

Abstract
Circular RNAs (circRNAs) are a newly recognized component of the transcriptome with critical roles in autoimmune diseases and viral pathogenesis. To address the importance of circRNA in RNA viral transcriptome, we systematically identified and characterized circRNAs encoded by the RNA genomes of betacoronaviruses using both bioinformatical and experimental approaches. We predicted 351, 224, and 2764 circRNAs derived from severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), SARS‐CoV, and Middle East respiratory syndrome coronavirus, respectively. We experimentally identified 75 potential SARS‐CoV‐2 circRNAs from RNA samples extracted from SARS‐CoV‐2‐infected Vero E6 cells. A systematic comparison of viral and host circRNA features, including abundance, strand preference, length distribution, circular exon numbers, and breakpoint sequences, demonstrated that coronavirus‐derived circRNAs had a spliceosome‐independent origin. We further showed that back‐splice junctions (BSJs) captured by inverse reverse‐transcription polymerase chain reaction have different level of resistance to RNase R. Through northern blotting with a BSJ‐spanning probe targeting N gene, we identified three RNase R‐resistant bands that represent SARS‐CoV‐2 circRNAs that are detected cytoplasmic by single‐molecule and amplified fluorescence in situ hybridization assays. Lastly, analyses of 169 sequenced BSJs showed that both back‐splice and forward‐splice junctions were flanked by homologous and reverse complementary sequences, including but not limited to the canonical transcriptional regulatory sequences. Our findings highlight circRNAs as an important component of the coronavirus transcriptome, offer important evaluation of bioinformatic tools in the analysis of circRNAs from an RNA genome, and shed light on the mechanism of discontinuous RNA synthesis.
Keywords: circular RNA, coronavirus, RNA biology, SARS‐CoV‐2, virology
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), SARS‐CoV, and Middle East respiratory syndrome coronavirus (MERS‐CoV) are closely related, single‐stranded, and positive sense RNA viruses belonging to the Betacoronavirus genus of the family of Coronaviridae. 1 They emerged within the last two decades and have posed major challenges to public health. Yet, we have limited knowledge of their pathogenicity factors. The genomes of SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV are ~30 kilo nucleotides (nt) in length and contains 11–14 open reading frames (ORFs). The first ORF, ORF1a/1b, is translated from the positive sense genomic RNA (gRNA) as polyproteins, which are then cleaved proteolytically into nonstructural proteins. Conserved structural proteins including spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins, and additional accessory proteins are encoded by ORFs located towards the 3′‐end of the genome. The structural and accessory proteins are translated from a set of subgenomic RNAs (sgRNAs). 2 Additional components have been identified in CoV transcriptomes. Recent transcriptome profiling of SARS‐CoV‐2 revealed the existence of noncanonical sgRNAs with coding potentials. 3 Small noncoding RNAs (ncRNAs) encoded by SARS‐CoV are found to contribute to lung pathology and inflammation in mice. 4 It is important to determine if CoV transcriptomes contain additional components that contribute to the exacerbated inflammatory responses seen in coronavirus disease 2019, SARS, and MERS patients.
Circular RNAs (circRNAs) are a class of single‐stranded ncRNA species with a covalently closed circular configuration. 5 The lack of 5′‐ and 3′‐ends makes circRNAs resistant to exonuclease‐mediated degradation and thus more stable than linear RNAs. 6 CircRNAs encoded by DNA genomes are produced during gene transcription and by the spliceosome either through back‐splicing of exons or from intron lariats by escaping debranching. 7 They can encode proteins 8 or function as decoys for microRNAs (miRNAs) and proteins. 9 Accumulating data show that circRNAs are important pathological biomarkers for cancers, neurological diseases, and autoimmune diseases. 10 , 11 , 12 Viral‐derived circRNAs have also been identified from several DNA viruses and are implicated in viral pathogenesis. 13 , 14 , 15 , 16 However, circRNAs encoded by RNA viral genomes remain uninvestigated. Thus, we used SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV as examples to assess circRNA expression potentials in RNA viruses.
Here we took both bioinformatical and experimental approaches to systematically identify circRNAs encoded by SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV. To understand circRNA biogenesis from RNA viral genomes, we compared viral and host circRNA features, including abundance, strand preference, length, circular exons, and splicing junction sequences, and demonstrated that CoV circRNAs are different in properties from circRNAs generated by the spliceosome. We further reported the experimental identification of 75 potential circRNAs. Based on sequencing data, RNase R sensitivity assay, northern blotting, amplified fluorescence in situ hybrodization (AmpFISH), and reverse‐transcription polymerase chain reaction (RT‐PCR) results, we concluded that not all back‐splice junction (BSJ)‐spanning sgRNA were circRNA. As we prepared this study for publication, Cai et al. 17 reported the computational prediction of CoV‐encoded circRNAs and their functions. Yet, their algorithms overlooked fundamental differences in circRNA biogenesis from DNA and RNA genomes, resulting in inaccurate predictions on the length, strandness, abundance, and distribution of CoV‐encoded circRNAs. With significant amount of experimental data supporting computational predictions, our work offers a systematic and solid evaluation of CoV circRNA expression landscape. We further provided insights into the biogenesis of discontinuous CoV transcripts.
2. MATERIALS AND METHODS
2.1. Data sets
The RNA‐sequencing (RNA‐Seq) data sets of total RNAs harvested from SARS‐CoV‐ or SARS‐CoV‐2‐ infected Vero E6 (African green monkey kidney) cells at 24 h postinfection (GSE153940 and GSE56193) 18 and RNase R‐treated total RNAs harvested from MERS‐CoV‐infected Calu‐3 (human lung adenocarcinoma) cells at 24 hours post infection (hpi) (GSE139516 19 ) were collected from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) and Sequence Read Archive (SRA: https://www.ncbi.nlm.nih.gov/sra/) using the NCBI SRA Toolkit (http://www.ncbi.nlm.nih.gov/books/NBK158900/). Biological triplicates were pooled to increase reconstruction accuracy. The host circRNA analysis was performed on the same data sets as viral circRNA analyses.
2.2. De novo circRNA identification and reconstruction
The analysis was performed on two Intel W‐3175X CPUs with 128 GB memory running Ubuntu system (version 18.04). 20 Adaptor trimmed reads were aligned with BWA Aligner 21 (BWA‐MEM version 0.7.17‐R1188) and Bowtie2 aligner 22 to host and viral reference genomes: ChlSab1.1.101, hg19, NC_045512.2, NC_004718.3, and NC_019843.3. Alignment statistics was performed with Qualimap2 (version 2.2.1). 23 CIRI2 (version v2.0.6) 24 and find_circ 25 were used for circRNA calling. Reconstruction of partial and full‐length circRNAs was performed with CIRI‐full (version 2.0). 26 Default setting was used.
2.3. SARS‐COV‐2 circRNAs competitive endogenous RNA (ceRNA) coregulatory network analysis
Human and African green monkey (Chlorocebus sabaeus) mature miRNAs were obtained from miRbase (http://www.mirbase.org/) GSE99198, respectively. RNA‐Seq reads from SARS‐CoV‐2‐infected Vero E6 cells (GSE153940) were aligned to the host genome with STAR 27 and counted with FeatureCounts. 28 Differentially expressed genes (DEGs; at least twofold change, false discovery rate cutoff at 0.05) upon SARS‐CoV‐2 infection were selected using DESeq.2. 29 The interactions between human/African green monkey miRNAs with SARS‐CoV‐2 full‐length circRNAs and host DEGs were predicted using miRanda (‐sc 150 ‐en −7). 30 Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of SARS‐CoV‐2 circRNA‐associated DEGs were performed and visualized with ClusterProfiler 31 using a corrected p cutoff of 0.05.
2.4. Sequence homology analysis
For each experimentally identified BSJ or forward‐splice junction (FSJ), 60 bp around the 5′‐ and 3′‐breakpoints were compared using blastall (BLAST) to identify homologous and reverse complementary sequences of 6 bp or longer.
2.5. RNA structure prediction
RNA structure was predicted with RNAcofold from The Vienna RNA Website. 32
2.6. Internal ribosome entry site (IRES) signal analysis
IRES signal strength was analyzed with IRESfinder. 33
2.7. Code accessibility
Source codes for data processing and analyses are available at: https://github.com/ShaominYang/SARS-CoV-2-SARS-CoV-and-MERS-CoV-encode-circular-RNAs-of-spliceosome-independent-origin
2.8. Cell culture, infection, and overexpression of circRNA 29122 | 28295
Vero E6 cells (American Type Culture Collection [ATCC] No. CRL‐1586) were mock‐treated or infected with SARS‐CoV‐2 USA‐WA1/2020 strain (BEI Resources, National Institute of Allergy and Infectious Diseases [NIAID], National Institute of Health [NIH]) at a multiplicity of infection (MOI) of 0.3, based on 50% tissue culture infectious dose, and cultured in Dulbecco's modified Eagle's medium (DMEM, Life Technologies) supplemented with 2% fetal bovine serum (FBS; Hyclone), penicillin (100 IU/ml)–streptomycin (100 µg/ml), and amphotericin B (2.5 µg/ml, Sigma) at 37°C, 5% CO2. The WA1 virus has been passaged once in Vero E6 cells to make virus stock. HEK 293T cells (ATCC No. CRL‐11268) were maintained in DMEM supplemented with 10% FBS, penicillin (100 IU/ml)–streptomycin (100 µg/ml), and amphotericin B (2.5 µg/ml, Sigma) at 37°C, 5% CO2. CircRNA 29122 | 28295 sequence was inserted into pcDNA3.1, pcD‐ciR (Geneseed Biotech Co 34 ) and pcDNA3.1 CircRNA Mini. 35 Transfection was performed with Lipofectamine 3000 (ThermoFisher).
2.9. RNase R treatment, reverse transcription, PCR, and quantitative PCR (qPCR)
Total RNA was extracted with Direct‐zol RNA miniprep kit (Zymo). RNA concentration was determined using Qubit RNA BR Assay kit (ThermoFisher). RNase R treatment was performed with 10 U RNase R (Lucigen) per µg RNA at 37°C for 45 min. Follow‐up purification (RNA Clean and Concentrator, Zymo) was performed. Five hundred micrograms of total RNA or equal amount of RNA treated by RNase R was reverse‐transcribed with Superscript IV (ThermoFisher) using random hexamers or gene‐specific primers. Convergence and inverse PCR primers and qPCR primers used in this study were summarized in Table 2. PCR was performed with GoTaq Master Mix (Promega). qPCR was performed with TransStart® Green qPCR SuperMix (Transgen).
Table 2.
