

Summary
Repeat antigens, such as the Plasmodium falciparum circumsporozoite protein (PfCSP), use both sequence degeneracy and structural diversity to evade the immune response. A few PfCSP-directed antibodies have been identified that are effective at preventing malaria infection, including CIS43, but how these repeat-targeting antibodies might be improved has been unclear. Here, we engineered a humanized mouse model in which B cells expressed inferred human germline CIS43 (iGL-CIS43) B cell receptors and used both vaccination and informatics to obtain variant CIS43 antibodies with improved protective capacity. One such antibody, iGL-CIS43.D3, was significantly more potent than the current best-in-class PfCSP-directed antibody. We found that vaccination with a junctional epitope peptide was more effective than full-length PfCSP at recruiting iGL-CIS43 B cells to germinal centers. Structure-function analysis revealed multiple somatic hypermutations that combinatorically improved protection. This mouse model can thus be used to understand vaccine immunogens and to develop highly potent anti-malaria antibodies.
Keywords: adoptively transferred B cells, antibody CIS43, circumsporozoite, epitope-focused immunization, in situ vaccination, junctional epitope, malaria, passive protective, PfCSP
Graphical Abstract
eTOC Blurb
CIS43 is a malaria-protective antibody targeting a junctional epitope of the Plasmodium falciparum circumsporozoite protein. Kratochvil et al. engineer a mouse model expressing inferred germline CIS43 antibodies and use an immunofocusing strategy, in tandem with bioinformatic sieving, to generate improved CIS43 antibodies, including iGL-CIS43.D3, a best-in-class malaria-protective antibody.
Introduction
Plasmodium falciparum (Pf) is the etiological agent of malaria, a vector-borne infectious disease with an estimated disease burden of 229 million cases and 409,000 deaths in 2019 alone (Global Malaria Programme, 2020). While both medical and non-medical interventions, such as artemisinin-based combination therapies and insecticide-treated nets, have proven to be highly effective, these measures are offset by the appearance of drug-resistant parasite strains and insecticide-resistant mosquitos (Phillips et al., 2017). Thus, there is an urgent need for interventions that can prevent malaria.
Malaria infection is initiated following a mosquito bite. Sporozoites (SPZ), the infectious form of Plasmodium, are injected into the skin and blood; they then travel to the liver to infect hepatocytes and initiate the infection. The Pf circumsporozoite protein (PfCSP), comprised of an N-terminus, a central repeating tetrapeptide region, and a C-terminus, is the most abundant protein on the SPZ surface and is required for motility and hepatocyte invasion (Cerami et al., 1992; Tewari et al., 2002).
Repeat antigens such as PfCSP can assume divergent structures, which confound both recognition and maturation of the humoral immune response. Nevertheless, PfCSP is currently the best target for neutralizing antibodies, and the most advanced malaria vaccine—RTS,S—presents a truncated portion of the PfCSP repeat containing only NANP and C-terminal regions (Partnership, 2012; RTS, 2015). RTS,S/AS01 trials have found vaccine efficacy of approximately 36% among 5–17-month-olds after 4 years, waning over time with decreasing antibody titers (RTS, 2015; White et al., 2014). Protection appears to require extraordinarily high titers of PfCSP-directed antibody, suggesting most elicited PfCSP-directed antibodies are poorly protective. An alternative approach that could enhance protection over a defined period is with long-lasting, highly potent monoclonal antibodies (mAbs). The protection offered by mAbs may be independent of any host-parasite factors that can limit the effectiveness of vaccines.
The majority of mouse and human neutralizing mAbs demonstrating protection in vivo bind the central repeat region, containing an immunodominant NANP sequence (Imkeller et al., 2018; Oyen et al., 2017; Zavala et al., 1983). We recently reported the isolation of a highly potent human mAb that preferentially binds a unique tetrapeptide at the junction of the N-terminus and repeat region, NPDP, identifying this subdominant “junctional epitope” as a site of vulnerability (Kisalu et al., 2018). CIS43, altered to express an LS mutation in its Fc region to increase its half-life (Kisalu et al., 2020), was found to prevent malaria infection in humans eight months after a single administration (Gaudinski et al., 2021). Given the potential clinical utility of CIS43, it is critical to understand how this antibody lineage developed and how similar or more potent antibodies might be induced or engineered.
To investigate these fundamental questions, we established a pre-clinical knock-in (KI) mouse model. Recently, we developed a one-step CRISPR/Cas9-induced homology-directed repair (HDR) approach (Lin et al., 2018; Wang et al., 2020b), and applied this technique to insert inferred human germline CIS43 (iGL-CIS43) IgH and IgK chains at their respective native mouse loci (Lin et al., 2018; Wang et al., 2020b). Using this KI model, we demonstrated the utility of immunofocusing: iGL-CIS43 B cell recruitment to germinal centers was greater in animals immunized with the junctional peptide rather than full-length PfCSP. Epitope-focused immunization induced diversification via somatic hypermutation (SHM) and facilitated the generation of an antibody library comprising over a hundred CIS43 variant antibodies, a subset of which was bioinformatically selected to assess biophysical and structural properties to characterize epitope-specificities and to measure their ability to protect against malaria parasite challenge in vivo. Correlation of antibody properties with in vivo protection enabled the engineering of CIS43-variant antibodies that are more protective against malaria than mature CIS43 and current best-in-class mAbs. This study demonstrates the utility of a KI-mouse model for optimizing the protective capacity of a mAb targeting the PfCSP-repeat antigen for antibody-mediated prevention and for guiding next-generation vaccines to induce similar protective responses.
Results
Generation of KI Mice Expressing Inferred Germline Sequences of CIS43
We used inferred germline-antibody sequences of the anti-malarial antibody CIS43 to generate a KI mouse model (Kisalu et al., 2018). CIS43 uses heavy chain (HC) variable genes VH1–3*03, DH4–23*01 and JH3*02 and the cognate light chain (LC) variable genes Vκ4–1*01 and Jκ4*01; it was originally isolated from a malaria-naive participant exposed to radiation-attenuated live SPZ (Seder et al., 2013).
Using our CRISPR/Cas9 protocols (Lin et al., 2018; Wang et al., 2020b), the iGL-CIS43 HC and variable LC regions were inserted into the respective native loci. Fertilized mouse oocytes were microinjected with (a) two donor plasmids, each containing pre-rearranged inferred germline CIS43 HC and LC sequences driven by the mouse VHJ558 or Vκ4–53 promoters, respectively; (b) four single-guided RNAs (sgRNAs)—with two sgRNAs targeting each H or κ locus; and (c) AltR-Cas9 (Wang et al., 2020b). After implantation of the injected zygotes into pseudopregnant C57BL/6J females, pups were genotyped; mouse lines expressing both iGL-CIS43 HC and LC are referred to as HiGL-CIS43κiGL-CIS43.
To determine whether iGL-CIS43 HC and/or LC were expressed in functional BCRs, we performed flow cytometry using a PfCSP-probe. Heterozygous HiGL-CIS43κiGL-CIS43 KI mice showed higher median PfCSP-binding of peripheral B cells (10.80%) than either wild type (WT) C57BL/6J mice (0.07%) (Figure 1A) or the HC-only HiGL-CIS43 KI model (1.20%) (Figure S1A and S1B). In homozygous HiGL-CIS43κiGL-CIS43 mice, ~90% of naïve B cells bind to PfCSP (Figure S1C); peripheral B cell populations of iGL-CIS43 mice were comparable to C57BL/6 controls (Figure S1A and S1B). To confirm that BCRs from the HiGL-CIS43κiGL-CIS43 line consist of both human iGL-CIS43 HC and LC, we performed antigen-agnostic (B220+) and antigen-specific (B220+PfCSP+) single-cell sorting and sequencing (Figure 1B, Figure S1D). While ~67% (6/9) of B220+-sorted B cells were positive for the iGL-CIS43 HC and LC, B220+PfCSP+-sorted B cells were 80% positive for both (16/20) (Figure 1B). Having confirmed that the KI produced functional PfCSP-binders at high frequency and did not disrupt B cell differentiation, we used heterozygous iGL-CIS43 mice in all subsequent experiments.
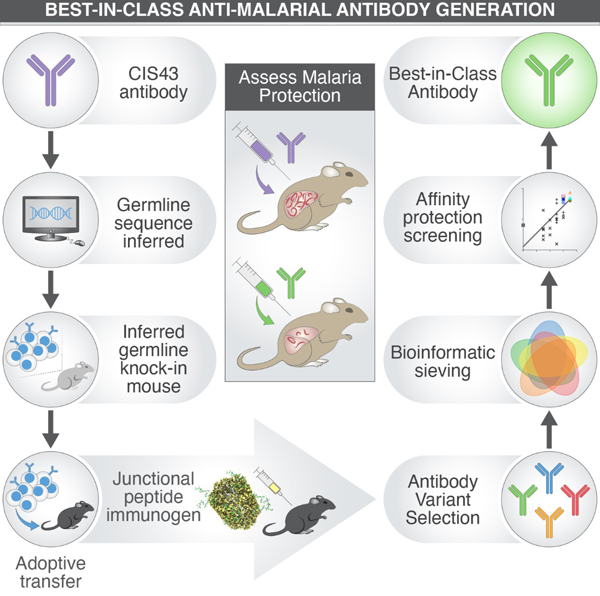
Figure 1: Activation of HiGL-CIS43κiGL-CIS43 B cells by PfCSP-immunization is dampened by host competitor B cells.
A. (Left) PfCSP-binding of peripheral B cells in naïve iGL-CIS43 HC and LC KI mouse models (HiGL-CIS43κiGL-CIS43). Events were pre-gated on lymphocytes/singlets/CD4-CD8-F4/80-Gr1-/B220+ B cells and C57BL/6J mice used as negative controls. (Right) Quantification of PfCSP-binding blood peripheral B cells from HiGL-CIS43κiGL-CIS43 KI model vs. C57BL/6J mice.
B. Human iGL-CIS43 HC (purple), human iGL-CIS43 LC (light purple) and murine LC (grey) sequences amplified from single-cell sorted (a) antigen-agnostic (left) or PfCSP-specific (right) naive B cells. The center number is sequence pairs amplified.
C. Titration of cell transfer model to generate precise HiGL-CIS43κiGL-CIS43 precursor frequencies at time of immunization (Day 0). See also Figure S3C-S3E for metrics of total B cells counted and recovered HiGL-CIS43κiGL-CIS43 B cells.
D. Precursor frequencies corresponding to number of B cells transferred (left), indicated on x-axis, and analysis of linearity of CD45.2 HiGL-CIS43κiGL-CIS43 B cells recovered 24 h post transfer (right).
E. HiGL-CIS43κiGL-CIS43 B cell transfer/immunization system used for (F-H).
F. Representative flow cytometry graphs at 7 DPI.
G. Quantification of B cell subsets (in order of left to right: total GCs, ratios of GC CD45.1+/CD45.2+ B cells, PfCSP-reactive CD45.2+ B cells and PfCSP-reactive CD45.1+ competitor B cells) responsive to PfCSP-immunization at 7 DPI.
P values were calculated by Mann-Whitney test (panels A and H). ***P < 0.001; ns, statistically non-significant differences. See also Figure S1.
KI B Cells are Outcompeted After PfCSP Immunization
The vaccine-mediated activation of specific B cell clones is dependent on their frequency in the B cell repertoire (Abbott et al., 2018; Dosenovic et al., 2018). Due to high precursor numbers in HiGL-CIS43κiGL-CIS43 KI mice (Figure 1A and 1B), we lowered the precursor frequency by adoptive transfer of CD45.2+ HiGL-CIS43κiGL-CIS43 B cells into CD45.1+ C57BL/6 recipient mice at three different concentrations (500,000, 100,000 and 10,000 naive B cells). To establish the absolute number of PfCSP-reactive HiGL-CIS43κiGL-CIS43 precursor B cells in the spleen, recipient mice were sacrificed at day 0 (24 h post transfer) for analysis (Figure 1C, Figure S1E-S1G). PfCSP-reactive CD45.2 B cell numbers are linearly dependent on the number of B cells transferred and were approximately 1:104, 1:105 and 1:106 at the time of immunization (Figure 1C and 1D).
Mice with rare precursor frequencies (1:104 and 1:105) were used, on the basis of prior studies (Abbott et al., 2018; Havenar-Daughton et al., 2018). Cohorts of recipient mice were immunized intraperitoneally with PfCSP (10 µg/mouse) in Alhydrogel 24 h after adoptive transfer, then sacrificed at 7 days post immunization (DPI) and their spleens harvested to measure GC formation (CD38lowCD95+) (Figure 1E). At 24 h, ~0.6% of the B cells were CD38lowCD95+, demonstrating GC formation. CD45.2+ HiGL-CIS43κiGL-CIS43 B cells accounted for ~4.3% and 0.6% of the total GC responses at precursor frequencies of 1:104 to 1:105, respectively, confirming that HiGL-CIS43κiGL-CIS43 B cells are competent to respond to antigen-challenge in vivo (Figure 1F). Though total GC responses were equivalent (and low in magnitude) at both 1:104 and 1:105, few HiGL-CIS43κiGL-CIS43 B cells entered GCs at the 1:105 frequency (Figure 1F and 1G). There was significant activation of host CD45.1 B cells with high PfCSP-specificity (Figure 1G, rightmost panel). This outgrowth among GC B cells by host CD45.1 B cells may be due to the immunodominant malaria NANP-repeat epitopes expressed in PfCSP (Foquet et al., 2014; Kisalu et al., 2018; McNamara et al., 2020; Oyen et al., 2017; Triller et al., 2017; Zavala et al., 1983). This suggests that alternative immunogens may be preferable in activating iGL-CIS43 B cells.
Immunofocusing Enhances Immune Responses by CIS43-precursor B Cells
Since PfCSP is a multi-epitope immunogen (Cockburn and Seder, 2018; Wardemann and Murugan, 2018), we hypothesized that immunofocusing could enhance the activation of iGL-CIS43 B cells and improve GC recruitment. The PfCSP-junctional region, which links the N-terminal and NANP-repeat region of PfCSP, is contained within 19 amino acids (KQPADGNPDPNANPNVDPN; corresponding to residues 95–115 of PfCSP). CIS43 preferentially targets peptide 21 (NPDPNANPNVDPNAN, (Kisalu et al., 2018)), while the mAb MGG4 targets NPDP19 (KQPADGNPDPNANPNVDPN, (Tan et al., 2018)) (Figure 2A, left panel). Since NPDP19 contains all tetrapeptide epitopes of interest and induces anti-PfCSP responses in BALB/c mice (Tan et al., 2018), we used it in our immunofocusing strategy.
