

Abstract
The c-Jun N-terminal kinases (JNKs) are evolutionary highly conserved serine/threonine kinases. Numerous findings suggest that JNK3 is involved in the pathogenesis of neurodegenerative diseases, so the inhibition of JNK3 may be a potential therapeutic intervention. The identification of novel compounds with promising pharmacological properties still represents a challenge. Fluorescence thermal shift screening of a chemically diversified lead-like scaffold library of 2024 pure compounds led to the initial identification of seven JNK3 binding hits, which were classified into four scaffold groups according to their chemical structures. Native mass spectrometry validated the interaction of 4 out of the 7 hits with JNK3. Binding geometries and interactions of the top 2 hits were evaluated by docking into a JNK3 crystal structure. Hit 5 had a Kd of 21 μM with JNK3 suggested scaffold 5-(phenylamino)-1H-1,2,3-triazole-4-carboxamide as a novel and selective JNK3 binder.
Introduction
The c-Jun N-terminal kinases (JNKs), also known as stress-activated protein kinases, belong to the mitogen activated protein kinases (MAPKs) family where serine and threonine are highly conserved.1,2 Human JNKs are encoded by three genes jnk1, jnk2, and jnk3. JNK1 and JNK2 are ubiquitously expressed, whereas JNK3 has much more limited expression, primarily confined to the nervous system.3 JNK3 is involved in the pathogenesis in neurodegenerative diseases, such as β-amyloid processing and neuronal apoptosis in Alzheimer’s disease4 as well as the mediation of neurotoxicity in the rodent models of Parkinson’s disease.5 In particular, JNK3 knockout mice exhibit amelioration of neurodegeneration and exhibit neuroprotective effects in animal models,6−8 making JNK3 an interesting therapeutic target for neurodegenerative disorders.9 During the past two decades, medicinal chemistry efforts have led to a number of JNK inhibitors showing reasonable in vitro profiles.10−15 However, due to the highly conserved adenosine 5′-triphosphate (ATP) binding site among JNKs and other MAPKs, especially p38α MAPK and extracellular-regulated kinase 2, highly selective JNK3 inhibitors are still rare.16,17 Examples of known ligands are summarized in Figure 1. Therefore, the identification of novel scaffolds with acceptable pharmacological properties still plays a significant role in JNK related drug discovery.
Figure 1.

Known JNK3 ligands.
The fluorescence thermal shift (FTS) assay is a fast, cost-effective, and easy-to-operate platform for studies of protein–ligand interactions.18 The FTS assay is based on the principle that a protein unfolds at a critical temperature and a protein’s thermal stability may increase when it binds to a ligand, resulting in a rise in the unfolding temperature. The temperature at which 50% of the protein has denatured is designated as the melting temperature (Tm). A fluorescence probe that binds to protein hydrophobic surfaces is used to monitor protein unfolding and give a thermal melting curve. In this manner, the changes in Tm are indicative of the ligand binding affinity. Due to the broad scale of applicability and combination with high throughput screening (HTS) techniques, the HTS–FTS assay has gained widespread popularity19−22 in the study of protein–ligand interaction, protein–DNA interaction, protein stability investigation, and membrane protein crystallography.
Native mass spectrometry (native MS) is a label-free, fast, and accurate method that permits the direct observation of noncovalent and covalent protein–ligand complexes.23−28 The technique relies on nondenaturing electrospray ionization (ESI) to generate multicharged proteins in their near-native states.
It has been widely recognized that in silico approaches are useful tools for drug discovery.29−32 Molecular docking can predict the binding affinity of small molecules into the protein binding site by evaluating ligand–receptor binding free energy.33 AutoDock Vina34 provides an accurate and fast way to visually examine predicted binding geometries that contribute crucially to the further development of a lead compound.
In this study, a chemically diversified assay-ready scaffold library of 2024 lead-like pure compounds was used for an HTS–FTS assay to explore novel JNK3 binding ligands. Furthermore, native mass spectrometry was used to validate the initial hits (1—7) and confirmed binding between compounds 2, 3, 5, and 6 to JNK3. The ligand binding geometries and interactions of the 2 strongest hits with JNK3 were visualized by molecular docking.