Primers and probes used in this study
| Primer set name | Primer name | Sequence (5′–3′) |
|---|---|---|
| Divergent primers | ||
| circHIPK3 | circHIPK3‐F | TTCAACATATCTACAATCTCGGT |
| circHIPK3‐R | ACCATTCACATAGGTCCGT | |
| Set‐1 | 29083‐F | AACACAAGCTTTCGGCAGAC |
| 27893‐R | GTTCGTTTAGGCGTGACAAGT | |
| Set‐2 | 29045‐F | CCTCGGCAAAAACGTACTGC |
| 28443‐R | GTGAGAGCGGTGAACCAAGA | |
| Set‐3 | 28829‐F | GCAGTCAAGCCTCTTCTCGT |
| 28494‐R | ATTGGAACGCCTTGTCCTCG | |
| Set‐4 | 28936‐F | GCTGCTGCTTGACAGATTGA |
| 28553‐R | TTCGTCTGGTAGCTCTTCGGT | |
| Set‐5 | 29570‐F | AACGTTTTCGCTTTTCCGTTT |
| 39‐R | TTGGTTGGTTTGTTACCTGGG | |
| Distant‐1 | 29045‐F | CCTCGGCAAAAACGTACTGC |
| 51‐R | AGAGATCGAAAGTTGGTTGGT | |
| Distant‐2 | 29083‐F | AACACAAGCTTTCGGCAGAC |
| 51‐R | AGAGATCGAAAGTTGGTTGGT | |
| Distant‐3 | 29230‐F | CATTGGCATGGAAGTCACAC |
| 51‐R | AGAGATCGAAAGTTGGTTGGT | |
| Distant‐4 | 29356‐F | AACATTCCCACCAACAGAGC |
| 51‐R | AGAGATCGAAAGTTGGTTGGT | |
| Distant‐5 | 29457‐F | TTCCTGCTGCAGATTTGGAT |
| 51‐R | AGAGATCGAAAGTTGGTTGGT | |
| Distant‐6 | 29572‐F | CGTTTTCGCTTTTCCGTTTA |
| 51‐R | AGAGATCGAAAGTTGGTTGGT | |
| Distant‐7 | 29668‐F | CACATAGCAATCTTTAATCAGTGTG |
| 51‐R | AGAGATCGAAAGTTGGTTGGT | |
| Distant‐8 | 28445‐F | CAACATGGCAAGGAAGACCT |
| 51‐R | AGAGATCGAAAGTTGGTTGGT | |
| Local‐1 | 28533‐F | ACCGAAGAGCTACCAGACGA |
| 27671‐R | ACTTCCTCTTGTCTGATGAACA | |
| Local‐2 | 28642‐F | TGGTGCTAACAAAGACGGCAT |
| 27671‐R | ACTTCCTCTTGTCTGATGAACA | |
| Local‐3 | 28936‐F | GCTGCTGCTTGACAGATTGA |
| 27671‐R | ACTTCCTCTTGTCTGATGAACA | |
| Local‐4 | 28533‐F | ACCGAAGAGCTACCAGACGA |
| 27816‐R | CAAGGAATAGCAGAAAGGCTAAA | |
| Local‐5 | 28642‐F | TGGTGCTAACAAAGACGGCAT |
| 27816‐R | CAAGGAATAGCAGAAAGGCTAAA | |
| Local‐6 | 28936‐F | GCTGCTGCTTGACAGATTGA |
| 27816‐R | CAAGGAATAGCAGAAAGGCTAAA | |
| ORF3a | 26198‐F | CGACTACTAGCGTGCCTTTG |
| 25546‐R | GTGCAACGCCAACAATAAGC | |
| E | 26457‐F | TGATCTTCTGGTCTAAACGAACT |
| 26217‐R | CAAAGGCACGCTAGTAGTCG | |
| M | 27148‐F | ACCATTCCAGTAGCAGTGACA |
| 26544‐R | TAGTACCGTTGGAATCTGCC | |
| ORF6 | 27413‐F | TGGCACTGATAACACTCGCT |
| 27186‐R | TGTCACTGCTACTGGAATGGT | |
| ORF7a | 27653‐F | TCATCAGACAAGAGGAAGTTCAA |
| 27432‐R | AGCGAGTGTTATCAGTGCCA | |
| ORF7b | 27878‐F | TCACGCCTAAACGAACATGA |
| 27816‐R | CAAGGAATAGCAGAAAGGCTAAA | |
| ORF8 | 28226‐F | TCATGACGTTCGTGTTGTTTT |
| 27893‐R | GTTCGTTTAGGCGTGACAAGT | |
| N | 29457‐F | TTCCTGCTGCAGATTTGGAT |
| 28341‐R | GAATCTGAGGGTCCACCAAA | |
| ORF10 | 29668‐F | CACATAGCAATCTTTAATCAGTGTG |
| 29593‐R | CGTAAACGGAAAAGCGAAAA | |
| Clone‐1 | 29045‐F | CCTCGGCAAAAACGTACTGC |
| 29134‐R | CCCAAAATTTCCTTGGGTTT | |
| Clone‐2 | 28445‐F | CAACATGGCAAGGAAGACCT |
| 28341‐R | GAATCTGAGGGTCCACCAAA | |
| circRNA_ | 29036‐F | TCTAAGAAGCCTCGGCAAAA |
| 29122 | 28295 | 28455‐R | TTGCCATGTTGAGTGAGAGC |
| Convergent primers | ||
| β‐Actin | β‐Actin‐F | CACACTGTGCCCATCTATGAGG |
| β‐Actin‐R | TCGAAGTCTAGGGCGACATAGC | |
| ORF3a | 25531‐F | ATTGTTGGCGTTGCACTTCT |
| 25711‐R | AGAGAAAAGGGGCTTCAAGG | |
| M | 26457‐F | TGATCTTCTGGTCTAAacgaact |
| 27168‐R | TGTCACTGCTACTGGAATGGT | |
| ORF7a | 27413 F | TGGCACTGATAACACTCGCT |
| 27671 R | ACTTCCTCTTGTCTGATGAACA | |
| N | 29083‐F | AACACAAGCTTTCGGCAGAC |
| 29249‐R | GTGTGACTTCCATGCCAATG | |
| ORF10 | 29572‐F | CGTTTTCGCTTTTCCGTTTA |
| 29692‐R | CACACTGATTAAAGATTGCTATGTGA | |
| Gene and strand specific RT‐inverse PCR primers | ||
| RT‐F1 | 28809 F | GCAGTCAAGCCTCTTCTCGT |
| RT‐R1 | 28494 R | ATTGGAACGCCTTGTCCTCG |
| Inverse‐F1 | 29045‐F | CCTCGGCAAAAACGTACTGC |
| Inverse‐R1 | 28443‐R | GTGAGAGCGGTGAACCAAGA |
| RT‐F2 | 29083‐F | AACACAAGCTTTCGGCAGAC |
| RT‐R2 | 51‐R | AGAGATCGAAAGTTGGTTGGT |
| Inverse‐F2 | 29356‐F | AACATTCCCACCAACAGAGC |
| Inverse‐R2 | 39‐R | TTGGTTGGTTTGTTACCTGGG |
| Inverse‐F3 | 29668‐F | CACATAGCAATCTTTAATCAGTGTG |
| Primers for in vitro transcription | ||
| N‐BSJ‐F | TAATACGACTCACTATAGGGCCTCGGCAAAAACGTACTGC | |
| N‐BSJ‐R | GTGAGAGCGGTGAACCAAGA | |
| Probes for AmpFISH | ||
| circRNA_29122 | 28295 (–) DP | GTTACAGACGACTCCCACAGTCC‐AATCAGCGAAATGCACCCCGCATT GGACT | |
| circRNA_29122 | 28295 (–) AP | AGACGTGGTCCAGAACAAACCCAA GGACTGTGGGAGTCGTCTGTAACTACTTCATGTTACAGACGACTCCCAC | |
| circRNA_29122 | 28295 (+) DP | GTTACAGACGACTCCCACAGTCC‐ TTGGGTTTGTTCTGGACCACGTCT‐ GGACT | |
| circRNA_29122 | 28295 (+) AP |
AATGCGGGGTGCATTTC GCTGATT GGACTGTGGGAGTCGTCTGTAACTACTTCATGTTACAGACGACTCCCAC |
|
| Primers for circRNA overexpression | ||
| LinearRNA_28295‐29122‐F | TTGGCAATGTTGTTCCTTGA | |
| LinearRNA_28295‐29122‐R | ACCGAAGAGCTACCAGACGA | |
| circRNA_29122 | 28295‐F | TTGCCATGTTGAGTGAGAGC | |
| circRNA_29122 | 28295‐R | TCTAAGAAGCCTCGGCAAAA | |
| Primers for qPCR | ||
| GPR115‐F | TTTAAGGACTCAACTGGTGCATC | |
| GPR115‐R | ACACTCTCAATGGTCTCTGGAG | |
| GAPDH‐F | CCAAGAGAAAGATGGACCCTG | |
| GAPDH‐R | TCGAACAGGAGGAGCAGAGAGCG | |
2.10. Cloning and identification of BSJs
Inverse RT‐PCR products were separated on 2% agarose gels. Candidate BSJ sequences were gel purified (Zymoclean Gel DNA Recovery kit, Zymo) and TA cloned (ThermoFisher) according to the manufacturers' instructions. At least eight colonies were checked for insertion of candidate PCR products by PCR with M13 universal primers. Following PCR purification (DNA clean and concentrator kit, Zymo), candidate BSJ sequences were Sanger‐sequenced (MCLAB) with M13 universal primers. Sequencing results were blasted against the SARS‐CoV‐2 reference genome (NC_045512.2). The 5′‐ and 3′‐breakpoints of BSJs and FSJs were manually curated so that if ambiguous nucleotides exist around the junction they are counted as the donor sequence. BSJs with breakpoints differing within 20 nt were considered as variants of one BSJ.
2.11. Northern blotting
A digoxin‐labeled RNA probe targeting the BSJ 29122 | 28295 was prepared using digoxigenin (DIG) Northern Starter Kit (Roche) with PCR product as the template. A sequenced colony containing 28809–29122 | 28295–28494 was used for PCR amplification with T7 sequence‐containing primers (Table 2). Northern blotting was performed with NorthernMax Kit (ThermoFisher). One microgram of total RNA per lane was loaded on 1% denaturing agarose gels from SARS‐CoV‐2‐infected Vero E6 cells with or without RNase R treatment. Gels were transferred to BrightStar‐Plus Positively Charged Nylon Membrane (ThermoFisher) and UV‐crosslinked. Hybridization was performed at 62°C overnight. Washing, staining, and imaging was performed as instructed by the manufacturer.
2.12. Detection of SARS‐CoV‐2 linear and circRNA by single‐molecule FISH (smFISH) and AmpFISH
Pairs of AmpFISH probes for circRNA 29122 | 28295 and smFISH probes for ORF1 were designed and purified as previously described. 36 To specifically amplify the circRNA but not sgRNAs carrying the N gene sequence, one probe targeted the donor sequence and the other targeted the acceptor sequence of the circRNA back‐splice junction. Hybridization chain reaction (HCR) hairpin sequences were added to the probes as previously described. 36 Probe sequences were provided in Table 2. RNA FISH was performed as described previously. 36 Briefly, Vero E6 cells were seeded on the glass coverslips (thickness 0.1 mm) coated with 0.1% gelatin and cultured in DMEM media with 10% FBS and antibiotics penicillin (100 IU/ml)–streptomycin (100 µg/ml). Cells were infected with SARS‐CoV‐2 at an MOI of 0.01 and coverslips were fixed at 24 h postinfection. For fixation, cells were washed with 1× phosphate‐buffered saline (PBS; pH 7.4) and fixed with 4% paraformaldehyde/PBS for 10 min at room temperature (RT). After being washed with 1× PBS, the cells were incubated with 70% ethanol for 10 min at RT. Then the cells were permeabilized with 0.05% Triton X‐100 at 4°C for 10 min. For the cells to be treated with RNase R, the cells were first equilibrated with 1x RNase R reaction buffer (Lucigen, Cat#RNR07250) for 30 min followed by RNAse R (Lucigen, cat#RNR07250) treatment at 37°C for 3 h. Cells were equilibrated with hybridization wash buffer (10% formamide/2× solution of sodium citrate [SSC]) for 5 min at RT, incubated with probes specific for positive‐ and negative‐stranded linear or circRNA at 37°C overnight in a hybridization buffer containing 10% formamide/2× SSC. 37 Coverslips were washed twice with hybridization wash buffer. Amplification reactions of hybridized probe was performed in HCR buffer containing 5× SSC and mounted as described previously. 36 RNase R treatment was performed at 37°C for 3 h followed by AmpFISH. Z‐stacks images were acquired using Axiovert 200M microscope using the same setting for all samples and were processed by maximum projection.