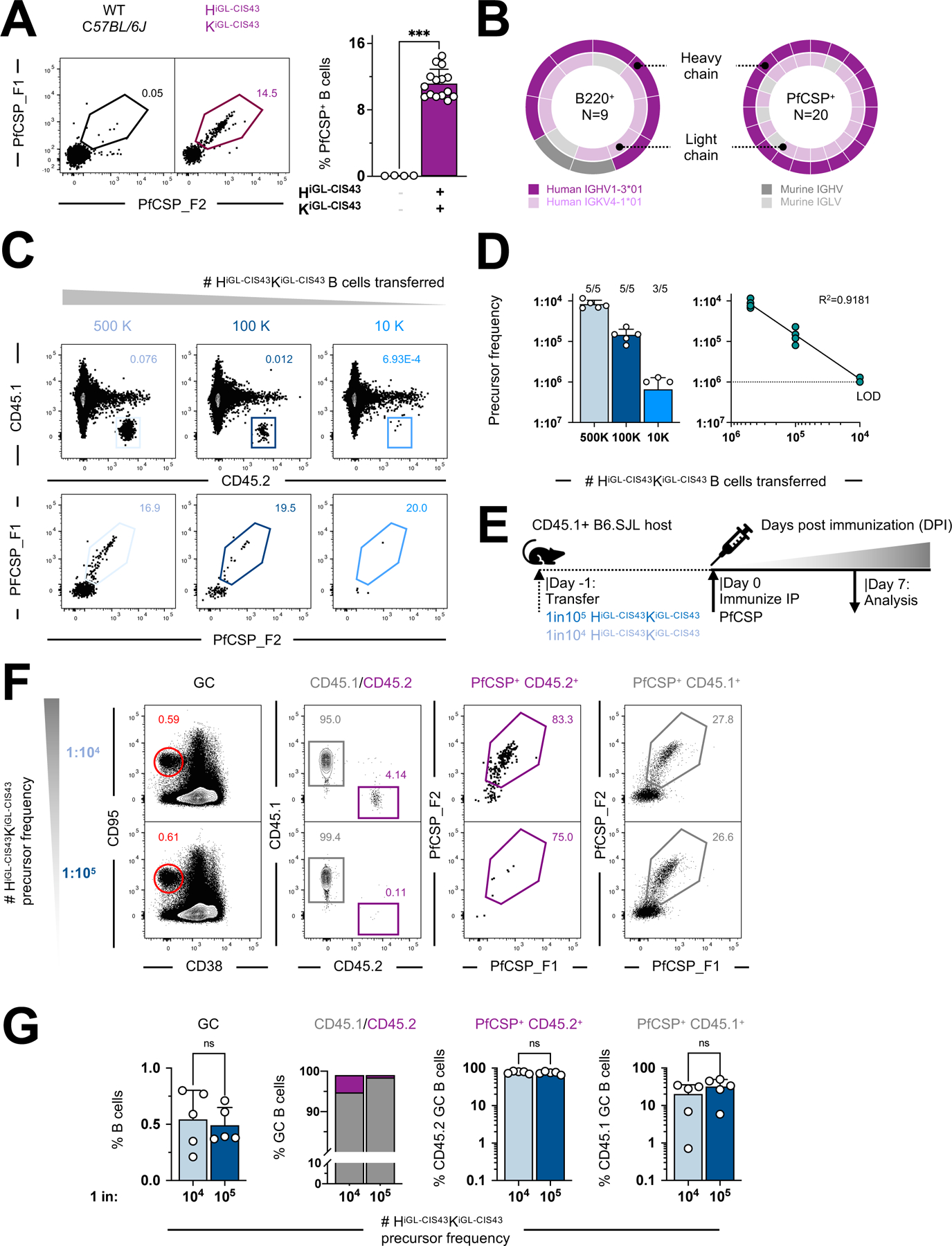
Figure 2: Immunization with NPDP19-KLH leads to specific and strong activation of HiGL-CIS43κiGL-CIS43 B cells.
A. (I) Schematic of PfCSP (NF54), with region 1 (R1) in red, NANP repeats in orange, NPDP/NVDP repeats in blue and region 2 (R2) thrombospondin repeats (TSR) in green. Junctional epitope NPDP19 included in our (II) KLH-based prototype malaria vaccine candidate and malaria peptide21 published (Kisalu et al., 2018) are indicated. Numbering (95–115) corresponds to PfCSP numbering. Schematic adapted from (Cockburn and Seder, 2018a; Kisalu et al., 2018).
B. ELISA to assess if NPDP19-peptide can be recognized by both iGL-CIS43 (purple) and mature CIS43 (black) antibodies in the context of KLH. The red line indicates HIV-1 bNAb VRC01, used as a negative control.
C. HiGL-CIS43κiGL-CIS43 B cell transfer system used for (D).
D. Representative flow cytometry graphs for recipient mice adoptively transferred to achieve precursor frequencies of 1:104, 1:105 and 1:106, immunized with PfCSP [50 µg/mouse]/Alhydrogel (n=5 per cell dilution) or NPDP19-KLH [50 µg/mouse]/Alhydrogel (n=5 per cell dilution). (Left to right) Graphs indicate total GC-responses, HiGL-CIS43κiGL-CIS43 CD45.2 B cell responses, and PfCSP-binding of CD45.2 B cells. Percentages based on parent populations.
E. Quantification of (D). Data normalized to B220+ B cells.
P values calculated by 2-way ANOVA for multiple comparisons (panel E). *P < 0.05; **P<0.01***; P<0.001; ****P<0.0001; ns, statistically non-significant differences.
See also Figure S2.
To enhance peptide immunogenicity, NPDP19 was covalently linked to KLH (Figure 2A, right panel); ELISA confirmed that the NPDP19 peptide is recognized by iGL-CIS43 in the context of the carrier protein KLH (~390 kDa) (Figure 2B).
HiGL-CIS43κiGL-CIS43 B cells were adoptively transferred into congenic mice to achieve precursor frequencies of 1:104, 1:105 and 1:106 for immunization with either PfCSP/Alhydrogel or NPDP19-KLH/Alhydrogel at 50µg/mouse. Splenocytes were analyzed by flow cytometry at 13 DPI (Figure 2C). While GC B cells (CD38lowCD95+) were detected in both immunization conditions, the number of GC B cells induced by PfCSP (at 1:106: N=0.003±0.004%; 1:105: N=0.043±0.014%; at 1:104: N=0.086±0.060%) were significantly lower than GCs induced by NPDP19-KLH (at 1:106: N=0.29±0.088%; 1:105: N=1.26±0.493%; at 1:104: N=1.67±0.47%). Significantly more transferred CD45.2 PfCSP-reactive B cells were recruited to GCs following NPDP19-KLH immunization relative to PfCSP-immunization in animals with higher precursor frequencies (Figure 2D and 2E). NPDP19-KLH immunization increased junctional epitope-specific activation while reducing the recruitment of PfCSP-specific competitor CD45.1 GC B cells into the GCs (Figure S2A). Staining NPDP19-specific B cells revealed a lower number of host CD45.1 GC B cells bound the junctional epitope (Figure S2B). These data suggest that other regions of the full-length PfCSP protein may divert or sterically hinder B cell responses to the junctional epitope (Kisalu et al., 2018; Zavala et al., 1983).
As most residues critical to CIS43 binding are on the HC (Kisalu et al., 2018), we also tested our PfCSP and NPDP19-KLH immunization strategies in the HiGL-CIS43 KI model, in which human HiGL-CIS43 HC pairs with endogenous murine LCs. NPDP19-KLH immunization increased specific activation of HiGL-CIS43 B cells (Figure S2C-S2H), further confirming our immunofocusing strategy. Corresponding murine LCs were enriched for IGVK8–30*01 (96.3% (53/55)), indicating its preferential usage in combination with human IGHV1–3*01 in BCRs capable of binding to PfCSP (Figure S2E). Murine IGKV8–30*01 exhibits a high degree of sequence homology to its human counterpart (Figure S2H), differing at only 16 amino acids.
Overall, these results indicate that using the minimal junctional epitope instead of full-length PfCSP enhanced HiGL-CIS43κiGL-CIS43 B cell responses while reducing undesired host B cell responses.
Junctional Peptide Immunization Recruits CIS43-precursors to Long-Lasting Germinal Centers
To determine whether NPDP19-KLH can induce durable responses and mature-CIS43-like antibodies (Kisalu et al., 2018), GC responses were assessed at 13 and 28 DPI, starting from a precursor frequency of 1:105. At 13 DPI we included sham and adjuvant-only control groups (Figure 3A). In both control groups CD45.2 HiGL-CIS43κiGL-CIS43 B cells could not be recruited to GCs (Figure 3B), while CD45.2 HiGL-CIS43κiGL-CIS43 B cells reached a mean peak frequency of ~42.3% at 13 DPI and ~13.8% at 28 DPI with NPDP19-KLH/Alhydrogel (Figure 3C). A substantial proportion of these HiGL-CIS43κiGL-CIS43 GC B cells bound to PfCSP (13 DPI: N=41.0±4.9%; 28 DPI: N=71.5±10.2%) and was predominantly class-switched to IgG1 (13 DPI: N=65.7±8.2%; 28 DPI: N=57.6±12.2%), confirming normal GC functionality. Parallel staining using PfCSP or NPDP19 as probes revealed high epitope-specificity in CD45.2 GC B cell responses at 13 DPI (PfCSP: N=44.0±4.0%; NPDP19: N=77.9±3.4%) and 28 DPI (PfCSP: N=92.5±1.5%; NPDP19: N=61.1±0.39%) (Figure S3A and S3B). Differences in PfCSP- and NPDP19-recognition were not significant (13 DPI: p=0.06, 28 DPI: p=0.13) and are likely attributable to the more complex nature of PfCSP, in which steric clashes with immunodominant NANP-repeats can restrict epitope-accessibility (Zavala et al., 1983).
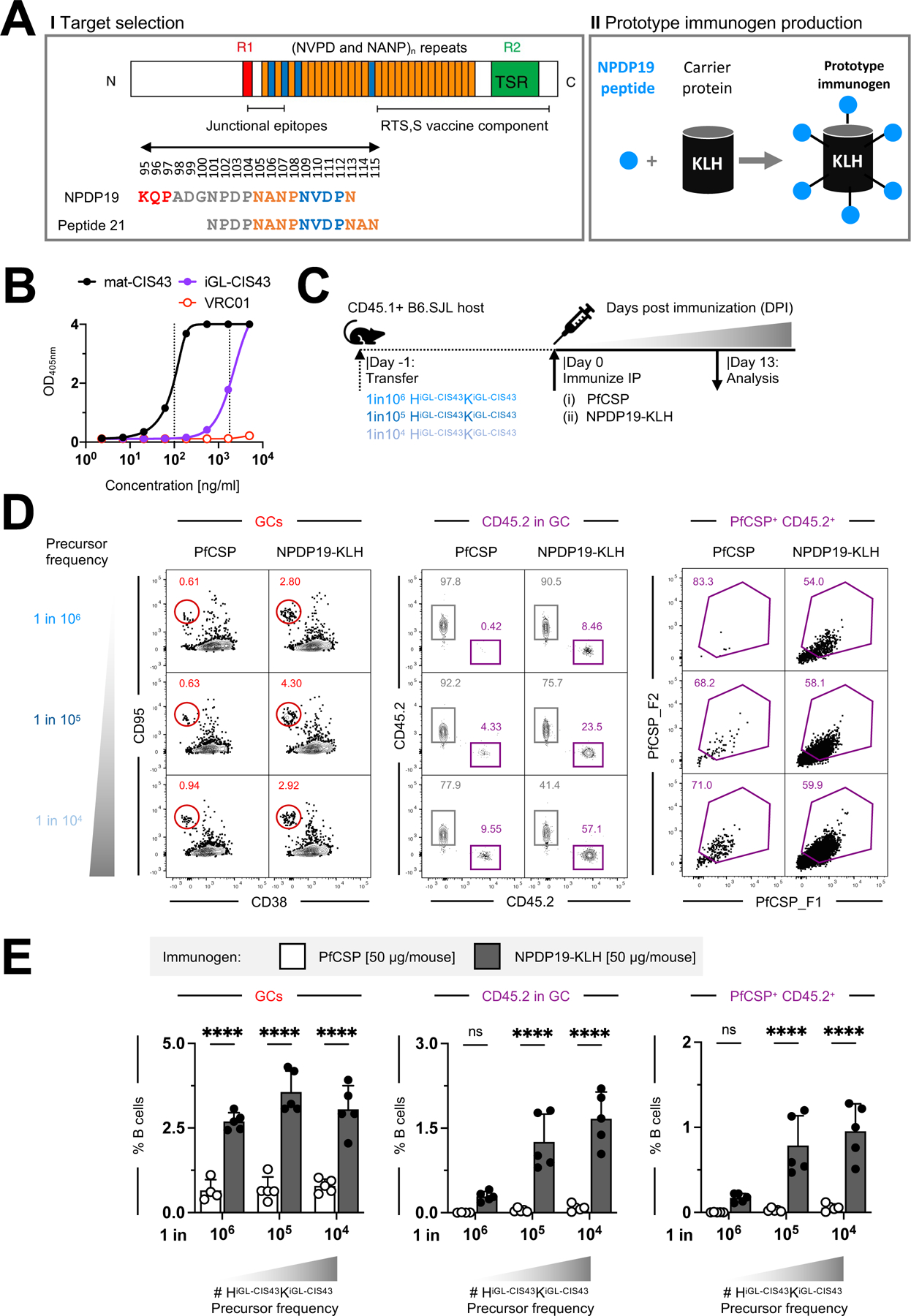
Figure 3: B cell kinetics of HiGL-CIS43κiGL-CIS43 B cells following immunization with NPDP19-KLH.
A. HiGL-CIS43κiGL-CIS43 B cell transfer system used for (B-D).
B. Gating strategy to identify PfCSP-specific HiGL-CIS43κiGL-CIS43 B cells. Representative flow cytometry plots shown for two time points, 13 and 28 DPI. For 13 DPI representative graphs for the two control groups (Sham-recipient mice were injected ip with 200 µl PBS; Alhydrogel only controls were injected ip with 200 µl of 1:1 PBS-Alhydrogel formulation) are shown.
C. Quantification of response by B cell subsets from (B). (+/−) on x-axis indicate whether Alhydrogel (Alum) and/or NPDP19-KLH were used for immunizations.