Results
The FTS Assay Is Effective for the JNK3-Ligand Screen
As not all proteins are amenable to FTS measurements, we first subjected purified JNK3 to FTS with the positive controls ATP and SP60012535 to evaluate the performance of the FTS assay. Both ATP and SP600125 dramatically increased the Tm of JNK3 protein. The maximum Tm shift induced by 500 μM ATP and SP600125 was about 4.6 and 12.7 °C, respectively, at a protein concentration of 2 μM. More interestingly, the involvement of cofactor Mg2+ significantly increased the temperature shift of ATP, indicating that Mg2+ is favorable for thermally stabilizing the transient complex conformation (Figure 2). We also measured the FTS denaturation profile at gradient concentrations of ATP and SP600125, respectively, and ensured they elicited a dose–response (Figure 2). Taken together, these findings established the assay’s tractability to perform thermal unfolding experiments to identify potential ligands for JNK3.
Figure 2.

ΔTm50 values of JNK3 against a gradient concentrations of SP600125 and ATP with/without MgCl2. The JNK3 protein denaturation profile showed a dose-dependent effect at concentrations of 50, 100, 200, and 500 μM. The ΔTm50 value induced by ATP significantly increased under MgCl2. Values shown are the mean ± SD (n = 3).
HTS–FTS Assay Yields Seven Compounds That Thermally Stabilize JNK3 with Higher Potency than ATP
Overall, 31 compounds (1.5% of the library) showed a binding profile in which the ΔTm50 value exceeded 1.0 °C through the two round HTS–FTS assay. As we aim to explore ligands possessing considerable binding potency, the ΔTm50 value induced by native ligand ATP was set as the cutoff value (2.73 °C). Eventually, seven compounds containing four new scaffolds (Figure 3) met the above selection criterion with the ΔTm50 values ranging from 2.99 to 4.77 °C (Figure 4A). The JNK selective inhibitor SP600125 displayed the highest temperature shift of 7.86 °C. It is also noted that when comparing the melting curve of SP600125 with all seven hit compounds, a steeper transition was shown for compound 1 by a greater ΔTm10 value than that of ΔTm50 (Figure 4B). As ΔTm10 gives more information about the change in cooperativity of the folding process, a steeper melting curve indicated the JNK3 protein underwent a different change in transition characteristics when binding with this ligand.
Figure 3.

Chemical structures of the 7 initial hits. Four scaffold skeletons are labeled (red).
Figure 4.

HTS–FTS assay result of JNK3. (A) Graph of ΔTm values of scaffold library hits determined at a compound concentration of 200 μM. Hit ligands are divided into four groups according to the scaffold structure. ΔTm50 values of ATP and SP600125 are also shown. Values shown are the mean ± SD (n = 3). (B) Normalized melting curves for JNK3 protein with hit compound 1 and controls. A steeper transition with 1 is shown by a greater Tm10 value than that of Tm.
Interestingly, N-(1H-pyrazol-4-yl)thieno[2,3-d]pyrimidin-4-amine (scaffold I) had four members (1–4) among the seven compounds and was found to induce the highest temperature shift compared to other compounds with the ΔTm values of 4.77, 4.25, 3.55, and 3.53 °C, respectively, indicating scaffold I may have very promising JNK3 ligand binding potency. When one compares the chemical structures in this group, it is suggested that the 2-morpholinomethyl and 5-(thiophen-2-yl) substitutions contribute more to binding potency while the substitution on position 6 counts little whether it is hydrogen or an alkyl group. The 5-(phenylamino)-1H-1,2,3-triazole-4-carboxamide (scaffold II, compound 5) also showed excellent binding potency with a ΔTm50 value of 3.38 °C. Indeed, there were two more compounds with this scaffold ranked in the top 31 (ΔTm50 ≥ 1 °C); however, due to their different substituent groups on the carboxamide nitrogen, the potencies were weaker than ATP and subsequently not selected here. The last two scaffold groups showed nearly equivalent potency with ΔTm50 values of 3.00 and 2.99 °C, respectively. Scaffold III (compound 6) has a triazolmethypiperidine moiety, and scaffold IV (compound 7) is a piperidinecarboxamide derivative. Although each scaffold has at least five different candidate compounds in the screening library, all of the other compounds induced only a slight Tm shift change. Thus, scaffolds III and IV probably may not be a priority for future lead optimization chemistry programs. In summary, the FTS assay result suggested that scaffold I and scaffold II are promising starting points for further medicinal chemistry campaigns to explore novel JNK inhibitors.