3. RESULTS
3.1. Bioinformatical identification and quantification of circRNAs encoded by SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV
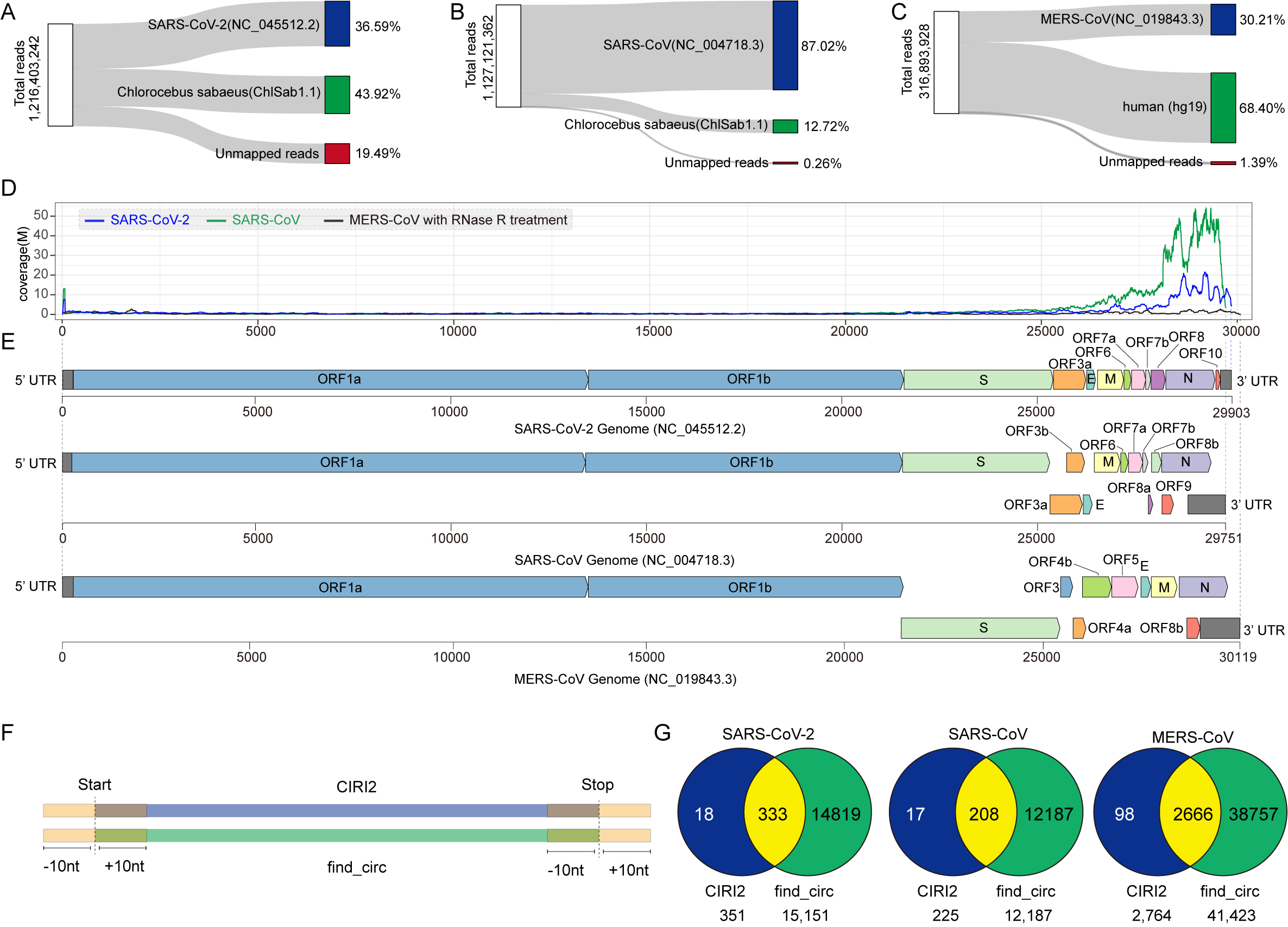
To identify circRNAs derived from CoVs, we performed de novo circRNA identification on publicly available deep RNA‐Seq data sets 17 , 18 of SARS‐CoV‐2 or SARS‐CoV‐infected Vero E6 (African green monkey kidney) cells and MERS‐CoV infected Calu‐3 (human lung) cells at 24 hpi. As CoVs synthesize gRNAs and sgRNAs in the cytoplasm of host cells, CoV circRNAs are likely to circularize in the cytoplasm independent of splicing. We thus excluded circRNA discovery algorithms with bias towards the GT | AG splicing signal and used exclusively the CIRI2 circRNA prediction pipeline 24 to unbiasedly identify gapped reads. FSJ reads and BSJ reads were determined based on whether the splice donor locates upstream or downstream in the reference genome (Figure 1A). The 5′‐ and 3′‐breakpoints were subsequently determined. After mapping with BWA‐MEM, 21 we obtained 1,216,403,242 total reads from the SARS‐CoV‐2 data set with 36.59% mapped to SARS‐CoV‐2 (Figures S1A), 1,127,121,362 total reads from the SARS‐CoV data set with 87.02% mapped to SARS‐CoV (Figure S1B), and 316,893,928 total reads from the MERS‐CoV data set with 30.21% mapped to MERS‐CoV (Figure S1C). The SARS‐CoV‐2 and SARS‐CoV data sets showed sharp peaks at the 5′‐leader sequence and high coverage towards the 3′‐end of the genome (Figure S1D). Genome coverage of the MERS‐CoV data set was substantially lower due to the removal of linear RNAs by RNase R digestion. We predicted 351, 224, and 2764 unique BSJs representing SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV circRNAs, respectively (Data S1). To assess the expression level of individual CoV circRNAs, we plotted BSJ‐spanning read counts of viral and host circRNAs identified from the same data sets against their ranked percentile. The expression level of viral circRNAs was higher than host circRNAs in the same ranked percentile (Figure 1B). The most abundant circRNA encoded by each CoV had over 10,000 BSJ‐spanning reads, comparable to the most abundant circRNAs identified in their hosts (Data S1). We conclude that circRNAs of SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV are highly expressed during infection.
Figure 1.
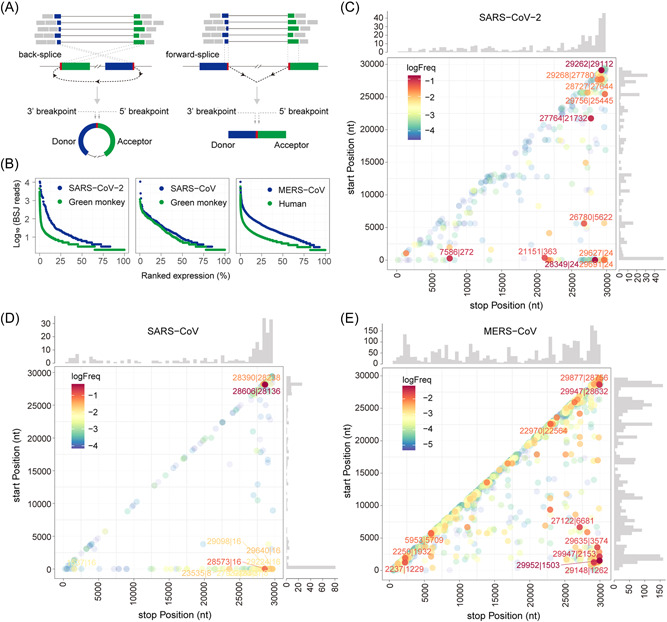
Predicted severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), SARS‐CoV, and Middle East respiratory syndrome coronavirus (MERS‐CoV) circular RNA (circRNA) abundance and landscapes. (A) Illustration of CIRI2‐based identification of circRNAs. Gapped reads are aligned to the donor and acceptor sequences. If the genomic location of the donor is downstream of the acceptor, it is considered as a back‐splice junction (BSJ). The 5′‐ and 3′‐breakpoints are then determined. (B) Ranked expression level of de novo identified circRNAs from CoV and host genomes. (C–E) Frequency of circularization events in SARS‐CoV‐2 (C), SARS‐CoV (D), and MERS‐CoV (E). Counts of BSJ‐spanning reads (starting from a coordinate in the x axis and ending in a coordinate in the y axis) indicated by color. The counts were aggregated into 500 nt bins for both axes. Distribution of start/end position was shown as histograms on the x and y axis.
We applied find_circ in parallel with CIRI2 and found that AG | GT signal‐biased algorithms, find_circ, 25 resulted in false positive reads (Figure S1F,G and Data S2). To determine the proportion of total BSJs to total CoV‐mapping reads, which were identified by CIRI2 and find_circ, we performed a statistic analysis of CoV BSJs. CIRI2 identified 0.01477%, 0.00296%, and 0.43997% of total CoV reads that were BSJs of SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV circRNAs, respectively (Table S1). Strikingly, find_circ found 0.36121%, 0.00733%, and 0.43962% of total CoV reads that were BSJs of SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV circRNAs, respectively. Therefore, CIRI2 is more reliable.
3.2. SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV circRNAs display common distant and local back‐splicing hotspots
To examine the circRNA landscape, we mapped all identified viral circRNAs by the 5′‐ and 3′‐breakpoints of the BSJs to their respective genomic locations and estimated the back‐splicing frequency by counting BSJ‐spanning reads (Figure 1C–E). We identified two frequent back‐splicing events shared by three CoVs: distant back‐splicing between the 3′‐ and the 5′‐ends of the genome and local back‐splicing in regions corresponding to the N gene and the 3′‐untranslated region (UTR) of CoVs. Additionally, we noticed a general enrichment of local BSJ‐spanning reads along the diagonal of the graph across three data sets. Although the local BSJ reads were low outside the 3′‐end of SARS‐CoV‐2 and SARS‐CoV genome (Figure 1C,D), a few local BSJs located in ORF1a/b, S, and between ORF3a and M of the MERS‐CoV‐2 genome had moderate to high read counts (Figure 1E). Interestingly, similar distribution patterns have been reported for SARS‐CoV‐2 FSJs in sgRNAs. 3 , 38 These results suggest that circRNAs may be conserved in betacoronaviruses.