D. IgG-binding profiles determined in ELISA to PfCSP and to malaria peptides NPDP19 and (NANP)5 at 13 and 28 DPI. mAb 2A10 with mouse variable and mouse IgG1 constant region was used as a standard to determine concentrations of antigen-specific IgG.
E. Left, binding to rPfCSP in the presence of varying concentrations of peptides of mature mAb CIS43 and polyclonal mouse sera from 13 and 28 days post NPDP19-KLH immunization (middle). Right, specified amino acid sequences numbered 19–29 are shown. Sequences are color-coded, representing overlapping peptides spanning the repeat region of PfCSP.
P values were calculated by Kruskal-Wallis test with Dunn’s correction.*P<0.05; **P<0.01.
See also Figure S3.
We also included WT controls for both timepoints, for which equivalent numbers of CD45.2 B cells from C57BL/6J mice (WT CD45.2) were transferred into CD45.1 recipient mice and immunized with NPDP19-KLH. In contrast to CD45.2 HiGL-CIS43κiGL-CIS43 B cells, which responded strongly to NPDP19-KLH immunization, WT CD45.2 cells transferred at rare frequencies were not recruited to GCs (Figure S3C and S3D).
To assess the amount of antibody produced in response to NPDP19-KLH immunization, IgG binding analysis was performed against PfCSP-, Junction- and (NANP)5. No antigen-specific serum IgG was detected in either control group, while high PfCSP- and Junction-specific IgG-levels were detected at both 13 DPI (PfCSP: N=25.8±3.2 µg/ml; Junction: N=53.8±8 µg/ml) and 28 DPI (PfCSP: N=44.7±30.5 µg/ml; Junction: N=42.8±42.0 µg/ml) in mice immunized with NPDP19-KLH/Alhydrogel; binding to the (NANP)5 repeat region was minimal (13 DPI: N=0.59±0.5 µg/ml; 28 DPI: N=2.4±1.8 µg/ml) (Figure 3D). These findings were further substantiated by epitope mapping of serum responses, with CIS43-like peptide-binding signatures detectable at 28 DPI (Figure 3E, S3G and S3H). Collectively, these data reveal that immunofocusing with the junctional epitope led to sustained, junction-specific antibody responses from CD45.2 HiGL-CIS43κiGL-CIS43 B cells.
Junctional Peptide Immunization Recapitulates Mature CIS43-like SHM
Mature CIS43 mAb exhibits a low level of SHM (~3–4%). To assess if iGL-CIS43 BCRs can undergo SHM and accumulate key mutations reported for mature CIS43 following immunization with NPDP19-KLH (Kisalu et al., 2018), paired sequence analysis was performed from antigen-specific HiGL-CIS43κiGL-CIS43 B cells at 13 and 28 DPI (Figure 4 and Figure S3E). Phylogenetic trees generated from bioinformatically assembled heavy-light chain sequence pairs revealed high sequence diversity in isolated CIS43-variant sequences (Figure 4A). Substantial SHM was detected in iGL-CIS43 HCs 13 DPI, with a maximum of 7 amino acid (aa) mutations and a median of 1 aa mutation. SHM was significantly (p<0.0001) increased 28 DPI to a maximum of 9 aa mutations and a median of 5 aa mutations. Corresponding iGL-CIS43 LCs accumulated SHM that significantly increased (p<0.0001) from a median of 2 aa mutations at 13 DPI to a median of 4 mutations at 28 DPI. Further, mean nucleotide (nt) mutations in iGL-CIS43 HCs (13 DPI: N=2.5±1.8; 28 DPI; N=6.4±2.5) and LCs (13 DPI: N=2.8±1.6; 28 DPI; N=5.7±2.3) were similar to the means of aa mutation in HCs (13 DPI: N=1.7±1.6; 28 DPI; N=4.9±1.9) and LCs (13 DPI: N=2.3±1.4; 28 DPI; N=4.5±1.8) at both time points (Figure 4B and 4C). Most nt mutations in the IGHV-region were non-silent (Figure S3F).
Figure 4: A single priming immunization with NPDP19-KLH induces antibodies with key CIS43-like mutations N52K and K59R (IGHV) and H89Q and T94S (IGKV).

Antigen-specific splenic CD95+CD38low CD45.2+ HiGL-CIS43κiGL-CIS43 B cells were sorted at day 13 and 28 post immunization (DPI) for single-cell BCR sequence analysis (see Figure S4E).
A. Clonal lineage trees generated from bioinformatically assembled heavy-light chain sequence pairs. Branch length is representative of sequence distance.
B. Total nucleotide (nt) and amino acid (aa) mutations acquired in iGL-CIS43 HCs at 13 and 28 DPI. The red line indicates the median number of mutations.
C. Total nt and aa mutations acquired in the iGL-CIS43 LC at 13 and 28 DPI. Red line indicates median number of mutations.
D. Accumulation of CIS43-like aa mutations shown for human HCs (left) and LCs (right) isolated at 13 and 28 DPI. Red (Briney et al., 2019) and black (Soto et al., 2019) stair step indicate calculated antigen-agnostic mutations. The numbers inside each square indicate the number of sequences that have the total aa mutations shown on the x-axis and the CIS43-like aa mutations shown on the y-axis.
E. Hotspot analysis shows frequency of observed HC mutations per residue at 13 (purple) and 28 (blue) DPI. HCDRs are highlighted in gray. Letters in red (only present in mature CIS43 HC) indicate key aa residues for the recognition of the junctional epitope. AA positions 52 and 58 were analyzed in (F). Kabat numbering was followed.
F. Distribution of select iGL-CIS43 B cell HC aa mutations in positions 52 and 58 over time.
G. Hotspot analysis shows frequency of observed LC mutations per residue at 13 (purple) and 28 (blue) DPI. LCDRs are highlighted in gray. Letters in red (only present in mature CIS43 LC) indicate key amino acid residues for the recognition of the junctional malaria epitope. AA positions 89 and 94 were analyzed in (H). Kabat numbering was followed.
H. Distribution of select iGL-CIS43 B cell LC aa mutations in positions 89 and 94 over time.
P values were calculated by unpaired Mann-Whitney test (panels B and C). ****P<0.0001.
See also Figure S3.
Next, we evaluated whether NPDP19-KLH immunization could induce an affinity maturation pathway with CIS43 antibody-like mutations (Figure 4D). At 13 DPI, 39% (45/114) of isolated HCs and 78.7% (89/114) of LCs exhibited at least one CIS43-like mutation, increasing to 95.2% (178/187) of HCs and 86.4% (121/140) of LCs at 28 DPI (Figure 4D). In iGL-CIS43 HC, aa mutations occurred at moderate frequencies in CDR-H2-proximal key residues N52K (1.8%) and K58R (13.2%) at 13 DPI and became more predominant (N52K: 28.6%; K58R: 77.8%) at 28 DPI (Figure 4E and 4F); these residues are important for binding to the junctional epitope (Kisalu et al., 2018). CIS43-like mutations in corresponding LCs were largely confined to the CDR-L3 (Figure 4G) and occurred at high frequencies in key residues Q89H (13 DPI: 44.7%; 28 DPI: 55.3%) and T94S (13 DPI: 69.3%; 28 DPI: 81.0%) (Figure 4H), highlighting the importance of CIS43-like LCs for junctional epitope recognition.
In sum, B cells bearing iGL-CIS43 BCRs can accrue significant SHM and CIS43-like mutations after a single-immunization with the junctional malaria epitope NPDP19.
Informatic Sieving Yields CIS43-Variant Antibodies with Improved Protection
The recapitulation of many mature CIS43 sequence features following adoptive transfer of HiGL-CIS43κiGL-CIS43 B cells and vaccination with NPDP19-KLH suggested their encoded antibodies might be as, or more, protective than the parental CIS43 antibody. To identify which of the 161 paired heavy-light chain sequences obtained at 13 DPI (114 sequences) and 28 DPI (47 sequences) might have improved efficacy, we sieved sequences using genetic features such as sequence identity to mature CIS43 or measures of SHM that might be expected a priori to correlate with increased malaria-protective efficacy (Figure 5A; Table S1).
Figure 5: Informatics-based analyses identifies affinity- and sequences-based correlates of improved protection.
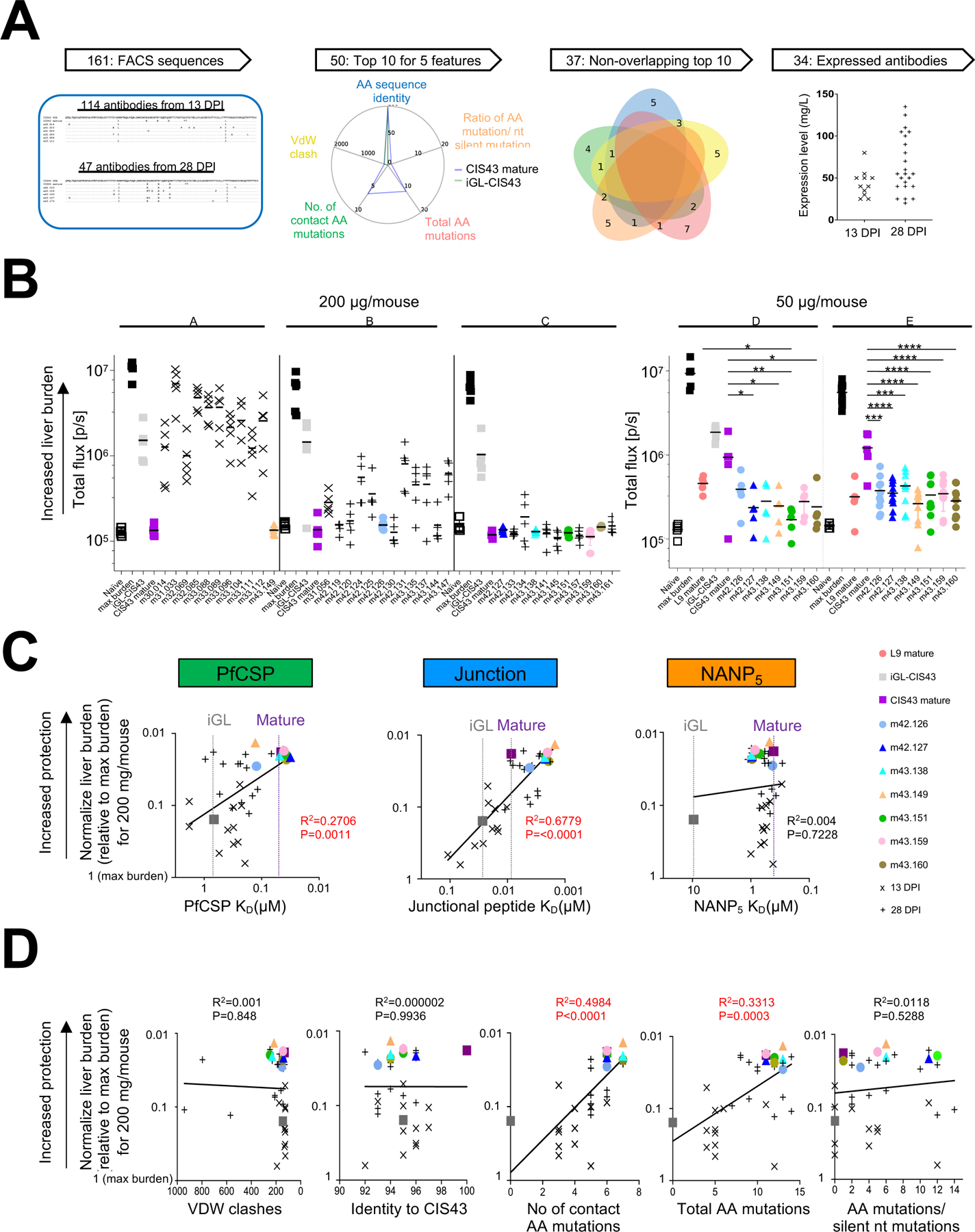
A. Sequence-based sieving of genetic features of 161 iGL-CIS43 B cells with heavy and light sequences (see Table S1), identified the top 10 sequences for 5 features, comprising 37 antibody sequences of which 34 expressed.
B. Mice were passively infused with either 200 µg antibody (left, experiments A, B and C) or 50 µg antibody (right, experiment D and E) before being challenged with transgenic P. berghei SPZ expressing PfCSP and a green fluorescent protein/luciferase fusion protein. Bioluminescent quantification of liver burden is shown 42 h post challenge, with each group of 11 antibodies assessed with controls: naïve (non-infected), max burden (no passively infected antibody), and both iGL and mature forms of CIS43. In experiments D and E, the L9 antibody was added as a control.
C. BLI affinity for 34 expressed antibodies was measured against PfCSP, junctional peptide and NANP5-repeat antigens (X-axis) and correlated to normalized liver burden (Y-axis), as assessed at 200 µg/ml.
D. Correlations between five genetic features chosen for sequence sieving and normalize protection, as assessed at 200 µg/ml.
First, as antibodies with van der Waals clashes with the CIS43-recognized epitope were unlikely to have high protective efficacy, we identified sequences that when threaded on the CIS43-peptide 21 structure (PDB-ID: 6B5M) had low van der Waals clash scores (Figure S4A and S4B leftmost panels). Second, we identified antibodies with high sequence identity to mature CIS43 (Figure S4A and S4B 2nd panels from left). Third, as germline-reverted CIS43 is poorly efficacious and SHM is required for increased efficacy, we quantified SHM in two ways: (i) the number of aa mutations within 5 Å of bound peptide in the threaded CIS43-peptide 21 structure, and (ii) the total number of aa mutations. With both SHM-related features, the 10 highest scoring variants were all from 28 DPI (Figures S4A and S4B, 3rd and 4th panels from left). Lastly, we analyzed the ratio of aa mutations to silent mutations, which can reflect the degree of selection. Roughly 10% of the sequences (especially from 13 DPI) had no silent mutations, while the highest ratios were observed more frequently at 28 DPI (Figure S4A and S4B far right panels).
