Hz) except when there is a nitro group in the central carbon [H–C–C(NO2)–C–H] where it attains 1.4–1.5 Hz; this effect of the nitro group has been reported for benzene derivatives.51,52
Solid-State Nuclear Magnetic Resonance (SSNMR) Results (CPMAS Experiments)
The chemical shifts of the four compounds, 2a–2d, whose X-ray structures have been determined in this work, are given in Table 2. As often happens in CPMAS, some signals are split, for instance, those of compound 2c. For this compound, when comparing its chemical shifts δExp (Table 2) with the calculated values δGIAO (Table 1), mean values have been used.
Table 2. 13C and 15N NMR Data of (1H-Indazol-1-yl)Methanol Derivatives in the Solid State (CPMAS).
| nuclei | 2a | 2b (4-NO2) | 2c (5-NO2) | 2d (6-NO2) |
|---|---|---|---|---|
| C3 | 133.5 | 134.3 | 136.7 | 135.3 |
| C3a | 126.0 | 120.2 | 123.3 | 125.9 |
| C4 | 119.4 | 138.8 | 118.9 | 124.0 |
| 121.2 | ||||
| C5 | 121.3 | 118.9 | 141.7 | 114.4 |
| C6 | 123.0 | 126.3 | 121.2 | 145.6 |
| 123.3 | ||||
| C7 | 109.0 | 115.3 | 109.2 | 103.8 |
| 109.7 | ||||
| C7a | 139.4 | 140.5 | 141.7 | 137.3 |
| CH2 | 69.3 | 70.7 | 70.1 | 69.8 |
| N1 | –173.4 | –171.7 | –172.9 | –173.4 |
| N2 | –68.2 | –66.6 | –63.3 | –65.4 |
| NO2 | –6.4 | –5.9 | –6.6 |
Comparing the values in Tables 1 and 2 results in the following regression equations
| 1 |
| 2 |
The largest residuals for 15N signals in the simple regression equation, eq 1, correspond to NO2 and N2. Including these effects as dummy variables, eq 2 was obtained with +9.7 and −16.0 ppm corrections for NO2 and N2, respectively. In any case, the 15N chemical shifts only can correspond to (indazol-1-yl)methanol isomers 2.
NMR in Solution
The experimental chemical shifts and SSCC in DMSO-d6 solution are reported in Table 3.
Table 3. 1H, 13C{1H}, and 15N NMR Data of (1H-Indazol-1-yl)Methanol Derivatives in DMSO-d6 Solutiona.
| 2ab | 2b | 3b | 2c | 2d | 1e–2H | 2e | 3e | |
|---|---|---|---|---|---|---|---|---|
| nuclei | H | 4-NO2 | 4-NO2 | 5-NO2 | 6-NO2 | 7-NO2 | 7-NO2 | 7-NO2 |
| 1H (ppm) | ||||||||
| H3 | 8.09 | 8.54 | 8.91 | 8.42 | 8.34 | 8.43 | 8.30 | 8.85 |
| H4 | 7.72 | NO2 | NO2 | 8.83 | 8.03 | 8.33e | 8.19 | 8.37 |
| H5 | 7.17 | 8.20 | 8.21 | NO2 | 7.99 | 7.37 | 7.38 | 7.28 |
| H6 | 7.41 | 7.68 | 7.40 | 8.27 | NO2 | 8.36e | 8.28 | 8.48 |
| H7 | 7.77 | 8.27 | 8.30 | 7.92 | 8.78 | NO2 | NO2 | NO2 |
| CH2 | 5.73 | 5.79 | 5.79 | 5.78 | 5.86 | 5.83 | 5.80 | |
| OH | 6.68 | 6.95 | 7.50 | 6.94 | 6.93 | 6.59 | 7.48 | |
| 13C (ppm) | ||||||||
| C3 | 134.2 | 132.1 | 124.8 | 136.4 | 134.0 | 136.2 | 135.5 | 120.4 |
| C3a | 126.0 | 116.6 | 113.9 | 123.3 | 127.4 | 127.6 | 130.6g | 125.7 |
| C4 | 121.6 | 140.6 | 143.0 | 118.9 | 122.2 | 130.4 | 128.8 | 126.7 |
| C5 | 121.7 | 118.7 | 120.7 | 141.4 | 115.3 | 120.7 | 121.3 | 120.4 |
| C6 | 127.0 | 126.0 | 123.8 | 121.0 | 145.9 | 124.0 | 124.8 | 125.7 |
| C7 | 111.0 | 118.3 | 126.6 | 112.0 | 107.2 | 132.6f | 138.2g | 137.4 |
| C7a | 139.8 | 139.7 | 149.2 | 140.8 | 137.6 | 132.4f | 130.8g | 140.1 |
| CH2 | 71.6 | 71.5 | 75.8 | 71.4 | 71.4 | 75.3 | 76.2 | |
| 15N (ppm) | ||||||||
| N1 | –180.8b | –173.7 | –90.2 | –174.3 | –173.0 | –170.1 | –86.5 | |
| N2 | –60.5b | –50.6 | –134.2 | –50.3 | –45.2 | –47.7 | –134.5 | |
| NO2 | –15.6 | –15.9 | –17.1 | –16.6 | –13.9 | –17.4 | ||
| SSCC (Hz) | ||||||||
| 3JCH2OH | –7.3c | –7.5c | –8.0c | –7.5c | –7.4c | –7.7c | –7.9c | |
| 3JH4H5 | 8.5 | NO2 | NO2 | NO2 | 8.8 | 7.9 | 7.9 | 8.2 |
| 3JH5H6 | 7.5 | 7.3 | 7.7 | NO2 | NO2 | 7.9 | 7.9 | 7.5 |
| 3JH6H7 | 8.0 | 7.6 | 8.4 | 9.2 | NO2 | 7.9 | NO2 | NO2 |
| 4JH4H6 | 1.0 | NO2 | NO2 | 2.2 | NO2 | 0.9 | 1.0 | 1.0 |
| 4JH5H7 | 0.9 | 0.9 | d | NO2 | 1.5 | NO2 | NO2 | NO2 |
| 5JH3H7 | 0.8d | 0.9 | 0.0 | 1.0 | 0.0 | NO2 | NO2 | NO2 |
In the 1H NMR spectrum of the reaction crude between protonated 7-nitroindazole (1eH+) and formaldehyde, we observed three triplets of the same intensity, 1:1:1, and approximately the same coupling constant, 8.0, 7.9, and 7.9 Hz for the 7.48, 7.38, and 7.28 ppm signals, respectively (Figure 5). By analogy to other compounds, these multiplets should correspond to three H5 protons coupled with H4 and H6.
Figure 5.

7.2–7.5 ppm region of the 1H NMR in DMSO-d6 at 400 MHz of the reaction product between 1eH+ and formaldehyde.
When the spectrum of 1e was recorded at 500 MHz in DMSO-d6, its 1H NMR spectrum shows some very unusual 1H–1H coupling constants (Figures 6 and 7).
Figure 6.

1H NMR spectrum (8.30–8.44 and 7.32–7.40 ppm regions) of 1e in DMSO-d6 at 500 MHz.
Figure 7.

1H–1H SSCCs of some indazoles and indoles first-order analyzed.