3.3. CoV circRNAs are different from DNA genome‐encoded circRNAs
To better characterize CoV circRNAs, we performed de novo reconstruction of full‐length SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV circRNAs using CIRI‐full. 26 We got 127, 122, and 836 full‐length circRNAs from SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV, respectively, and additional partially assembled viral circRNAs (Figure S2A–C and Data S3). Reconstruction of host circRNAs resulted in 4815 full‐length monkey circRNAs and 31,807 full‐length human circRNAs (Data S3). Comparison of host and viral circRNA features shows that CoV circRNAs are distinct from host circRNAs in several aspects. First, the length distribution of CoV circRNAs are different from that of the host (Figure 2A). Fifteen percent SARS‐CoV‐2 circRNAs and 19% MERS‐CoV circRNAs were between 500 and 1500 nt in length, whereas 92% host circRNAs were below 500 nt, consistent with previous reports. 26 This resulted in an increase in the average length of SARS‐CoV‐2 and MERS‐CoV circRNAs from host circRNAs (Figure 2A). Interestingly, SARS‐CoV circRNAs were extremely short, with a mean length of 187 nt. Our results disputed the length distribution predicted by Cai et al. 17 Their analysis overlooked gaps within circRNAs, thus overestimated the length of CoV circRNAs. Second, although circRNAs generated by both monkey and human genomes showed no strand preference, CoV circRNAs tend to be negative‐stranded (Figure 2B), which is opposite to the prediction of strand preference by Cai et al. 17 Third, CoVs tend to produce single‐exon circRNAs, whereas host circRNAs undergo further intron excision, 6 resulting in multiple FSJs in the circRNAs (Figure 2C). Our analysis predicts that 12%–35% of CoV circRNAs contain an FSJ, further supporting the existence of gaps in CoV circRNAs. Alternative intron inclusion of DNA genome‐encoded circRNAs gives rise to diverse circular isoforms, 39 which share the same BSJ but differ in FSJs and length. Although CoV circRNAs contain FSJs, we only predicted one full‐length MERS‐CoV circRNA with two circular isoforms (MERS‐CoV_29148 | 1262, 1051 and 155 nt, Data S3). Taken together, our results suggest that CoV circRNAs are different in properties from known circRNAs encoded by DNA genomes.
Figure 2.
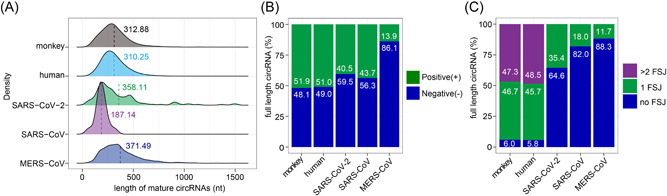
Comparison of predicted full‐length coronavirus (CoV) circular RNAs (circRNAs) and host circRNAs. (A) Length distribution of circRNAs derived from host genomes and CoVs. Average length indicated by dashed lines. (B) Strand distribution of host and viral circRNAs. (C) Statistics of forward‐splice junctions (FSJs) in host and viral circRNAs. Number of full‐length circRNAs used in the analysis: severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), 127; SARS‐CoV, 122; Middle East respiratory syndrome coronavirus (MERS‐CoV), 836; Monkey, 4815; Human, 31,807.
3.4. Systematic capture and identification of SARS‐CoV‐2 BSJs
Next, we systematically validated SARS‐CoV‐2 BSJ hotspots predicted by our bioinformatic analyses. We extracted total RNA from Vero E6 cells mock‐treated or infected with SARS‐CoV‐2 at 24 hpi. Forward and reverse inverse PCR primers were designed in such a way that all donor or acceptor sequences in each hotspot will be picked up (Figure 3A,B). To validate the two major back‐splicing events, we performed RT‐PCR with divergent primer sets targeting the distant BSJ hotspot 29001~29903 | 1~500 (Figure S3A) and the local BSJ hotspot 28501~29500 | 27501~28500 (Figure S3B). We also performed inverse RT‐PCR with five sets of primers targeting abundant SARS‐CoV‐2 circRNAs predicted by CIRI2 (Figure 3C). Some predicted full‐length CoV circRNAs contain ORFs and thus potentially encode proteins (Figure S2–4). We used divergent primers flanking individual ORFs to validate these circRNAs (Figure S3C). Most of the inverse RT‐PCR reactions using complementary DNA (cDNA) from the infected cells produced bands ranging from 200 to 800 bp, whereas no amplification was seen from mock samples (Figure 3C and S3A–C). Moreover, the band intensity of candidate BSJs was much higher than the abundant host circRNA circHIPK3 40 (Figure 3C and S3A–C).
Figure 3.
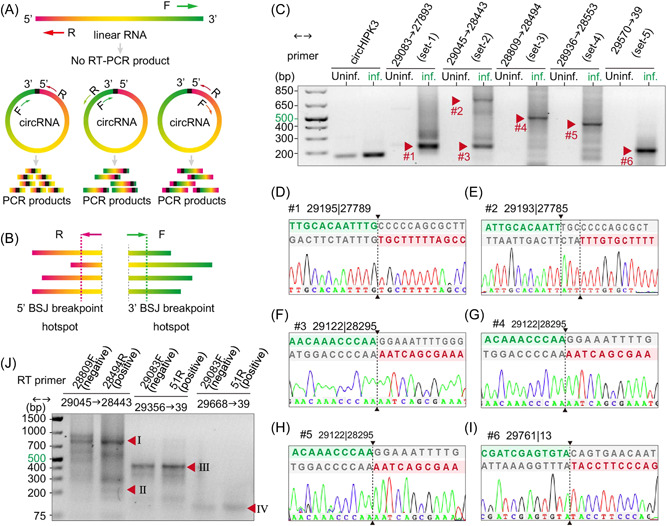
Experimental identification of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) circular RNAs (circRNAs) in Vero E6 cells. (A) Illustration of inverse reverse‐transcription polymerase chain reaction (RT‐PCR) with divergent primers would selectively amplify different regions of circRNAs but not linear RNAs. (B) Schematic diagram showing divergent primers were designed to amplify as much predicted back‐splice junctions (BSJs) in a given hotspot as possible. (C–I) Inverse RT‐PCR result with selected primer sets were shown in C. Bands indicated by red arrows were sequenced. Representative Sanger sequencing results were shown in D–I. BSJ breakpoints were indicated by dashed lines. Donor (green) and acceptor (red) sequences and downstream/upstream sequences (gray) flanking the junction were aligned to the BSJ sequence. (J) Strand‐specific RT inverse PCR result. Bands corresponding to BSJ 29122 | 28295 (225 bp), BSJ 29195 | 27789 (804 bp), and BSJ 29761 | 13 (431 and 119 bp) were indicated by red arrowheads.
To determine whether the inverse PCR products were BSJs rather than nonspecific PCR product, we gel‐purified candidate BSJ amplicons based on the molecular weight (Figure 3C and S3A–C, red arrowheads), subcloned, and Sanger‐sequenced at least eight colonies for each candidate. Using this pipeline, we identified 75 BSJs from 169 clones (Table 1 and Data S4). Six BSJs, namely 28576 | 27703 (#10), 29195 | 27789 (#15), 29122 | 28295 (#32), 29122 | 28320 (#33), 29085 | 28321 (#41), and 29761 | 13 (#60), were independently identified by at least two primer sets. Using overlapping amplicons carrying the same BSJs, we reconstructed circRNA 29122 | 28295(#32), circRNA 29122 | 28320 (#33), and circRNA 29085 | 28321 (#41; Table 1). The high detection rates of BSJ 29195 | 27789 (#15), 29122 | 28295 (#32), and 29761 | 13 (#60) by multiple primer sets and from subclones validated the frequent distant and local back‐splicing events predicted from the RNA‐Seq data (Figure 3D,F–I and Table 1).
Table 1.
Summary of experimentally identified SARS‐CoV‐2 circRNAs
| Primer range | Subclone no. | circRNA no. | Representative circRNAs | Occurrence |
|---|---|---|---|---|
| 26198–>25546 | 1 | 1 | 26254 | 46, 77 | 25393 (#1)a | 1/1 |
| 27148–>26544 | 19 | 4 | 27283 | 47, 76 | 26480 (#5)a | 11/19 |
| 28533–>27671 | 3 | 3 | 28576 | 27555 (#7) | 1/3 |
| 28533–>27816 | 9 | 7 | 29195 | 27789 (#15) | 3/4c |
| 28576 | 27703 (#10) | 1/5d | |||
| 28642–>27816 | 12 | 6 | 29195 | 27789 (#15) | 6/6c |
| 28936–>27816 | 7 | 4 | 29195 | 27789 (#15) | 3/7 |
| 28576 | 27703 (#10) | 1/7 | |||
| 29083–>27893 | 10 | 3 | 29195 | 27789 (#15) | 6/10 |
| 28445–>28341 | 24 | 10 | 29122 | 28295 (#32)b | 12/24 |
| 29122 | 28320 (#33)b | 1/24 | |||
| 29045–>28443 | 9 | 4 | 29122 | 28295 (#32)b | 5/7d |
| 29122 | 28320 (#33)b | 2/7d | |||
| 28809–>28494 | 16 | 4 | 29122 | 28295 (#32)b | 3/4c |
| 28855 | 28434 (#38) | 7/11d | |||
| 28853 | 28467 (#37) | 4/11d | |||
| 28936–>28553 | 5 | 5 | 29122 | 28295 (#32)b | 1/5 |
| 29085 | 28321 (#41)b | 1/5 | |||
| 29045–>28953 | 21 | 20 | 29085 | 28321 (#41)b | 2/21 |
| 29122 | 28295 (#32)b | 1/21 | |||
| 29570–>39 | 3 | 1 | 29761 | 13 (#60) | 3/3 |
| 28445–>51 | 9 | 8 | 29015 | 13 (#65) | 2/9 |
| 29356–>51 | 5 | 2 | 29378 | 9 (#69) | 4/5 |
| 29761 | 13 (#60) | 1/5 | |||
| 29457–>51 | 3 | 3 | 29761 | 13 (#60) | 1/3 |
| 29572–>51 | 5 | 3 | 29761 | 13 (#60) | 2/5 |
| 29664 | 8 (#72) | 2/5 | |||
| 29668–>51 | 8 | 3 | 29761 | 13 (#60) | 6/8 |
Note: 169 subclones and 75 circRNAs were identified in total. Refer to Data S4 for details.
Abbreviations: circRNA, circular RNA; ORF, open reading frame; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
ORF‐containing circRNAs.
Fully assembled circRNAs.
From bands of low molecular weight.
From bands of high molecular weight.
From the collection of experimentally identified BSJs we found the following characteristics. First, multiple BSJs were identified in almost all gel‐purified bands (Data S4), confirming our predictions that many circRNAs had overlapping sequences flanking the BSJs (Figure S2A). Second, SARS‐CoV‐2 circRNAs are not encoded by individual ORF. We identified distant fusion from ORF1a/1b, E, ORF6, N, ORF10, and the 3′‐UTR to the 5′‐UTR, as well as local fusion within N, from N to ORF7a, ORF7b, and ORF8, and from ORF6 to M (Data S4). Third, we found the breakpoints of a given circRNA unexpectedly flexible. For example, circRNA 29761 | 13 had seven variants with the 3′‐breakpoints ranging from genomic location 29759 to 29767 and the 5′‐breakpoints from 5 to 19 (Table 3). Insertion of additional nucleotides between the breakpoints was also observed (Figure 3D,E and Table 3). This is in sharp contrast to the accurate GU | AG back‐splice breakpoints seen in circRNAs reported thus far. It further suggests the mechanism driving CoV RNA back‐splicing is error‐prone.
Table 3.
