

Abstract
Recently infectious diseases caused by the increased emergence and rapid spread of drug-resistant bacterial isolates have been one of the main threats to global public health because of a marked surge in both morbidity and mortality. The only phosphonate antibiotic in the clinic, fosfomycin, is a small broad-spectrum molecule that effectively inhibits the initial step in peptidoglycan biosynthesis by blocking the enzyme, MurA in both Gram-positive and Gram-negative bacteria. As fosfomycin has a novel mechanism of action, low toxicity, a broad spectrum of antibacterial activity, excellent pharmacodynamic/pharmacokinetic properties, and good bioavailability, it has been approved for clinical use in the treatment of urinary tract bacterial infections in many countries for several decades. Furthermore, its potential use for difficult-to-treat bacterial infections has become promising, and fosfomycin has become an ideal candidate for the effective treatment of bacterial infections caused by multidrug-resistant isolates, especially in combination with other therapeutic drugs. Here we aim to present an overview of the biology and chemistry of fosfomycin including isolation and characterization, pharmacology, biosynthesis and chemical synthesis since its discovery in order to not only help scientists reassess the role of this exciting drug in fighting antibiotic resistance but also build the stage for discovering more novel phosphonate antibiotics in the future.
Recently infectious diseases caused by the increased emergence and rapid spread of drug-resistant bacterial isolates have been one of the main threats to global public health because of a marked surge in both morbidity and mortality.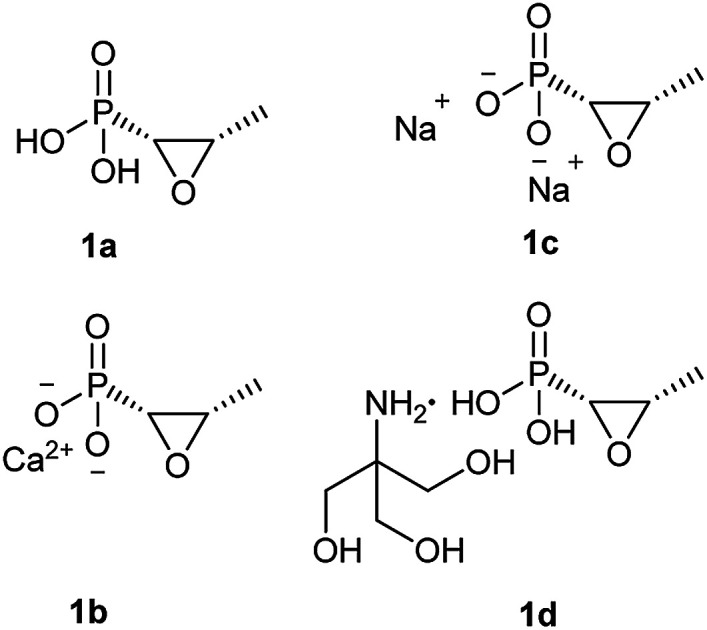
1. Introduction
The discovery of the first antibiotic, penicillin, in 1928 by Alexander Fleming, was one of the significant breakthroughs and the most successful chemotherapy in the history of medicine, saving many millions of lives in wartime. Since then, a panel of antibiotics effectively controls the rapid dissemination of bacterial infections that plagued human history for several centuries, resulting in a dramatic decrease in both morbidity and mortality caused by bacterial pathogens. However, the overuse and misuse of a variety of antibiotics in the medical field over the past 70 years has caused the increased emergence and global spread of multidrug-resistant (MDR) bacteria, leading to more than 750 000 death every year.1 Therefore, the World Health Organization (WHO) has recognized antibiotic drug resistance as one of the three main threats to global public health.
With rapid resistance to current antibiotics becoming common, there is a higher call for new medicines, but new antibacterial drug discovery and development is becoming more difficult for treating bacterial infections associated with MDR pathogens.
To solve this problem, one of the alternative strategies is the re-introduction and re-evaluation of “old” antibiotics which have been underexploited for a long time for the treatment of bacterial infections.2 One type of such “old” antibiotic is phosphonates containing at least one carbon–phosphorus (C–P) bond which have been extensively and widely recognized in the world.3 Though bond dissociation energy (BDE) of a C–P bond is weaker than that of an O–P bond, the C–P bond is remarkably more stable than the O–P bond for heterolytic cleavage.4–6 Therefore, they are highly similar to the phosphate esters and anhydrides but are quite stable and resistant to chemical hydrolysis, including a strong acid or base, thermal decomposition, photolysis, enzymatic degradation of phosphatases and phosphodiesterases.7 C–P compounds are stable isosteric for the labile phosphate esters, carboxylate, or anhydrides, which are omnipresent in the metabolic network so that they can be inhibitors of some critical enzymes occurring in primary metabolisms by mimicking their real substrates. These intriguing chemical stability and structural features attribute advantageous biological properties to many C–P compounds. The past century has experienced extensive use of C–P compounds in a variety of fields such as agriculture, chemical, and pharmaceutical industries.8 For example, since tabun, the first C–P nerve agent known, was discovered in the 1930s, a series of C–P containing chemicals exemplified by sarin and VX have been used as weapons of chemical warfare due to their extreme toxicity as nerve agents.9,10 They mimic the neurotransmitter acetylcholine and therefore seriously interferes with the function of the nervous system by irreversibly inhibiting the enzyme acetylcholinesterase because they form covalent bonds with serine residues at the active site.11 The active ingredient in the herbicide Roundup, glyphosate, the most heavily-used agricultural chemical in the history of the world, kills plants by blocking the biosynthesis of the aromatic amino acids in the host.12 It mimics the substrate phosphoenolpyruvate (PEP) and then effectively inhibits the function of the enzyme 5-enolpyruvylshikimate-3-phosphate (EPSP) synthase catalyzing the reaction of shikimate-3-phosphate (S3P) and PEP to form EPSP, which is a precursor for aromatic amino acids mentioned above.13 Acyclic nucleotide phosphonates such as tenofovir, cidofovir, and adefovir are inhibitors of viral reverse transcriptase because they mimic deoxynucleotide triphosphates and incorporate into a growing DNA strand, preventing viral DNA replication.14 So far, about 40 natural C–P small molecules (Fig. 3 and 4) have been discovered, and given the fact that several hundred various C–P natural products are likely to be present in nature,15 C–P natural products are believed to be a treasure trove of antibacterial drugs. However, only one phosphonate antibiotic, fosfomycin, has been in the clinic for more than 20 years in many countries, including USA, Japan, Germany, for urinary tract infections (UTIs) and was initially developed about 50 years ago.16 Furthermore, it exhibits both in vitro and in vivo activity against a wide range of MDR bacteria, fosfomycin is potentially an ideal candidate for treating bacterial infections caused by those bacteria.17,18 Recently a panel of clinical trials have been conducted to assess the potential clinical use of fosfomycin in the fight against multidrug-resistant bacterial isolates, revealing promising future for the revival of this old antibiotic.19
Fig. 3. Chemical structures of naturally occurring C–P small molecules as bioactive metabolites.

Fig. 4. Chemical structures of naturally occurring C–P small molecules as intermediates.

Therefore, we believe that it is essential and useful for scientists in various fields who are eager to deeply exploit more phosphonate antibiotics and chart a bright pathway from bench to bedside to comprehensively understand fosfomycin. In this review, we aim to provide an overview of the chemistry and biology of fosfomycin, with a focus on the discovery, pharmacology, clinical use, biosynthesis, and chemical synthesis of fosfomycin.