Multiple sequences were among the top 10 in several categories (Figure S4C). Indeed, 37 sequences covered the top 10 sequences of all 5 features (Figure 4A). Thirty four of these expressed sufficiently (Table S2) to enable assessment of in vivo protective efficacy in a mouse model of malaria infection (Raghunandan et al., 2020). The ability of these 34 antibodies to reduce parasite liver burden following passive transfer at 200 μg/mouse was determined in mice challenged intravenously with transgenic P. berghei SPZ expressing PfCSP and a green fluorescent protein/luciferase fusion protein (Pb-PfCSP-GFP/Luc-SPZ; hereafter Pb-PfCSP-SPZ) (Flores-Garcia et al., 2019). Bioluminescent quantification of liver burden was carried out 42 h post challenge with all 34 antibodies, benchmarked against mature and inferred germline versions of CIS43. Mature CIS43 confers roughly a 2-log reduction in liver parasite burden at this dose (Kisalu et al., 2018), while the same dose of the inferred germline version was significantly less protective. At a dose of 200 µg/mouse, multiple variant antibodies showed protective efficacy that was statistically indistinguishable from mature CIS43 (Figure 4B, left panel). To better differentiate their potency, we assessed seven of the most protective antibodies, all from 28 DPI, at a dose of 50 µg/mouse, adding as an additional benchmark the recently identified L9 antibody (Wang et al., 2020), which has ~3-fold higher protective efficacy than CIS43. In two independent experiments, five of the antibodies were significantly more protective than mature CIS43, and similar to L9, suggesting at 3-fold or higher efficacy relative to mature CIS43 (Table S3).
Delineation of CIS43 Features that Correlate with Protective Efficacy
The differential protection by the 34 CIS43 variants as well as by mature and iGL-versions of CIS43 provided an opportunity to determine how antibody properties relate to protective efficacy. We used bilayer surface interferometry to measure the affinity of these CIS43 variants to PfCSP, junctional peptide, and NANP repeats (Table S4). Binding to PfCSP was fit using a 2-component binding model, and the primary binding KD correlated moderately (R=0.52) with protection (Figure 5C, left panel). Binding to the junctional peptide fit well to a single component binding model, and junctional peptide affinity correlated more strongly (R=0.82) with protection (Figure 5C, middle panel). However, binding to the penta-NANP repeat did not correlate with protection (Figure 5C, right panel). In all three cases, the difference in affinity between iGL and mature CIS43 was similar, between 10- to 30-fold, to differences in correlation reflecting the distribution of the sequence-feature-selected antibodies. Affinity of mature CIS43 to PfCSP and NANP5 rivaled the most protective variants; affinity of mature CIS43 to junctional peptide was substantially less than the most protective variants. Overall, these correlations reflect the expected binding properties of CIS43 (Kisalu et al., 2018), with junctional affinity being the most critical property of CIS43 in vivo protective potency.
We also analyzed the correlation of protective efficacy with the five sequence-related features used in our genetic sieving. Notably, only the two properties associated with SHM, contact aa mutations and total aa mutations, correlated significantly with protection. Of these, contact mutations (R=0.71) correlated more strongly than total mutations (R=0.58) (Figure 5D). The lack of correlation with van der Waals clash scores, with CIS43 identity, or with the ratio of amino acid versus silent mutations suggested that there were multiple ways to satisfy these features, some with limited improvement efficacy.
Binding and Structural Basis of Improved CIS43 Antibodies
To gain insight into the binding and structural properties of the improved CIS43 variants, we analyzed the most protective antibodies. Antibody iGL-CIS43-HL.K28.m43.151, named for transferred B cells (HiGL-CIS43KiGL-CIS43), immunization with junctional peptide-coupled to KLH (K), isolation date (28 DPI), mouse (43), and a unique identifying number (151) (we refer to antibodies by ‘mouse.number’ hereafter and in figures) showed the greatest overall reduction in liver parasite burden, comparable to L9 and ~3-fold improved over mature CIS43 (Figure 5B, Table S3). Three other antibodies, m42.127, m43.149, and m43.160, also showed statistically significant reduced liver parasite burden versus mature CIS43 with the lower dose of antibody transferred, whereas three others, m42.126, m43.138, and m43.159 were statistically superior only in the 2nd experiment (Figure 5B).
We used isothermal titration calorimetry to characterize the interactions of all seven of these antibodies with a mutant form of PfCSP (PfCSPm) with four amino acid mutations that removed processing sites and prevented dimerization upon solubilization to increase yield and to facilitate consistent analyses (Wang et al., 2020a) (Figure 6A and S5A). Mature CIS43 showed the characteristic two-step recognition (Kisalu et al., 2018), with binding affinities of 18 nM and 63 nM and with stoichiometries of 2.4 and 5.8 respectively. Notably, m43.151 showed 4–5-fold affinity increases to both sites (3.7 and 14 nM). Other variants also showed higher affinities to both primary (junctional) and secondary (non-junctional) sites. The primary affinity correlated with protection, with the correlation increasing if the number of sites were taken into consideration (Figure S5B and S5C). The secondary affinity did not correlate with protection; indeed, it trended negative, suggesting that increased secondary affinity might correlate with off-target responses. To provide insight into the atomic-level characteristics of binding by these variant antibodies, we determined their structures in complex with peptide 21 (residues 101–115 of PfCSP) for two CIS43 variants: the most potent variant from mouse 43, iGL-CIS43-HL.K28.m43.151, and the most potent variant from mouse 42, iGL-CIS43-HL.K28.m42.127 to resolutions of 2.0 and 1.8 Å, respectively (Figure 6B, Table S5). Superimposition of the two structures with mature CIS43 revealed conformational conservation, with m43.151 and m42.127 structures differing from mature CIS43 by 0.60 Å and 0.29 Å root-mean square deviation (RMSD) (all atoms) in antibody variable regions respectively; and 0.156 Å and 0.158 Å RMSD (all atoms) in the antibody-recognized junctional peptide, respectively. A substantial proportion of SHM in these two antibodies—6 of 12 for m43.151 and 6 of 11 for m42.127—was shared with mature CIS43 (Figure 6B, left panels). Thus, the maturation pathway from germline CIS43 to mature antibodies with protective efficacy appeared to require a restricted set of SHM and a diverse set of changes.
Figure 6: Thermodynamic and structural basis of improved CIS43 antibodies from iGL-CIS43 mice.

A. Isothermal calorimetry titrations of PfCSPm Isothermal titration calorimetry of PfCSPm with various iGL-CIS43-dervied antibodies at pH 7.4 and 25 °C. The affinities and stoichiometry are shown for both KD1 and KD2.
B. Crystal structures of junctional peptide (peptide 21) in complex with the most potent iGL-CIS43 derived antibodies from m42 and m43, highlighting similarity in SHM (left) and variation in bound epitope – especially in relation to isoleucine or leucine at HC position 98 (right). Numbering on peptide 21 corresponds to the PfCSP numbering.
C. Sequences of iGL-CIS43-derived antibodies statistically superior to mature CIS43 are shown with that of mature and iGL-CIS43 along with the heavy (top) and light (bottom) V-gene mutational profiles.
Defining SHM observed in most of the CIS43-improved antibodies as the restricted set of SHM identified four residues on the HC (M34IH, N52KH, K58RH and V98IH) and four residues on the LC (S27ANL, V27BIL, Q89HLand T94SL) (Figure 6C). Residue G65DH in the HC was a near miss, observed in three of the improved antibodies and in CIS43. The presence of these restricted “Core8” set SHM as assessed on the 34 ‘top-10’ antibodies correlated highly with malaria protection (R=0.73, p<0.0001). Sequence-feature analysis indicated K52H to be associated with PfCSP binding, and both K52H and R58H to be associated with peptide 21 binding and significantly associated with liver burden protection (Table S6). Many of these SHM were prevalent V-gene substitutions (Guo et al., 2019), though the mutational specificity was high, with even single atom changes observed reproducibly. Three mutations of the restricted Core8 set were not found in mature CIS43, including the Asn-Ile dipeptide in the LC CDR-L1, observed in five of the seven improved antibodies and attractively positioned near the N-terminus of peptide 21 (Figure 6B, upper left panels) to interact with PfCSP, as well as Val98H in the CDR-H3, which changed to Ile in all improved variants, but to Leu in mature CIS43.
We investigated the impact of a Leu to Ile change at residue 98H both mutationally and computationally. For the former, L98IH in mature CIS43 increased affinity to PfCSP by 1.6-fold (to 11 nM), whereas mutation of I98LH in m43.151 decreased the affinity to PfCSP by 1.3-fold (to 4.9 nM); ITC analysis indicated the change in ΔG to be 0.2 kcal/mol (Figure S5D). Computationally, the reduction in binding energy from changing Leu to Ile was found to be −0.16 kcal/mol (CIS43 L98IH) and for Ile to Leu was 0.18 kcal/mol (Figure S5E).
Structural analysis revealed Ile98H to stabilize the antigen-binding pocket by enhancing the heavy-light chain contacts through hydrophobic interactions with Y49L and W50L; the Ile change also enables additional contact with the C-terminus of junctional region including residue Val110 on PfCSP (Figure 6B, right panel). Overall, these results confirmed the ability of single atom changes to affect CIS43 antibody function, potentially explaining the repeated selection of the exact same SHMs in the restricted set of mutations (Figure 6C and S5F). The higher prevalence of the restricted Core8 mutations in the improved antibodies may explain their increased protective capacity.
Improved CIS43 Antibody “D3” Is Best-In-Class
Several variant CIS43 antibodies were as potent for protection as antibody L9, a benchmark for the best-in-class malaria-protective antibody (Wang et al., 2020a). Thus, we sought to obtain a further improved CIS43 antibody by exploiting the high correlation between protective function and both contact and total aa mutations. For contact mutations, we used an expanded definition of contact residues to include direct neighbors (with the added requirement that they were observed in more than one mouse); we used this definition to sieve the database of CIS43-SHM obtained from adoptively transferred immunized mice to identify five potential beneficially mutations, S31RH and L95MH on the HC, and L27cFL, Y27dFL, and Y92FL on the LC, and added these in various combinations to m43.151 (design D1-D6). For total mutations, we identified aa mutations that occurred in the top clones, identifying 8 on HC and 11 on LC, and added these to m43.151 (design D7-D11) (Figure 7A).
Figure 7: Design based on iGL-CIS43 mice information yields best in class antibody iGL-CIS43.D3.
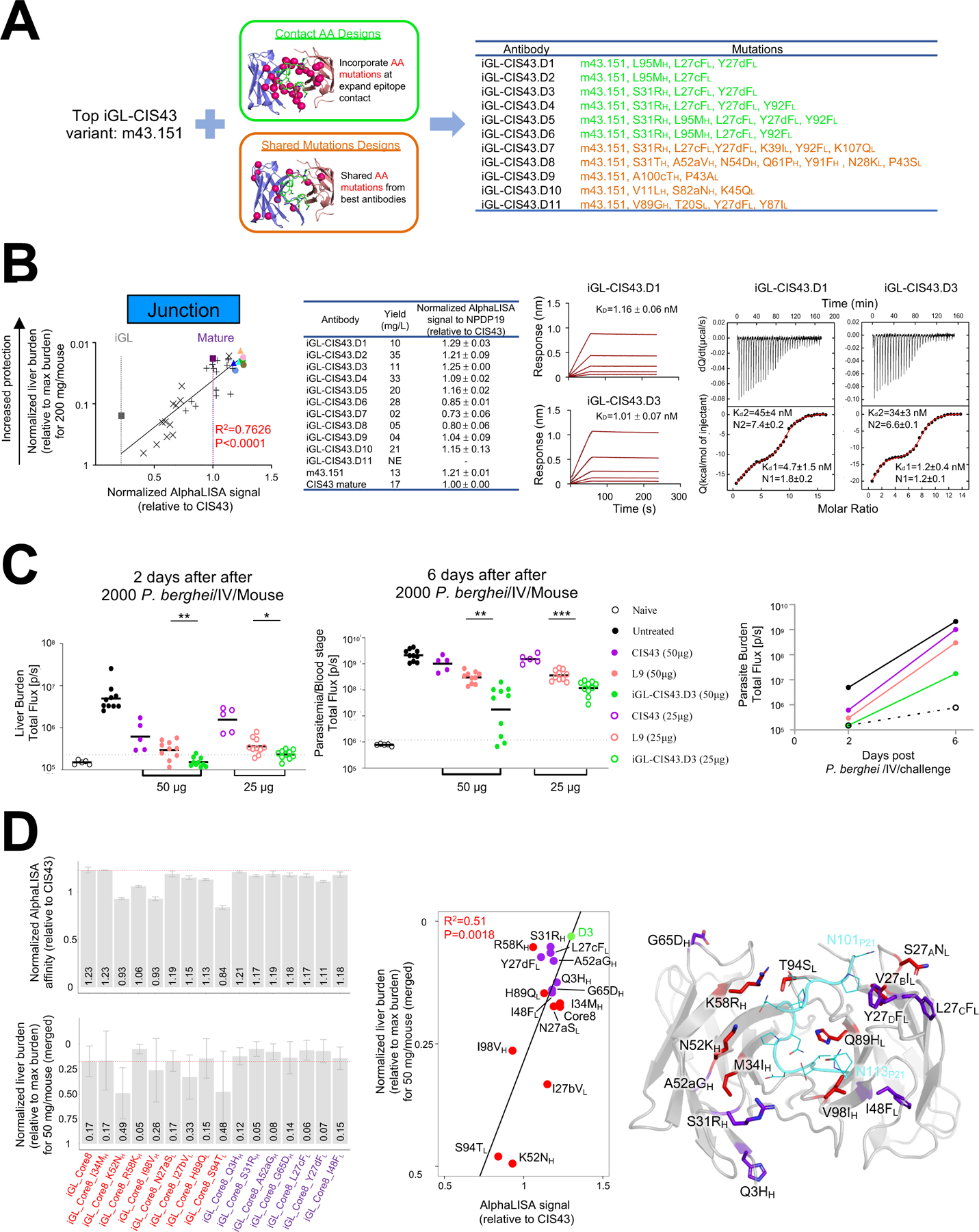
A. m43.151 variants designed using (i) expanded amino acid contact mutations (green color) and (ii) mutations on top antibodies (orange color).
B. AlphaLISA-measured apparent affinity to NPDP19 correlates strongly with protection (R=0.873) for 34 genetically identified antibodies analyzed in Fig. 5 (leftmost panel; antibodies are color coded as in Fig 5C). BLI and ITC affinities for iGL-CIS43.D1 and iGL-CIS43.D3 are shown for NPDP19 and PfCSPm, respectively (right panels).
C. Mice were passively infused with either 50 µg (filed circle) or 25 µg (empty circle) antibody before being challenged with transgenic P. berghei SPZ expressing PfCSPm and a green fluorescent protein/luciferase fusion protein. Bioluminescent quantification was done for each mouse on day 2 and day 6.