BSJ breakpoint flexibility of circRNA 29761 | 13
| Clone # | Length | BSJ | Primer range | BSJ sequence (5′–3′) |
|---|---|---|---|---|
| 153 | 441 | 29759 ∣ 15 | 29356–>51 | GAGTACGATCGAGTG | CCTTCCCAGGTAACA |
| 155 | 350 | 29759 ∣ 5 | 29457–>51 | GAGTACGATCGAGTG | AAGGTTTATACCTTC |
| 160 | 232 | 29759 ∣ 8 | 29572–>51 | GAGTACGATCGAGTG | GTTTATACCTTCCCA |
| 161 | 227 | 29762 ∣ 16 | 29572–>51 | TACGATCGAGTGTAC | CTTCCCAGGTAACAA |
| 164 | 119 | 29750 ∣ 10 | 29668–>51 | GGCCACGCGGAGTAC | TTATACCTTCCCAGG |
| 165 | 126 | 29759 ∣ 19 | 29668–>51 | CGAGTACGATCGAGT | CCCAGGTAACAAACC |
| 166 | 135 | 29760 ∣ 10 | 29668–>51 | AGTACGATCGAGTGT | TTATACCTTCCCAGG |
| 167 | 133 | 29761 ∣ 13 | 29668–>51 | GTACGATCGAGTGTA | TACCTTCCCAGGTAA |
| 168 | 133 | 29761 ∣ 13 | 29668–>51 | GTACGATCGAGTGTA | TACCTTCCCAGGTAA |
| 169 | 148 | 29767 ∣ 10 | 29668–>51 | ATCGAGTGTACAGTG | CAGCGTG | TTATACCTTCCCAGG |
Abbreviations: BSJ, back‐splice junction; circRNA, circular RNA.
Taken together, we experimentally confirmed that the SARS‐CoV‐2 transcriptome contains abundant sgRNA with distant and local back‐splicing junctions and further revealed the diversity of SARS‐CoV‐2 circRNAs at the genome scale and the junction sequence scale.
3.5. Experimentally captured SARS‐CoV‐2 BSJ‐spanning transcripts contain FSJs and repetitive BSJs
Next, we tried to validate our bioinformatic predictions of SARS‐CoV‐2 circRNA length and composition with experimental data. About 30% of sequenced BSJ‐spanning amplicons were over 500 nt, indicating that full‐length circRNAs were even longer (Table 4). This result supports our prediction that SARS‐CoV‐2 circRNAs are longer than host circRNAs. Although 81.7% of sequenced amplicons carried two fragments separated by a BSJ, we found 16% amplicons with three fragments and 2%, which were even more complex (Table 4). FSJs were identified in 30 (18%) clones. Interestingly, most FSJs represent the leader‐to‐body fusion in canonical sgRNAs (77 | 25393, 76 | 26480, and 75 | 28266) and were paired with a distant BSJ to the 5′‐UTR (Data S4). These sgRNA‐like circRNAs displayed little flexibility in the FSJ breakpoint but great variation in the BSJ breakpoints. The 3′‐BSJ breakpoints range from genomic location 28465–29271 and the 5′‐breakpoints ranged from genomic location 3–40. We also identified 10 BSJs with noncanonical FSJs 3 paired with BSJs (Data S4). Strikingly, we identified 4 out of the 75 cricRNAs with multiple BSJs: Clone #63, #140, or #162 had an FSJ flanked by two BSJs as shown in Figure S3E, and clone #62 contained three repetitive BSJs with slight variations in the breakpoints as shown in Figure S3F (Data S4). Figures S3E,F also illustrated how and where the multiple or repetitive BSJs formed.
Table 4.
Length and junction distribution in experimentally identified circRNAs
| Fragments | 2 | 3 | 4 | 6 | |
|---|---|---|---|---|---|
| Count (percentage) | 138 (81.7%) | 27 (16.0%) | 3 (1.8%) | 1 (0.6%) | |
| BSJ | 1 | 2 | 3 | ||
| Count (percentage) | 165 (97.6%) | 3 (1.8%) | 1 (0.6%) | ||
| FSJ | 0 | 1 | 3 | ||
| Count (percentage) | 139 (82.2%) | 29 (17.2%) | 1 (0.6%) | ||
| Length (nt) | 0–100 | 101–300 | 301–500 | 501–700 | >700 |
| Count (percentage) | 11 (6.5%) | 79 (46.7%) | 23 (13.6%) | 26 (15.4%) | 30 (17.8%) |
Abbreviations: BSJ, back‐splice junction; circRNA, circular RNA; FSJ, forward‐splice junction.
The identification of distant BSJs coupling with canonical and noncanonical FSJs confirmed our prediction of SARS‐CoV‐2 circRNAs with more than one circular exons (Figure S2A). Our experimental data set further revealed rare occurrence of repetitive BSJs in SARS‐CoV‐2 transcripts. It is possible that sgRNA‐like circRNAs are generated from canonical sgRNAs through one back‐splicing event. However, the coupling of BSJs with noncanonical FSJs and the existence of repetitive BSJs suggest that BSJs can be formed during transcription and may not equal to RNA circularization.
3.6. SARS‐CoV‐2 circRNAs can exist as both sense and antisense forms
To verify the strandness of circRNAs, we performed strand‐specific reverse transcription followed by inverse PCR. Region‐specific and strand‐specific reverse transcription and inverse PCR was designed to target three major BSJs: 29195 | 27789 (#15), 29122 | 28295 (#32), and 29761 | 13 (#60). For the first two BSJs, forward primer 28809‐F and reverse primer 28494‐R targeting the N gene were used for reverse transcription from antisense and sense RNA, respectively. For 29761 | 13, cDNA was synthesized with either 29083‐F (N) or 51‐R (5′‐UTR). Inverse PCR result showed that a band corresponding to the 225 bp amplicon containing BSJ 29122 | 28295 (Arrowhead Ⅱ) was obtained only from cDNA of sense‐stranded RNA (Figure 3J). This result suggests that circRNA 29122 | 28295 exists in the positive stranded form. On the other hand, the 804 bp amplicon containing BSJ 29195 | 27789 (Arrowhead Ⅰ) was obtained with cDNA from both RNA strands. Similarly, BSJ 29761 | 13 (Arrowhead Ⅲ) amplified by two different sets of primers was detected using cDNA from either strand. It is worthwhile to note that both BSJ 29195 | 27789 and BSJ 29761 | 13 amplified from the cDNA of positive‐stranded RNA were more abundant than that from the cDNA of negative‐stranded RNA, suggesting that these circRNAs were preferentially in the sense form. Lastly, our strand‐specific RT inverse PCR result also showed the existence of BSJs that were exclusively negative‐stranded (Figure 3J, Arrowheads Ⅰ, Ⅲ, and Ⅳ). We concluded that SARS‐CoV‐2 BSJs can either be strand‐specific or exist as both sense and antisense RNA. Our results indicate that bioinformatic prediction of strandness could be unreliable with circRNAs from an RNA genome.
3.7. SARS‐CoV‐2 produces circRNAs that are resistant to RNase R treatment
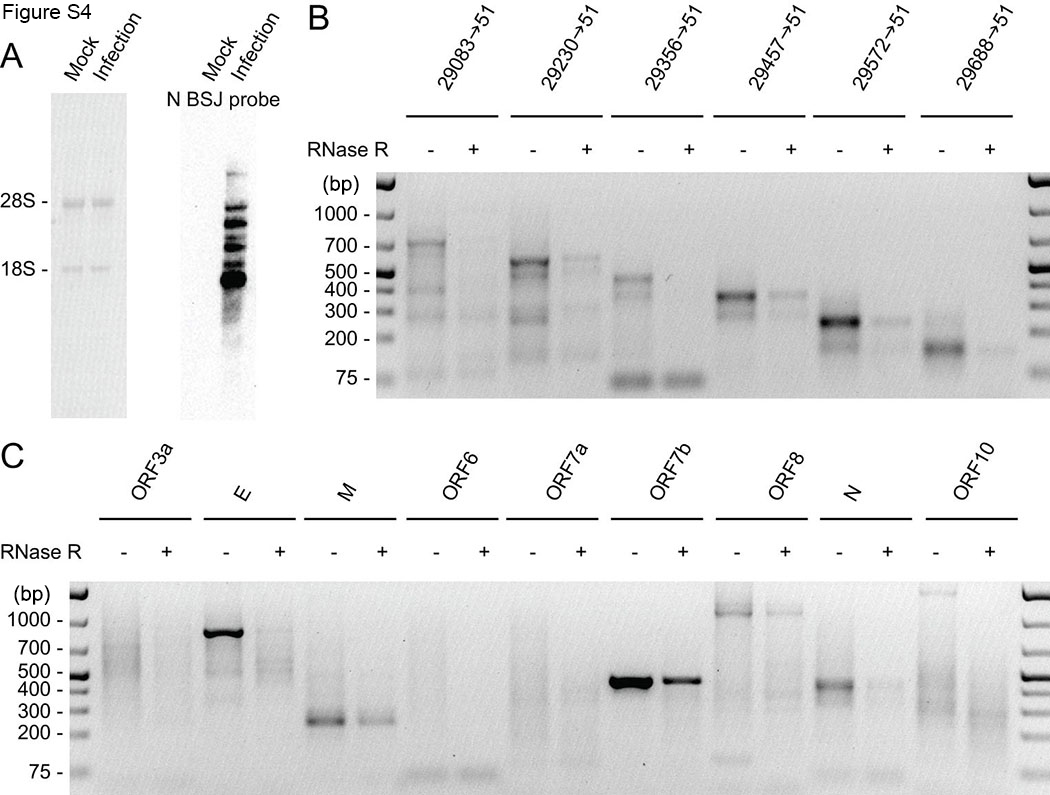
RNase R is a 3′–5′ exoribonuclease that digests all linear RNAs except lariat or circRNA structures. As our experimental data set suggests BSJ‐spanning transcripts may not be circularized, we performed RNase R sensitivity assays to determine whether BSJ‐containing sgRNAs were truly circular. We first examined the genome‐wide resistance of SARS‐CoV‐2 RNA to RNase R treatment. Agarose gel electrophoresis of total RNA extracted from SARS‐CoV‐2‐infected Vero E6 cells without and with RNase R treatment showed that ribosomal RNAs were completely degraded after 45 min of RNase R treatment (Figure 4A). Northern blotting with a DIG‐labeled host β‐actin probe confirmed the degradation of actin messenger RNA (mRNA) by RNase R (Figure 4B). We further showed with a BSJ‐spanning probe targeting SARS‐CoV‐2 N gene that gRNA and canonical sgRNAs containing the N gene sequence were efficiently removed by RNase R (Figure 4C). 41 As no signal was detected in the mock sample with the N BSJ probe (Figure S4A), the smear signal around and below canonical sgRNAs bands were likely noncanonical sgRNAs detected by the N BSJ probe (Figure S4A). RT‐PCR with convergent primers showed that SARS‐CoV‐2 RNAs were not completely degraded by RNase R, suggesting that some RNA components are resistant to RNase R (Figure 4E). Furthermore, different regions of SARS‐CoV‐2 genome exhibited varied degrees of RNase R resistance. It is expected that ORF10, which locates most close to the 3′‐end of gRNA and sgRNAs, is more likely to be degraded by RNase R. However, RNA located in N and ORF7a are more resistant to RNase R than RNA in ORF3a and M. gRNA and sgRNA cannot be detected after RNase R treatment by northern blotting, we concluded that the RT‐PCR revealed abundant circRNAs carrying sequences from N and ORF7. This result is consistent with our bioinformatic prediction (Figure 1C) and experimentally identified BSJs (Table 1).