2. Isolation and characterization
Fosfomycin (originally phosphonomycin) was isolated, characterized and developed in a joint program of Merck and Spain's Compañía Española de Penicilina de Antibióticos (CEPA).20 It was initially isolated by screening a strain database composed of actinomyces, including Streptomyces fradiae (ATCC 21096) for detecting the potential ability to inhibit the biosynthesis and formation of cell wall of growing bacteria. In this morphological spheroplast assay discovering novel antibiotics, including cephamycin C, thienamycin, and carbapenems, Gram-negative bacteria are streaked and grown in one particular medium, which is designed to be osmotically protective. Inhibition of any steps in the biosynthesis of peptidoglycan will avoid the production of cell walls and lead to the formation of spheroplasts, which are less refractile, evidenced by microscope.21
A combination of ion-exchange chromatography, gel filtration, and adsorption chromatography was used to isolate and purify fosfomycin from fermentation broth. Fosfomycin is confirmed to be also produced by several other strains including Streptomyces viridochromogenes (ATCC21240), Streptomyces wedmorensis (ATCC 21239), Pseudomonas viridiflava PK-5, and Pseudomonas syringae PB-5123.22,23
Fosfomycin (1a, Fig. 1) [(2R,3S-3-methyloxiran-2-yl) phosphonic acid] is a natural phosphonic acid containing an epoxide ring, which is structurally strained and thus chemically reactive, which lend good antibacterial activity to this molecule. Furthermore, fosfomycin, with a low molecular weight of 138.06 g mol−1, is different from any other antibiotic family, indicating no cross resistance for this drug in the clinical application.
Fig. 1. Chemical structure of fosfomycin (1a), fosfomycin calcium (1b), fosfomycin, disodium (1c), and fosfomycin trometamol (1d).

Fosfomycin was initially formulated as both a disodium salt (fosfomycin disodium, 1c, Fig. 1) for parenteral administration and a more hydrophobic calcium salt (fosfomycin calcium, 1b, Fig. 1) for oral administration. Later, fosfomycin trometamol (1d, Fig. 1) showed enhanced bioavailability and became a significant formulation for oral use.
3. Biological activities
Fosfomycin exhibits a promising bactericidal activity against a variety of Gram-negative and Gram-positive bacteria, including clinical isolates of Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, Pseudomonas aeruginosa, Proteus vulgaris, Salmonella schottmuelleri, Serratia marcescens, Salmonella typhi, Citrobacter spp. Enterococcus faecalis, Staphylococcus aureus (including methicillin-resistant S. aureus [MRSA]), Staphylococcus epidermidis and Streptococcus pyogenes.20 Whereas Pseudomonas aeruginosa exhibits moderate susceptibility, fosfomycin shows improved efficacy, especially in combinations with other antibiotics, including cefepime, aztreonam, and meropenem.16 The strains which are resistant to fosfomycin includes some isolates of Acinetobacter baumannii, Vibrio fischeri, Chlamydia trachomatis, and Bacteroides species.24,25
Fosfomycin has been confirmed to be biologically active against multiple drug-resistant (MDR), extensively drug-resistant (XDR), and pan-drug-resistant (PDR) bacteria. For example, fosfomycin shows both in vitro and in vivo antibacterial activities against a wide arrange of MDR and XDR species of Enterobacteriaceae, notably including species associated with extended-spectrum-lactamases (ESBL) and metallo-lactamases (MBL), indicating that fosfomycin can effectively eradicate more than 90% of Escherichia coli and 80% of Klebsiella pneumoniae isolates.26
4. Mechanism of action
As Fig. 2 shows, fosfomycin (1a) can be imported into the cytoplasm by the transporters GlpT and UhpT because it mimics the chemical structure of both glucose-6-phosphate (G6P) and glycerol-3-phosphate (G3P). As a result, fosfomycin (1a) may be shipped into the bacterial cytoplasm via two specific transport system induced by both G6P and G3P.27,28 After it goes into the cells, fosfomycin (1a), a chemical mimic of PEP analog, is bactericidal by inactivating specifically bacterial cell wall biogenesis through irreversible inhibition of MurA (UDP-N-acetylglucosamine-3-enolpyruvyltransferase) which catalyzes the ligation of PEP (31) to UDP-N-acetylglucosamine in the first biosynthesis step in the peptidoglycan formation.29 Thio group of the cysteine 115 residue of Escherichia coli MurA is an electron-rich nucleophile, attacking partially positive carbon on the epoxide of fosfomycin in a time-dependent reaction,30,31 evidenced by crystal structure of Escherichia coli MurA which is complexed with UDP-GlcNAc and fosfomycin. The Cys115-bound fosfomycin is tightly positioned between MurA and UDP-GlcNAc, resulting in generating electrostatic interactions between three conserved positively charged residues Lys22, Arg120, and Arg397 of the enzyme MurA and the negatively charged phosphonate group of fosfomycin.32 Fosfomycin's mechanism of action is unprecedented and totally different from other bacterial cell wall inhibitors as well as other types of antibiotics indicating minimal possibility of cross-resistance to other antibiotics.33
Fig. 2. Mechanism of action of fosfomycin. Chemical structure of fosfomycin mimics both glycerol-3-P (G3P) and glucose-6-P (G6P), which are transported by transporters GlpT and UhpT, respectively. MurA catalyzes the formation of UDP-GlcNac-3-O-enolpyruvate, a peptidoglycan precursor, from UDP-GlcNAc and PEP during the first step of peptidoglycan biosynthesis. Once fosfomycin (F) is present, it is transported inside the cell by GlpT and UhpT, blocking the UDP-GlcNac-3-O-enolpyruvate synthesis by mimicking the original substrate of MurA, PEP, avoiding cell wall synthesis and leading to cell death.

5. Pharmacology
5.1. Pharmacodynamics
Some studies have shown that fosfomycin exhibits a concentration-dependent antibacterial activity against some isolates of the Gram-negative bacteria Escherichia coli and Proteus mirabilis in vitro as well as some strains of the Gram-positive bacteria Streptococcus pneumoniae in vivo with complete sterilization at concentrations of more than 4× MIC.34–36 However, other relevant experiments have revealed a time-dependent activity of fosfomycin to destroy clinical isolated of Staphylococcus aureus and pyogenes in vitro. Moreover, the drug may exhibit an in vitro concentration-dependent post-antibiotic effect (PAE) (between 3.2 and 4.7 h) against isolates of Escherichia coli and Proteus mirabilis. However, these above studies cannot provide decisive conclusions on the concentration- or time-depending antibacterial activity of fosfomycin,36,37 which becomes a significant obstacle that must be figured out in order to reach the best therapy for further clinical trials.
5.2. Pharmacokinetics
Fosfomycin disodium salt was initially formulated for parenteral administration and is currently clinically used in only several European countries, including France, Germany, Spain, the United Kingdom, the Netherlands. Hydrophilicity and low molecular weight permits favorable tissue availability and high diffusibility of the drug.38 With considerable tissue penetration, fosfomycin disodium has become a potential chemotherapy for treating bacterial infections at different body regions including the central nervous system (CNS), bone, lungs, soft tissues, muscle, appendix, gallbladder, common bile duct, heart valves, and abscesses. For example, fosfomycin disodium can penetrate extensively into the interstitial fluid in soft tissues, achieving sufficient concentration to eradicate bacterial pathogens. Furthermore, fosfomycin disodium is also a suitable candidate for treating severe infections associated with the osseous matrix.
After intravenous injection or infusion, fosfomycin in the blood experiences a fast disposition and then a decelerating distribution phase. Biological half-life (t1/2) of fosfomycin disodium is measured to be 1.5–2 hours.39,40 The maximum serum concentration (Cmax) that fosfomycin achieves in the blood obtained with the standard intravenous administration reaches 200–644 mg L−1, which is much higher than the oral administration. The volume of fosfomycin distribution can arrive at most 27 L at a steady state.41 About 93% of an administered dose of fosfomycin is excreted unchanged into the urine after the glomerular filtration in the kidney.