Native Mass Spectrometry
An independent biophysical technique was used to validate the binding of the compounds to JNK3. The seven hits were tested against protein JNK3 (9 μM) by native MS, and protein–ligand complexes were detected from 4 compounds with an affinity order of 5 > 2 > 3 > 6 (Figure S1). The binding affinity of the best compound 5 (scaffold II) was further investigated by a titration experiment. Twelve concentrations of hit 5 at 0.01, 0.03, 0.1, 0.3, 1, 3, 10, 30, 50, 100, 200, and 500 μM were incubated with JNK3 (9 μM), and protein–ligand complexes were detected and quantified by native MS (Figure 5A). The percentages of the ligand-bound complex within the sample were used to plot a dissociation curve from which a Kd of 21.0 ± 3.4 μM was calculated (Figure 5B). Compound 2 (scaffold I) also showed a moderate binding affinity to JNK3 with 68.6% protein occupation with 100 μM ligand addition. Another native MS screening has been conducted for compounds 1–7 against protein STING (stimulator of interferon genes), which is known to control the induction of the JNK pathway.36 No binding was observed, indicating hits 2 and 5 may target the JNK level directly (Figure S2).
Figure 5.

Determination of Kd between 5 and JNK3. (A) Overlay of the 12 mass spectra of JNK3 at a concentration of 9 μM incubated with increasing concentrations of 5 (0.01–500 μM). (B) Plot of [P–L]/[P] + [P–L] versus ligand concentrations for the titration of JNK3 with 5. The Kd was calculated as 21.0 ± 3.4 μM.
We also calculated the molecular properties of hits 2 and 5 as listed in Table 1. The calculated total polar surface area (TPSA) value of compound 2 was 66.5. As the TPSA value ranging from 60 to 70 is a parameter index that tends to be identified as central nervous system (CNS) active compounds,37,38 compound 2 may be a likely a candidate as a neurodegenerative disease therapeutic agent. The calculated partition coefficient (clogP) values of compounds 2 and 5 are 2.72 and 2.97, respectively. Usually, a clogP value of less than 3 suggests a probability of good intestinal permeability; thus, these three compounds may possess better metabolism and pharmacokinetic properties. Hit 5 has a TPSA value of 95.8, which may indicate a minor CNS penetration property. It is worth paying more attention to how to improve and optimize pharmaceutical properties of hit 5 in future studies.
Table 1. Calculated Parameters of Hits.
Predicted clogP and TPSA values were calculated by Molinspiration Cheminformatics free web services: https://www.molinspiration.com/.
Molecular Docking of the Two Confirmed Strong Hits against JNK3
Docking studies were pursued in order to gain insight into ligand interactions with JNK3 kinase using AutoDock Vina as described in the Experimental Section. The crystal structure of JNK339 (PDB code: 1PMV with the ligand SP600125 removed) was used for the docking study. First of all, the docking quality was evaluated by comparison of the docked pose and experimental pose of SP600125 in the JNK3 crystal structure. The analysis of the best predicted conformation of SP600125 (Figure 6) showed the ligand made hydrogen bonds with the carbonyl oxygen of Glu147 (3.4 Å) and the main chain nitrogen of Met149 (3.1 Å), which was in accordance with the experimental data from the PDB structure (2.8 Å each). The calculated binding energy of SP600125 was the lowest (−9.2 kcal/mol) among all hit compounds, which is in good accordance with the experimental data of the FTS assay. Furthermore, the alignment of the best-docked conformation with the crystal conformation yielded a root-mean-square deviation (RMSD) of 0.8 Å, indicating the docking simulation was able to reproduce the X-ray structure.
Figure 6.