Figure 4.
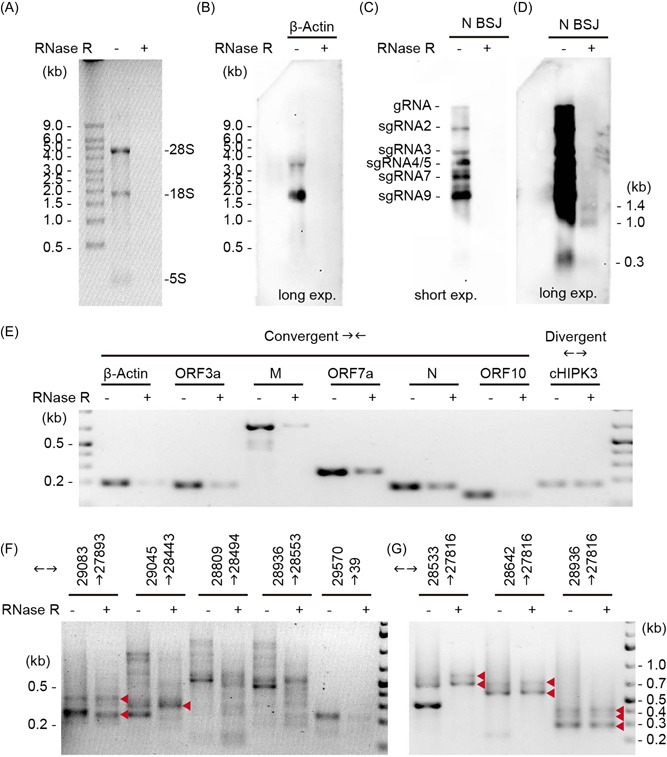
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) back‐splice junctions (BSJs) are resistant to RNase R treatment. (A) Gel electrophoresis of total RNA with and without RNase R treatment. (B) Northern blotting of host actin mRNA. (C,D) Northern blotting with a BSJ‐spanning N probe (28809–29122 | 28295–28494) under short (C) and long (D) exposure conditions. The size of three circular RNA (circRNA) bands were marked. (E) Reverse‐transcription polymerase chain reaction (RT‐PCR) with convergent primers targeting action and different regions of SARS‐CoV‐2 genome. Divergent primers targeting circHIPK3 was used as positive control for RNase R treatment. (F,G) Inverse RT‐PCR on RNA treated with and without RNase R. Red arrowheads indicate RNase‐R resistant bands.
Next, we performed inverse RT‐PCR on total RNA with or without RNase R treatment. Our result showed that some bands are resistant to RNase R whereas others are susceptible (Figure 4F,G and S4B,C). Specifically, bands corresponding to BSJ 29195 | 27789 (Figure 4E,F) were resistant to RNase R whereas bands corresponding to BSJ 29122 | 28295 and BSJ 29761 | 13 were more susceptible. These results suggest that not all BSJ‐containing RNAs were circularized.
Lastly, using the BSJ‐spanning probe targeting 29122 | 28295, we identified three distinct bands at 0.3, 1.0, and 1.5 kb after RNase R treatment (Figure 4D). This is consistent with our bioinformatic prediction that the length of SARS‐CoV‐2 circRNAs fall into three groups, one below 0.5 kb, one near 1 kb, and one around 1.5 kb (Figure 2A). As our inverse RT‐PCR suggest that BSJ 29122 | 28295 is sensitive to RNase R treatment, the bands we detected using the BSJ‐spanning probe were likely due to partial hybridization to the donor sequence (28809–29122) or the acceptor sequence (28295–28494) in N. It is possible that the 1.5 kb band correspond to circRNA 29195 | 27789, which should be 1406 nt in length without FSJ.
In conclusion, using RNase R treatment followed by northern blotting and RT‐PCR, we confirmed that SARS‐CoV‐2 contained abundant circRNAs, and that not all BSJs‐containing sgRNAs were circularized.
3.8. SARS‐CoV‐2 circRNA 29122 | 28295 localized in the cytoplasm
Next, we examined the distribution of circRNA 29122 | 28295 in the host cells. We utilized smFISH 37 to detect linear RNAs corresponding to SARS‐CoV‐2 ORF1 and AmpFISH 36 to detect SARS‐CoV‐2 circRNA 29122 | 28295. For the detection of circRNAs, we designed two pairs of donor and acceptor probes for the sequences that are juxtaposed in the circRNAs. Each pair of probes corresponded to the positive and negative strand polarities of the expected circRNA. As depicted in Figure 5A, only when the pair of target sequences are juxtaposed, an amplified signal will be produced in AmpFISH. 36 We found that that in addition to the linear ORF1 RNA, the positive‐stranded circRNA 29122 | 28295 was abundantly present and was localized in the cytoplasm of SARS‐CoV‐2‐infected cells, whereas the negative‐stranded circRNA 29122 | 28295 was not detected (Figure 5B). To confirm that the signals stem from circRNAs and not linear RNAs, we treated the fixed and permeabilized cells (before hybridization) with RNase R, which degrades linear RNAs but spares circRNAs. This treatment lead to a reduction in linear RNAs corresponding to ORF1 but not in circRNA 29122 | 28295, confirming its presence in the infected cells. A quantification of decrease on average fluorescence intensity in infected cells of signal upon RNase R treatment was conducted as shown in Figure 5C. The treatment of RNase R significantly decreased the levels of linear RNA (upper panel), while it has no effects on circRNA (lower panel). Therefore, we designed specific juxtaposed primers to amplify only circRNA that is resistant to RNase R.
Figure 5.
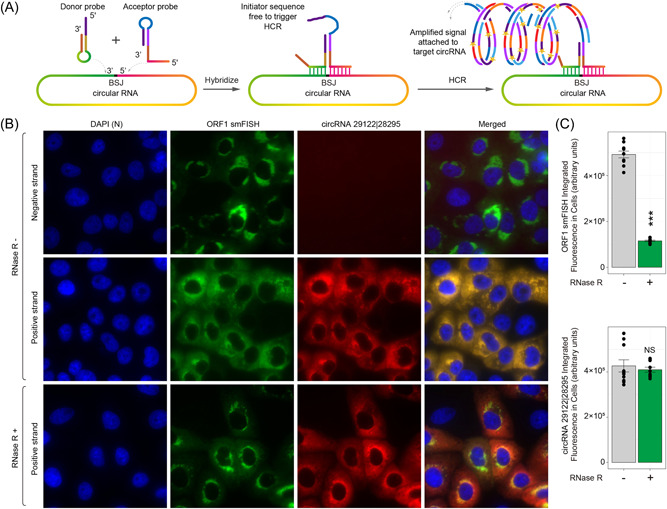
Localization of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) circular RNAs (circRNAs) in infected Vero E6 cells. (A) Design of amplified fluorescence in situ hybridaization (AmpFISH) probes for the detection of a juxtaposed pair of sequences in the circRNA 29122 | 28295. Only when the two sequences are next to each other, which occurs in the circular RNA but not in linear RNA, the donor and acceptor probes can interact with each other and give rise to an amplified hybridization chain reaction (HCR) signal. (B) Multiplex imaging of the linear RNA corresponding to ORF1 obtained by single‐moleclule FISH (smFISH; green) and circRNA 29122 | 28295 (green) obtained from AmpFISH in the same set of cells. Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI). RNAse R treatment is expected to degrade linear but not the circular forms of RNAs. (C) Mean fluorescence intensity in single infected cells that were either treated with RNAse R (green) or left untreated (gray). Linear RNA was detected using ORF1‐specific smFISH probe sets (top) and the circRNA was detected by back‐splice junction (BSJ)‐specific AmpFISH probes pair (bottom).
3.9. SARS‐CoV‐2 BSJs were flanked by homologous and reverse complementary sequences
Repetitive intronic elements, including the primate‐specific Alu elements, enables intramolecular RNA looping, thereby promoting cellular circRNA biogenesis in cis. 35 , 42 , 43 As for RNA recombination in CoVs, the prevailing model predicts that discontinuous RNA synthesis is mediated by homologous motifs called transcription‐regulatory sequences (TRSs). There is a leader TRS (TRS‐L) located in the 5′‐UTR that is identical or highly homologous to the different body TRSs (TRS‐Bs) located in front of each ORF. During negative‐strand RNA synthesis, the RNA‐dependent RNA polymerase (RdRP) pauses when it reaches a TRS‐B, then can either read through or switch to the TRS‐L based on the binding affinity between the leader and body TRSs, resulting in canonical sgRNAs. We proposed that homologous sequences like TRSs exist across CoV genomes to enable long‐range and short‐range RNA–RNA interaction, thereby promoting bidirectional template‐switching to generate “fused” transcripts with either FSJs or BSJs. Based on our finding that canonical FSJs exhibit less junction diversity than BSJs, we further predicted that the degree of homology determines the frequency and the accuracy of “fusion.” To test our hypothesis, we compared sequences flanking the 5′‐ and 3′‐breakpoints of experimentally identified BSJs and FSJs. Among 185 unique BSJs, we found that 88 BSJs had homology (6–12 nt) and 75 BSJs had reverse complementarity (6–10 nt) between sequences around the 5′‐ and 3′‐breakpoints (Data S5). Homologous and reverse complementary sequences (6–12 nt) were found in 6 of 11 unique FSJs. Notably, sequences flanking the 5′‐ and 3′‐breakpoints can have both homology and reverse complementarity. RNA structure prediction showed that a stable stem is formed between genomic loci 28285–28297 and 29127–29139 of the N gene (Figure 6A). This configuration brings the 5′‐ and 3′‐breakpoints of circRNA 29122 | 28295 to proximity (Figure 6B). Due to the strong CG pairing in the stem, the transcription may pause to allow alternative base‐pairing of the nascent strand to the 3′‐breakpoint, thus resulting in template switching (Figure 6C,D). In support of our prediction, a recent RNA–RNA interactome study of SARS‐CoV‐2 provided evidence for the physical interaction between genomic loci 28260–28300 and 29125–29180. 44
Figure 6.
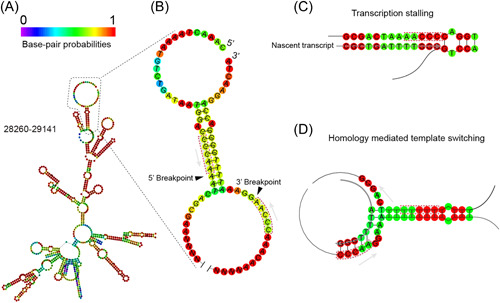
Predicted secondary structure in N gene and the possible formation of back‐splice junction (BSJ) 29122 | 28295. (A) RNA structure prediction for genomic region 28260–29141 of the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) N gene. (B) Stem structure is predicted near the BSJ of circular RNA (circRNA) 29122 | 28295. Short homology (CCCAA) immediately upstream of the 5′‐ and 3′‐breakpoints (black arrowheads) are highlighted. (C,D) Proposed model of SARS‐CoV‐2 back‐splicing using BSJ 29122 | 28295 as an example. (C) Strong RNA–RNA basepairing causes RNA‐dependent RNA polymerase (RdRp) to stall. (D) Inverse homology near the base of the stem allows nascent transcript to bypass the stem and switch template.