For severe systemic bacterial infections, a fosfomycin disodium adult dose of 12–24 g per day is usually injected in at most four separate infusions. Reduction of daily dose of fosfomycin is necessary to clean creatinine with the clearance of 0.40 mL min−1.42
6. Clinical applications
Fosfomycin was discovered and developed in Europe by CEPA and has been in clinical use since the 1970s, firstly as an intravenous use formulation of fosfomycin disodium salt (1c) and later as an oral formulation of calcium salt (1b) and fosfomycin trometamol (1d). Its primary use in many countries, including Germany, France, Spain, South Africa, Japan, and Brazil, has been marketed for urinary tract infections (UTIs). Fosfomycin was registered for clinical use in the United States (as Monurol, 1d) in 1996 for the treatment of uncomplicated UTIs (acute cystitis) in women caused by Escherichia coli and Enterococcus faecalis. With the emergence and spread of increasing resistance to other antibiotics, parenteral administration of fosfomycin has been extensively analyzed in combination with other antibiotics in the treatment of patients suffering from a variety of infections including serious infections, sepsis or nosocomial infections.43
Recently, the promising in vitro and in vivo antibacterial activity and good pharmacological profile of fosfomycin has attracted renewed attention for treating severe systemic infections caused by multidrug-resistant bacteria.19
7. Fosfomycin resistance
There are several mechanisms responsible for fosfomycin resistance:
7.1. Decreased fosfomycin penetration
Decreased fosfomycin penetration is the most common mechanism of fosfomycin resistance in clinical isolates.43 It results from mutations (point mutations, insertions, deletions) of any of genes encoding fosfomycin permeases, including the glycerol-3-phosphate transporter (GlpT) and a hexose phosphate transporter, the glucose-6-phosphate transporter (UhpT), and their local regulators, uhpA-C.44 The complete expression of the fosfomycin transporters, GlpT and UhpT, depends on high levels of cAMP, which is highly regulated by adenyl cyclase CyaA or the phosphotransferase enzyme, PtsI, a component of the PEP sugar phosphotransferase transport system. Mutations in CyaA or PstI can downregulate the expression of GlpT and UhpT and contribute to fosfomycin resistance.45
7.2. Mutation of the target MurA
The mutation of the antibiotic target represents one of the most common mechanisms for bacterial resistance to antibiotics. In E. coli mutation of Cys115, the fosfomycin-binding site in MurA, results in resistance to this antibiotic. Interestingly, the Cys115 to Asp mutation in the E. coli MurA generates a completely active enzyme, yet completely insensitive to fosfomycin, while the Cys115 to Glu mutant shows no enzymatic activity.46,47 Furthermore, mutations in the MurA sequence of clinical isolates of E. coli, Asp369 to Asn and Leu370 to Ile, have been confirmed to contribute to the development of fosfomycin resistance in vivo as both highly conserved residues could be crucial for PEP substrate binding and affect interaction between the MurA and fosfomycin.48
7.3. Overproduction of the target MurA
Bacterial resistance to a specific antibiotic can often be acquired by the overproduction of a drug's molecular target.49 Although overproduction of the target MurA in fosfomycin resistant bacteria is rare, the comprehensive analysis of 5272 chromosomal genes in an E. coli ASKA library has indicated that murA is the only overexpressed gene in the entire E. coli genome capable of conferring clinical levels of antibiotic resistance. Higher MurA levels in E. coli correlate with higher levels of fosfomycin resistance, reaching clinical resistance levels (32 μg mL−1) at a low fitness cost.50 Furthermore, it has been confirmed that the enhanced transcription of the murA gene is responsible for the acquisition of fosfomycin resistance in several Shiga-like toxin-producing E. coli clinical isolates.51
7.4. Fosfomycin modification by fosfomycin-inactivating enzymes
Several fosfomycin-modifying enzymes have been found that inactivate fosfomycin. Three types of metalloenzymes (FosA, FosB, or FosX) have been characterized to date in fosfomycin-resistant bacteria. These enzymes catalyze the opening of the oxirane ring of the antibiotic by the addition of different substrates (glutathione, bacillithiol, or H2O) to the C1 of the oxirane ring, respectively. FosA is a K+- and Mn2+-dependent glutathione S-transferase encoded by fosA-like genes, catalyzing the addition of glutathione to the oxirane ring of fosfomycin.52 FosB is an Mg2+ and Mn2+-dependent thiol S-transferase encoded by fosB-like genes, catalyzing the addition of a thiol group using bacillithiol, the α-anomeric glycoside of l-cysteinyl-d-glucosamine with l-malic acid as a donor substrate.53 FosX is an Mn2+-dependent epoxide hydrolase that catalyzes the hydration of fosfomycin, breaking the oxirane ring and then generating a diol product.54,55
Fosfomycin producers, including some strains of Streptomyces and Pseudomonas syringae, keep resistant genes be close to biosynthetic genes in the fosfomycin gene cluster and biologically synthesize several antibiotic kinases that phosphorylate fosfomycin inside the cells in order to protect cells from the antibacterial effect of fosfomycin.56 In Streptomyces spp., two fosfomycin kinases sequentially modify the antibiotic in the presence of ATP and Mg2+. FomA converts fosfomycin to fosfomycin monophosphate, while FomB produces fosfomycin diphosphate using the monophosphate form as a substrate.57,58 In Pseudomonas syringae, FosC is an ortholog of FomA.56
8. Biosynthesis
Since the first naturally occurring C–P compound, 2-aminoethylphosphonic acid (2-AEP) was identified and characterized from rumen protozoa in 1959,59 a new chapter has been opening up in the biology and chemistry of C–P compounds since nature has long time on perfecting various chemistry to build limitlessly complicate scaffolds present in secondary metabolites primarily using limited building blocks and enzymes in a combinatorial style. However, this field is still in its initial stage of development.60 Only about 40 natural C–P small molecules (Fig. 3 and 4) have been discovered, and many of them exhibit exciting biological activities.
With the exception of three related phosphonates K-4 (12a), K-26 (12b), I5B1(12c), I5B2 (12d), SF2513B (12e) and SF2513C (12f),60,61 the first step of biosynthetic pathways of all known C–P natural product is the reversible conversion of PEP (31) to phosphonopyruvate (PnPy) (18a) catalyzed by PepM. Because the isomerization equilibrium greatly favors PEP (31) by a factor of at least 500, PnPy (18a) formation is always coupled to an essentially irreversible reaction. Several subsequent reactions have been discovered, including decarboxylation, transamination, aldol reactions, and phosphorylation, which all, in turn, give rise to some intermediates (Fig. 4) that can be used in various ways to produce a wide array of C–P natural products.
Fosfomycin was produced by several bacterial strains belonging to the genus Streptomyces or Pseudomonas.20,22,23,62 Although the complete pathway and some relevant enzymatic activities remain elusive, it is unequivocally evident that those strains from Streptomyces synthesize the FDA-approved phosphonic acid differently from those from Pseudomonas.56
8.1. Biosynthesis of fosfomycin in Streptomyces
The biosynthesis of fosfomycin in Streptomyces consists of seven chemical steps.63,64 Heterologous expression and genetic inactivation experiments indicated that the gene cluster is composed of fom1–4 and fomA–D.65 Most of these steps (Fig. 5) have been reconstituted and characterized in vitro.
Fig. 5. Proposed pathways for fosfomycin biosynthesis in Streptomyces (left pathway) and Pseudomonas (right pathway).