D. Structure-function analysis. Initial core 8 mutation were transplanted onto iGL-CIS43 backbone sequence (iGL_Core8) and structure-function analysis was performed, which included the reversion of single mutations and addition of mutations from iGL-CIS43.D3 onto iGL_Core8 construct. AlphaLISA-measured apparent affinities to NPDP19 were determined (D3 has AlphaLISA value 5.53×106), and liver protection was measured (D3 has normalized liver burden 0.03) (left panels). These measurements correlated (middle panel), and the location of each alteration is depicted on the iGL-CIS43.D3 structure (right panel).
As the affinity for NPDP19 showed high correlation with function, we used this biochemical property to select which of the 11 antibodies to assess functionally. We used AlphaLISA apparent affinity, measured in the context of full antibody, as it demonstrated higher correlation with function (R2 = 0.7626) than the Fab-derived BLI KD (R2 = 0.6779) (Figure 7B, left panels and S6A). Designs iGL-CIS43.D1 (D1) and iGL-CIS43.D3 (D3) yielded the highest AlphaLISA signals. The BLI-measured affinity for NPDP19 was 1.16 nM for D1 and 1.01 nM for D3, while ITC-measured affinity for PfCSPm was 4.7 nM for D1 and 1.2 nM for D3 (Figure 7B, right panels and S6B). Because of the virtually equivalent affinity of D1 and D3 for NPDP19 and the much higher affinity of D3 for PfCSPm, we assessed the ability of D3 to reduce parasite liver burden following passive transfer of 25 or of 50 μg/mouse (Figure 7C). In both cases, D3 significantly reduced liver burden versus L9.
Structure-Function Assessment and Crystal Structure of iGL-CIS43.D3
To provide insight into the specific alterations that improved iGL-CIS43.D3 function, we investigated the 15 aa alterations in D3 versus iGL-CIS43. We used Core8 mutations as a reference, either removing or adding single changes onto Core8 (Figure 7D, left panel and S6C-D). Core8 showed an AlphaLISA-measured apparent affinity equivalent to or higher than each of the mutated forms (though ~10% lower than D3). Reversion of HC I34MH, R58KH, or I98VH or of LC S94TL significantly reduced affinity. In terms of protective function, the normalized liver burden was slightly less for Core8 versus the single added mutations from D3, and with the two most substantial reduction in protective efficacy (K52NH and S94TL) trending with reduced affinity. Each of the seven additional mutations showed affinities and protective function that were very similar to Core8, which were substantially below the affinity and protective function of D3, suggesting the seven additional mutations act collectively to improve D3 affinity and function.
Overall, AlphaLISA-measured apparent affinity correlated with reduction in liver burden (Figure 7D, middle panel). We determined the crystal structure of D3 in complex with junctional peptide (Figure 7D, right panel). Half of the alterations found in D3 were distal from the junctional peptide, though still on the exposed portion of the Fab, suggesting the interactions between D3 and PfCSP involve more than those observed with the junctional peptide; indeed, the three mutations which improve D3 relative to m43.151, S31RH, L27cFL, and Y27dFL, were each located on the face of the antibody that would be expected to contact PfCSP, but distal from bound peptide, consistent with D3 having ~3-folder higher affinity for PfCSP, but similar junctional peptide affinity as m43.151.
Discussion
Initially, we assessed how HiGL-CIS43κiGL-CIS43 B cells responded to immunization with full-length PfCSP. This strategy was recently highlighted by McNamara et al., who used an immunoglobulin (Ig) KI mouse model bearing the germline-reverted HC from a murine PfCSP-neutralizing mAb to demonstrate that epitope masking hinders B cell recall responses to malaria vaccination (McNamara et al., 2020). However, our data show HiGL-CIS43κiGL-CIS43 B cells responded poorly to single PfCSP-immunization and were outcompeted by endogenous B cells, suggesting that a peptide-based vaccine may better direct the immune response to target the junctional epitope. While it has been suggested that the malaria NANP-repeat can elicit antibodies that cross-react with the junction (Oyen et al., 2020), the outgrowth of GC B cells by host B cells observed in this study may be attributable to overabundant immunodominant NANP-repeat epitopes detracting from more junction-specific responses (Oyen et al., 2017, 2020).
To address this, we produced a prototypical immunogen using an approach from the field of HIV-1 vaccine research (Xu et al., 2018) and found that a junctional epitope-focused immunization strategy induced a strong response from HiGL-CIS43κiGL-CIS43 B cells. This is in agreement with two recent studies showing that the junctional epitope, when displayed on alphavirus virus-like particle (VLP) (Francica et al., 2021) or Qß bacteriophage VLPs (Jelínková et al., 2021), elicits partial protection against intravenous sporozoite challenge. Calvo-Calle et al. furthermore demonstrated that immunization with a tetra-branched version of the junctional epitope region leads to a reduction in liver burden following mosquito bite challenge (Calvo-Calle et al., 2021). A comparable vaccination strategy using NPDP19-KLH induced anti-PfCSP responses in BALB/c mice, but the mouse antibodies generated failed to inhibit PfSPZ invasion of a hepatocyte cell line (Tan et al., 2019). Atcheson et al. hypothesized that the inclusion of the ‘KQP’ at the beginning of the NPDP19-KLH immunogen could negatively impact the elicitation of neutralizing antibodies (Atcheson et al., 2021). However, our findings demonstrate that immunization with NPDP19-KLH effectively activates malaria-specific germline-precursor B cells and elicits neutralizing CIS43-variants that offer up to 3-fold better protection than mature CIS43, confirming that the induction of protective CIS43-variant antibodies is achievable with epitope-based vaccine design (Kisalu et al., 2018).
A central finding of our study is that epitope-based immunization induced CIS43 variants with greater potency than mature CIS43. This was established by correlating protective function with genetic properties, identifying contact amino acid changes and total SHM as correlating highly with protective function. Mutations defined by these genetic properties were further tested to identify D3, which we found to be more protective than L9—the current best-in-class (Wang et al., 2020a). Anti-malarial antibodies with improved potency and duration have great promise for the prevention of malaria infection via passive transfer for a variety of clinical use cases which range from travelers to seasonal control and ultimately elimination (Gaudinski et al., 2021). In terms of durability, we recently showed that incorporating an LS mutation into CIS43 increases the half-life of mature CIS43 from 22 to 39 days in rhesus macaques (Kisalu et al., 2020). CIS43LS conferred protection against malaria eight months after administration in a phase I clinical trial (Gaudinski et al., 2021). More potent CIS43-variants could lower the threshold required for protection, reducing the cost of this intervention. The most protective antibody isolated in this study, D3, reduced parasite burden 5–10-fold more than mature CIS43, providing direct evidence that existing human anti-malarial antibodies can be improved via our KI mouse/adoptive transfer system. Overall, this humanized mouse B cell platform has the potential to contribute to both passive prevention and vaccine-based approaches for preventing malaria.
Limitations of the study
In this study the iGL sequence of CIS43 was used to generate a KI mouse model to assess vaccine immunogens and develop highly potent anti-malaria antibodies. The prediction of germline BCR sequences is challenging as these develop stochastically, and it is possible that iGL sequences do not adequately represent sequences in the human B cell repertoire (Huang et al., 2020). Recent HIV-vaccine studies have highlighted that this can be overcome through the identification and use of authentic germline BCR sequences (Huang et al., 2020; Wang et al., 2020b). Studies are needed to identify authentic germline sequences of anti-malarial antibodies, particularly as many potent malaria antibodies depend on germline-encoded residues for epitope-binding (Julien and Wardemann, 2019).
We do not rule out that alternative peptide-immunogens and sequential immunizations, potentially using different vaccine modalities such mRNA and/or virus-like particles, could induce longer lasting GCs and give rise to even more improved CIS43-variants. Additional studies are needed to assess if epitope-focusing can also activate B cells that are cross-reactive to the NANP-repeat (such as 317) and induce variants of anti-malarial antibodies with enhanced protection (Imkeller et al., 2018; Murugan et al., 2020).
Lastly, we envision that future work will combine this experimental system with malaria infection studies to assess the protective capacity of malaria vaccine candidates and to select the most promising regimens for clinical studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by the Lead Contact, Facundo D. Batista (fbatista1@mgh.harvard.edu).
Materials Availability
The datasets and/or materials generated and/or analyzed during the current study are available from the corresponding authors under standard material transfer agreements.
Data and Code Availability
The crystal structures reported in this manuscript have been deposited in the Protein Data Bank, www.rcsb.org. The 161 CIS43 variant antibody nucleotide sequences are deposited in GenBank. Accession codes are listed in the KRT.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
For experiments male B6.SJL-Ptprcapepcb/BoyJ mice (CD45.1+/+) between 7–12 weeks were purchased from The Jackson Laboratory (Bar Harbor ME). F0-mice from the inferred germline CIS43 KI mouse (CD45.2+/+) colony were bred at the animal facility of the Gene Modification Facility (Harvard University) and breeding for colony expansion and experimental procedures was subsequently performed at the Ragon Institute of MGH, MIT and Harvard. Ear or tail snips from CIS43-germline KI mice were used for genotyping by TaqMan assay for a fee for service agreement (TransnetYX). TaqMan probes for the genotyping assay were developed by TransnetYX. CD45.2+ B cells from iGL-CIS43 donor KI mice were enriched using the Pan B Cell Isolation Kit II (Miltenyi Biotec), enumerated, diluted to desired cell numbers in PBS and adoptively transferred into CD45.1+ recipient mice as reported previously (Abbott et al., 2018). Preparations of immunogens (PfCSP at 5 µg/mouse (or 50 µg/mouse) and/or NPDP19-KLH at 50 µg/mouse) were diluted in PBS at a volume of 100 µl/mouse and mixed at a 1:1 ratio with 100 µl/mouse Alhydrogel 2 % (Invivogen) for at least 25 min, and then injected intraperitoneally (i.p.) (total volume of 200 µl/mouse). All experiments were done with approval by the Institutional Animal Care and Use Committee (IACUC) of Harvard University and the Massachusetts General Hospital and conducted in accordance with the regulations of the American Association for the Accreditation of Laboratory Animal Care (AAALAC).
Female 6–8-week-old B6(Cg)-Tyrc-2J/J albino mice were obtained from The Jackson Laboratory. All animals were cared for in accordance with American Association for Accreditation of Laboratory Animal Care standards in accredited facilities. All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committees of the National Institute of Allergy and Infectious Diseases, National Institutes of Health, specifically: Animal Study Protocol VRC-17–702.
Generation of CIS43-germline knock-in (KI) mice
Inferred germline CIS43 KI mice were generated following published protocols (Lin et al., 2018; Wang et al., 2020b). In brief, the targeting vector 4E10 (Ota et al., 2013) was modified by the incorporation of human rearranged CIS43-germline VDJ (heavy chain construct) or VJ (light chain construct) sequences downstream of the promoter region and by elongation of the 5’ and 3’ homology regions utilizing the Gibson assembly method (NEB). The targeting vector DNA was confirmed by Sanger sequencing (Eton Bioscience Inc.). sgRNAs used here were identical to sgRNAs previously validated for BG18gH and PGT121 κ (Lin et al., 2018).
Next, an injection mix containing both heavy and light chain DNA constructs described above (200 ng/µl), Cas9 protein (50 ng/µl), the corresponding sgRNAs (25 ng/µl) and injection buffer was prepared for microinjecting 200 fertilized oocytes. Following culture, resulting zygotes were implanted into the uteri of pseudopregnant surrogate mothers.
Cell lines
Expi293F human female kidney cell line was obtained from ThermoFisher Scientific Inc (Thermo Fisher, Catalog number: A14527). The cell line was used directly from the commercial sources.
METHOD DETAILS
Immunogen and FACS probe production
Full length recombinant Plasmodium falciparum circumsporozoite protein (rPfCSP) was generated as previously described (Kisalu et al., 2018). The peptide-based prototypical immunogen was generated by conjugating NPDP19 (KQPADGNPDPNANPNVDPN) via a maleimide linker to KLH that had been equipped with free -SH groups via Trauts reagents (GenScript). For flow cytometric probe binding rPfCSP was biotinylated by BirA enzymatic reaction (Avidity, Inc) according to the manufacturer’s protocol. The junctional peptide (NPDP19) was synthetically made and biotinylated at its N terminus (GenScript). Biotinylated rPfCSP and NPDP19 peptide were pre-reacted in independent tubes for at least 30 min in a 4:1 molar ratio with fluorescently labeled streptavidin (SA-A488 and/or SA-647). Reagents were then combined with fluorescently labeled antibodies for FACS-staining.
ELISA
96-well plates were coated overnight at 4°C with one of the following: NPDP19 at 50 ng per well, NANP5 at 125 ng per well or PfCSP 25 ng per well. Plates were washed 5 times with 0.05% Tween 20 in PBS, blocked with 100 µl of 3% BSA in PBS for 1 h at room temperature (RT), and washed again prior to incubation with 1:3 or 1:5 serially diluted mouse serum samples for 1 h at RT. Wells were washed and incubated with Alkaline Phosphatase AffiniPure Goat Anti-Mouse IgG (Jackson Immuno Research) at 1:1,000 in PBS with 0.5% BSA for 1h at RT. p-Nitrophenyl phosphate dissolved in ddH2O (50 µl/well, RT, 25 min) was used for detection. A chimeric version of the anti-PfCSP antibody 2A10 (Fisher et al., 2017; Hollingdale et al., 1984) with human Ig heavy and Ig kappa and the fully human mature anti-PfCSP CIS43 antibody were used as standard reference materials. ELISA curves were calculated and analyzed using GraphPad Prism 8.4.3 (GraphPad).
AlphaLISA
AlphaLISA® (Perkin-Elmer) is a bead-based proximity assay in which singlet oxygen molecules, generated by high energy irradiation of Donor beads, transfer to Acceptor beads, which are within a distance of approximately 200 nm. It is a sensitive high throughput screening assay that does not require washing steps. A cascading series of chemical reactions results in a chemiluminescent signal. Purified antibodies were diluted to 100 nM in AlphaLISA® buffer (PBS + 0.05% Tween-20 + 0.5 mg/mL BSA). Subsequently, 5 μL of the IgGs were transferred to an OptiPlate-384 assay plate (white opaque, PerkinElmer), mixed with 10μL (10 nM final conc.) of biotinylated peptide probe and 10 uL (10 μg/mL final conc.) of Anti-human IgG (Fc specific; Perkin-Elmer) acceptor beads. After an hour of incubation at RT, non-shaking, 25 uL (40 μg/mL final conc.) of streptavidin donor beads (Perkin-Elmer) were added. The plate was then incubated for 30 min at RT in the dark before the AlphaLISA signal was detected using a SpectraMax® i3x multi-mode microplate reader (Molecular Devices).