3.10. SARS‐COV‐2 circRNAs as cellular miRNA sponges regulated host genes
CircRNAs function as miRNA sponges, involved in the ceRNA regulatory network. 45 To investigate potential gene expression regulated by SARS‐COV‐2 circRNAs via the miRNA sponging pathway, we performed a circRNA–miRNA–mRNA network analysis (Figure 7A). Briefly, first, 671 green monkey miRNAs from total 1360 green monkey miRNAs were predicted to interact with 126 highly expressed full‐length SARS‐COV‐2 circRNAs, named as SARS‐COV‐2–circRNAs–miRNAs. Second, 486 SARS‐COV‐2–circRNAs–miRNAs were predicted to interact with 1064 upregulated genes and 137 downregulated genes, named as SARS‐COV‐2–circRNAs–miRNAs‐regulated genes. Finally, SARS‐COV‐2–circRNAs–miRNAs‐regulated genes were analyzed by GO and KEGG pathways functional enrichment analyses.
Figure 7.
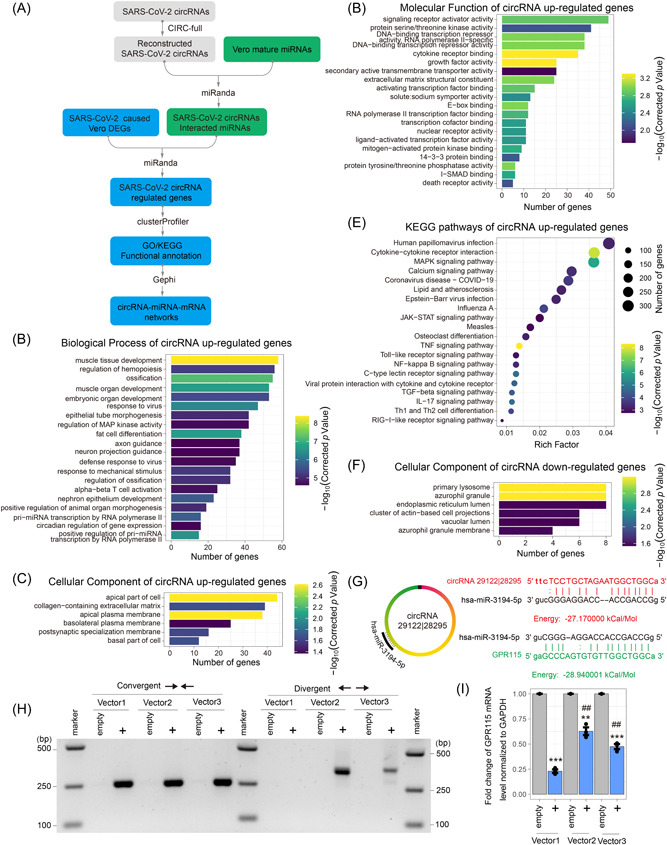
Potential severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) circular RNAs (circRNAs) function in the competitive endogenous RNA (ceRNA) coregulatory network. (A) Schematic diagram of potential SARS‐CoV‐2 circRNAs ceRNA coregulatory network analysis method. (B–D) The Gene Ontology (GO) enrichment of SARS‐CoV‐2 circRNAs competitive differentially expressed genes (DEGs). It shows the top 20 significantly enriched GO terms including Biological Process of SARS‐CoV‐2 circRNAs associated upregulated genes (B), cellular component of SARS‐CoV‐2 circRNAs associated upregulated genes (C), and molecular function of SARS‐CoV‐2 circRNAs associated upregulated genes (D), and cellular component of SARS‐CoV‐2 circRNAs associated downregulated genes (E). (F) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of SARS‐CoV‐2 circRNAs associated upregulated genes. (G) Schematic of hsa‐miR‐3194‐5p‐binding sites and the sequences of SARS‐CoV‐2 circRNA 29122 | 28295 and GPR115. (H) Inverse reverse‐transcription polymerase chain reaction (RT‐PCR) with divergent primers targeting SARS‐CoV‐2 circRNA 29122 | 28295 and convergent primers and targeting both linear RNA 28295–29122and circRNA 29122 | 28295. (I) The messenger RNA (mRNA) expression levels of GPR115 using quantitative real‐time PCR. Vector1, pcDNA3.1 (linear RNA‐overexpressed Vector); Vector2, pcD‐ciR (circRNA‐overexpressed Vector); Vector3, pcDNA3.1 CircRNA mini (circRNA‐overexpressed Vector); empty, empty Vector. Two‐tailed unpaired t test. **p < 0.01, ***p < 0.001 circRNA 29122 | 28295 versus empty Vector; ##p < 0.01 pcD‐ciR or pcDNA3.1 CircRNA mini versus pcDNA3.1.
The GO Biological Processes analysis showed that SARS‐COV‐2–circRNAs–miRNAs‐upregulated host genes were mainly associated with “muscle tissue development,” “ossification,” and “response to virus” (Figure 7B). GO Cellular Component analysis revealed the enrichment of “apical part of cell” and “apical plasma membrane” genes (Figure 7C). Molecular function of the candidate genes fell into the classifications of “cytokine receptor binding” and “growth factor activity” (Figure 7D). A few cellular genes were downregulated by the viral circRNAs (Figure 5E) probably indirectly. In addition, KEGG pathways analysis showed that SARS‐COV‐2–circRNAs–miRNAs‐upregulated genes were involved in “Tumor necrosis factor signaling pathway,” “Cytokine−cytokine receptor interaction,” and “Mitogen‐activated protein kinase signaling pathway” (Figure 7F). On the other hand, SARS‐COV‐2–circRNAs–miRNAs‐downregulated genes were mainly associated with “primary lysosome” and “azurophil granule.”
SARS‐CoV‐2 circRNAs–miRNAs–mRNAs network analyses showed that SARS‐CoV‐2 circRNA 29122 | 28295 contained the binding site of hsa‐miR‐3194‐5p, which targeted a downregulated gene, GPR115 (Figure 7G). To test the effect of circRNA 29122 | 28295 on GPR115 by sponging miRNA, we overexpressed circRNA 29122 | 28295 using circRNA overexpression vectors pcD‐ciR 34 and pcDNA3.1 CircRNA mini 35 in 293 T cells. Inverse RT‐PCR with divergent primers shown that circRNA 29122 | 28295 was more efficiently circulized by pcD‐ciR in comparison with pcDNA3.1 CircRNA mini (Figure 7H). GPR115 was significantly downregulated with overexpressing linear RNA 28295–29122 (Figure 7I). However, the downregulation of GPR115 was rescued by the circularization of 29122 | 28295. These results suggested that SARS‐COV‐2 circRNAs as cellular miRNA sponges regulated host genes involved in widely cellular function and signaling pathway regulation.
4. DISCUSSION
CircRNAs have been widely observed in animals and plants. They are recently recognized as an important group of ncRNA transcripts with versatile functions. However, only a handful of viral circRNAs have been identified from DNA viruses. 13 , 14 , 15 , 16 The biogenesis of circRNA is thought to be conserved depending on RNA polymerase II‐mediated transcription and back‐splicing of pre‐mRNA. No circRNA had been reported from pathogens with an RNA genome, except for the circRNA genome of hepatitis delta virus. 46 This study provided the first line of evidence that circRNAs are an important component of the transcriptome of betacoronaviruses, and that circRNAs can be produced independent of RNA polymerase II‐mediated transcription and pre‐mRNA splicing. SgRNA transcripts are a common feature of viruses in the order of Nidovirales. 47 A more thorough analysis of viral transcriptomes would be needed to determine if circRNAs are universally encoded by large RNA viral genomes.
Previous work, either on the function or on the biogenesis of host and viral circRNAs, were mostly case studies. We were the first to take a systematic approach to evaluate circRNA expression landscape and identify circRNAs encoded by SARS‐CoV‐2. Our experimental data set in combination with computational analyses provided helpful evaluations on de novo circRNA prediction pipelines that could be used for future RNA viral circRNA detection. Although several de novo circRNA discovery algorithms have been developed, we found that AG | GT signal‐biased algorithms, such as find_circ, 25 resulted in a high false positive rate (Figure S1F,G). This might explain the opposite conclusions on strand preference from Cai et al. 17 Thus, an unbiased algorithm like CIRI2 is preferred for de novo circRNA discovery. As BSJs could be coupled with FSJs in SARS‐CoV‐2 circRNAs (Data S4), circRNA reconstruction is critical for downstream analyses of viral circRNA features. We also demonstrated that gapped read count is a more reasonable method for circRNA abundance prediction than the transcripts per million method used by Cai et al. 17 Our data further pinpoint inadequacy of current circRNA prediction algorithms. Repetitive sequences and reverse complementary sequences in CoV genomes are likely to cause a high false positive rate when calling gapped reads. In support of this concern, we found that quite a few reconstructed circRNAs contained circular exons of <10 nt and can be mapped to multiple loci in both positive and negative gRNAs (Data S4). Further, FSJs and BSJs of our experimentally identified circRNAs frequently contained nucleotides that can be either allocated to the donor sequence or the acceptor sequence. This characteristic makes prediction of circRNA 5′‐ and 3′‐breakpoints inaccurate. Lastly, experimental determination of circRNA strandness demonstrated that circRNA strand prediction could be unreliable when dealing with RNA‐Seq data from RNA viruses (Figure 3J). In conclusion, current de novo computational circRNA discovery tools are helpful in the initial evaluation of circRNA landscape and prediction of back‐splicing hotspots in RNA genomes, but downstream analyses would depend heavily on experimental data.
Spliceosome‐mediated pre‐mRNA splicing is not the only mechanism known to produce discontinuous RNA transcripts. Group I introns are one ancient type of self‐splicing introns found in the genomes of some bacteria, bacteriophages, mitochondria, and chloroplasts, and in the ribosome RNA genes of eukaryotic microorganisms. 48 RNA recombination occurs at a high rate in positive‐sense single‐stranded RNA viruses and some retroviruses. 49 The prevailing model of viral RNA recombination predicts that the RdRP switches from a donor template to an acceptor template during RNA synthesis, while still bound to the nascent transcript, thereby generating an RNA molecule with mixed ancestry. Several factors have been shown to influence template switching in CoVs, including the extent of local sequence identity between the RNA templates, the kinetics of transcription, and secondary structure in the RNA. 49 Our finding that SARS‐CoV‐2 sgRNAs contain both FSJs and BSJs suggests that template‐switching is bidirectional. Our junction sequence analysis and RNA structure prediction further offer a more plausible model for TRS‐independent discontinuous SARS‐CoV‐2 RNA transcription than the current model, which predicts that stem‐loop structures in TRSs induce RdRp stalling yet lacks details in the mechanism of template‐switching. We propose that reverse complementarity bring distant genomic loci to physical proximity, and that local homologous sequences enable template‐switching. When the 5′‐ and the 3′‐end of nascent transcript are close enough, RNA circularization occurs. We found that the recently published RNA–RNA interactome of SARS‐CoV‐2 gRNA and sgRNA highly correlated with our identified short‐ and long‐range BSJ hotspots. 44 The identified local (<1000 nt) RNA–RNA interaction also explained the enrichment of BSJ‐ and FSJ‐spanning reads along the diagonal of junction plots (Figure 1C–E). 3 , 38
Our work provides insights into the understanding of CoV gene function during viral propagation, immune evasion, and pathogenesis. The functions of ncRNAs encoded by CoV genomes remain largely unclear. Circularization could be a mechanism utilized by CoVs to extend mRNA stability, thereby providing a more stable way to synthesize important and abundant viral proteins. Our RNase R assay demonstrated that circRNAs containing N and ORF7 were abundant compared to host circHIPK3 and host actin mRNA level. Yet, circRNAs only represent a small fraction of the viral transcriptome. We identified two potentially translatable SARS‐CoV‐2 circRNA containing ORF3a and M. Sequence analysis showed that they contained strong IRES signals than the protein‐expressing circ‐FBXW7 (Figure S5A–C). The SARS‐CoV‐2 genome exhibits strong affinity to host miRNAs 17 and RNA‐binding proteins. 50 CoV circRNAs may act as decoys to indirectly regulate host gene expression. Recent studies have showed that foreign circRNAs activate innate immunity through the nucleic acid senor RIG‐I, 51 and that RNA circularization diminishes immunogenicity compared to the linear form. 52 Our future plan is to investigate the biological functions of SARS‐CoV‐2 circRNAs. Moreover, we will perform a deep circRNA sequencing for SARS‐CoV‐2 variants, including Alpha, Beta, Delta, Gamma, and Omicron to see whether the circRNAs in SARS‐CoV‐2 are conserved.