Those measured in Figure 6 are reported on the left side in Figure 7. Because prototropy couplings with the NH are very rare and have been observed only on 3-azido-1H-indazole, we assigned this to the azido group blocking the tautomerism of indazole.53 Indazole tautomer 1H resembles 1H-indole where H1 is coupled, besides to H2, to H3 and H4.54 The calculated SSCCs of 1e–1H are given on the right side in Figure 7. The strong HB between H1 and one oxygen atom of the nitro group prevents the prototropy and allows the SSCCs with H1 to be observed. Note that the 1H NMR spectrum of 1e–1H has been described several times but these small couplings were never reported.47,55,56
The spectrum of 1e–1H in the region of the NH proton (DMSO-d6 at 500 MHz) shows two signals, a large one (13.95 ppm, 94%) and a small one (14.83, 6%), as shown in Figure 8. We assign the small signal to tautomer 1H by analogy with the GIAO calculations, 10.14 and 10.81 ppm. The differences are 0.88 ppm, experimental, and 0.67 ppm, calculated, and the shift produced by the solvent is about 3.9 ppm. The other signals of the minor tautomer are not observed except that of H6 that appears at 8.58 ppm (3J56 = 7.9, 4J46 = 0.9 Hz) due to the spinning side bands and the big signals of the 1e–2H tautomer. An equation relies on experimental and calculated values if the effect of DMSO on NH signal is taken into account: Exp. = (0.95 ± 0.16) Calc. + (3.8 ± 0.4) NH, n = 7, R2 = 0.998.
Figure 8.

1H NMR complete spectrum (0–16 ppm) of indazole 1e at 500 MHz in DMSO-d6. The inset corresponds to the zone of NH protons.
Actually, the triplets of Figure 5 correspond to a 1:1 mixture of 1e–1H (H5 at 7.38 ppm) and 3e (H5 at 7.28 and OH at 7.48 ppm). The A2X system of the methanol part appears well resolved in some 1H NMR spectra in DMSO-d6 (3JHH ∼7.5 Hz) (Figure 9), which is not always the case for common alcohols.
Figure 9.

Appearance of the CH2OH group in 1H NMR spectra in DMSO-d6 at 400 MHz of the derivative of 6-nitro-1H-indazole 2d; 3JCH2OH = 7.5 Hz.
Note that the 3JH4H5 and 3JH5H6 are identical for 2e and different for 3e: this is characteristic of 1- and 2-substituted indazoles.47 COSY experiments correlate OH → CH2 → H3 → H4 → H5 → H6 in the case of 2e and 3e with some exceptions when signals are under the larger signals of 1e.
To compare the experimental values of Table 3 (DMSO-d6 solution) with the calculated chemical shifts in Table 1 (gas phase), we have used simple regressions between both values, except in two cases. First, in 1H NMR chemical shifts, the OH signal is systematically underestimated because our equations relating absolute shieldings in the gas phase to chemical shifts in solution correct general solvent effects and not the hydrogen bond between the OH and DMSO.57,58 Second, in the 3JCH2OH SSCC, the same happens for the same reason. To correct these deviations, an additional variable (1 if OH was present and 0 if it was absent) was added.59−61 In any case, the intercept was not significant and the regressions were repeated imposing intercept = 0, but the squared correlation coefficient, R2, was that of the regression with the intercept because imposing the intercept to be 0 increased considerably the R2 value (Table 4).
Table 4. Results of the Slopes of the Five Regression Equations: Experimental Values = a Calculated Values + b OH Protons.
| eq | no. of points | a calc. | b OH | R2 | RMS error | |
|---|---|---|---|---|---|---|
| 1 | 1H chemical shifts | 44 | (1.02 ± 0.01) | (5.1 ± 0.1) | 0.947 | 0.23 ppm |
| 2 | 13C chemical shifts | 53 | (0.996 ± 0.002) | 0.994 | 1.7 ppm | |
| 3 | 15N chemical shifts | 6 | (0.986 ± 0.005) | 1.000 | 1.7 ppm | |
| 4 | 1H, 13C, and 15N | 103 | (0.994 ± 0.001) | (5.1 ± 0.5) | 1.000 | 1.2 ppm |
| 5 | 1H–1H SSCC | 25 | (1.08 ± 0.02) | –(3.5 ± 0.3) | 0.962 | 0.6 Hz |
The slopes are close to 1.0; the experimental 1H chemical shifts of the OH are 5.1 ppm higher on average than the calculated ones, while the SSCCs involving the OH group, 3JCH2OH, are 3.5 Hz lower.
The most interesting 1H NMR spectra are those of the crude of 2b (Figure 10, neutral solid in DMSO-d6 solution) and those of the crude of 3e freshly prepared (Figure 11, filtered solid in DMSO-d6 solution) and after a week in the NMR tube.
Figure 10.

1H NMR spectrum of crude of 2b in DMSO-d6 solution containing 95% of 2b and 5% of 3b.
Figure 11.

Top: 1H NMR spectrum of the crude of 3e in DMSO-d6 solution freshly prepared; bottom: 1H NMR spectrum of the crude of 3e in DMSO-d6 solution after a week in the NMR tube (both at 400 MHz). The signals in the 4.5–5.0 ppm region and a doublet in the 5.5–6.0 region are not indazole derivatives but most probably formaldehyde short polymers.
After crystallization (see the Supporting Information), the 5% amount of 3b has been eliminated.
Figure 11 (top) corresponds to a mixture of starting 7-nitro-1H-indazole 1e and its 2-methanol derivative, 3e. After a week, Figure 11 (bottom), 3e (neutral) has decomposed into 1e and a small quantity of another compound that we have identified as isomer 2e.
This behavior (Scheme 4) will be explained by the theoretical calculations in the following section.
Scheme 4. Decomposition of 3e into 1e (Major) and 2e (Minor).

Reaction Mechanism: DFT Calculations
According to the conclusions of the NMR analyses, the five indazoles regroup in three cases: obtaining in HCl (aq) 1-methanol derivatives (unsubstituted a, 5-nitro c, and 6-nitro d), obtaining a mixture of 1- and 2-methanol derivatives (95/5 4-nitro b), and obtaining a 2-methanol derivative 3e that in DMSO-d6 decomposes and partly isomerizes into 1-methanol derivative 2e (7-nitro e).
In Table 5 are the energies corresponding to the equilibria between 2 and 3 isomers; in all cases, the 1-CH2OH isomer is more stable than the 2-CH2OH one, similarly to what happens for the NH tautomers; note that the values are similar except in the case of the e series where the difference is much larger, about 3.5 times. This is due to a strong hydrogen bond between the N–H and O=N–O– bonds (Figure 12) that disappears in the N-1-substituted derivative, confirming the NMR discussion about the HB (Figures 6 and 7). An analogous HB is present in 3,7-dinitro-1H-indazole.62 Protonation on N2 must reinforce the strength of the HB, being now N1(+)–H···O. The X-ray distances of the atoms involved in the hydrogen bond are the mean of two very similar structures;63,64 the only difference between the experimental and the calculated geometry lies in the N–H distance, which is underestimated by X-ray crystallography;65 this in turn affects the O···H distance.