Flow Cytometry
At select time points following immunization, whole spleens were mechanically dissociated using 5 ml syringe plungers to generate single-cell suspensions. ACK lysis buffer was used to remove red blood cells and splenocytes were then resuspended in FACS buffer (2% FBS/PBS), Fc-blocked (clone 2.4G2, BD Biosciences) and stained for viability with Live/Dead Blue (Thermo Fisher Scientific) for 20 min at 4°C. For surface staining tetramer rPfCSP and/or NPDP19 probes (described above), as well as antibodies against CD4-APC-eF780, CD8-APC-eF780, Gr-1-APC-eF780, F4/80-APC-eF780, B220-B510, CD95-PE-Cy7, CD38-A700, CD45.1-PerCP-Cy5.5, CD45.2-PE, IgD-BV786, IgM-BUV395 and IgG1-BV421, were used. Cells were acquired by a BD FACSymphony (BD Biosciences) for flow cytometric analysis and sorted using a BD FACS Aria II instrument (BD Biosciences). Data was analyzed using FlowJo software (Tree Star). The gating strategy is shown in Figure S4E. CD95+CD38+CD45.2+IgG1+ B cells were single-cell dry-sorted into 96-well PCR plates, rapidly frozen on dry ice and stored at −80°C until processing.
BCR sequencing
Following single-cell sorting of antigen-specific B cells, the genes encoding the variable region of the heavy and light chains of IgG were amplified through RT-PCR. In brief, first strand cDNA synthesis was carried out using SuperScript III Reverse Transcriptase (Invitrogen) according to manufacturer’s instructions. Nested PCR reactions consisting of PCR-1 and PCR-2 were performed as 25 µl reactions using HotStarTaq enzyme (QIAGEN), 10 mM dNTPS (Thermo Fisher Scientific) and cocktails of IgG- and IgK-specific primers and thermocycling conditions described previously (von Boehmer et al., 2016). PCR products were run on precast E-Gels 96 2% with SYBR Safe (Thermo Fisher Scientific) and wells with bands of the correct size were submitted to GENEWIZ company for Sanger sequencing. HC products were sequenced using the HC reverse primer from PCR-2 (5’ GCTCAGGGAARTAGCCCTTGAC 3’) and the LC was sequenced using the LC reverse primer (5’ TGGGAAGATGGATACAGTT 3’) from PCR-2. Reads were quality-checked, trimmed, aligned and analyzed using the Geneious software (Geneious). IMGT/V-QUEST (http://www.imgt.org) was used for mouse/Human Ig gene assignments. CIS43-like mutation calculation (Figure 4D) were done as described previously (Briney et al., 2019; Soto et al., 2019).
Epitope mapping and competition ELISAs
Competitive ELISAs using overlapping linear PfCSP peptides (peptides 16–61; Genscript) that span the R1 and repeat region of PfCSP were performed on the Meso Scale Discovery (MSD) U-Plex Assay platform. Peptides were all 15 amino acids in length, overlapping by 11 residues, and numbered according to their position on the protein. Briefly, streptavidin-coated plates (Meso Scale Discovery, MSD) were blocked with 5% BSA in PBS for 30 min at room temperature (RT), washed five times (wash buffer, 0.05% Tween-20 in PBS), then coated with biotinylated-recombinant PfCSP (0.2μg/mL, Genscript) in PBS with 1% BSA, and allowed to incubate for 1h at RT. Either PfCSP specific monoclonal antibodies (all at 10 ng/mL except iGL-CIS43 at 100ng/mL), or polyclonal mouse sera (pooled per group then diluted 1:250) were preincubated with varying concentrations (0 – 1,000 mg/mL) of selected PfCSP peptides in PBS with 1% BSA/0.05% Tween-20 for 2hrs at 37C, then added onto the rPfCSP- coated plates. Plates were incubated for 1 h at RT, washed five times, then incubated for an additional 1h at RT with 1 μg/mL of appropriate secondary (either anti-human or anti-mouse) IgG SULFO-TAG (Meso Scale Discovery) in PBS with 1% BSA/0.05% Tween-20. After washing, plates were read using 1X MSD Read T Buffer (Meso Scale Discovery) on an MSD SECTOR © Imager 6000 instrument.
Sporozoites
Transgenic P. berghei (strain ANKA 676m1c11, MRA-868) expressing full-length P. falciparum PfCSP and a green fluorescent protein/luciferase fusion protein (Pb-PfCSP-GFP/Luc-SPZ) were obtained as previously described (Flores-Garcia et al., 2019).
IV challenge and quantification of protection
IV challenges were performed as previously described (Wang et al., 2020a). Briefly, mAbs were diluted in sterile PBS (pH 7.4) to give a final dose of 50–200 μg, as indicated, in a total volume 200 μl/mouse) and were injected IV via the tail vein. Approximately 4 hours later, mice were then intravenously challenged in the tail vein with 2,000 freshly harvested Pb-PfCSP-GFP/Luc-SPZ in Leibovitz’s L-15 Medium (Gibco). 40–42 h post-challenge, mice were injected intraperitoneally with 150 μL of D-luciferin (PerkinElmer; 30 mg/mL), anesthetized with isoflurane and imaged with the IVIS® Spectrum in vivo imaging system (PerkinElmer) 10 min after luciferin injection. Liver burden was quantified by analyzing a region of interest (ROI) in the upper abdominal region; the total flux (p/s) was measured using the manufacturer’s software (Living Image 4.5, PerkinElmer). To measure parasitemia, luciferin was re-injected 7 days post-challenge and quantification was performed with an ROI encompassing the whole body.
For the correlation of mAb sequence characteristics with protection, percent (%) protection for each mouse was calculated based on the liver burden flux (p/s) data. % protection = [100 – ((antibody-treated mouse flux / mean flux of untreated mice)*100)].
Bioinformatics
Accumulation of CIS43-like mutation
Non-duplicated VH1–3 sequences were obtained from 13 healthy donors (PRJNA511481, (Soto et al., 2019), PRJNA406949, (Briney et al., 2019)), and non-duplicated VK4–1 sequences from 3 healthy donors (PRJNA511481). We calculated per donor mutation profile for VH1–3 and VK4–1 germline. In brief, we aligned the heavy and light chain protein sequences to VH1–3 and VK4–1 germline protein sequence, respectively, and calculated the probability distributions: 1.the number of mutations, 2. per site mutation rate, and 3. per site amino acid frequency. Based on per donor probability distribution, we generate synthetic VH1–3 and VK4–1 antibody sequences. The CIS43-like mutations were defined as all the SHM on CIS43 V germline gene. The mean frequency of randomly having CIS43 mutation in synthetic VH1–3/VK4–1 sequences and the 95% confidence intervals were calculated by using Python script.
Sequence-based sieving
Based on the antibody sequence, we defined five properties, VdW clashes, sequence identity to mature CIS43, SHM on peptide contact residues, total SHM, and total SHM divided by silent mutations. We used RosettaRemodel to predict the structure model of CIS43 like variants (Huang et al., 2011). CIS43 in complexed with peptide21 (PDB ID 6B5M) was used as template for model building of CIS43 like variants and defined peptide contact residues (Kisalu et al., 2018). The value of fa_rep reported by RosettaRemodel used as the VdW clash score. The sequences of CIS43 like variants were aligned with CIS43 iGL or CIS43 mature sequence to obtain CIS43 sequence identity, SHM peptide on contact residues, total SHM and Total A.A. mutations divided by silent mutations.
Gene-specific substitution profile
Per residue mutation profiles of IGHV1–3 and IGKV4–1 germline genes were obtained from cAb-Rep server, and the substitution frequency less than 0.5% was defined as rare mutation (Sheng et al., 2017).
Informatic analysis on binding energy of WT, CIS43_L98I and I98L mutations
Van der Waals (VdW) pairwise binding energy between peptide and Fab residues were calculated upon mutation to determine the importance of the L98I mutation. The mature CIS43 antibody (PDB: 6B5M) and m43.151 complex structures were used as templates to model the 34 CIS43-variant antibodies by using the FoldX software (http://foldxsuite.crg.eu/). The variants were then minimized using YASARA (http://www.yasara.org/). The reference structures were also modeled and minimized by making VH1V and QH1Q identity mapping mutations. A single DCD trajectory frame was generated with VMD’s Autopsf software (http://www.ks.uiuc.edu/Research/vmd/). The residue interaction energies were calculated using gRINN (https://grinn.readthedocs.io/en/latest/index.html) and NAMD_2.14_Linux-x86_64-multicore with CHARMM36 force field (Best et al., 2012; Guvench et al., 2011) a default NAMD solute dielectric of 1, non-bonded cutoff 12 Å, filtering cutoff of 15. The resulting VdW energy matrix between peptide 21 and m heavy/light chain residues within the cutoffs was generated for all 34 variants. The energy differences were calculated by subtracting the variant matrix from the aligned reference matrix. Only mutated entries strictly greater than zero were selected and summed across peptide positions. Finally, ΔE values were grouped by mutation position and type and averaged across variants. The CIS43_L98I and m43.151_I98L variants were also generated and analyzed by following the same procedure.
Production of antibodies and antigen-binding fragments (FABs)
Antibody heavy and light chain genes were synthesized (Gene Universal Inc, Newark DE) and subcloned into corresponding pVRC8400 vectors. To express the antibodies, equal amounts of heavy and light chain plasmids were transfected into Expi293F cells (Life Technology) by using Turbo293 transfection reagent (Speed BioSystems). Transfected cells were cultured in shaker incubator at 120 rpm, 37 °C, 9% CO2 for 5 days. Culture supernatants were harvested and purified over Protein A (GE Health Science) resin in columns. Each antibody was eluted with IgG elution buffer (Pierce), immediately neutralized with one tenth volume of 1M Tris-HCl pH 8.0. The antibodies were then buffer exchanged in PBS by dialysis.
Fabs containing a 6xHis-tag on heavy chain were expressed as above. On day 6 post transfection, culture supernatants were harvested and incubated with cOmplete His-Tag Purification resin. After washing with PBS containing 20 mM imidazole, Fabs were eluted in 50 mM sodium phosphate pH 8.0, 300 mM NaCl, and 250 mM imidazole. The protein was further purified by size exclusion chromatography (SEC) on a Superose 6 10/300 GL column in PBS.
Affinity measurements by BLI
Antibody Fab binding affinity to various ligands were measured using biolayer interferometry on an Octet Red384 instrument (fortéBio) with streptavidin capture biosensors (fortéBio) in solid black tilt-well 96-well plates (Geiger Bio-One). Assays were performed with agitation at 25°C. Immobilization of biotinylated rPfCSP, peptides 21 or peptide NANP5 was performed for 60s, followed by a 60s baseline in buffer (PBS + 1% BSA). Association with Fab (serially diluted from 1000 to 62.5 nM) was done for 60s, followed by a dissociation step in buffer for 120s. In all Octet measurements, parallel correction to subtract systematic baseline drift was carried out by subtracting the measurements recorded for a loaded sensor incubated in PBS. Data analysis was carried out using Octet software, version 9.0. Experimental data were fitted globally with a 1:1 Langmuir model of binding.
ITC
A stabilized version of PfCSP with increased expression (3D7 clone of the NF54 strain (PlasmoDB ID: PF3D7_0304600.1)) was used for the ITC experiments. This construct, termed PfCSP_SAmut_C5S (Wang et al., 2020a) was modified from rPfCSP (Kisalu et al., 2018) by introducing four amino acid mutations in the N-terminal domain that removed processing sites and prevented dimerization upon solubilization to increase yield and facilitate consistent analyses. The protein was expressed through transient transfection in 293F cells (Thermo Fisher Scientific) and purified from culture supernatants through polyhistidine-tag affinity chromatography followed by size-exclusion chromatography (GE Healthcare). Monomer-containing fractions were pooled, concentrated, snap frozen, and stored at −80°C.
Calorimetric titrations of full-length rPfCSP with selected antibodies, m42.126, m42.127, m43.138, m43.149, m43.151, m43.159, m43.160 and CIS43 mature, were made using a MicroCal VP-ITC from Malvern Panalytical (Northampton, MA, USA). rPfCSP and the antibodies were prepared in PBS, pH 7.4, and all the titrations were performed at 25 °C. The concentration of rPfCSP in the calorimetric cell (~1.4 mL) was 0.20 – 0.25 µM and antibody solution at a concentration of 23 – 28 µM antigen binding sites was added in 7-µL aliquots until saturation was reached. The injections were made at 300 s intervals. The exact concentration of the experimental solutions was determined from the absorbance at 280 nm. The heat produced upon each injection was obtained by integration of the calorimetric signal and the heat associated with antibody binding to rPfCSP was obtained after subtraction of the heat of dilution from the heat of reaction. The individual heats of binding were expressed as a function of the molar ratio and the association constant, Ka = 1/Kd, the enthalpy, ΔH, and the stoichiometry, N, were obtained by nonlinear regression of the data to a sequential binding model to two sets of sites with different binding energetics and stoichiometries (Freire et al., 2009).
Crystallization and structural analysis
Antibody Fab and peptide 21 (PfCSP residues 101–115) complexes were prepared by mixing 1:2 molar ration to a concentration of 15 mg/ml. Crystallization conditions were screened in Hampton Research screening kits, Wizard screening kits, Precipitant Synergy screening kits, JCSG1–4 screening kits using a mosquito robot. Crystals initially observed from the wells were manually reproduced. The m42.127 : P21 complex crystal grew in 0.1 M Sodium acetate trihydrate pH 4.5 and 30% w/v polyethylene glycol 1,500; the m43.151 : P21 complex crystal grew in 0.1 M Citric acid pH 3.5 and 25% w/v polyethylene glycol 3,350; the m43.160:P21 complex crystal grew in 0.1 M BIS-TRIS pH 5.5, 25% w/v Polyethylene glycol 3,350; the m42.126:P21 complex crystal grew in 0.1 M Sodium citrate tribasic dihydrate pH 5.5, 18% w/v Polyethylene glycol 3,350; the m43.149:P21 complex crystal grew in 0.2 M Sodium formate, 20% w/v Polyethylene glycol 3,350; the m43.159:P21 complex crystal grew in 0.1 M Sodium acetate trihydrate pH 4.0, 10% w/v Polyethylene glycol 4,000; the m43.138:P21 complex crystal grew in 0.1 M Sodium citrate tribasic dihydrate pH 5.0, 10% w/v Polyethylene glycol 6,000; the iGL-CIS43.D3 crystals grew in 0.2 M Zinc acetate, 0.1 M MES pH 6.0, and 20% w/v polyethylene glycol 8,000. Crystals were cryoprotected in 25% glycerol and flash-frozen in liquid nitrogen. Data were collected at a temperature of 100 K and a wavelength of 1.00 Å at the SER-CAT beamline ID-22 (Advanced Photon Source, Argonne National Laboratory). Diffraction data were processed with the HKL2000 suite (Otwinowski and Minor, 1997). Structure solution was obtained by molecular replacement with Phaser using CIS43 Fab structures (PDB ID: 6B5L) as a search model. Model building was carried out with Coot (Emsley and Cowtan, 2004). Refinement was carried out with Phenix (Liebschner et al., 2019). Ramachandran statistical analysis indicated that the final structures contained no disallowed residues or no more than 0.11% disallowed residues. Data collection and refinement statistics are shown in Table S5.