AUTHOR CONTRIBUTIONS
S.Y and H.Z. designed the experiments, S.Y, H.Z., R.C., Mingde Liu, Jiayu Xu, Xiaoyu Niu, Qiyi Tang, performed the experiments, Shaomin Yang, H.Z., H.Z., Q.T, analyzed the data, H.Z., H.Z., Qiyi Tang, Qiuhong Wang wrote the paper, Yaolan Li, L.X, Q.W, H.Z., Q.T, supervised the study
CONFLICTS OF INTEREST
The authors declare noconflicts of interest.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
{kind=link}
Supporting information.
{kind=link}
Supporting information.
{kind=link}
Supporting information.
{kind=link}
Supporting information.
{kind=link}
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We acknowledge the original SRAS‐CoV‐1/WA1 strain from BEI [NR‐52281 Source: Centers for Disease Control and Prevention]. We thank Dr. Juliette Hanson and Kaitlynn Starr for BSL3 training and assistance in BSL3‐related work at The Ohio State University. This study was supported by National Institute on Minority Health and Health Disparities of the National Institutes of Health under Award Number G12MD007597 (Qiyi Tang), an NIH/NIAID grant SC1AI112785 (Qiyi Tang), and an NIH/NCI grant CA227291 (Sanjay Tyagi). Qiyi Tang and her group were supported by Center for Food Animal Health, and state and federal funds appropriated to College of Food, Agricultural, & Environmental Sciences, The Ohio State University.
Yang S, Zhou H, Liu M, et al. SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV encode circular RNAs of spliceosome‐independent origin. J Med Virol. 2022;94:3203‐3222. 10.1002/jmv.27734
Shaomin Yang and Hong Zhou are the co‐first authors and contributed equally to this work.
Contributor Information
Qiuhong Wang, Email: wang.655@osu.edu.
Hua Zhu, Email: zhuhu@njms.rutgers.edu.
Qiyi Tang, Email: qiyi.tang@howard.edu.
DATA AVAILABILITY STATEMENT
All data are available in the manuscript or the Supporting information. The following reagent was deposited by the Centers for Disease Control and Prevention, and was obtained through BEI Resources, NIAID, NIH: SARS‐CoV‐2, Isolate USA‐WA1/2020, NR‐52281.
REFERENCES
- 1. Petrosillo N, Viceconte G, Ergonul O, Ippolito G, Petersen E. COVID‐19, SARS and MERS: are they closely related? Clin Microbiol Infect. 2020;26(6):729‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92(4):418‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H. The architecture of SARS‐CoV‐2 transcriptome. Cell. 2020;181(4):914‐921.e910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morales L, Oliveros JC, Fernandez‐Delgado R, tenOever BR, Enjuanes L, Sola I. SARS‐CoV‐encoded small RNAs contribute to infection‐associated lung pathology. Cell Host Microbe. 2017;21(3):344‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Salzman J. Circular RNA expression: its potential regulation and function. Trends Genet. 2016;32(5):309‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Han B, Chao J, Yao H. Circular RNA and its mechanisms in disease: from the bench to the clinic. Pharmacol Ther. 2018;187:31‐44. [DOI] [PubMed] [Google Scholar]
- 8. Li X, Yang L, Chen LL. The biogenesis, functions, and challenges of circular RNAs. Mol Cell. 2018;71(3):428‐442. [DOI] [PubMed] [Google Scholar]
- 9. Li Y, Zheng F, Xiao X, et al. CircHIPK3 sponges miR‐558 to suppress heparanase expression in bladder cancer cells. EMBO Rep. 2017;18(9):1646‐1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vo JN, Cieslik M, Zhang Y, et al. The landscape of circular RNA in cancer. Cell. 2019;176(4):869‐881.e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang L, Luo T, Bao Z, Li Y, Bu W. Intrathecal circHIPK3 shRNA alleviates neuropathic pain in diabetic rats. Biochem Biophys Res Commun. 2018;505(3):644‐650. [DOI] [PubMed] [Google Scholar]
- 12. Hao Y, Luo X, Ba X, et al. Huachansu suppresses TRPV1 up‐regulation and spinal astrocyte activation to prevent oxaliplatin‐induced peripheral neuropathic pain in rats. Gene. 2019;680:43‐50. [DOI] [PubMed] [Google Scholar]
- 13. Ungerleider N, Concha M, Lin Z, et al. The Epstein Barr virus circRNAome. PLoS Pathog. 2018;14(8):e1007206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abere B, Li J, Zhou H, Toptan T, Moore PS, Chang Y. Kaposi's sarcoma‐associated herpesvirus‐encoded circRNAs are expressed in infected tumor tissues and are incorporated into virions. mBio. 2020;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng SR, Zhang HR, Zhang ZF, et al. Human papillomavirus 16 E7 oncoprotein alters the expression profiles of circular RNAs in Caski cells. J Cancer. 2018;9(20):3755‐3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nahand JS, Jamshidi S, Hamblin MR, et al. Circular RNAs: new epigenetic signatures in viral infections. Front Microbiol. 2020;11:1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cai Z, Lu C, He J, et al. Identification and characterization of circRNAs encoded by MERS‐CoV, SARS‐CoV‐1 and SARS‐CoV‐2. Brief Bioinform. 2021;22(2):1297‐1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Riva L, Yuan S, Yin X, et al. Discovery of SARS‐CoV‐2 antiviral drugs through large‐scale compound repurposing. Nature. 2020;586(7827):113‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang X, Chu H, Wen L, et al. Competing endogenous RNA network profiling reveals novel host dependency factors required for MERS‐CoV propagation. Emerg Microbes Infect. 2020;9(1):733‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang S, Wu S, Yu Z, et al. Transcriptomic analysis reveals novel mechanisms of SARS‐CoV‐2 infection in human lung cells. Immun Inflamm Dis. 2020;8(4):753‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25(14):1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Langmead B, Salzberg SL. Fast gapped‐read alignment with Bowtie 2. Nat Methods. 2012;9(4):357‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Okonechnikov K, Conesa A, Garcia‐Alcalde F. Qualimap 2: advanced multi‐sample quality control for high‐throughput sequencing data. Bioinformatics. 2016;32(2):292‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform. 2018;19(5):803‐810. [DOI] [PubMed] [Google Scholar]
- 25. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333‐338. [DOI] [PubMed] [Google Scholar]
- 26. Zheng Y, Ji P, Chen S, Hou L, Zhao F. Reconstruction of full‐length circular RNAs enables isoform‐level quantification. Genome Med. 2019;11(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 29(1), 2013:15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923‐930. [DOI] [PubMed] [Google Scholar]
- 29. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq. 2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila . Genome Biol. 2003;5(1):R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36(Web Server issue):W70‐W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao J, Wu J, Xu T, Yang Q, He J, Song X. IRESfinder: identifying RNA internal ribosome entry site in eukaryotic cell using framed k‐mer features. J Genet Genomics. 2018;45(7):403‐406. [DOI] [PubMed] [Google Scholar]
- 34. Zhao Q, Liu J, Deng H, et al. Targeting mitochondria‐located circRNA SCAR alleviates NASH via reducing mROS output. Cell. 2020;183(1):76‐93.e22. [DOI] [PubMed] [Google Scholar]
- 35. Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014;28(20):2233‐2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marras SAE, Bushkin Y, Tyagi S. High‐fidelity amplified FISH for the detection and allelic discrimination of single mRNA molecules. Proc Natl Acad Sci USA. 2019;116(28):13921‐13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5(10):877‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang D, Jiang A, Feng J, et al. The SARS‐CoV‐2 subgenome landscape and its novel regulatory features. Mol Cell. 2021;81(10):2135‐2147.e2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang J, Chen S, Yang J, Zhao F. Accurate quantification of circular RNAs identifies extensive circular isoform switching events. Nat Commun. 2020;11(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zheng Q, Bao C, Guo W, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hou YJ, Okuda K, Edwards CE, et al. SARS‐CoV‐2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. 2020;182(2):429‐446.e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence‐mediated exon circularization. Cell. 2014;159(1):134‐147. [DOI] [PubMed] [Google Scholar]
- 43. Ivanov A, Memczak S, Wyler E, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10(2):170‐177. [DOI] [PubMed] [Google Scholar]
- 44. Ziv O, Price J, Shalamova L, et al. The short‐ and long‐range RNA‐RNA interactome of SARS‐CoV‐2. Mol Cell. 2020;80(6):1067‐1077.e1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17(5):272‐283. [DOI] [PubMed] [Google Scholar]
- 46. Kos A, Dijkema R, Arnberg AC, van der Meide PH, Schellekens H. The hepatitis delta (delta) virus possesses a circular RNA. Nature. 1986;323(6088):558‐560. [DOI] [PubMed] [Google Scholar]
- 47. Di H, McIntyre AA, Brinton MA. New insights about the regulation of Nidovirus subgenomic mRNA synthesis. Virology. 2018;517:38‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Haugen P, Simon DM, Bhattacharya D. The natural history of group I introns. Trends Genet. 2005;21(2):111‐119. [DOI] [PubMed] [Google Scholar]
- 49. Simon‐Loriere E, Holmes EC. Why do RNA viruses recombine? Nat Rev Microbiol. 2011;9(8):617‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Srivastava R, Daulatabad SV, Srivastava M, Janga SC. SARS‐CoV‐2 contributes to altering the post‐transcriptional regulatory networks across human tissues by sponging RNA binding proteins and micro‐RNAs. bioRxiv. 2020. 10.1101/2020.07.06.190348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen YG, Kim MV, Chen X, et al. Sensing Self and foreign circular RNAs by intron identity. Mol Cell. 2017;67(2):228‐238.e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wesselhoeft RA, Kowalski PS, Parker‐Hale FC, Huang Y, Bisaria N, Anderson DG. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol Cell. 2019;74(3):508‐520.e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
All data are available in the manuscript or the Supporting information. The following reagent was deposited by the Centers for Disease Control and Prevention, and was obtained through BEI Resources, NIAID, NIH: SARS‐CoV‐2, Isolate USA‐WA1/2020, NR‐52281.