Table 5. Energies (kJ·mol–1) Corresponding to Scheme 3 Calculated at the DLPNO/CCSD(T)/def2-TZVP//B3LYP/6-311++G(d,p) Levela.
| parent | 4-NO2 | 5-NO2 | 6-NO2 | 7-NO2 | |
|---|---|---|---|---|---|
| a | b | c | d | e | |
| 2-isomer 1-CH2OH (reference) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 3 isomer 2-CH2OH | 18.6 | 12.6 | 16.6 | 16.9 | 13.3 |
| 1-1H (reference) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 1-2H | 18.3 | 10.5 | 16.3 | 16.6 | 42.9 |
The values are given relative to reference compounds.
Figure 12.

Experimental (adapted) and calculated structures of 1e–1H.
The differences decrease in the order a > d > c > e > b. The formation of 2e from 3e is not related to the ΔΔE value (13.7 kJ·mol–1) but simply that it is only in the e series that 2-isomer 3 is formed since in all cases the 2 isomers are more stable than the 3 isomers.
The mechanism for the unsubstituted indazole, a series, is represented in a simplified way in Scheme 5 and in a more realistic way, including TSs and IRCs (see the Supporting Information), in Figure 13.
Scheme 5. Proposed Mechanism Illustrated for a Series.

The indazolium rings are rotated in the right part of the figure to keep formaldehyde, water molecules, and the hydroxymethyl group at the same position.
Figure 13.

The differences in stability of the five pairs of indazolium salts are reported in Scheme 6 and Table 6. In this table, N1 and N2 indicate the position of the CH2OH group and complex, TS, and adduct corresponds to the complex, transitions state, and adduct in Scheme 5.
Scheme 6. Relative Stability of Water-Solvated 1- and 2-Methanol Indazolium Salts.

Table 6. Energies (kJ·mol–1) Corresponding to Scheme 6; x = a, b, c, d, ea.
| DLPNO + PCM | ||||||
|---|---|---|---|---|---|---|
| water | parent a | 4-NO2 b | 5-NO2 c | 6-NO2 d | 7-NO2 e | mean a–d |
| N1–complex 1xH+·CH2O·H2O | 0.0 | 0.0 | 0.0 | 0.0 | 0.0b | 0.0 |
| N1–TS | 82.2 | 67.9 | 69.7 | 70.1 | b | 72.5 |
| N1–adduct 2xH+·OH2 | –54.6 | –49.3 | –48.9 | –46.9 | –29.7 | –49.9 |
| N2–complex 1xH+·CH2O·H2O | –2.3 | –2.7 | –1.4 | 0.1 | 0.0 | –1.6 |
| N2–TS | 74.6 | 57.8 | 60.2 | 61.0 | 61.8 | 63.4 |
| N2–adduct 3xH+·OH2 | –38.1 | –55.9 | –54.9 | –53.1 | –52.3 | –50.5 |
| N2–adduct – N1–adduct | 16.5 | –6.6 | –6.0 | –6.2 | –22.6 | –0.6 |
The differences between the N-complexes and between the N-adducts are also reported. The energies correspond to DLPNO/CCSD(T) single-point calculations including the contribution of PCM–water. The gas phase values are reported in the Supporting Information.
See the comment below.
Although there are some differences in Table 6, the behavior of the a, b, c, and d series is similar (see mean a–d): in 1-series, a barrier of about 72 kJ·mol–1, the adduct being more stable than the complex by about 52 kJ·mol–1; in 2-series, a barrier of about 63 kJ·mol–1, the adduct being more stable than the complex by about 2 kJ·mol–1. The differences between both series, bottom of Table 6, are very small, ±1.6 kJ·mol–1. The e series is very different; when reacting by N2–H, far from the nitro group, the behavior is near identical, 52.3/50.5 and 61.8/63.4 kJ·mol–1, but when reacting N1–H, hydrogen-bonded to the nitro group, the complex spontaneously isomerizes to the complex formed by N2–H, which leads to 3eH+·OH2 (Scheme 6). This explains why this isomer reacts differently from all of the other indazoles.
Conclusions
We have demonstrated that the reaction of NH-indazoles with formaldehyde, previously reported to yield exclusively 1-CH2OH derivatives, gives rise in some cases to 2-CH2OH indazoles, as found for 4-nitro-1H-indazole (1b) and 7-nitro-1H-indazole (1e). This result is important when hydroxymethyl-indazoles are used as intermediates without isolating them.
The structure, tautomerism, and reactivity of 1e are of interest because of its unique ability to inhibit both MAO-B and nNOS, two biologically important enzyme systems. Furthermore, its general use as an investigative drug to study the inhibition of nNOS makes the structural study of this molecule very relevant.12,13 This compound is the first reported indazole where both tautomers have been observed and the second in which spin–spin coupling constants with H1 have been observed observed and determined.
The X-ray structures of four 1-CH2OH indazoles, (1H-indazol-1-yl)methanol (2a), (4-nitro-1H-indazol-1-yl)methanol (2b), (5-nitro-1H-indazol-1-yl)methanol (2c), and (6-nitro-1H-indazol-1-yl)methanol (2d), were solved, offering a solid ground for NMR spectra in the solid state, and, in turn, these spectra were used for assigning the NMR spectra in DMSO-d6 solution.
Theoretical calculations at the B3LYP/6-311++G(d,p) level have been used to understand the reaction mechanism and, in particular, the different behavior of 1e. Besides, GIAO calculations based on the optimized geometries proved an excellent tool to identify indazole isomers.
Experimental Section
General Methods
Acetonitrile, nitromethane, dioxane, heptane, hydrochloric acid, and indazoles were purchased from Merck without further purification. Melting points were determined by a capillary method in a Metler Toledo scientific melting point apparatus (MP760) at a heating rate of 1 °C/min. A PerkinElmer Spectrum Two, fitted with a diamond single-bounce ATR, was used to collect the IR spectra at 4 cm–1 spectral resolution with four co-adds (i.e., the number of averaged replicate spectra). The compound was pressed on the diamond crystal and measured directly without any further sample. For 1H and 13C NMR spectra, see below. Reactions heated under microwave irradiation were carried out for 60 min at 80 °C in sealed reaction vessels of a Biotage Initiatior microwave oven reactor (frequency: 2045 GHz). Analytical HPLC was performed with a SunFire C18, 3.5 μm column (4.6 mm × 50 mm). Mobile phase A was CH3CN + 0.08% formic acid, and mobile phase B was H2O + 0.05% formic acid. The gradient was from 10 to 95% of acetonitrile. UV diode array detection was carried out from 190 to 440 nm.
General Synthesis of Indazolyl-N-Methanol Derivatives
All of the indazolyl-N-methanol derivatives were synthesized using the method reported in the literature15 with some differences: the reactions were stirred overnight at room temperature to ensure that all final products were obtained and no crystallization from water was used (because the starting products were obtained in this solvent). Indazoles (42 mmol) are suspended in 30 mL of 30% hydrochloric acid and then 3.85 mL of a 30% aqueous solution of formaldehyde (42 mmol) was added. After 1 h, 30 mL of water was added and the mixture was kept overnight at room temperature. The precipitate was collected by filtration to give a solid. To obtain crystals, the solid was suspended in the solvent specified for each compound and heated and the solution was filtered to remove undesirable products. By slow cooling, crystals were precipitated and removed from the solvent to give the desired compound. Crystallization solvents were specified for each compound. Compound 3b (4-NO2) was obtained as a minor product and could not be isolated and was only observed by NMR. Compound 2e and 3e (7-NO2) could not be isolated due to decomposition but could be detected by NMR.
