

Abstract
This report describes a case of a 15-year-old male with cystic fibrosis caused by N1303K and Q493X cystic fibrosis transmembrane conductance regulator (CFTR) protein variants. In this case, CFTR modulators including tezacaftor-ivacaftor and subsequently elexacaftor-tezacaftor-ivacaftor were used and resulted in clinical stability and improvement.
Keywords: case reports, cystic fibrosis, cystic fibrosis transmembrane conductance regulator
Introduction
The use of cystic fibrosis transmembrane conductance regulator (CFTR) modulators has expanded in the last decade. The CFTR modulators target the underlying defect of cystic fibrosis (CF). Classified as potentiators or corrector molecules, these medications work independently or in tandem to modify the CFTR protein and improve chloride transport. This improved CFTR function results in increased lung function, decreased pulmonary exacerbations, improved growth, and quality of life. The most used CFTR modulator regimen in clinical practice includes combination of 1 or 2 correctors (e.g., lumacaftor, tezacaftor, elexacaftor) with ivacaftor, with the latter being the lone available potentiator agent. In 2019, elexacaftortezacaftor-ivacaftor (ETI) was approved for individuals with CF and at least 1 F508del CFTR variant. Because labeling has expanded, now more than 90% of individuals with CF will qualify for CFTR modulator therapy.1
For those who do not qualify for CFTR modulator treatment, based on their CF variants, other treatments to repair, restore, and fix or replace the CFTR protein continue to be evaluated. In addition to alternative treatment strategies, n-of-1 studies have been completed to demonstrate in vitro and in vivo responses including changes in sweat chloride concentration and improvement in pulmonary function testing.2,3 In this report, we describe the case of a 15-year-old male with CF heterozygous for a N1303K (class II variant) and Q493X (class I variant) variant and receiving ETI.
Case
A 15-year-old male was diagnosed with CF in infancy subsequent to meconium ileus, with CF genetic analysis demonstrating the presence of N1303K and Q493X CF genetic variants. He had mild CF lung disease and pancreatic insufficiency. He was prescribed respiratory and oral medications as recommended by Cystic Fibrosis Foundation guidelines including bronchodilator, mucolytics with airway clearance, oral pancreatic enzyme replacement therapy, and fat-soluble vitamin supplementation. However, in 2016, he began to harbor the non-tuberculous mycobacterium (NTM) Mycobacterium abscessus. Despite aggressive airway clearance and treatment of his other chronic infections of methicillin-susceptible Staphylococcus aureus and Stenotrophomonas maltophilia, he experienced a slow decline in pulmonary function testing, increased symptoms, and changes on computed tomography scan imaging. Therefore, in late 2016, he initiated the intensification phase of NTM treatment per the Cystic Fibrosis Foundation and European Cystic Fibrosis Society guidelines.4 Despite aggressive management with 4 to 5 intravenous, oral, and inhaled antimicrobials, he continued to intermittently be smear and culture positive for his NTM infection.
In 2019, based on in vitro evidence of benefit for the N1303K variant and clinical deterioration, the CF care team decided to pursue off-label use of ivacaftortezacaftor (IVA-TEZ).5 The patient began treatment with IVA-TEZ in February 2019 administered by mouth as 1 combination tablet of ivacaftor 150 mg–tezacaftor 100 mg in the morning and 1 tablet of ivacaftor 150 mg in the evening with fat-containing foods. The initiation of IVA-TEZ resulted in stabilization of his clinical status such as stable lung health parameters (predicted FEV1 [forced expiratory volume during the first second], pulmonary exacerbation frequency), growth trends (height, weight, body mass index [BMI]), and improved quality of life indicated by improvement in his Cystic Fibrosis Questionnaire Revised (CFQ-R) from pretreatment. Repeated sweat chloride testing was completed after 1 year of treatment with IVA-TEZ and results remained elevated at 115 mmol/L and 110 mmol/L, which was consistent with the most recent testing done in 2014, namely 100 mmol/L and 97 mmol/L. Primarily based on patient-reported improvement in quality of life and stabilization of clinical outcome measures, IVA-TEZ therapy was continued.