Redocked mode of SP600125 superimposed with the cocrystallized ligand (PDB code: 1PMV). Left: Overview. The predicted ligand pose (yellow) agrees very well with the ligand pose in the crystal structure (cyan) at an RMSD of 0.8 Å. The ligand is shown as a stick model, and the residues, which form hydrogen bonds, are shown as stick a model (tints). Right: Hydrogen bond interactions. The predicted hydrogen bonds are shown in dotted lines (wheat) with Met149 and Glu147 of 3.1 and 3.4 Å, respectively. The similar pairs of hydrogen bonds in the crystal structure are shown in dotted lines (cyan) with a distance of 2.8 Å each. Figures were generated using PyMOL.
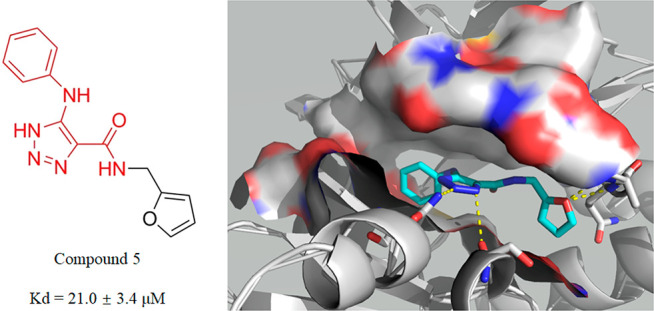
The analysis of the best docking poses of the two confirmed strong hits 5 and 2 (Figure 7) revealed that they both docked into the ATP binding pocket and formed hydrogen bonds with some of the conserved residues in the JNK3 active pocket, suggesting they are ATP-competitive ligands. The predicted binding energy of each compound is relatively low (−8.0 kcal/mol of hit 5 and −6.8 kcal/mol of hit 2), indicating comparatively strong binding. The best docking pose (Figure 7A) of 5 (scaffold II) was predicted to make four hydrogen bonds with JNK3: two with the glycine-rich region (main chain nitrogen of Ala74 and Gln75 with a distance of 3.0 and 3.2 Å, respectively), one with the side chain nitrogen of Asn152 in the hinge region of 3.1 Å, and one with the carbonyl oxygen of Ser193 in the c-lobe of 3.4 Å. Moreover, the phenyl substituent group was predicted to make hydrophobic interactions with residues of Val78, Val196, and Leu206. Scaffold I hit 2 was predicted to form two hydrogen bonds with conserved residues of JNK3 of Asn152 and Glu147 of 3.4 and 3.2 Å, respectively. It is also predicted to interact with conserved residues of Ala91, Val196, Met149, and Leu206.
Figure 7.

Simulated binding mode for hit compounds in the ATP-binding domain of JNK3 (PDB code: 1PMV). (A) Scaffold II: hit 5. (B) Scaffold I: hit 2. Ligand structures are shown as stick models (cyan). The residues predicted to form interactions are shown as stick models (gray). Nitrogen, oxygen, and sulfur atoms are shown in blue, red, and yellow, respectively. Predicted hydrogen bonds are shown in dotted lines; cutoff value = 3.4 Å. Figures were generated using PyMOL.
Those residues that contribute to binding potency are highly conserved in the ATP binding pocket. (Ile70, Ala74, Gln75, and Val78 are seated in glycine-rich loops at the top of the pocket; Ser193 and Val196 are seated in the C-lobe, and Leu206 is located in the hydrophobic region. The most important Glu147, Met149, and Asn152 are seated in the hinge reign, which to a great degree determines the inhibitor’s potency on selectivity and affinity.) Note that residues Val196, Asn152, Glu147, Gln75, and Ile70 are not conserved in other MAPKs12 (JNK3-V196 vs p38a-A157, JNK3-N152 vs p38a-D112, JNK3-E147 vs p38a-H107, JNK3-Q75 vs p38a-Y35, and JNK3-I70 vs p38a-V30); hence, this difference may contribute to the selectivity for JNKs over other kinases. The molecular docking result was essentially in agreement with experimental data of FTS and native MS and further confirmed the binding potency of two hits against the JNK3 protein in vitro.
The crystal structures of JNK1 and JNK2 (PDB codes: 1UKH and 4W4W, respectively) were used for a docking study with hit 5. The calculated free binding energies were −6.3 and −5.9 kcal/mol for JNK1 and JNK2, respectively. Since the free binding energy of hit 5 was significantly stronger at −8.0 kcal/mol for JNK3, the docking result suggests hit 5 has predicted JNK3 selectivity compared to that of JNK1 and JNK2. There were no significant predicted interactions in the best docking pose of hit 5 with either JNK1 or JNK2 (Figure S3).