(1H-indazol-1-yl)methanol (2a)
Yield 98% (6.15 g), white crystalline solid; mp: 110.5–111.5 °C (heptane); 1H NMR (400 MHz, DMSO-d6): δ 8.09 (s, 1H, H3), 7.77 (d, 1H, J = 8.0 Hz, H7), 7.72 (d, 1H, J = 8.5 Hz, H4), 7.41 (t, 1H, J = 8.0 Hz, H6), 7.17 (t, 1H, J = 8.5 Hz, H5), 6.68 (t, 1H, J = 7.3 Hz, OH), 5.73 (d, 2H, J = 7.3 Hz, CH2) ppm. 13C{1H} NMR (100 MHz, DMSO-d6): δ 139.8 (C7a), 134.2 (C3), 127.0 (C6), 126.0 (C3a), 121.7 (C5), 121.6 (C4), 110.1 (C7), 71.6 (CH2) ppm. MS (ES+): m/z: calcd for [M + H]+ C8H9N2O: 149.07, found: 148.94 (3.168 min).
(4-nitro-1H-indazol-1-yl)methanol (2b)
Yield 92% (7.52 g), yellow crystalline solid; mp: 168–170 °C (acetonitrile); 1H NMR (400 MHz, DMSO-d6): δ 8.54 (s, 1H, H3), 8.27 (d, 1H, J = 7.6 Hz, H7), 8.20 (d, 1H, J = 7.3 Hz, H5), 7.68 (t, 1H, J = 7.3 Hz, H6), 6.95 (t, 1H, J = 7.5 Hz, OH), 5.79 (d, 2H, J = 7.5 Hz, CH2) ppm. 13C{1H} NMR (100 MHz, DMSO-d6): δ 140.6 (C4), 139.7 (C7a), 132.1 (C3), 126.0 (C6), 118.7 (C5), 118.3 (C7), 116.6 (C3a), 71.5 (CH2) ppm. MS (ES+): calcd for [M + H]+ C8H8N3O3: 194.06, found: 194.05 (4.162 min).
(4-nitro-2H-indazol-2-yl)methanol (3b)
(4-NO2, minor product, NMR tube). Mp: 1H NMR (400 MHz, DMSO-d6): δ 8.91 (s, 1H, H3), 8.30 (d, 1H, J = 8.4 Hz, H7), 8.21 (d, 1H, J = 7.7 Hz, H5), 7.50 (t, 1H, J = 8 Hz, OH), 7.40 (t, 1H, J = 8.4 Hz, H6), 5.79 (d, 2H, J = 8.0 Hz, CH2) ppm. 13C{1H} NMR (100 MHz, DMSO-d6): δ 149.2 (C7a), 143.0 (C4), 126.6 (C7), 124.8 (C3), 123.8 (C6), 120.7 (C5), 113.9 (C3a), 75.8 (CH2) ppm.
(5-nitro-1H-indazol-1-yl)methanol (2c)
Yield 96% (7.85 g), light yellow crystalline solid; mp: 155.5–156.5 (dioxane) °C; 1H NMR (400 MHz, DMSO-d6): δ 8.83 (s, 1H, H4), 8.42 (s, 1H, H3), 8.27 (d, 1H, J = 9.2, H6), 7.92 (d, 1H, J = 9.2 Hz, H7), 6.94 (t, 1H, J = 7.5 Hz, OH), 5.78 (d, 2H, J = 7.5 Hz, CH2) ppm. 13C{1H} NMR (100 MHz, DMSO-d6): δ 141.4 (C5), 140.8 (C7a), 136.4 (C3), 123.3 (C3a), 121.0 (C6), 118.9 (C4), 112.0 (C7), 71.4 (CH2) ppm. MS (ES+): calcd for [M + H]+ C8H8N3O3: 194.06, found: 193.95 (4.250 min).
(6-nitro-1H-indazol-1-yl)methanol (2d)
Yield 94% (7.69 g), brown crystalline solid; mp: 142.5–143.5 °C (nitromethane); 1H NMR (400 MHz, DMSO-d6): δ 8.78 (s, 1H, H7), 8.34 (s, 1H, H3), 8.03 (d, 1H, J = 8.8 Hz, H4), 7.99 (dd, 1H, J = 8.8, 1.5 Hz, H5), 6.93 (t, 1H, J = 7.4 Hz, OH), 5.86 (d, 2H, J = 7.4 Hz, CH2) ppm. 13C{1H} NMR (100 MHz, DMSO-d6): δ 145.9 (C6), 137.6 (C7a), 134.0 (C3), 127.4 (C3a), 122.2 (C4), 115.3 (C5), 107.2 (C7), 71.4 (CH2) ppm. MS (ES+): calcd for [M + H]+ C8H8N3O3: 194.06, found: 193.98 (4.520 min).
(7-nitro-1H-indazol-1-yl)methanol (2e)
(NMR tube). Yellow solid. 1H NMR (500 MHz, DMSO-d6): δ 8.30 (s, 1H, H3), 8.28 (d, 1H, H6), 8.19 (d, 1H, J = 7.9 Hz, H4), 7.38 (t, 1H, J = 7.9 Hz, H5), 6.59 (t, 1H, J = 7.7 Hz, OH), 5.83 (d, 2H, J = 7.7 Hz, CH2) ppm. 13C{1H} NMR (125 MHz, DMSO-d6): δ 138.2 (C7), 135.5 (C3), 130.8 (C7a), 130.6 (C3a), 128.8 (C4), 124.8 (C6), 121.3 (C5), 75.3 (CH2) ppm.
(7-nitro-2H-indazol-2-yl)methanol (3e)
(NMR tube). 1H NMR (500 MHz, DMSO-d6): δ 8.85 (s, 1H, H3), 8.48 (d, 1H, H6), 8.37 (d, 1H, J = 8.2 Hz, H4), 7.28 (t, 1H, J = 7.9, 2 Hz, H5), 7.48 (t, 1H, J = 7.9 Hz, OH), 5.80 (d, 2H, J = 7.9 Hz, CH2) ppm. 13C{1H} NMR (125 MHz, DMSO-d6): δ 140.1 (C7a), 137.4 (C7), 126.7 (C4), 125.7 (C3a), 125.7 (C6), 120.4 (C3), 120.4 (C5), 76.2 (CH2) ppm.