8.1.1. The conversion of PEP to PnPy
Bioinformatic analysis showed that Fos1 is involved in the first step of fosfomycin (1a) biosynthesis in Streptomyces because Fos1 encompasses PEP mutase domain, which was confirmed by relevant genetic inactivation experiments.65,66 However, Fom1 is a bifunctional PepM which is CyTase-fused and identified only in the gene cluster of fosfomycin (1a) from Streptomyces strains.63
8.1.2. The conversion of 2-HEP to HEP-CMP
This step was not proposed and confirmed until the cytidylyltransferase (CyTase) domain in Fom1 was characterized.63,64 Fom3 was previously believed to stereospecifically methylate at C2 of 2-HEP (19a) to yield S-2-hydroxypropylphosphonate (2-HPP) (26). However, enzymatic activity and mechanism of Fom3 was not completed elucidated,67,68 because incubation of the reconstituted Fom3 with 2-HEP (19a) generated an uncharacterized product.69 To uncover the pathway producing 2-HPP (26) from 2-HEP (19a), Kuzuyama et al.63,64 extensively analyzed the gene cluster (fom) in Streptomyces strains and found that besides its C-terminal PepM domain, in its N-terminus Fom1 has an additional CyTase domain which is characteristic of nucleotidyltransferases. In vitro enzymatic experiments unambiguously verified that the Fom1 CyTase domain combines 2-HEP (19a) with cytidine monophosphate (CTP) to produce HEP-CMP (32). The cytidylylation of an intermediate in the metabolism has been addressed in the biosynthesis of FR-900098 (2a).70 FrbH, which contains both a cytidylyltransferase domain and a decarboxylase domain, catalyzes the cytidylylation and the decarboxylation to yield CMP-3-aminobutylphosphonate.71 The Fom1 has the same tertiary structures as other CyTases so that it has a similar enzymatic mechanism. However, some residues in the active and binding sites of the Fom1 CyTase domain are exclusive, which was demonstrated by X-ray structure indicating high substrate specificities of the Fom1 CyTase domain.63
8.1.3. The conversion of HEP-CMP to HPP-CMP
Previously it was reported that trace amount of (S)-2-HPP (26) was obtained from 2-HEP (19a) with the purified Fom3,69 which belongs to B-class of radical SAM methyltransferases because of a C-terminal radical SAM domain and an N-terminal cobalamin binding domain.72–74 This class of radical SAMs, known as the largest subfamily, is capable of methylating sp3, sp2 carbons, and the phosphorus atom of phosphinates.75 However, 2-HEP (19a) is not a real substrate of Fom3, and the CMP group of HEP-CMP (32) is required for substrate recognition by Fom3.63,76 Fom3 needs two equivalents of SAM for each catalysis: one for producing a 5′-deoxyadenosyl radical (5′-dA˙) and another for providing the CH3 group. Furthermore, the reconstituted Fom3 yielding equal (S)- and (R)-HPP-CMP indicated that methylation catalyzed by Fom3 is not stereoselective, which is different from C-methylations in other methylcobalamin (MeCbl)-dependent radical SAM enzymes including GenK.77 Fom3 is a member of MeCbl-dependent radical SAM C-methyltransferases, and therefore the enzymatic mechanism of Fom3 is believed to agree with those of other radical enzymes such as Genk and CysS. Two possible mechanisms (Fig. 6) can be proposed for the Fom3-catalyzed conversion of HEP-CMP (32) to HPP-CMP (33).64,78 Like other radical SAM enzymes, Fom3 has a CX3CX2C sequence motif, which can be used to bind a [4Fe–4S] cluster. Three irons of a [4Fe–4S] cluster were accommodated by the three cysteines, while the fourth ion has two free ligand sites used for binding SAM. The reduced [4Fe–4S]+ cluster can homolytically break the S–C bond of SAM, furnishing l-methionine and a 5′-dA˙.75 In the first proposed pathway (Fig. 6A), the 5′-dAdo˙ radical abstracts one hydrogen atom at C2 of HEP-CMP (32) to furnish a radical intermediate that obtains a methyl radical from MeCbl to produce HPP-CMP (33). The way of transferring a radical is unusual among the MeCbl-dependent methyltransferases. However, the way of transferring a methyl cation from MeCbl is also envisioned. The early steps of the second mechanism (Fig. 6B) are identical to those of Fig. 6A. However, the radical intermediate is deprotonated to generate the ketyl radical, which initiates nucleophilic attack on MeCbl and therefore give rise to the transfer of a methyl cation, producing an alkoxide product radical which could finally be quenched. In both mechanisms, the oxidized [4Fe–4S]2+ cluster is reduced by the NADP+/NADPH system and the produced cob(ii)alamin is reduced by DTT to cob(i)alamin, which is methylated to yield MeCbl.
Fig. 6. Proposed mechanism of the Fom3-catalyzed reaction. (A) HEP-CMP radical (41) is quenched by the transfer of methyl radical from Me-Cbl(iii) to give Cbl(ii), which is reduced to Cbl(i) in order to accept a new methyl group from SAM. (B) Transfer of methyl cation to HEP-CMP ketyl radical (47) followed by reduction and protonation of product radical (48).

8.1.4. The conversion of HPP-CMP to (S)-HPP
(S)-HPP-CMP (33) from HEP-CMP (32) seems to be the possible intermediate in the fosfomycin pathway. (S)-HPP-CMP (33) cannot be oxidized by Fom4 because of the CMP moiety. Therefore, the CMP group of (S)-HPP-CMP (33) has to be cleaved to yield (S)-HPP (26), which then furnish fosfomycin (1a) through an oxidation reaction catalyzed by Fom4. A possible enzyme catalyzing this reaction is FomD, which shows a 34% identity to a putative phosphatase SCO5041 in Streptomyces coelicolor A3(2) and is a DUF402 domain-containing protein. Currently, the crystal structure of FomD has been elucidated.64,65
8.1.5. The conversion of (S)-HPP to fosfomycin
The 2-hydroxypropylphosphonate epoxidase Fom4, known as HppE, has been extensively studied and is confirmed to meditate an unprecedented 1,3-dehydrogenation of the alcohol in (S)-2-HPP (26) to generate the C–O bond of the epoxide ring of fosfomycin.79 The structure of HppE, a non-hem-iron(ii) enzyme, was further confirmed by its X-ray crystal structure, revealing that HppE exists as a homotetramer containing one ferrous ion in each monomer. Same as other members of cupin family, each HppE monomer is composed of an α-domain that is a whole α-helix, and a β-domain consisting of anti-parallel β-strands in a jellyroll β-barrel motif. However, the enzymatic mechanism of HppE and the nature of the oxidant remain a mystery. HppE was regarded as an oxidase that took advantage of Fe(iii)-superoxo for hydrogen radical abstraction. However, Liu and co-workers demonstrated that the best cosubstrate of HppE is H2O2 instead of O2 because the presence of H2O2 is crucial for high turnover during catalysis.79 Therefore, HppE is supposed to be a non-heme iron peroxidase.
The reaction catalyzed by HppE is intriguing because the oxygen atom in the three-membered ring is derived from the hydroxyl group at C2-of (S)-2-HPP (26). HppE was verified to catalyze two fascinating reactions when stereo- and structural isomers of (S)-2-HPP (26) are used as substrates: both (R)-1-hydroxypropyl-1-phosphonate and (S)-1-hydroxypropyl-1-phosphonate are converted to the corresponding ketone and aldehyde, respectively via oxidative 1,2-phosphonate migration. However, (R)-2-hydroxypropyl-1-phosphonate is converted to the corresponding ketone via C2 dehydrogenation (Fig. 7).79,80
Fig. 7. Reactions catalyzed by HppE with substrate analogs.

Two possible mechanisms have been proposed (Fig. 8).79–83 The substrate (S)-2-HPP (26) is first accommodated to the active site FeII and chelates the cofactor through the C2 hydroxyl group and one oxygen atom of phosphonate moiety, followed by reduction of the preferred cosubstrate H2O2 to H2O giving rise to the high-valent FeIV-oxo (ferryl) complex, which abstracts a pro-(R) hydrogen atom (H˙) from the C1 methylene to generate the FeIV–OH. An ensuing one-electron oxidation furnishes a C1 carbocation that can be quenched by the oxygen at C2, producing a new C–O bond and ultimately closing the epoxide ring in fosfomycin (1a) (Fig. 8A).
Fig. 8. Two possible reaction mechanisms for HppE with (S)-HPP. (A) A high-valent FeIV-oxo (ferryl) complex involved; (B) a highly reactive HO˙ radical as the only oxidant in the process of C–H activation in HppE.