Conclusion
Many ATP-competitive JNK small molecule inhibitors have been described in the last two decades; however, JNK3 inhibitors with high selectivity and considerable affinity are still rare. Here, we presented an HTS–FTS assay platform for fast identification of novel scaffolds and identified four new scaffold series containing seven compounds that potentially bind against JNK3 protein. Aligned with our primary goal to identify chemical starting points, we identified compounds that elicited a ΔTm greater than that of ATP. Secondary screening by an independent binding assay using native mass spectrometry identified that four of the seven compounds (scaffold I and scaffold II) formed ligand–protein complexes. Hit 5 (scaffold II) ranked the top at a Kd of 21.0 μM, and the molecular docking results suggested an ATP-competitive ligand with JNK3 selectivity.
Experimental Section
Cloning, Expression, and Purification of Human JNK3
A 1089-base pair DNA fragment encoding human JNK3 (40–402 amino acids) was amplified from JNK3 synthetic construct GenBank ABK42248.1 using forward primer 5′-TACTTCCAATCCAATGCTATGAGCAAAAGCAAGGTGGACAACCA-3′ and reverse primer 5′-TTATCCACTTCCAATGCTATTCTGAGTTCATTACCTCCTTGTAGA-3′. The resulting DNA fragment was cloned into vector pMCSG7, which incorporates an N-terminal his-tag followed by a Tobacco Etch Virus (TEV) protease recognition site using ligase-independent cloning (LIC), and transformed into E. coli BL21 (DE3) cells. The cells were cultured at 37 °C in Luria–Bertani (LB; Oxoid Ltd., Yeast Extract cat. LP0021, Tryptone cat. LP0042) medium containing 100 μg/mL ampicillin until the OD600nm reached 0.8. The target protein was induced with 0.2 mM isopropyl-b-d-thiogalactoside (IPTG; Sigma cat. I5502) at 16 °C overnight. The cells were harvested by centrifugation at 4680g for 30 min. The pellets were resuspended in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 50 mM Na2HPO4, 10 mM KH2PO4, pH 7.4) and lysed by sonication. Cell debris was removed by centrifugation at 38 900g for 20 min. The clarified supernatant was loaded onto a Ni-NTA column (GE LifeSciences cat. 17-5248-01), which was pre-equilibrated with PBS. After washing with 100 mL of binding buffer (PBS) followed by 50 mL of washing buffer (PBS containing 20 mM imidazole), the target protein was eluted with 10 mL of elution buffer (PBS containing 300 mM imidazole, Sigma cat. I5513). The target proteins collected above were then concentrated and loaded on a Superdex G75 size exclusion chromatography column (GE LifeSciences cat. 17-1068-01) equilibrated with 20 mM N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES; Sigma-Aldrich cat. 83264), pH 7.5, and 150 mM NaCl. Fractions containing the protein were pooled, concentrated, aliquoted, and stored at −80 °C.
Fluorescence Thermal Shift Assay
The HTS–FTS assay of JNK3 was conducted on a 2024-member compound library that was provided by Compounds Australia, Griffith University. Cluster analysis of a 34 000-member library containing 1200 unique nonflat scaffolds was used to provide the representative subset. The compounds were purchased from ChemDiv and Enamine. Each compound was supplied at 5 mM concentration in dimethyl sulfoxide (DMSO; Sigma cat. D9170), and 1 μL was added to separate wells of 96-well PCR plates (Bio-Rad Laboratories cat. HSP9601). After the first round of the HTS–FTS assay, the initial hit compounds were supplied and prepared as above but screened in triplicate in the second round of HTS in order to test the reproducibility. All controls were set up in triplicate for every run. Native MAPK family substrates ATP (Sigma cat. A2383) and JNK selective inhibitor SP600125 (Sigma cat. 420119) were used as positive controls.