QUANTIFICATION AND STATISTICAL ANALYSIS
For immunization studies, statistical analysis was performed in Prism 9.01 (GraphPad) using either two-tailed Mann–Whitney test assuming non-normal distribution, Wilcoxon matched pairs signed rank test or Kruskal-Wallis test with Dunn’s correction, as described in the figure legends. One-way ANOVA test with Dunnett’s multiple comparisons was used to calculate the statistical differences of log10(normalized liver burden) between CIS43 or LS wildtype and each of the six CIS43 variants in the low-dose protection study. The correlation between the normalized liver burden and various kinetics, sequence, and structural properties were calculated using two-tailed Pearson’s correlation method. To compare the liver burden between different studies, the liver burden of each group was normalized based on the geometric mean of the liver burden values from the untreated mice in the same experiment. In terms of Feature-Sequence Associations, an in-house version of SeqFeatR was used to down-select antibody sequence position-amino acid combinations associated with peptide21 and PfCSP binding or liver burden function (Budeus et al., 2016). Further, we used Fisher’s exact test to determine the significance of the association between amino acid frequencies and binding or liver burden. The peptide21 binding, PfCSP binding, and liver burden data was divided into two classes by using the geometric mean as split point, hereby generating low and high binding, or low and high liver burden classes. P values less than 0.05 were considered significant (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001) as indicated in the figure legends.
Supplementary Material
Supplemental Figure S1. Characterization of inferred germline CIS43 heavy antibody chain KI mouse, related to Figure 1.
Supplemental Figure S2. Immunization with NPDP19-KLH is more specific and leads to strong activation of HiGL-CIS43 B cells, related to Figures 1 and 2.
Supplemental Figure S3. Quantification of C57BL/6J control groups, validation of PfCSP fluorescent probe and/or NPDP19-fluorescent probe staining and silent/non-silent mutation analysis, related to Figures 3 and 4.
Supplemental Figure S4. Informatic analyses of elicited heavy-light sequenced antibodies, related to Figure 5.
Supplemental Figure S5. Isothermal titration calorimetry measurements and structure of variant CIS43 recognition of PfCSP, related to Figure 6.
Supplemental Figure S6. Analyses of AlphaLISA readout, ITC and Core8 variants mediated liver burden protection, related to Figure 7.
Supplemental Figure S7. A mouse model to improve repeat-antigen-targeting antibodies, related to Fig. 7.
Table S1. Sequences of 161 CIS43 variants, 114 from 13 DPI and 47 from 28 DPI, related to Figure 5 (Supplied as an excel file).
Table S2. Production yields of 37 ‘top 10’-selected antibodies, related to Figure 5.
Table S3. Normalized malaria liver burden protection versus antibodies CIS43 and L9, related to Figure 5.
Table S4. BLI-measured affinities of ‘top 10’-selected antibodies, related to Figure 5.
Table S5. Crystallographic data collection and refinement statistics, related to Figure 6 and Figure 7.
Table S6. Sequence feature analysis related to restricted set of 8 SHM, observed in a majority of the improved antibodies, related to Figure 6.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-mouse-CD16/32 purified (clone 2.4G2) | BD Biosciences | Cat#:553142 |
| Rat monoclonal anti-mouse CD4 APC-eF780 (clone: RM4–5) | Invitrogen | CAT#:47–0042-80 |
| Rat monoclonal anti-mouse CD8 APC-eF780 (clone: 53–6.7) | Invitrogen | CAT#:47–0081-80 |
| Rat monoclonal anti-mouse F4/80 APC-eF780 (clone: BM8) | Invitrogen | CAT#:47–4801-80 |
| Rat monoclonal anti-mouse Ly-6G APC-eF780 (clone: RB6–8C5) | Invitrogen | CAT#:47–5931-80 |
| Rat monoclonal anti-mouse B220 BV510 (clone: RA3–6B2) | BD Biosciences | CAT#:563103 |
| Hamster monoclonal anti-mouse CD95 PE-Cy7 (clone: Jo2) | BD Biosciences | CAT#:557653 |
| Rat monoclonal anti-mouse CD38 A700 or A488 (clone: 90) | Invitrogen, Biolegend | CAT#:56–0381-82, 102714 |
| Mouse monoclonal anti-mouse CD45.2 PE (clone: 104) | Biolegend | CAT#:109808 |
| Mouse monoclonal anti-mouse CD45.1 PerCP Cy5.5 (clone: A20) | Biolegend | CAT#:110728 |
| Rat monoclonal anti-mouse GL7 A647 (clone: GL7) | Biolegend | CAT#:144606 |
| Rat monoclonal anti-mouse IgM BUV395 or BV421 (clone: Il/41) | BD Biosciences | CAT#:743329, 743323 |
| Rat monoclonal anti-mouse IgD BV786 (clone: 11–26c.2a) | BD Biosciences | CAT#:563618 |
| Rat monoclonal anti-mouse IgD PE-Cy7 (clone: 11–26c.2a) | Biolegend | CAT#:405720 |
| Rat monoclonal anti-mouse IgG1 BV421 (clone: A85–1) | BD Biosciences | CAT#:562580 |
| Rat monoclonal anti-mouse Ig, κ light chain BUV395 (clone: 187.1) | BD Biosciences | CAT#:742839 |
| Goat Anti-Mouse IgG Fcγ ALP | Jackson Immuno Research | CAT#:115–055-071 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant PfCSP | Produced in house | N/A |
| Biotinylated recombinant PfCSP | Produced in house | N/A |
| Biotinylated junctional peptide NPDP19 | Genscript | Order ID:# U134AFB120 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit, for UV excitation | Thermo Fisher Scientific | Cat#: L34962 |
| Streptavidin-A488 | Biolegend | CAT#:405235 |
| Streptavidin-647 | Biolegend | CAT#:405237 |
| Streptavidin-PE | Biolegend | CAT#:405204 |
| BD HORIZON BRILLIANT STAIN BUFFER | BD Biosciences | CAT#:566349 |
| SIGMAFAST™ p-Nitrophenyl phosphate Tablets | Sigma | CAT#: N2770–50SET |
| SuperScript™ III Reverse Transcriptase | Thermo Fisher Scientific | CAT#:18080085 |
| RNasin® Ribonuclease Inhibitors | Promega | CAT#:N2515 |
| dNTP Mix (10 mM each) | Thermo Fisher Scientific | CAT#: R0193 |
| HotStarTaq DNA Polymerase | QIAGEN | CAT#:203209 |
| Critical Commercial Assays | ||
| CountBright™ Absolute Counting Beads, for flow cytometry | Thermo Fisher Scientific | CAT#:C36950 |
| UltraComp eBeads™ Compensation Beads | Thermo Fisher Scientific | CAT#:01–2222-42 |
| Pan B Cell Isolation Kit II, mouse | Miltenyi Biotec | CAT#:130–104-443 |
| Deposited Data | ||
| PDB file | This paper. | PDB ID 7LKB |
| PDB file | This paper. | PDB ID 7LKG |
| PDB file | This paper | PDB ID 7RD3 |
| PDB file | This paper | PDB ID 7RDA |
| PDB file | This paper | PDB ID 7RD4 |
| PDB file | This paper | PDB ID 7RD9 |
| PDB file | This paper | PDB ID 7RCS |
| PDB file | This paper | PDB ID 7RAJ |
| Nucleotide sequences of iGL-CIS43 variant antibodies | This paper | GenBank: OK422520 - OK422843 |
| Experimental Models: Cell Lines | ||
| Human (female): Expi293 cell | Thermo Fisher Scientific | Cat#A14527 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.SJL-Ptprcapepcb/BoyJ | The Jackson Laboratory | JAX:002014 |
| Mouse: B6(Cg)-Tyrc-2J/J albino | The Jackson Laboratory | JAX:000058 |
| Mouse: C57BL/6J | The Jackson Laboratory. | JAX:000664 |
| Mouse: HiGL-CIS43 | This paper | N/A |
| Mouse: κiGL-CIS43 | This paper | N/A |
| Mouse: HiGL-CIS43κiGL-CIS43 | This paper | N/A |
| Sporozoite: P. berghei sporozoite expressing PfCSP, GFP, and luciferase | (Flores-Garcia et al., 2019) | N/A |
| Oligonucleotides and/or other sequence-based reagents | ||
| sgRNA | (Lin et al., 2018; Wang et al., 2020b)(Lin et al., 2018; Wang et al., 2020b) | N/A |
| Recombinant DNA | ||
| Plasmodium falciparum circumsporozoite protein (clone 3D7) | PlasmoDB | PF3D7_0304600.1 |
| pVRC8400 huIgG1 | Genscript | N/A |
| pVRC8400 huIgK | Genscript | N/A |
| Software and Algorithms | ||
| FlowJo 10.7.1 | Treestar | https://www.flowjo.com/ |
| Prism 9.0.1 | GraphPad | https://www.graphpad.com/ |
| Microsoft Office | Microsoft | https://www.office.com/ |
| IMGT/V-quest | (Lefranc et al., 2015) | http://www.imgt.org/IMGTindex/V-QUEST.php |
| Geneious 2020.2 | Biomatters | https://www.geneious.com |
| The PyMOL Molecular Graphics System | Schrodinger | https://pymol.org |
| RosettaRemodel | Rosetta | https://www.rosettacommons.org/docs/latest/application_documentation/design/Remodel |
| foldx5Linux64 | FoldX | http://foldxsuite.crg.eu/ |
| YASARA | YASARA | http://www.yasara.org/ |
| NAMD_2.14_Linux-x86_64-multicore | NAMD | https://www.ks.uiuc.edu/Development/Download/download.cgi?PackageName=NAMD |
| gRINN v1.1.0.hf1 | gRINN | https://grinn.readthedocs.io/en/latest/index.html |
| Gene-specific substitution profile | (Sheng et al., 2017) | https://cab-rep.c2b2.columbia.edu/ |
| Other | ||
| Armadillo PCR Plate, 96-well, clear, clear wells | Thermo Scientific | CAT#:AB2396 |
| Eppendorf twin.tec PCR Plate 96, semi-skirted, yellow | Genesee/Eppendor f | CAT#:951020320 |
| Eppendorf twin.tec® PCR Plate 96, skirted, clear wells, blue | Genesee/Eppendor f | CAT#:951020460 |
| Adhesive Sealing Sheets | Thermo Scientific | CAT#: AB-0558 |
| Microseal® ‘F’ PCR Plate Seal, foil, pierceable #msf1001 | Bio-Rad | CAT#: MSF1001 |
| E-Gel 96 2% with SYBR Safe | Thermo Fisher Scientific | CAT#: G720802 |
| E-Gel 96 Low Range Quantitative DNA Ladder | Thermo Fisher Scientific | CAT#: 12373–031 |
| BD FACSymphony™ | BD Biosciences | N/A |
| BD FACS Aria II cell sorter | BD Biosciences | N/A |
| VP-ITC microcalorimeter | Malvern Panalytical | N/A |
| IVIS® Spectrum In Vivo Imaging System | Perkin Elmer | N/A |
| Eppendorf® Mastercycler® | Eppendorf | N/A |
Highlights.
Generated a knock-in mouse with B cells expressing the inferred germline of CIS43
Vaccination with junctional-epitope peptide is more effective than full-length PfCSP
Engineered a best-in-class malaria-protective antibody, iGL-CIS43.D3
Structure and informatics analyses revealed alterations improving protective efficacy
Acknowledgments
We thank the Batista Lab members at the Ragon Institute, in particular Stephanie R. Weldon, for critically reading, editing and discussing the manuscript. We thank the members of the Ragon Institute FACS facility for their technical advice and expertise. The project was made possible by the Flexible Ragon Funds provided as part of the FDB start-up. We also thank V. Kholodenko for assistance with ITC, M. Sastry for assistance with peptide probes for AlphaLISA, J. Stuckey for assistance with figures, and members of the Virology Laboratory and Vector Core, Vaccine Research Center for discussion and comments. Support for this work was also provided by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases (NIAID). Use of sector 22 (Southeast Region Collaborative Access team) at the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science (contract W-31–109-Eng-38).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Ms Flynn and Drs. Seder, Idris and Kisalu hold patents on CIS43 (International Application No. PCT/US2018/017826; US Patent Application No. 16/485,354; issued June 1, 2021). Mr. Wang, and Drs. Seder and Francica, have submitted a US Provisional Patent Application describing mAb L9 (62/842,590; filed 3 May 2019).