Shaik and his co-workers tried to elucidate the enzymatic mechanism of HppE and ruled out the possibility of the ferryl FeIV-oxo and FeIII–O˙ complex as the intermediates in the abstraction of the pro-(R) hydrogen from (S)-2-HPP (26) and indicated the role of a highly reactive HO˙ radical as the only oxidant in the process of C–H activation in HppE by taking advantage of quantum mechanical/molecular mechanical (QM/MM) calculations. A new mechanism was proposed based on QM/MM calculations (Fig. 8B). The homolytic O–O cleavage of the FeII–H2O2 complex gives rise to a FeIII–OH intermediate that accommodates the HO˙ radical, which is accommodated by a hydrogen bond to the FeIII–OH species. Abstracting pro-(R) hydrogen from C1 and proton from O2–H with the help of Glu142 generate this species. A facile conformational change happens and then proceed to the cis epoxide formation of this species.82
8.2. Biosynthesis of fosfomycin in Pseudomonas
The study on the biosynthetic pathway of fosfomycin (1a) in Pseudomonas started from the identification of homologous enzymes in P. syringae PB-5123 of the methyltransferase Fom3, which was poorly soluble when it was reconstituted in vitro. Genomic sequencing of P. syringae PB-5123 and heterologous expression identified and confirmed a possible phosphonate gene cluster that encodes a putative PepM (called Psf1) and a characteristic HppE (termed Psf4). However, the gene cluster includes several genes that are not homologs of enzymes responsible for any other steps in the biosynthetic pathway of fosfomycin (1a) Streptomyces. Bioinformatic analysis of each gene led to the proposed biosynthetic pathway (Fig. 5) of fosfomycin (1a) in P. syringae PB-5123 including the isomerization of PEP (31) to PnPy (18a) catalyzed by Psf1, the addition of an acetyl (Ac) group to the carbonyl of PnPy (18a) to generate 2-phosphonomethylmalate (PMM) (22) catalyzed by Psf2, decarboxylation of the C-3 carboxyl group of PMM (22) and concomitant elimination of the hydroxyl group at C-1 catalyzed by Psf7, tautomerization, decarboxylation to produce 2-oxopropionyl phosphonate (2-OPP) (37), stereospecific reduction of the C2 ketone of 2-OPP (37) to produce (S)-2-HPP (26) catalyzed by Psf3, and ring closure of (S)-2-HPP (26) to yield the final product fosfomycin (1a) catalyzed by Psf4. So far, the enzymatic activity of only Psf2, Psf3, and Psf4 have been extensively studied though Psf5, Psf6, and Psf7 were successfully reconstituted in vitro.56In vitro reconstitution of Psf2, a homolog of citrate synthase-like enzyme FrbC from the FR-900098 pathway, confirmed that it catalyzes an aldol-like condensations of acetyl-CoA (Ac-CoA) and PnPy (18a), followed by hydrolysis to furnish PMM (22).56 Psf3, an NAD(P)H-dependent dehydrogenase, carries out both the reduction of 2-OPP (37) to (S)-2-HPP (26) with NADPH as the cofactor and the oxidation of only (S)-2-HPP (26) to 2-OPP (37). In the Psf3 reaction, a hydride is delivered from the Si face of NADPH to C2 of 2-OPP (37). Although Psf4 shares only 27% identity with Fom4 in amino acid sequence, the essential residues for Fos4 catalysis are conserved in Psf4.84,85 However, Psf4 is composed of a C-terminal cupin fold similar to that observed in HppE. Psf4 and HppE differ in the N-terminal domain, and Psf4 holds an α/β-fold which is built by two β-strands flanked by two α-helices, which is different from a structurally unrelated helical bundle in the N-terminal domain of HPPE.86
9. Chemical synthesis
The first chemical synthesis was described by Christensen et al.62 Since several chemical synthesis of fosfomycin (1a) have been reported. Currently, the syntheses generally are divided into three categories: epoxidation of (Z)-1-propenylphosphonates, 1,2-dihydroxylpropylphosphonate ring closure, or halohydrinphosphonate ring closure.
9.1. Epoxidation of (Z)-1-propenylphosphonates
The first chemical synthesis of fosfomycin (1a) reported in 1969 was accomplished based on epoxidation of the (Z)-1-propenylphosphonic acid (Fig. 9A).62 The (Z)-1-propenylphosphonic acid (58) was obtained by stereospecific reduction of the dibutyl 1-propynylphosphonate (56) to dibutyl (Z)-1-propenylphosphonate (57) followed by removal of protecting groups with concentrated hydrochloric acid to produce racemic (±)-(Z)-1,2-epoxypropylphosphonic acid, which finally generated enantiomerically pure fosfomycin (1a) by using the quinine salt.
Fig. 9. Total synthesis of fosfomycin. (A) and (B) Epoxidation of (Z)-1-propenylphosphonates; (C) and (D) 1,2-dihydroxylpropylphosphonate ring closure; (E–G) base-catalyzed halohydrinphosphonate ring closure.

The kind of synthesis was improved by converting propargyl alcohol into (Z)-1-propenyl-phosphonic acid (58) (Fig. 9B). Furthermore, acid-sensitive t-butyl alcohol was used as a protecting group instead of n-butyl alcohol because acid-catalyzed cleavage of t-butyl groups is clean and rapid. Di-t-butyl-2-propynylphosphite (61), prepared from di-t-butyl phosphorochloridate (60) and propargyl alcohol, was unstable thermally and rearranged to the propadienylphosphonate (62). Selectively stereospecific hydrogenation in benzene with Pd/C to 62 followed by deprotection (81% yield) produced (Z)-1-propenylphosphonic acid (58), which was converted to fosfomycin (1a) by using (+)-α-phenethylamine. Optically pure fosfomycin salt was obtained from a single recrystallization (33% yield).87
9.2. 1,2-Dihydroxylpropylphosphonate ring closure
Fosfomycin (1a) was synthesized from Sharpless asymmetric dihydroxylation of (E)-1-propenyl-phosphonate (64) by using an enriched AD-mix-α (Fig. 9C). The modified reagent not only accelerated the reaction but also proceeded the reaction at a 0 °C, obtaining dibenzyl (1S,2S)-1,2-dihydroxypropylphosphonate (65) in 65% yield with >99% ee. Regioselective sulfonylation of diol by using nosyl chloride in the presence of triethylamine led to monosulfonate, followed by treatment with K2CO3 to generate the dibenzyl epoxide (67) in 67% yield. Hydrogenolysis of 67 to produce fosfomycin (1a).88
Another approach (Fig. 9D) started with the production of a protected 1,2-dihydroxyphosphonate (67) through stereoselective addition of trimethylsiyldibenzylphosphite to the (S)-triisopropylsiyloxylacetaldehyde (68) at −78 °C to yield protected α-hydroxyphosphonates (70), which reacted with methanesulfonyl chloride to give methanesulfonate, followed by deprotection and ring closure with tetra-n-butylammonium fluoride (TBAF). The diastereoisomerically pure epoxide was deprotected to produce fosfomycin (1a) in 76% yield.89
9.3. Base-catalyzed halohydrinphosphonate ring closure
(Z)-1-Propenyl-phosphonate (58) was treated with sodium hypochlorite to afford threo-1-chloro-2-hydroxypropylphosphonic acid (72), which was resolved with (−)-α-phenylethylamine to produce (+)-chlorohydrin in 80% yield. Finally, fosfomycin (1a) was achieved by putting chlorohydrin into 10 M aqueous NaOH in 85–90% yield (Fig. 9E).90
Another asymmetric synthesis of fosfomycin (1a) started with tartaric acid as a chiral auxiliary to direct diastereoface selection when cis-1-propenylphosphonic acid dichloride (73) was bromohydroxylated (Fig. 9F). (Z)-1-Propenyl-phosphonic dichloride reacted with tartaric acid derivatives in DCM at −10 °C to afford the cyclic phosphonates, followed by ring opening and crystallization to produce the monoesters in 70% yield. The bromohydroxylation at 15 °C catalyzed by N-bromoacetamide (NBA) in water produced (1R, 2S)-bromohydrin (76) in a highly chemoselective, regiospecific, and stereospecific manner. After resolution via crystallization, (1R, 2S)-bromohydrin in diastereomerically pure form was quickly converted to enantiomerically pure fosfomycin (1a) in the presence of MeONa in MeOH at 40 °C for 3 h.89,91,92
A large-scale chemical synthesis of fosfomycin (1a) was achieved based on the production of the (1R, 2S)-threo-bromohydrin (76) through enantioselective hydrogenation of β-oxophosphonates (Fig. 9G). Dimethyl 2-oxopropylphosphonate reacted with HBr and H2O2 in THF to produce racemic protected α-bromo-β-oxophosphonates (78), which was enatioselectively hydrogenated to protected (1R,2S)-1-bromo-2-hydroxypropylphosphonate in 98% ee in the presence of (S)-BINAP-Ru(ii) complex as an active catalyst. Fosfomycin (1a) was obtained by acid-catalyzed hydrolysis and treatment with NaOH.90,93,94
10. Summary and outlook
The World Health Organization currently recognizes that antibiotic drug resistance caused by over-reliance on and misuse of antibiotics is one of the significant threats to global public health with the increased emergence and rapid dissemination of multidrug-resistant bacteria. Fatal bacteria resistant to more than 100 various antibiotics in clinic are killing 700 000 people every year, with the global deaths from antibiotic-resistant infections rising to 10 000 000 per year by 2050 if no urgent action is taken for inhibiting the rapid spread of superbugs. Although genomics and bioinformatic analysis indicated that a majority of new antibiotics from bacteria and fungi are awaiting discovery, the number of antibacterial agents from bench to bedside is dramatically decreasing. In this respect, reassessing “old” antibiotics such as phosphonates has been emerged as a feasible and applicable strategy in treating drug-resistant bacterial infections. Phosphonates exhibit exciting biological activities by mimicking phosphate esters or anhydrides or carboxylate groups in the enzyme–substrate complex occurring in the cell primary metabolism. However, the type of small molecules is underexploited in fighting bacterial infections because, so far, there is only one phosphonate antibiotic, fosfomycin, which was marketed in the world. Fosfomycin is a broad-spectrum phosphonate antibiotic and has been in the clinic for more than 20 years. Recently fosfomycin was confirmed to exhibit promising in vivo and in vitro antibacterial activity against a wide range of bacteria, including MDR, XDR, and PDR bacteria.