ETI was approved in 2019 and with in vitro evidence of some improvement in CFTR protein function in individuals with N1303K, the CF care team decided to pursue off-label use for this treatment.6–8 ETI was approved by insurance, and the patient transitioned to this treatment in January 2021. Two weeks after ETI therapy initiation, he presented to the CF clinic for a standard follow-up visit. There were no other changes to his medication regimen. From his previous clinic visit 6 weeks prior, his weight had increased 2.9 kg (10th up to 42nd percentile) and his BMI improved from the 29th to the 38th percentile. His lung function improved from 2.98 L (84.3% predicted FEV1) to 3.41 L (93.3% predicted FEV1) and from 3.85 L (93.7% predicted forced vital capacity [FVC]) to 4.20 L (99% predicted FVC). He reported some increased cough and mucus production during the first week of treatment with ETI, which resolved without intervention and otherwise felt a subjective improvement in his exercise tolerance. He was not able to produce expectorated sputum, therefore the CF care team was unable to assess for the presence of NTM infection at that visit, but at subsequent visits he continued to have intermittent positive NTM cultures. In addition, he completed a CFQ-R prior to initiation of ETI and within 12 weeks of receiving treatment, which demonstrated improvements in the physical, vitality, and health domains. Interestingly, his repeated sweat chloride values 2 months after initiation of ETI remained elevated at 113 mmol/L and 111 mmol/L. At his clinic visit around the same time, he had stable lung function (FEV1 3.39 L, 90.2% predicted), inability to produce sputum and normal flora on oropharyngeal swab, and increased weight up to 56.2 kg (43rd percentile for his BMI). A summary of his clinical outcome measures and CFTR modulator utilization is featured in the Figure.
Figure.
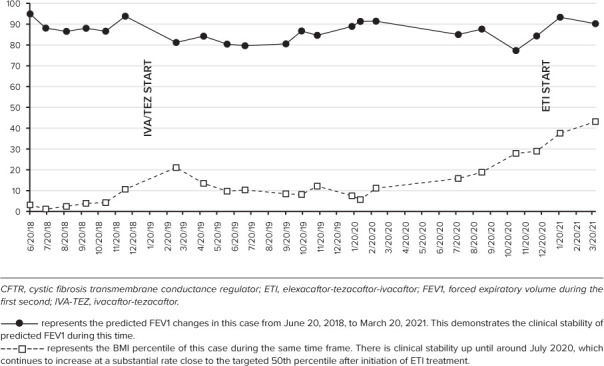
Clinical outcome measures and CFTR modulator use.
For safety monitoring, he received annual ophthalmology examinations and routine hepatic function monitoring. He had intermittently increased transaminase levels while taking IVA-TEZ, which was believed to be secondary to his NTM regimen (baseline aspartate aminotransferase [AST], 76 units/L and alanine transaminase [ALT], 119 units/L; with IVA-TEZ: AST range, 30–163 units/L and ALT range, 26–137 units/L). He had hepatic function testing prior to initiation of ETI that was within normal limits (AST, 31 units/L and ALT, 26 units/L). A repeated hepatic function panel was done 8 weeks after initiation that showed a mild increase in transaminase levels but less than 2 times the upper limit of normal (AST, 54 units/L and ALT, 88 units/L). These will continue to be monitored. He reported no other adverse reactions with initiation and continued treatment with ETI.