X-ray Crystallographic Methods
Colorless parallel pipe-shaped crystals of 2a, 2b, 2c, and 2d were selected under a polarizing optical microscope. Data were collected at 250 K on a Bruker X8 four circle kappa-diffractometer equipped with a Cu Incoatec microsource operating at 50 W power (50 kV, 1.0 mA) to generated Cu Kα radiation (λ = 1.54178 Å) and a Bruker VANTEC 500 area detector (microgap technology). Diffraction data were collected exploring over a hemisphere of the reciprocal space in a combination of φ and ω scans to reach a resolution of around 0.85 Å, using the Bruker APEX21 software suite (each exposure, depending on ω, was of 10, 30, or 60 s covering 1° in ω or φ). Unit cell dimensions were determined by a least-squares fit of reflections with I > 2 σ(I). Data were integrated and scaled using the SAINTplus program.66 A semiempirical absorption and scale correction based on equivalent reflection was carried out using SADABS.67 Space group determination was carried out using XPREP.69 The structure was solved by direct methods using SHELXT,68 showing all no-hydrogen atoms. Additional cycles of refinement and electron difference maps show the rest of hydrogen atoms. The hydrogen atoms were refined riding on the coordinates of the respectively C-bonded atoms. The OH hydrogen atoms were allowed to ride on the O atom and rotate about the C–O bond. All calculations were performed using APEX3 software for data collection and OLEX2-1.369 and SHELXTL69 to resolve and refine the structure. Mercury70 was used for structural figures and supramolecular packing studies. The final structure was examined and tested using PLATON.71 A summary of the main crystallographic data is shown in Table S1, and ORTEP representations of the asymmetric units are shown in Figure S19a–d.
NMR Spectroscopy
Solution spectra were recorded either on three spectrometers, a Bruker DRX-400 (9.4 Tesla, 400.13 MHz for 1H, 100.62 MHz for 13C and 40.54 MHz for 15N), a Bruker Avance III HD-400 (1H 399.86 MHz, 13C 100.55 MHz), and a Varian SYSTEM 500 NMR (1H 499.81 MHz, 13C 125.69 MHz) equipped with a 5 mm HCN cold probe. Chemical shifts (δ in ppm) are given from the internal solvent: DMSO-d6, 2.49 for 1H and 39.5 for 13C. Nitromethane was used as an external reference for 15N. For 13C, WALTZ-16 was used for broadband proton decoupling and 15N NMR spectra were acquired using 2D (1H–15N) gradient-selected heteronuclear multiple bond correlation by means of standard pulse sequences and in absolute mode.
Typical parameters: for 1H spectra, spectral width of 5200 Hz, acquisition time of 6.3 s, digital resolution of 0.41 Hz per point, and pulse width of 7.6 μs at an attenuation level of −1 dB; for 13C spectra, spectral width of 20.2 kHz, acquisition time of 1.6 s, digital resolution of 1.12 Hz per point, pulse width of 14.5 μs at an attenuation level of −4 dB, and relaxation delay of 2 s; the FIDS were multiplied by an exponential weighting (lb = 1 Hz) before Fourier transformation.
Solid-state 13C (100.73 MHz) and 15N (40.60 MHz) CPMAS NMR spectra were obtained on a Bruker WB 400 spectrometer at 300 K using a 4 mm DVT probehead. Samples were carefully packed in a 4 mm diameter cylindrical zirconia rotor with Kel-F end caps. 13C spectra were originally referenced to a glycine sample, and then the chemical shifts were recalculated to the Me4Si [for carbonyl atom (glycine) δ = 176.1 ppm] and 15N spectra to 15NH4Cl and then converted to the nitromethane scale using the following relationship: δ15N (nitromethane) = δ15N (ammonium chloride) −338.1 ppm. Typical acquisition parameters for 13C CPMAS are as follows: 3.2 μs 90° 1H pulses and decoupling SPINAL 6472 sequence spectral width, 40 kHz; recycle delay, 5–120 s; acquisition time, 30 ms; contact time, 2–4 ms; and spin rate, 12 kHz. Typical acquisition parameters for 15N CPMAS are as follows: 3.2 μs 1H 90° pulses (SPINAL 64) spectral width, 40 kHz; recycle delay, 5–120 s; acquisition time, 35 ms; contact time, 7–9 ms; and spin rate, 6 kHz.
Abbreviations for multiplicity are as follows: d indicates doublet, t indicates triplet, m indicates multiplet, bs indicates broad singlet, bd indicates broad doublet, dd indicates double doublet, dt indicates double triplet. Chemical shifts are reported in ppm referenced to DMSO-d6 at 2.50 ppm for 1H NMR and at 39.5 ppm for 13C NMR, and coupling constants in hertz (Hz).
The assignment of the signals in solution is based on conventional 2D techniques, 1H–1H COSY, HMBC, and HSQC, and by comparisons with calculated values.
Computational Details
All of the calculations were carried out using the Gaussian-16 package.73 In all cases, we used the B3LYP/6-311++G(d,p) method;74,75 frequency calculations were carried out to verify that the structures obtained correspond to energetic minima (I = 0) or to transition states (TS, I = 1). These geometries were used for the calculation of the absolute chemical shieldings with the GIAO method76 and the SSCC.
Equations 3–5 were used to transform absolute shieldings into chemical shifts46
| 3 |
| 4 |
| 5 |
To locate the intermediates at either sites of the TS point, we followed the vibrational mode of the imaginary frequency, forward and backward, along the intrinsic reaction coordinate (IRC)77,78 and relaxed the geometry for searching an energy (local) minimum. Although all of the stationary points were calculated at the B3LYP/6-311++G(d,p) level, they were recalculated at the 6-31G* level79 to calculate the IRCs.
To have a better description of the energy, domain-based local pair natural orbital coupled cluster method with single, double, and perturbative triple excitations, DLPNO-CCSD(T),80,81 with the def2-TZVP basis set82 has been carried out on the B3LYP/6-311++G(d,p) geometries with the Orca program (Version 5.0.1).83 The effect of the solvent has been taken into account by optimizing the structures using the polarizable continuum model (PCM)84 with the water parameters at the B3LYP/6-311++G(d,p) level.
Acknowledgments
This work was carried out with financial support from the Ministerio de Ciencia, Innovación y Universidades (Projects PGC2018-094644-B-C2 and 5931125495-125495-4-21) and Dirección General de Investigación e Innovación de la Comunidad de Madrid (PS2018/EMT-4329 AIRTEC-CM). Thanks are given to the CTI (CSIC) and to the Irish Centre for High-End Computing (ICHEC, Dublin) for their continued computational support. Thanks are given to Dr. María Ángeles García Fernández, responsible of the NMR Service at the UNEDLAB for technical assistance and to Dr. J. Jonathan Nué Martínez for the HPLC determinations.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c00154.
Reproduction of the 1H and 13C NMR spectra of all novel compounds; details of computational methods and details of the crystal structure determinations of 2a (2113708), 2b (2113709), 2c (2113710), and 2d (2113711) (PDF)
Accession Codes
CCDC 2113708–2113711 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- SciFinder; Chemical Abstracts Service: Columbus, OH, https://scifinder.cas.org (Accessed on Feb. 1, 2022).
- ScienceDirect, Elsevier, https://www.sciencedirect.com/ (Accessed on Feb. 1, 2022).
- Clarivate Web of Science, 2021https://clarivate.com/webofsciencegroup/web-of-science-journal-citation-reports-2021-infographic/ (Accessed on Feb. 1, 2022).