Furthermore, several relevant clinical trials have recently been conducted to assess the potential application of fosfomycin in the treatment of bacterial infections caused by multidrug-resistant isolates, highlighting the enormous potential of natural phosphonates as ideal candidates in the fight against antimicrobial resistance. Furthermore, recent advances in chemical synthesis and protein engineering have enabled medicinal chemists to modify natural product-based scaffolds with maximum flexibility to provide a chemical library for further high-throughput screening. Although limited functionalities are present in the structure of fosfomycin, many fosfomycin analogs have been successfully synthesized 95−97and more analogs will be added to the fosfomycin library for further screening. Therefore, a more in-depth knowledge of biology and chemistry of fosfomycin could enhance the successful use of the underexplored phosphonate antibiotics for the treatment of bacterial infections. Given the current commercial use of phosphonate in medicine and agriculture, we believe that the success of the only marketed phosphonate antibiotic will help the academy and pharmaceutical industry discover more new phosphonate antibiotics for the effective treatment of the current antibiotic crisis.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31870050), Minjiang Scholar Project grants from Fujian Provincial Government and Fujian Agriculture and Forestry University (Grant No. 114-118360030).
References
- Laxminarayan R. Duse A. Wattal C. Zaidi A. K. Wertheim H. F. Sumpradit N. Vlieghe E. Hara G. L. Gould I. M. Goossens H. Lancet Infect. Dis. 2013;13:1057–1098. doi: 10.1016/S1473-3099(13)70318-9. [DOI] [PubMed] [Google Scholar]
- Bergen P. J. Landersdorfer C. B. Lee H. J. Li J. Nation R. L. Curr. Opin. Infect. Dis. 2012;25:626. doi: 10.1097/QCO.0b013e328358afe5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman L. D. Doak G. Chem. Rev. 1957;57:479–523. doi: 10.1021/cr50015a003. [DOI] [Google Scholar]
- Hemelsoet K. Van D. F. Van S. V. Reyniers M. F. Waroquier M. J. Phys. Chem. A. 2010;114:2864. doi: 10.1021/jp908502d. [DOI] [PubMed] [Google Scholar]
- Ashkenazi N. Segall Y. Chen R. Sod-Moriah G. Fattal E. J. Org. Chem. 2010;75:1917. doi: 10.1021/jo9026325. [DOI] [PubMed] [Google Scholar]
- Westheimer F. H. Acc. Chem. Res. 2002;1:70–78. doi: 10.1021/ar50003a002. [DOI] [Google Scholar]
- Horiguchi M., Biochemistry of natural CP compounds, 1984, pp. 24–52 [Google Scholar]
- Kafarski P. Lejczak B. Curr. Med. Chem. Anti Cancer Agents. 2001;1:301–312. doi: 10.2174/1568011013354543. [DOI] [PubMed] [Google Scholar]
- Martin T. Lobert S. Crit. Care Nurse. 2003;23:15–20. [PubMed] [Google Scholar]
- Hörnberg A. Tunemalm A.-K. Ekström F. Biochemistry. 2007;46:4815–4825. doi: 10.1021/bi0621361. [DOI] [PubMed] [Google Scholar]
- Millard C. B. Kryger G. Ordentlich A. Greenblatt H. M. Harel M. Raves M. L. Segall Y. Barak D. Shafferman A. Silman I. Biochemistry. 1999;38:7032–7039. doi: 10.1021/bi982678l. [DOI] [PubMed] [Google Scholar]
- Schönbrunn E. Eschenburg S. Shuttleworth W. A. Schloss J. V. Amrhein N. Evans J. N. Kabsch W. Proc. Natl. Acad. Sci. 2001;98:1376–1380. doi: 10.1073/pnas.98.4.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinrücken H. Amrhein N. Biochem. Biophys. Res. Commun. 1980;94:1207–1212. doi: 10.1016/0006-291X(80)90547-1. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Biochem. Pharmacol. 2007;73:911–922. doi: 10.1016/j.bcp.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Yu X. Doroghazi J. R. Janga S. C. Zhang J. K. Circello B. Griffin B. M. Labeda D. P. Metcalf W. W. Proc. Natl. Acad. Sci. U. S. A. 2013;110:20759. doi: 10.1073/pnas.1315107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falagas M. E. Giannopoulou K. P. Kokolakis G. N. Rafailidis P. I. Clin. Infect. Dis. 2008;46:1069–1077. doi: 10.1086/527442. [DOI] [PubMed] [Google Scholar]
- Falagas M. E. Maraki S. Karageorgopoulos D. E. Kastoris A. C. Mavromanolakis E. Samonis G. Int. J. Antimicrob. Agents. 2010;35:240–243. doi: 10.1016/j.ijantimicag.2009.10.019. [DOI] [PubMed] [Google Scholar]
- Falagas M. E. Kastoris A. C. Kapaskelis A. M. Karageorgopoulos D. E. Lancet Infect. Dis. 2010;10:43–50. doi: 10.1016/S1473-3099(09)70325-1. [DOI] [PubMed] [Google Scholar]
- Hashemian S. M. R. Farhadi Z. Farhadi T. Ther. Clin. Risk Manag. 2019;15:525. doi: 10.2147/TCRM.S199119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendlin D. Stapley E. O. Jackson M. Wallick H. Miller A. K. Wolf F. J. Miller T. W. Chaiet L. Kahan F. M. Foltz E. L. Science. 1969;166:122. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- Gadebusch H. H. Stapley E. O. Zimmerman S. B. Crit. Rev. Biotechnol. 1992;12:225–243. doi: 10.3109/07388559209069193. [DOI] [PubMed] [Google Scholar]
- Katayama N. Tsubotani S. Nozaki Y. Harada S. Ono H. J. Antibiot. 1990;43:238–246. doi: 10.7164/antibiotics.43.238. [DOI] [PubMed] [Google Scholar]
- Shoji J. Kato T. Hinoo H. Hattori T. Hirooka K. Matsumoto K. Tanimoto T. Kondo E. J. Antibiot. 1986;39:1011–1012. doi: 10.7164/antibiotics.39.1011. [DOI] [PubMed] [Google Scholar]
- Karageorgopoulos D. E. Wang R. Yu X.-h. Falagas M. E. J. Antimicrob. Chemother. 2011;67:255–268. doi: 10.1093/jac/dkr466. [DOI] [PubMed] [Google Scholar]