References
- Abbott RK, Lee JH, Menis S, Skog P, Rossi M, Ota T, Kulp DW, Bhullar D, Kalyuzhniy O, Havenar-Daughton C, et al. (2018). Precursor Frequency and Affinity Determine B Cell Competitive Fitness in Germinal Centers, Tested with Germline-Targeting HIV Vaccine Immunogens. Immunity 48, 133–146.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atcheson E, Hill AVS, and Reyes-Sandoval A (2021). A VLP for validation of the Plasmodium falciparum circumsporozoite protein junctional epitope for vaccine development. Npj Vaccines 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, and MacKerell AD (2012). Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ 1 and χ 2 Dihedral Angles. J. Chem. Theory Comput 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Boehmer L, Liu C, Ackerman S, Gitlin AD, Wang Q, Gazumyan A, and Nussenzweig MC (2016). Sequencing and cloning of antigen-specific antibodies from mouse memory B cells. Nat. Protoc 11, 1908–1923. [DOI] [PubMed] [Google Scholar]
- Briney B, Inderbitzin A, Joyce C, and Burton DR (2019). Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566, 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budeus B, Timm J, and Hoffmann D (2016). SeqFeatR for the Discovery of Feature-Sequence Associations. PLoS One 11, e0146409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo-Calle JM, Mitchell R, Altszuler R, Othoro C, and Nardin E (2021). Identification of a neutralizing epitope within minor repeat region of Plasmodium falciparum CS protein. Npj Vaccines 6, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami C, Frevert U, Sinnis P, Takacs B, Clavijo P, Santos MJ, and Nussenzweig V (1992). The basolateral domain of the hepatocyte plasma membrane bears receptors for the circumsporozoite protein of plasmodium falciparum sporozoites. Cell 70, 1021–1033. [DOI] [PubMed] [Google Scholar]
- Cockburn IA, and Seder RA (2018). Malaria prevention: from immunological concepts to effective vaccines and protective antibodies. Nat. Immunol 19, 1199–1211. [DOI] [PubMed] [Google Scholar]
- Dosenovic P, Kara EE, Pettersson AK, McGuire AT, Gray M, Hartweger H, Thientosapol ES, Stamatatos L, and Nussenzweig MC (2018). Anti–HIV-1 B cell responses are dependent on B cell precursor frequency and antigen-binding affinity. Proc. Natl. Acad. Sci. U. S. A 115, 4743–4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fisher CR, Sutton HJ, Kaczmarski JA, McNamara HA, Clifton B, Mitchell J, Cai Y, Dups JN, D’Arcy NJ, Singh M, et al. (2017). T-dependent B cell responses to Plasmodium induce antibodies that form a high-avidity multivalent complex with the circumsporozoite protein. PLoS Pathog 13, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Garcia Y, Herrera SM, Jhun H, Pérez-Ramos DW, King CR, Locke E, Raghunandan R, and Zavala F (2019). Optimization of an in vivo model to study immunity to Plasmodium falciparum pre-erythrocytic stages. Malar. J 18, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foquet L, Hermsen CC, van Gemert G-J, Van Braeckel E, Weening KE, Sauerwein R, Meuleman P, and Leroux-Roels G (2014). Vaccine-induced monoclonal antibodies targeting circumsporozoite protein prevent Plasmodium falciparum infection. J. Clin. Invest 124, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francica JR, Shi W, Chuang GY, Chen SJ, Da Silva Pereira L, Farney SK, Flynn BJ, Ou L, Stephens T, Tsybovsky Y, et al. (2021). Design of alphavirus virus-like particles presenting circumsporozoite junctional epitopes that elicit protection against malaria. Vaccines 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire E, Schön A, and Velazquez‐ Campoy A (2009). Chapter 5 Isothermal Titration Calorimetry: General Formalism Using Binding Polynomials. Methods Enzymol 455, 127–155. [DOI] [PubMed] [Google Scholar]
- Gaudinski MR, Berkowitz NM, Idris AH, Coates EE, Holman LA, Mendoza F, Gordon IJ, Plummer SH, Trofymenko O, Hu Z, et al. (2021). A Monoclonal Antibody for Malaria Prevention. N. Engl. J. Med 385, 803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Malaria Programme, W.G. (2020). World Malaria Report 2020 [Google Scholar]
- Guo Y, Chen K, Kwong PD, Shapiro L, and Sheng Z (2019). cAb-Rep: A Database of Curated Antibody Repertoires for Exploring Antibody Diversity and Predicting Antibody Prevalence. Front. Immunol 10, 2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvench O, Mallajosyula SS, Raman EP, Hatcher E, Vanommeslaeghe K, Foster TJ, Jamison FW, and MacKerell AD (2011). CHARMM Additive All-Atom Force Field for Carbohydrate Derivatives and Its Utility in Polysaccharide and Carbohydrate–Protein Modeling. J. Chem. Theory Comput 7, 3162–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havenar-Daughton C, Abbott RK, Schief WR, and Crotty S (2018). When designing vaccines, consider the starting material: the human B cell repertoire. Curr. Opin. Immunol 53, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingdale MR, Nardin EH, Tharavanij S, Schwartz AL, and Nussenzweig RS (1984). Inhibition of entry of Plasmodium falciparum and P. vivax sporozoites into cultured cells; an in vitro assay of protective antibodies. J. Immunol 132, 909–913. [PubMed] [Google Scholar]
- Huang D, Abbott RK, Havenar-Daughton C, Skog PD, Al-Kolla R, Groschel B, Blane TR, Menis S, Tran JT, Thinnes TC, et al. (2020). B cells expressing authentic naive human VRC01-class BCRs can be recruited to germinal centers and affinity mature in multiple independent mouse models. Proc. Natl. Acad. Sci 117, 22920–22931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P-S, Ban Y-EA, Richter F, Andre I, Vernon R, Schief WR, and Baker D (2011). RosettaRemodel: a generalized framework for flexible backbone protein design. PLoS One 6, e24109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imkeller K, Scally S, Bosch A, Pidelaserra Martí G, Costa G, Triller G, Murugan R, Kremsner P, Hoffman S, Mordmüller B, et al. (2018). Anti-idiotope affinity maturation improves human B cell responses against a repetitive parasite antigen. Revis 5304, 1–10. [Google Scholar]
- Jelínková L, Jhun H, Eaton A, Petrovsky N, Zavala F, and Chackerian B (2021). An epitope-based malaria vaccine targeting the junctional region of circumsporozoite protein. Npj Vaccines 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien J-P, and Wardemann H (2019). Antibodies against Plasmodium falciparum malaria at the molecular level. Nat. Rev. Immunol 19. [DOI] [PubMed] [Google Scholar]
- Kisalu NK, Idris AH, Weidle C, Flores-Garcia Y, Flynn BJ, Sack BK, Murphy S, Schön A, Freire E, Francica JR, et al. (2018). A human monoclonal antibody prevents malaria infection by targeting a new site of vulnerability on the parasite. Nat. Med 24, 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisalu NK, Pereira LDS, Ernste KJ, Flores-Garcia Y, Idris AH, Asokan M, Dillon M, MacDonald S, Shi W, Chen X, et al. (2020). Enhancing durability of CIS43 monoclonal antibody by Fc mutation or AAV delivery for malaria prevention. JCI Insight [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M-P, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, et al. (2015). IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res 43, D413–D422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. D, Struct. Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Pecetta S, Steichen JM, Kratochvil S, Melzi E, Arnold J, Dougan SK, Wu L, Kirsch KH, Nair U, et al. (2018). One‐ step CRISPR/Cas9 method for the rapid generation of human antibody heavy chain knock‐ in mice. EMBO J e99243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara HA, Idris AH, Sutton HJ, Bonsignori M, Seder RA, Cockburn Correspondence IA, Vistein R, Flynn BJ, Cai Y, Wiehe K, et al. (2020). Antibody Feedback Limits the Expansion of B Cell Responses to Malaria Vaccination but Drives Diversification of the Humoral Response. Cell Host Microbe 28, 572–585.e7. [DOI] [PubMed] [Google Scholar]
- Murugan R, Scally SW, Costa G, Mustafa G, Thai E, Decker T, Bosch A, Prieto K, Levashina EA, Julien J-P, et al. (2020). Evolution of protective human antibodies against Plasmodium falciparum circumsporozoite protein repeat motifs. Nat. Med 1–11. [DOI] [PubMed] [Google Scholar]
- Ota T, Doyle-Cooper C, Cooper AB, Doores KJ, Aoki-Ota M, Le K, Schief WR, Wyatt RT, Burton DR, and Nemazee D (2013). B Cells from Knock-in Mice Expressing Broadly Neutralizing HIV Antibody b12 Carry an Innocuous B Cell Receptor Responsive to HIV Vaccine Candidates. J. Immunol 191, 3179–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Oyen D, Torres JL, Wille-Reece U, Ockenhouse CF, Emerling D, Glanville J, Volkmuth W, Flores-Garcia Y, Zavala F, Ward AB, et al. (2017). Structural basis for antibody recognition of the NANP repeats in Plasmodium falciparum circumsporozoite protein. Proc. Natl. Acad. Sci. U. S. A 114, E10438–E10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyen D, Torres Id JL, Aoto Id PC, Flores-Garcia Y, Pela Binter Id, S.ˇ, Pholcharee Id T, Carroll S, Reponen S, Wash R, Liang Q, et al. (2020). Structure and mechanism of monoclonal antibody binding to the junctional epitope of Plasmodium falciparum circumsporozoite protein [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partnership SCT (2012). A Phase 3 Trial of RTS,S/AS01 Malaria Vaccine in African Infants. N. Engl. J. Med 367, 2284–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MA, Burrows JN, Manyando C, van Huijsduijnen RH, Van Voorhis WC, and Wells TNC (2017). Malaria. Nat. Rev. Dis. Prim 3, 17050. [DOI] [PubMed] [Google Scholar]
- Raghunandan R, Mayer BT, Flores-Garcia Y, Gerber MW, Gottardo R, Jhun H, Herrera SM, Perez-Ramos DW, Locke E, King CR, et al. (2020). Characterization of two in vivo challenge models to measure functional activity of monoclonal antibodies to Plasmodium falciparum circumsporozoite protein. Malar. J 19, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RTS SCTP (2015). Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: Final results of a phase 3, individually randomised, controlled trial. Lancet 386, 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seder RA, Chang L-J, Enama ME, Zephir KL, Sarwar UN, Gordon IJ, Holman LA, James ER, Billingsley PF, Gunasekera A, et al. (2013). Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 341, 1359–1365. [DOI] [PubMed] [Google Scholar]
- Sheng Z, Schramm CA, Kong R, NISC Comparative Sequencing Program, M., Mullikin JC, Mascola JR, Kwong PD, and Shapiro L (2017). Gene-Specific Substitution Profiles Describe the Types and Frequencies of Amino Acid Changes during Antibody Somatic Hypermutation. Front. Immunol 8, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C, Bombardi RG, Branchizio A, Kose N, Matta P, Sevy AM, Sinkovits RS, Gilchuk P, Finn JA, and Crowe JE (2019). High frequency of shared clonotypes in human B cell receptor repertoires. Nature 566, 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Sack BK, Oyen D, Zenklusen I, Piccoli L, Barbieri S, Foglierini M, Fregni CS, Marcandalli J, Jongo S, et al. (2018). A public antibody lineage that potently inhibits malaria infection through dual binding to the circumsporozoite protein. Nat. Med 24, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Piccoli L, and Lanzavecchia A (2019). The Antibody Response to Plasmodium falciparum: Cues for Vaccine Design and the Discovery of Receptor-Based Antibodies. Annu. Rev. Immunol 37, 225–246. [DOI] [PubMed] [Google Scholar]
- Tewari R, Spaccapelo R, Bistoni F, Holder AA, and Crisanti A (2002). Function of Region I and II Adhesive Motifs ofPlasmodium falciparum Circumsporozoite Protein in Sporozoite Motility and Infectivity. J. Biol. Chem 277, 47613–47618. [DOI] [PubMed] [Google Scholar]
- Triller G, Scally SW, Levashina EA, Julien J-P, Wardemann H, Costa G, Pissarev M, Kreschel C, Bosch A, Marois E, et al. (2017). Natural Parasite Exposure Induces Protective Human Anti-Malarial Antibodies Article Natural Parasite Exposure Induces Protective Human Anti-Malarial Antibodies. Immunity 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LT, Pereira LS, Flores-Garcia Y, O’Connor J, Flynn BJ, Schön A, Hurlburt NK, Dillon M, Yang ASP, Fabra-García A, et al. (2020a). A Potent Anti-Malarial Human Monoclonal Antibody Targets Circumsporozoite Protein Minor Repeats and Neutralizes Sporozoites in the Liver. Immunity 53, 733–744.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ray R, Kratochvil S, Melzi E, Lin Y, Giguere S, Xu L, Warner J, Cheon D, Liguori A, et al. (2020b). Multiplexed CRISPR/CAS9‐ mediated engineering of pre‐ clinical mouse models bearing native human B cell receptors. EMBO J [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardemann H, and Murugan R (2018). From human antibody structure and function towards the design of a novel Plasmodium falciparum circumsporozoite protein malaria vaccine. Curr. Opin. Immunol 53, 119–123. [DOI] [PubMed] [Google Scholar]
- White MT, Bejon P, Olotu A, Griffin JT, Bojang K, Lusingu J, Salim N, Abdulla S, Otsyula N, Agnandji ST, et al. (2014). A combined analysis of immunogenicity, antibody kinetics and vaccine efficacy from phase 2 trials of the RTS,S malaria vaccine [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Acharya P, Kong R, Cheng C, Chuang GY, Liu K, Louder MK, O’Dell S, Rawi R, Sastry M, et al. (2018). Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat. Med 24, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavala F, Cochrane AH, Nardin EH, Nussenzweig RS, and Nussenzweig V (1983). Circumsporozoite proteins of malaria parasites contain a single immunodominant region with two or more identical epitopes. J. Exp. Med 157, 1947–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Characterization of inferred germline CIS43 heavy antibody chain KI mouse, related to Figure 1.
Supplemental Figure S2. Immunization with NPDP19-KLH is more specific and leads to strong activation of HiGL-CIS43 B cells, related to Figures 1 and 2.
Supplemental Figure S3. Quantification of C57BL/6J control groups, validation of PfCSP fluorescent probe and/or NPDP19-fluorescent probe staining and silent/non-silent mutation analysis, related to Figures 3 and 4.
Supplemental Figure S4. Informatic analyses of elicited heavy-light sequenced antibodies, related to Figure 5.
Supplemental Figure S5. Isothermal titration calorimetry measurements and structure of variant CIS43 recognition of PfCSP, related to Figure 6.
Supplemental Figure S6. Analyses of AlphaLISA readout, ITC and Core8 variants mediated liver burden protection, related to Figure 7.
Supplemental Figure S7. A mouse model to improve repeat-antigen-targeting antibodies, related to Fig. 7.
Table S1. Sequences of 161 CIS43 variants, 114 from 13 DPI and 47 from 28 DPI, related to Figure 5 (Supplied as an excel file).
Table S2. Production yields of 37 ‘top 10’-selected antibodies, related to Figure 5.
Table S3. Normalized malaria liver burden protection versus antibodies CIS43 and L9, related to Figure 5.
Table S4. BLI-measured affinities of ‘top 10’-selected antibodies, related to Figure 5.
Table S5. Crystallographic data collection and refinement statistics, related to Figure 6 and Figure 7.
Table S6. Sequence feature analysis related to restricted set of 8 SHM, observed in a majority of the improved antibodies, related to Figure 6.
Data Availability Statement
The crystal structures reported in this manuscript have been deposited in the Protein Data Bank, www.rcsb.org. The 161 CIS43 variant antibody nucleotide sequences are deposited in GenBank. Accession codes are listed in the KRT.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.