- Gaikwad D. D.; Chapolikar A. D.; Devkate C. G.; Warad K. D.; Tayade A. P.; Pawar R. P.; Domb A. J. Synthesis of Indazole Motifs and their Medicinal Importance: An Overview. Eur. J. Med. Chem. 2015, 90, 707–731. 10.1016/j.ejmech.2014.11.029. [DOI] [PubMed] [Google Scholar]
- Polo E.; Trilleras J.; Ramos J.; Galdámez A.; Quiroga J.; Gutierrez M. Efficient MW-Assisted Synthesis, Spectroscopic Characterization, X-ray and Antioxidant Properties of Indazole Derivatives. Molecules 2016, 21, 903. 10.3390/molecules21070903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denya I.; Malan S. F.; Joubert J. Indazole Derivatives and their Therapeutic Applications: A Patent Review (2013-2017). Expert Opin. Ther. Pat. 2018, 28, 441–453. 10.1080/13543776.2018.1472240. [DOI] [PubMed] [Google Scholar]
- Zhang S.-G.; Liang C.-G.; Zhang W.-H. Recent Advances in Indazole-Containing Derivatives: Synthesis and Biological Perspectives. Molecules 2018, 23, 2783. 10.3390/molecules23112783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W.; Liu Z.; Wu X.; Gan C.; Su Z.; Liu H.; Que H.; Zhang Q.; Xue Q.; Yue L.; Yu L.; Ye T. Synthesis and Biological Evaluation of Indazole Derivatives as Anti-Cancer Agents. RSC Adv. 2021, 11, 15675–15687. 10.1039/D1RA01147B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.; Zhang S.; Guo L.; Feng L.; Tan B. Experimental and Theoretical Studies on the Corrosion Inhibition of Carbon Steel by Two Indazole Derivatives in HCl Medium. Materials 2019, 12, 1339. 10.3390/ma12081339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A. A.; Aly A. A.; Tawfeek H. N. Indazoles: Synthesis and Bond-Forming Heterocyclization. Adv. Heterocycl. Chem. 2018, 125, 235–300. [Google Scholar]
- Förstermann U.; Sessa W. C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837. 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claramunt R. M.; López C.; Lopez A.; Pérez-Medina C.; Pérez-Torralba M.; Alkorta I.; Elguero J.; Escames G.; Acuña-Castroviejo D. Synthesis and biological evaluation of indazole derivatives. Eur. J. Med. Chem. 2011, 46, 1439–1447. 10.1016/j.ejmech.2011.01.027. [DOI] [PubMed] [Google Scholar]
- Claramunt R. M.; Santa María D.; Alkorta I.; Elguero J. The Structure of N-phenyl-pyrazoles and Indazoles: Mononitro, Dinitro, and Trinitro Derivatives. J. Heterocycl. Chem. 2018, 55, 44–64. 10.1002/jhet.3026. [DOI] [Google Scholar]
- Ooms F.; Norberg B.; Isin E. M.; Castagnoli N.; Van der Schyf J.; Wouters J. 7-Nitroindazole. Acta Crystallogr., Sect. C 2000, 56, e474–e475. 10.1107/S0108270100011975. [DOI] [Google Scholar]
- Elguero J.; Fruchier A.; Jacquier R. Recherches dans la Série de Azoles. XLII. Utilisation de la Résonance Magnétique Nucléaire pour l’Identification des Dérivés de l’Indazole. Bull. Soc. Chim. France 1969, 2064–2076. [Google Scholar]
- Elguero J.; Martínez-Martorell C.; Elguero J. A Selective Synthesis of Unsymmetrical 1,1’-Methyenebisazoles bu Solid-Liquid Phase Transfer Catalysis. Heterocycles 1986, 24, 2233–2237. 10.3987/R-1986-08-2233. [DOI] [Google Scholar]
- Alkorta I.; Elguero J.; Jagerovic N.; Fruchier A.; Yap G. P. A. Study of the Structure of 1-Hydroxymethylindazole and 1-Hydroxymethylbenzotriazole by X-ray crystallography, Multinuclear NMR in Solution and DFT Calculations. J. Heterocycl. Chem. 2004, 41, 285–289. 10.1002/jhet.5570410223. [DOI] [Google Scholar]
- Dvoretzky I.; Richter G. H. Formaldehyde Condensation in the Pyrazole Series. J. Org. Chem. 1950, 15, 1285–1288. 10.1021/jo01152a026. [DOI] [Google Scholar]
- Katritzky A. R.; Lue P.; Akutagawa K. Formaldehyde: A Reagent for Simultaneous Protection of Heterocyclic NH and Activation of Alternative Locations to Electrophilic Attack. Part II. A New Synthetic Method for the 5(3)-Substitution of N-Unsubstituted Pyrazoles. Tetrahedron 1989, 45, 4253–4262. 10.1016/S0040-4020(01)81320-X. [DOI] [Google Scholar]
- Dunlop P.; Marini M. A.; Fales H. M.; Sokoloski E.; Martin C. J. NMR Studies of the Reaction of Formaldehyde with the Imidazole Side Chain of Histidine: A Reactive Amino Acid in Enzyme Catalysis. Bioorg. Chem. 1973, 2, 235–247. 10.1016/0045-2068(73)90026-6. [DOI] [Google Scholar]
- Alley P. W. Imidazole-Formaldehyde Reaction. Formation of 1-Imidazolemethanol. J. Org. Chem. 1975, 40, 1837–1838. 10.1021/jo00900a035. [DOI] [Google Scholar]
- Vlasova L. A.; Shamaeva E. M.; Afanas’eva G. B.; Postovskii I. Y. Synthesis of Hydroxymethyl Compounds of 1,2,4-Triazole and Investigation of their Antitumorogenic Activity. Pharm. Chem. J. 1971, 5, 473–477. 10.1007/BF00771732. [DOI] [Google Scholar]
- Kalisiak J.; Sharpless E. B.; Fokin V. V. Efficient Synthesis of 2-Substituted-1,2,3-triazoles. Org. Lett. 2008, 10, 3171–3174. 10.1021/ol8006748. [DOI] [PubMed] [Google Scholar]
- Yu F.-l.; Zhang R.-l.; Xie C.-x.; Yu S.-t. Synthesis of Thermoregulated Phase-Separable Triazolium Ionic Liquid Catalysts and Application for Stetter Reaction. Tetrahedron 2010, 66, 9145–9150. 10.1016/j.tet.2010.09.094. [DOI] [Google Scholar]
- Tselinskii I. V.; Mel’nikov A. A.; Varyagina L. G.; Zhigadlova I. G. Synthesis of N-Hydroxymethyl Derivatives of 3-Substituted Tetrazoles. Chem. Heterocycl. Compd. 1983, 19, 341–343. 10.1007/BF00513275. [DOI] [Google Scholar]
- Katritzky A. R.; Akutagawa K. Formaldehyde: A Reagent for the Simultaneous Protection of Nucleophilic Centers and the Activation and Stabilization of Alternative Locations to Electrophilic Attack. I. A New Synthetic Method for the 2-Substitution of N-Unsubstituted Benzimidazoles: Formaldehyde As A Versatile Protecting Agent For Heterocyclic NH. J. Org. Chem. 1989, 54, 2949–2952. [Google Scholar]
- Burckhalter J. H.; Stephens V. C.; Hall L. A. R. Proof of Structures Derived from the Hydroxy- and Amino-methylation of Benzotriazole. J. Am. Chem. Soc. 1952, 74, 3868–3870. 10.1021/ja01135a044. [DOI] [Google Scholar]
- Gaylord N. G. Displacement Reactions in the Benzotriazole Series. J. Am. Chem. Soc. 1954, 76, 285–287. 10.1021/ja01630a081. [DOI] [Google Scholar]