- Lu C.-L. Liu C.-Y. Huang Y.-T. Liao C.-H. Teng L.-J. Turnidge J. D. Hsueh P.-R. Antimicrob. Agents Chemother. 2011;55:4295–4301. doi: 10.1128/AAC.00349-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuner E. A. Sekeres J. Hall G. S. van Duin D. Antimicrob. Agents Chemother. 2012;56:5744–5748. doi: 10.1128/AAC.00402-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkmans A. C. Zacarias N. V. O. Burggraaf J. Mouton J. W. Wilms E. B. van Nieuwkoop C. Touw D. J. Stevens J. Kamerling I. M. C. Antibiotics. 2017;6:24. doi: 10.3390/antibiotics6040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falagas M. E. Giannopoulou K. P. Kokolakis G. N. Rafailidis P. I. Chin. J. Infect. Chemother. 2010;46:1069–1077. doi: 10.1086/527442. [DOI] [PubMed] [Google Scholar]
- Kahan F. M. Kahan J. S. Cassidy P. J. Kropp H. Ann. N. Y. Acad. Sci. 1974;235:364–386. doi: 10.1111/j.1749-6632.1974.tb43277.x. [DOI] [PubMed] [Google Scholar]
- Marquardt J. L. Brown E. D. Lane W. S. Haley T. M. Ichikawa Y. Wong C.-H. Walsh C. T. Biochemistry. 1994;33:10646–10651. doi: 10.1021/bi00201a011. [DOI] [PubMed] [Google Scholar]
- Brown E. D. Marquardt J. L. Lee J. P. Walsh C. T. Anderson K. S. Biochemistry. 1994;33:10638–10645. doi: 10.1021/bi00201a010. [DOI] [PubMed] [Google Scholar]
- Skarzynski T. Mistry A. Wonacott A. Hutchinson S. E. Kelly V. A. Duncan K. Structure. 1996;4:1465–1474. doi: 10.1016/S0969-2126(96)00153-0. [DOI] [PubMed] [Google Scholar]
- Zhanel G. G. Walkty A. J. Karlowsky J. A. Can. J. Infect Dis. Med. Microbiol. 2016;2016:2082693. doi: 10.1155/2016/2082693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribes S. Taberner F. Domenech A. Cabellos C. Tubau F. Linares J. Viladrich P. F. Gudiol F. J. Antimicrob. Chemother. 2006;57:931–936. doi: 10.1093/jac/dkl047. [DOI] [PubMed] [Google Scholar]
- Mazzei T. Cassetta M. I. Fallani S. Arrigucci S. Novelli A. Int. J. Antimicrob. Agents. 2006;28:35–41. doi: 10.1016/j.ijantimicag.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Pfausler B. Spiss H. Dittrich P. Zeitlinger M. Schmutzhard E. Joukhadar C. J. Antimicrob. Chemother. 2004;53:848–852. doi: 10.1093/jac/dkh158. [DOI] [PubMed] [Google Scholar]
- Grif K. Dierich M. P. Pfaller K. Miglioli P. A. Allerberger F. J. Antimicrob. Chemother. 2001;48:209–217. doi: 10.1093/jac/48.2.209. [DOI] [PubMed] [Google Scholar]
- Saiprasad P. Krishnaprasad K. Indian J. Med. Microbiol. 2016;34:416. doi: 10.4103/0255-0857.195379. [DOI] [PubMed] [Google Scholar]
- Duez J.-M. Mousson C. Siebor E. Péchinot A. Freysz M. Sixt N. Bador J. Neuwirth C. Clin. Med. Rev. Ther. 2011;3:123–142. [Google Scholar]
- Joukhadar C. Klein N. Dittrich P. Zeitlinger M. Geppert A. Skhirtladze K. Frossard M. Heinz G. Müller M. J. Antimicrob. Chemother. 2003;51:1247–1252. doi: 10.1093/jac/dkg187. [DOI] [PubMed] [Google Scholar]
- Reffert J. L. Smith W. J. Pharmacotherapy. 2014;34:845–857. doi: 10.1002/phar.1434. [DOI] [PubMed] [Google Scholar]
- Falagas M. E. Vouloumanou E. K. Samonis G. Vardakas K. Z. Clin. Microbiol. Rev. 2016;29:321–347. doi: 10.1128/CMR.00068-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver L. L. Cold Spring Harbor Perspect. Med. 2017;7:a025262. doi: 10.1101/cshperspect.a025262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Gutiérrez G. Docobo-Pérez F. Rodriguez-Beltrán J. Rodríguez-Martínez J.-M. Aznar J. Pascual A. Blázquez J. Antimicrob. Agents Chemother. 2018;62:e01899-17. doi: 10.1128/AAC.01899-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y. Furukawa S. Ogihara H. Yamasaki M. Biosc. Biotech. Biochem. 2003;67:2030–2033. doi: 10.1271/bbb.67.2030. [DOI] [PubMed] [Google Scholar]
- Kim D. H. Lees W. J. Kempsell K. E. Lane W. S. Duncan K. Walsh C. T. Biochemistry. 1996;35:4923–4928. doi: 10.1021/bi952937w. [DOI] [PubMed] [Google Scholar]
- Eschenburg S. Priestman M. Schönbrunn E. J. Biol. Chem. 2005;280:3757–3763. doi: 10.1074/jbc.M411325200. [DOI] [PubMed] [Google Scholar]
- Takahata S. Ida T. Hiraishi T. Sakakibara S. Maebashi K. Terada S. Muratani T. Matsumoto T. Nakahama C. Tomono K. Int. J. Antimicrob. Agents. 2010;35:333–337. doi: 10.1016/j.ijantimicag.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Palmer A. C. Kishony R. Nat. Commun. 2014;5:4296. doi: 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couce A. Briales A. Rodríguez-Rojas A. Costas C. Pascual Á. Blázquez J. Antimicrob. Agents Chemother. 2012;56:2767–2769. doi: 10.1128/AAC.06122-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horii T. Kimura T. Sato K. Shibayama K. Ohta M. Antimicrob. Agents Chemother. 1999;43:789–793. doi: 10.1128/AAC.43.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beharry Z. Palzkill T. J. Biol. Chem. 2005;280:17786–17791. doi: 10.1074/jbc.M501052200. [DOI] [PubMed] [Google Scholar]
- Roberts A. A. Sharma S. V. Strankman A. W. Duran S. R. Rawat M. Hamilton C. J. Biochem. J. 2013;451:69–79. doi: 10.1042/BJ20121541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillgrove K. L. Pakhomova S. Newcomer M. E. Armstrong R. N. J. Am. Chem. Soc. 2003;125:15730–15731. doi: 10.1021/ja039307z. [DOI] [PubMed] [Google Scholar]
- Kerry L. F. Svetlana P. Matthew R. S. Newcomer A. M. E. Richard N. A. Biochemistry. 2007;46:8110. doi: 10.1021/bi700625p. [DOI] [PubMed] [Google Scholar]
- Kim S. Y. Ju K. S. Metcalf W. W. Evans B. S. Kuzuyama T. Wa V. D. D. Antimicrob. Agents Chemother. 2012;56:4175. doi: 10.1128/AAC.06478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzuyama T. Kobayashi S. O'hara K. Hidaka T. Seto H. J. Antibiot. 1996;49:502–504. doi: 10.7164/antibiotics.49.502. [DOI] [PubMed] [Google Scholar]
- Kobayashi S. Kuzuyama T. Seto H. Antimicrob. Agents Chemother. 2000;44:647–650. doi: 10.1128/AAC.44.3.647-650.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi M. Kandatstu M. Nature. 1959;184:901–902. doi: 10.1038/184901b0. [DOI] [PubMed] [Google Scholar]
- Metcalf W. W. Donk W. A. V. D. Annu. Rev. Biochem. 2009;78:65. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsman G. P. Zechel D. L. Chem. Rev. 2016;117:5704–5783. doi: 10.1021/acs.chemrev.6b00536. [DOI] [PubMed] [Google Scholar]