Purified concentrated JNK3 protein was appropriately diluted in assay buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, and 1000 diluted SYPRO Orange dye (Sigma cat. S5692) to a resultant protein concentration of 2 μM. The 24 μL protein–dye mixture was dispensed into each well of the PCR 96-well plates (preformatted with 1 μL of compound per well as described above) with a Biomek FXP liquid handling platform (Beckman Coulter Life Sciences). The final reaction volume was 25 μL per well with a final DMSO concentration of 4% v/v, and the compound–protein ratio was approximately 100:1. The plates were sealed and then shaken at room temperature for 30 min away from light and centrifuged before the assay.
The FTS assay was performed on a real-time PCR instrument (Bio-Rad Laboratories Type CFX96). The fluorescence signals as a function of temperature were recorded in the fluorescence resonance energy transfer (FRET) mode in which the fluorescence intensity was measured with excitation/emission of 450–490/560–580 nm. The temperature gradient was set in the range of 20–75 °C with a ramp of 0.5 °C over the course of 15 s. The Boltzmann model was used for plotting melting curves of JNK3 protein to obtain the midpoint of the thermal unfolding value for JNK3 using the curve-fitting software XLfit5 (ID Business Solutions Ltd.). Compounds with a positive ΔTm higher than 1.0 °C in the first round were moved to the second round assay. Eventually, compounds with a positive ΔTm higher than that induced by ATP were considered as final hits for further analysis.
Native Mass Spectrometry Assay
The JNK3 protein (41.9 kDa) was buffer-exchanged into ammonium acetate buffer (200 mM) under nearly physiological conditions (pH 7.0) using size exclusion chromatography prior to the ESI–MS analysis. Protein concentration after buffer exchange was 10 μM.
Experiments were performed on a Bruker SolariX XR 12 T Fourier transform ion cyclotron resonance mass spectrometer (ESI-FT-ICR-MS) (Bruker Dal-tonics Inc., Billerica, MA) equipped with an automated chip-based nanoelectrospray system (TriVersa NanoMate, Advion Biosciences, Ithaca, NY, USA). Mass spectra were recorded in positive ion and profile modes with a mass range from 50 to 6000 m/z. Each spectrum was a sum of 16 transients (scans) composed of 1 M data points. All aspects of pulse sequence control and data acquisition were controlled by Solarix control software in a Windows operating system.
Kd Study
One μL of compound 5 (concentrations of 0.1 μM, 0.3 μM, 1 μM, 3 μM, 10 μM, 30 μM, 100 μM, 300 μM, 500 μM, 1 mM, 2 mM, and 5 mM in methanol) was mixed with 9 μL of JNK3 (10 μM in 200 mM ammonium acetate buffer) and tested by native mass spectrometry after a 30 min incubation. The final concentrations of ligand were 0.01, 0.03, 0.1, 0.3, 1, 3, 10, 30, 50, 100, 200, and 500 μM, and the final concentration of protein was 9 μM. The percentage of ligand-bound protein was determined using the following equation: % ligand-bound protein = [P–L]/([P] + [P–L]), where [P–L] is the total intensity of the protein–ligand complex and [P] is the total intensity of the apoprotein for a single charged state. A binding curve was generated (ligand concentration against the percentage of ligand-bound protein), and nonlinear regression using the following equation was fit in GraphPad Prism: Y = Bmax × X/(Kd + X).
Molecular Docking
Docking was performed using AutoDock Vina.34 The three-dimensional crystal structure of complex JNK3–SP600125 was downloaded from the Protein Data Bank (https://www.rcsb.org; PDB ID: 1PMV). The grid size for docking measured 22 × 22 × 22 Å to cover all binding possibilities inside the ATP-binding pocket. The x, y, and z dimensions of the center grid box are center_x = 19.9 Å, center_y = 28.3 Å, and center_z = 19.6 Å. The docking method was first verified by the location of the redocked X-ray ligand SP600125. For hit ligand docking, the flexibility ligands were applied at their torsional angle using Python Molecular Viewer with the Molecular Graphics Laboratory (MGL) tools. Each docking was run individually with ten poses, and the best docking pose was selected on the basis of both the binding affinity score and the binding orientation. The crystal structures of JNK1 and JNK2 (PDB codes: 1UKH and 4W4W, respectively) were used for docking as above. Figures were generated using PyMOL (www.pymol.org).