- Katritzky A. R.; Palenik G. J.; Anders E.; Tropsch J. G.; Vanden Eynde J. J.; Zhang Z. N-[(2-Naphthyloxy)methyl]benzazoles: Synthesis and Investigation by X-ray Analysis and by Semiempirical MO Calculations. Chem. Ber. 1990, 123, 1185–1191. 10.1002/cber.19901230536. [DOI] [Google Scholar]
- Pozharskii F. T.; Kazanbieva M. A.; Tertov B. A. 1-Hydroxymethyl Derivatives of Indazoles. Zh. Obsh. Khim. 1964, 34, 3367–3370. [Google Scholar]
- Seeman J. I. The Curtin-Hammett Principle and the Winstein-Holnes Equation. J. Chem. Educ. 1986, 63, 42–48. 10.1021/ed063p42. [DOI] [Google Scholar]
- Catalán J.; de Paz J. L. G.; Elguero J. Importance of Aromaticity on the Relative Stabilities of Indazole Annular Tautomers: An ab initio Study. J. Chem. Soc., Perkin Trans. 2 1996, 115, 57–60. 10.1039/P29960000057. [DOI] [Google Scholar]
- Anandan K.; Kolandaivel P.; Kumaresan R. Ab initio and DFT Studies on Tautomerism of Indazole in Gaseous and Aqueous Phases. J. Mol. Struct.: THEOCHEM 2004, 686, 83–89. 10.1016/j.theochem.2004.08.014. [DOI] [Google Scholar]
- McTigue P. T.; Sime J. M. The Basic Strength of Formaldehyde. Aust. J. Chem. 1963, 16, 592–595. 10.1071/CH9630592. [DOI] [Google Scholar]
- Catalán J.; Abboud J. L. M.; Elguero J. Basicity and Acidity of Azoles. Adv. Heterocycl. Chem. 1987, 41, 187–274. [Google Scholar]
- Katritzky A. R.; Kuzmierkiewicz W.; Rachwal B.; Rachwal S.; Thomson J. The Chemistry of N-Substituted Benzotriazoles. Part 5. Reactions of Benzotriazole with Aldehydes and Thionyl Chloride--Formation of (Benzotriazol-1-yl)-1-chloroalkanes and Bis(benzotriazolyl) alkanes. J. Chem. Soc., Perkin Trans. 1 1987, 811–817. 10.1039/P19870000811. [DOI] [Google Scholar]
- Nesmeyanov A. N.; Zavelovich E. B.; Babin V. N.; Kochetkova N. S.; Fedin E. I. H and C NMR Study of Tautomerism of Azoles. II. Proton Transfer in Ketone Solvents. Tetrahedron 1975, 31, 1463. 10.1016/0040-4020(75)87081-5. [DOI] [Google Scholar]
- Katritzky A. R.; Perumal S.; Savage G. P. An NMR Study of the Equilibria Involved with Benzotriazole, Carbonyl Compounds, and Their Adducts. J. Chem. Soc., Perkin Trans. 2 1990, 921–924. 10.1039/p29900000921. [DOI] [Google Scholar]
- Feldmann M. T.; Widicus S. L.; Blake G. A.; Kent D. R.; Goddard W. A. Aminomethanol Water Elimination: Theoretical Examination. J. Chem. Phys. 2005, 123, 034304 10.1063/1.1935510. [DOI] [PubMed] [Google Scholar]
- Nomura A.; Jones C. W. Enhanced Formaldehyde-Vapor Adsorption Capacity of Polymeric Amine-Incorporated Aminosilicas. Chem. – Eur. J. 2014, 20, 6381–6390. 10.1002/chem.201304954. [DOI] [PubMed] [Google Scholar]
- Ćmikiewicz A.; Gordon A. J.; Berski S. Characterization of the Reaction Mechanism between Ammonia and Formaldehyde from the Topological Analysis of ELF and Catastrophe Theory Perspective. Struct. Chem. 2018, 29, 243–255. 10.1007/s11224-017-1024-x. [DOI] [Google Scholar]
- a Allen F. H. The Cambridge Structural Database: A Quater of a Million Crystal Structures and Rising. Acta Crystallogr. 2002, 58, 380–388. 10.1107/S0108768102003890. [DOI] [PubMed] [Google Scholar]; b Allen F. H.; Motherwell W. D. S. Applications of the Cambridge Structural Database in Organic Chemistry and Crystal Chemistry. Acta Crystallogr. 2002, 58, 407–422. 10.1107/S0108768102004895. [DOI] [PubMed] [Google Scholar]
- Rivera A.; Maldonado M.; Ríos-Motta J.; Fejfarova K.; Dusek M. (1H-Benzimidazol-1-yl)methanol. Acta Crystallogr., Sect. E 2012, 68, o615. 10.1107/S1600536812004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov Y.; et al. On the HALA Effect in the NMR Carbon Shielding Constants of the Compounds Containing Heavy p-Elements. Int. J. Quantum Chem. 2016, 116, 1404–1412. 10.1002/qua.25196. [DOI] [Google Scholar]
- Marín-Luna M.; Sánchez-Andrada P.; Alkorta I.; Elguero J.; Torralba M. C.; Delgado-Martínez P.; Santa María D.; Claramunt R. M. A Structural Analysis of 2,5-Diaryl-4H-2,4-dihydro-3H-1,2,4-triazol-3-ones: NMR in the Solid State, X-Ray Crystallography, and GIPAW Calculations. Magn. Reson. Chem. 2021, 59, 423–438. 10.1002/mrc.5107. [DOI] [PubMed] [Google Scholar]
- Blanco F.; Alkorta I.; Elguero J. Spectral Assignments and Reference Data. Statistical Analysis of 13C and 15N Chemical Shifts from GIAO/B3LYP/6-311++G** Calculated Absolute Shieldings. Magn. Reson. Chem. 2007, 45, 797–800. 10.1002/mrc.2053. [DOI] [PubMed] [Google Scholar]
- Fruchier A.; Pellegrin V.; Schimpf R.; Elguero J. NMR Studies in the Heterocyclic Series. XXIV. 1H, 13C and 15N Study of 15N Labelled Indazoles. Org. Magn. Reson. 1982, 18, 10–13. 10.1002/mrc.1270180103. [DOI] [Google Scholar]
- Alam R. M.; Keating J. J. Regioselective N-Alkylation of the 1H-Indazole Scaffold; Ring Substituent and N-Alkylating Reagent Effects on Regioisomeric Distribution. Beilstein J. Org. Chem. 2021, 17, 1939–1951. 10.3762/bjoc.17.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elguero J.; Fruchier A.; Jacquier R. Recherches dans la Série des Azoles. V. Étude RMN dans la Série de l’Indazole. Bull. Soc. Chim. France 1966, 2075–2084. [Google Scholar]
- Constantino M. G.; Lacerda V.; da Silva G. V. J.; Tasic L.; Rittner R. Principal Component Anaklysis of Long-Range ’W’ Coupling Constants of Some Cyclic Compounds. J. Mol. Struct. 2001, 597, 129–136. 10.1016/S0022-2860(01)00591-9. [DOI] [Google Scholar]
- Chen H. E.Substituent Effects on the Magnetic Resonance Spectra of 1,4-Disubstituted Benzenes, Thesis, University of Missouri, 1972. [Google Scholar]
- Pretsch E.; Bühlmann P.; Badertscher M.. Structure Determination of Organic Compounds, 4th ed.; Springer-Verlag: Berlin, 2009. [Google Scholar]
- Fruchier A.; Alcalde E.; Elguero J. NMR Studies in the Heterocyclic Series. XVI. Determination of the Structure of 3-Azidoindazole by 1H and 13C NMR Spectroscopy. Org. Magn. Reson. 1977, 9, 235–236. 10.1002/mrc.1270090412. [DOI] [Google Scholar]