- Christensen B. G. Leanza W. J. Beattie T. R. Patchett A. A. Arison B. H. Ormond R. E. Kuehl Jr F. A. Albers-Schonberg G. Jardetzky O. Science. 1969;166:123. doi: 10.1126/science.166.3901.123. [DOI] [PubMed] [Google Scholar]
- Cho S.-H. Kim S.-Y. Tomita T. Shiraishi T. Park J.-S. Sato S. Kudo F. Eguchi T. Funa N. Nishiyama M. ACS Chem. Biol. 2017;12:2209–2215. doi: 10.1021/acschembio.7b00419. [DOI] [PubMed] [Google Scholar]
- Sato S. Kudo F. Kim S.-Y. Kuzuyama T. Eguchi T. Biochemistry. 2017;56:3519–3522. doi: 10.1021/acs.biochem.7b00472. [DOI] [PubMed] [Google Scholar]
- Woodyer R. D. Shao Z. Thomas P. M. Kelleher N. L. Blodgett J. A. Metcalf W. W. Wa V. D. D. Zhao H. Chem. Biol. 2006;13:1171–1182. doi: 10.1016/j.chembiol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Hidaka T. Goda M. Kuzuyama T. Takei N. Hidaka M. Seto H. Mol. Gen. Genet. MGG. 1995;249:274–280. doi: 10.1007/BF00290527. [DOI] [PubMed] [Google Scholar]
- Woodyer R. D. Li G. Zhao H. Wa V. D. D. Chem. Commun. 2007;4:359. doi: 10.1039/B614678C. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T. Hidaka T. Kamigiri K. Imai S. Seto H. J. Antibiot. 1992;45:1812–1814. doi: 10.7164/antibiotics.45.1812. [DOI] [PubMed] [Google Scholar]
- Allen K. D. Wang S. C. Arch. Biochem. Biophys. 2014;543:67. doi: 10.1016/j.abb.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliot A. C. Griffin B. M. Thomas P. M. Johannes T. W. Kelleher N. L. Zhao H. Metcalf W. W. Chem. Biol. 2008;15:765–770. doi: 10.1016/j.chembiol.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes T. W. DeSieno M. A. Griffin B. M. Thomas P. M. Kelleher N. L. Metcalf W. W. Zhao H. Chem. Biol. 2010;17:57–64. doi: 10.1016/j.chembiol.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerle M. R. Schwalm E. L. Booker S. J. J. Biol. Chem. 2015;290:3995. doi: 10.1074/jbc.R114.607044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia H. J. Chen G. Hetzler B. G. Reyesspindola J. F. Miller N. E. Nucleic Acids Res. 2001;29:1097. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q. Wa V. D. D. Liu W. Acc. Chem. Res. 2012;45:555–564. doi: 10.1021/ar200202c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick J. B. Duffus B. R. Duschene K. S. Shepard E. M. Chem. Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweifer A. Hammerschmidt F. Biochemistry. 2018;57:2069–2073. doi: 10.1021/acs.biochem.8b00264. [DOI] [PubMed] [Google Scholar]
- Kim H. J. McCarty R. M. Ogasawara Y. Liu Y.-n. Mansoorabadi S. O. LeVieux J. Liu H.-w. J. Am. Chem. Soc. 2013;135:8093–8096. doi: 10.1021/ja312641f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J. McCarty R. M. Ogasawara Y. Liu Y. Mansoorabadi S. O. Levieux J. Liu H. J. Am. Chem. Soc. 2013;135:8093–8096. doi: 10.1021/ja312641f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. Chang W. Guo Y. Huang H. Peck S. C. Pandelia M. E. Lin G. Liu H. Krebs C. Jr J. M. B. Science. 2013;342:991. doi: 10.1126/science.1240373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W. C. Mansoorabadi S. O. Liu H. W. J. Am. Chem. Soc. 2013;135:8153–8156. doi: 10.1021/ja403441x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. Chang W.-c. Pai P.-J. Romo A. Mansoorabadi S. O. Russell D. H. Liu H.-w. J. Am. Chem. Soc. 2012;134:16171–16174. doi: 10.1021/ja3078126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. Lu J. Dubey K. D. Dong G. Lai W. Shaik S. J. Am. Chem. Soc. 2016;138:8489–8496. doi: 10.1021/jacs.6b03555. [DOI] [PubMed] [Google Scholar]
- Huang H. Chang W.-C. Lin G.-M. Romo A. Pai P.-J. Russell W. K. Russell D. H. Liu H.-W. J. Am. Chem. Soc. 2014;136:2944–2947. doi: 10.1021/ja4100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munos J. W. Moon S. J. Mansoorabadi S. O. Chang W. Hong L. Yan F. Liu A. Liu H. W. Biochemistry. 2008;47:8726–8735. doi: 10.1021/bi800877v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzuyama T. Seki T. Kobayashi S. Hidaka T. Seto H. Biosc. Biotech. Biochem. 1999;63:2222–2224. doi: 10.1271/bbb.63.2222. [DOI] [PubMed] [Google Scholar]
- Olivares P. Ulrich E. C. Chekan J. R. van der Donk W. A. Nair S. K. ACS Chem. Biol. 2017;12:456–463. doi: 10.1021/acschembio.6b00939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glamkowski E. J. Gal G. Purick R. Davidson A. J. Sletzinger M. J. Org. Chem. 1970;35:3510–3512. doi: 10.1021/jo00835a070. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y. William A. D. Tokoro Y. J. Org. Chem. 2001;66:7903–7906. doi: 10.1021/jo010701u. [DOI] [PubMed] [Google Scholar]
- Bandini E. Martelli G. Spunta G. Panunzio M. Tetrahedron: Asymmetry. 1995;6:2127–2130. doi: 10.1016/0957-4166(95)00281-S. [DOI] [Google Scholar]
- Girotra N. Wendler N. Tetrahedron Lett. 1969;10:4647–4650. doi: 10.1016/S0040-4039(01)88772-4. [DOI] [Google Scholar]
- Giordano C. Castaldi G. J. Org. Chem. 1989;54:1470–1473. doi: 10.1021/jo00267a050. [DOI] [Google Scholar]
- Castaldi G. and Giordano C., Eur. Pat., EP0251324A2, 1988
- Inukai N. Iwamoto H. Tamura T. Yanagisawa I. Ishii Y. Murakami M. Chem. Pharm. Bull. 1976;24:820–822. doi: 10.1248/cpb.24.820. [DOI] [PubMed] [Google Scholar]
- Kitamura M. Tokunaga M. Noyori R. J. Am. Chem. Soc. 1995;117:2931–2932. doi: 10.1021/ja00115a030. [DOI] [Google Scholar]
- Wróblewski A. E. Bąk-Sypień I. I. Tetrahedron: Asymmetry. 2007;18:520–526. doi: 10.1016/j.tetasy.2007.02.006. [DOI] [Google Scholar]
- Du F. Zhou J. Peng Y. Org. Lett. 2017;19:1310–1313. doi: 10.1021/acs.orglett.7b00128. [DOI] [PubMed] [Google Scholar]
- Wróblewski A. E. Bąk-Sypień I. I. Tetrahedron: Asymmetry. 2007;18:2218–2226. doi: 10.1016/j.tetasy.2007.09.003. [DOI] [Google Scholar]