Discussion
The patient's genetic variants (Q493X and N1303K) are classified as class I and class II variants, respectively. The Q493X variant is a nonsense variant resulting in decreased quantity of CFTR. The N1303K variant is a class II missense variant with decreased quantity and function of CFTR and is in the same class as the most common CFTR variant, F508del. Individuals with 1 copy of F508del qualify for ETI treatment. In 2018, an in vitro study5 demonstrated 8-time improvement in the open probability with ivacaftor and 3-time improvement in biogenesis with corrector molecules (tezacaftor, lumacaftor). A more recent study6 in 2021 found improvement in CFTR in 2 individuals homozygous for N1303K and found a significant improvement in CFTR measured by transepithelial current from nasal epithelial cells within 48 hours of treatment. The authors6 did note there did not appear to be a significant change in CFTR processing and theorized the benefit was primarily related to potentiation of membrane N1303K proteins. Other in vitro studies for homozygous N1303K variant found improvements in CFTR with CFTR modulators.7 These data from in vitro analysis are consistent with our case report demonstrating significant clinical benefit in an individual heterozygous for the N1303K variant regarding pulmonary function, growth parameters, and quality of life scores. This therapy was well tolerated with mild increases in hepatic function. It is worthwhile to note that despite these clinical parameters for improvement, the patient's sweat chloride values remained unchanged with both IVA-TEZ and ETI treatment. Huang et al8 describe a similar case in an individual with CF caused by N1303K and E193X who started receiving ETI treatment. This individual also had significant clinical improvement, however minimal change in sweat chloride values from 108 mmol/L to 95 mmol/L.8 It is unclear why individuals with N1303K treated with ETI have a less robust sweat chloride change but it could be secondary to a more modest change in CFTR conduction, differences between target organs, differences between secretory and non-secretory epithelial cells, or other epithelial channels that may be linked.
Conclusion
This case further demonstrates clinical benefit from ETI in an individual with a N1303K genetic variant. Because of the numerous CFTR genetic variants identified and associated with CF, this case demonstrates the importance of n-of-1 study in individuals with CF and rare variants to ensure all patients that could benefit from CFTR modulator therapy may receive treatment.
ABBREVIATIONS
- ALT
alanine transaminase
- AST
aspartate aminotransferase
- BMI
body mass index
- CF
cystic fibrosis
- CFQ-R
Cystic Fibrosis Questionnaire Revised
- CFTR
cystic fibrosis transmembrane conductance regulator
- ETI
elexacaftor-tezacaftor-ivacaftor
- FEV1
forced expiratory volume during the first second
- FVC
forced vital capacity
- IVA-TEZ
ivacaftor-tezacaftor
- NTM
non-tuberculous mycobacterium
Footnotes
Disclosures. The authors declare no conflicts or financial interest in any product or service mentioned in the manuscript, including grants, equipment, medications, employment, gifts, and honoraria. The authors had full access to all patient information in this report and take responsibility for the integrity and accuracy of the report.
Ethical Approval and Informed Consent. The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national guidelines. Given the nature of this study, IRB approval was not required by our institution; however, this individual did complete an authorization for presentation of case report.
References
- 1.Gramegna A, Contarini M, Aliberti S et al. From ivacaftor to triple combination: a systematic review of efficacy and safety of CFTR modulators in people with cystic fibrosis. Int J Mol Sci . 2020;21(16):5882. doi: 10.3390/ijms21165882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGarry ME, Illek B, Ly NP et al. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr Pulmonol . 2017;52(4):472–479. doi: 10.1002/ppul.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nick JA, St Clair C, Jones MC;, VX12-770-113 Study Team Ivacaftor in cystic fibrosis with residual function: lung function results from an N-of-1 study. J Cyst Fibros . 2020;19(1):91–98. doi: 10.1016/j.jcf.2019.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Floto RA, Olivier KN, Saiman L;, US Cystic Fibrosis Foundation and European Cystic Fibrosis Society US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. Thorax . 2016;71(suppl 1):i1–i22. doi: 10.1136/thoraxjnl-2015-207360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeStefano S, Gees M, Hwang TC. Physiological and pharmacological characterization of the N1303K mutant CFTR. J Cyst Fibros . 2018;17(5):573–581. doi: 10.1016/j.jcf.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laselva O, Bartlett C, Gunawardena TNA et al. Rescue of multiple class II CFTR mutations by elexacaftor+ tezacaftor+ivacaftor mediated in part by the dual activities of Elexacaftor as both corrector and potentiator. Eur Respir J . 2021;57(6):2002774. doi: 10.1183/13993003.02774-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veit G, Roldan A, Hancock MA et al. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight . 2020;5(18):e139983. doi: 10.1172/jci.insight.139983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang Y, Paul G, Lee J et al. Elexacaftor/tezacaftor/ivacaftor improved clinical outcomes in an N1303KCFTR patient based on in vitro experimental evidence. Am J Respir Crit Care Med . 2021;204(10):1231–1235. doi: 10.1164/rccm.202101-0090LE. [DOI] [PMC free article] [PubMed] [Google Scholar]