- Black P. J.; Heffernan M. L. The Analysis of the Proton Magnetic Resonance Spectra of Heteroaromatic Systems. IV Benzofuran, Indole, and Related Compounds. Aust. J. Chem. 1965, 18, 353–361. 10.1071/CH9650353. [DOI] [Google Scholar]
- Bouissane L.; El Kazzouli S.; Léger J.-M.; Jarry C.; Rakib E. M.; Khouili M.; Guillaumet G. New and Effocient Synthesis of Bi- and Trisubstituted Indazoles. Tetrahedron 2005, 61, 8218–8225. 10.1016/j.tet.2005.06.038. [DOI] [Google Scholar]
- Bouissane L.; El Kazzouli S.; Léonce S.; Pfeiffer B.; Rakib E. M.; Khouili M.; Guillaumet G. Synthesis and Biological Evaluation of N-(7-indazolyl)benzenesulfonamide Derivatives as Potent Cell Cycle Inhibitors. Bioorg. Med. Chem. 2006, 14, 1078–1088. 10.1016/j.bmc.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Siskos M. G.; Choudhary M. I.; Gerothanassis I. P. Hydrogen Atomic Positions of O–H···O Hydrogen Bonds in Solution and in the Solid State: The Synergy of Quantum Chemical Calculations with 1H-NMR Chemical Shifts and X-ray Diffraction Methods. Molecules 2017, 22, 415. 10.3390/molecules22030415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siskos M. G.; Kontogianni V. G.; Tsiafoulis C. G.; Tzakos A. G.; Gerothanassis I. P. Investigation of Solute–Solvent Interactions in Phenol Compounds: Accurate ab initio Calculations of Solvent Effects on 1H NMR Chemical Shifts. Org. Biomol. Chem. 2013, 11, 7400–7411. 10.1039/c3ob41556b. [DOI] [PubMed] [Google Scholar]
- Free S. M. J.; Wilson J. W. A Mathematical Contribution to Structure-Activity Studies. J. Med. Chem. 1964, 7, 395–399. 10.1021/jm00334a001. [DOI] [PubMed] [Google Scholar]
- Kubinyi H.QSAR: Hansch Analysis and Related Approaches. Methods and Principles in Medicinal Chemistry; In Mannhold R.; Krogsgaard-Larsen P.; Timmerman H., Eds.; VCH: Weinheim, 1993; Vol. 1. [Google Scholar]
- Cativiela C.; García J. I.; Elguero J.; Mathieu D.; Phan Tan Luu R. Description of Heterocyclic Substituents: A Free-Wilson Type Approach Using D-Optimal Designs. Quant. Struct.–Act. Relat. 1987, 6, 173–178. 10.1002/qsar.19870060405. [DOI] [Google Scholar]
- Cabildo P.; Claramunt R. M.; López C.; García M. A.; Pérez-Torralba M.; Pinilla E.; Torres M. R.; Alkorta I.; Elguero J. Crystal and Molecular Structure of Three Biologically Active Nitroindazoles. J. Mol. Struct. 2011, 985, 75–81. 10.1016/j.molstruc.2010.10.023. [DOI] [Google Scholar]
- Sopková-de Oliveira Santos J.; Collot V.; Rault S. 7-Nitro-1H-indazole, an Inhibitor of Nitric Oxide Synthase. Acta Crystallogr., Sect. C 2000, 56, 1503–1504. 10.1107/S0108270100012956. [DOI] [PubMed] [Google Scholar]
- Ooms F.; Norberg B.; Isin E. M.; Castagnoli N.; Van der Schyf C. J.; Wouters J. 7-Nitroindazole. Acta Crystallogr., Sect. C 2000, 56, e474–e475. 10.1107/S0108270100011975. [DOI] [Google Scholar]
- Wanat M.; Malinska M.; Hoser A. A.; Wozniak K. Further Validation of Quantum Crystallography Approaches. Molecules 2021, 26, 3730. 10.3390/molecules26123730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAINTplus Package; Bruker AXS Inc.: Madison, WI, 2006.
- Sheldrick G. M.SADABS Program Integrated in APEX3 Package 2016, Bruker AXS Inc.: Madison, WI.
- Sheldrick G. M.XPREP, SHELXT and SHELXL Programs Integrated in APEX3 Package 2016, Bruker AXS Inc.: Madison, WI.
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Macrae C. F.; Sovago I.; Cottrell S. J.; Galek P. T. A.; McCabe P.; Pidcock E.; Platings M.; Shields G. P.; Stevens J. S.; Towler M.; Wood P. A. Mercury 4.0: from visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. 10.1107/S1600576719014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek A. L.PLATON, A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, Holland, 2015.
- Fung B. M.; Khitrin A. K.; Ermolaev K. An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids. J. Magn. Reson. 2000, 142, 97–101. 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, revision A.03; Gaussian, Inc.: Wallingford, CT, 2016.
- a Becke A. D. Density-functional Exchange-energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]; b Becke A. D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; c Lee C.; Yang W.; Parr R. G. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- a Ditchfield R.; Hehre W. J.; Pople J. A. Self-Consistent Molecular Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. 10.1063/1.1674902. [DOI] [Google Scholar]; b Frisch M. J.; Pople J. A.; Binkley J. S. Self-Consistent Molecular Orbital Methods. 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. 10.1063/1.447079. [DOI] [Google Scholar]
- a London F. Quantum Theory of Interatomic Currents in Aromatic Compounds. J. Phys. Radium 1937, 8, 397–409. 10.1051/jphysrad:01937008010039700. [DOI] [Google Scholar]; b Ditchfield R. Self-consistent Perturbation Theory of Diamagnetism. I. A Gauge-Invariant LCAO (Linear Combination of Atomic Orbitals) Method for NMR Chemical Shifts. Mol. Phys. 1974, 27, 789–807. 10.1080/00268977400100711. [DOI] [Google Scholar]
- Fukui K. The Path of Chemical Reactions – The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. 10.1021/ar00072a001. [DOI] [Google Scholar]
- Hratchian H. P.; Schlegel H. B.. Theory and Applications of Computational Chemistry: the First 40 Years; In Dykstra C. E.; Frenking G.; Kim K. S.; Scuseria G., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp 195–249. [Google Scholar]
- Hariharan P. C.; Pople J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. 10.1007/BF00533485. [DOI] [Google Scholar]
- Riplinger C.; Neese F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106 10.1063/1.4773581. [DOI] [PubMed] [Google Scholar]
- Riplinger C.; Sandhoefer B.; Hansen A.; Neese F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101 10.1063/1.4821834. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Neese F.; Wennmohs F.; Becker U.; Riplinger C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108 10.1063/5.0004608. [DOI] [PubMed] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.