Acknowledgments
We are grateful to Compounds Australia, Griffith Institute for Drug Discovery for providing the compound library. We thank Dr. Ngoc Pham who prepared all assay-ready plates. We also thank Dr. Ouyang Songying and Dr. Vanja for constructive discussion.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00340.
Figure S1, native MS spectra of JNK3 and hits (1–7); Figure S2, native MS of STING and hits (1–7); Figure S3, docking of hit 5 into JNK3 and JNK1 and JNK2; experimental details for STING (PDF)
Author Contributions
C.C. conceived the experiment, which was designed with input from Z.L., Z.X., and R.J.Q. X.G. and D.W. performed the gene cloning, and C.C. and J.M. performed protein expression and purification. C.C. set up the FTS assay. M.P. processed the docking experiment. M.L. performed the native MS experiment. C.C., M.L., and R.J.Q. wrote the manuscript with discussion and improvements from all authors.
This study was supported by The National Health and Medical Research Council of the Australian Government Early Career Fellowship Grant ID 1054172, the Australian Research Council Discovery Grant DP130102400 and Linkage Grant LE120100170, and the National Natural Science Foundation of China Grant ID 31330019.
The authors declare no competing financial interest.
Supplementary Material
References
- Chang L.; Karin M. Mammalian MAP kinase signalling cascades. Nature 2001, 410 (6824), 37–40. 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Davis R. J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103 (2), 239–52. 10.1016/S0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Mohit A. A.; Martin J. H.; Miller C. A. p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 1995, 14 (1), 67–78. 10.1016/0896-6273(95)90241-4. [DOI] [PubMed] [Google Scholar]
- Yoon S. O.; Park D. J.; Ryu J. C.; Ozer H. G.; Tep C.; Shin Y. J.; Lim T. H.; Pastorino L.; Kunwar A. J.; Walton J. C.; Nagahara A. H.; Lu K. P.; Nelson R. J.; Tuszynski M. H.; Huang K. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron 2012, 75 (5), 824–37. 10.1016/j.neuron.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J. W.; Pachori A.; Howard S.; Ganno M.; Hansen D. Jr.; Kamenecka T.; Song X.; Duckett D.; Chen W.; Ling Y. Y.; Cherry L.; Cameron M. D.; Lin L.; Ruiz C. H.; Lograsso P. Small Molecule c-jun-N-terminal Kinase (JNK) Inhibitors Protect Dopaminergic Neurons in a Model of Parkinson’s Disease. ACS Chem. Neurosci 2011, 2 (4), 198–206. 10.1021/cn100109k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D. D.; Kuan C. Y.; Whitmarsh A. J.; Rincon M.; Zheng T. S.; Davis R. J.; Rakic P.; Flavell R. A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389 (6653), 865–870. 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Raina A. K.; Rottkamp C. A.; Aliev G.; Perry G.; Boux H.; Smith M. A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem 2001, 76 (2), 435–41. 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- Hunot S.; Vila M.; Teismann P.; Davis R. J.; Hirsch E. C.; Przedborski S.; Rakic P.; Flavell R. A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (2), 665–70. 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick L.; Fennell M. Targeting JNK3 for the treatment of neurodegenerative disorders. Drug Discov Today 2004, 9 (21), 932–9. 10.1016/S1359-6446(04)03251-9. [DOI] [PubMed] [Google Scholar]
- Gehringer M.; Muth F.; Koch P.; Laufer S. A. c-Jun N-terminal kinase inhibitors: a patent review (2010 - 2014). Expert Opin. Ther. Pat 2015, 25 (8), 849–72. 10.1517/13543776.2015.1039984. [DOI] [PubMed] [Google Scholar]
- Zheng K.; Iqbal S.; Hernandez P.; Park H.; LoGrasso P. V.; Feng Y. Design and synthesis of highly potent and isoform selective JNK3 inhibitors: SAR studies on aminopyrazole derivatives. J. Med. Chem. 2014, 57 (23), 10013–30. 10.1021/jm501256y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch P.; Gehringer M.; Laufer S. A. Inhibitors of c-Jun N-terminal kinases: an update. J. Med. Chem. 2015, 58 (1), 72–95. 10.1021/jm501212r. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch M. A.; Arthur P. G. Inhibitors of c-Jun N-terminal kinases: JuNK no more?. Biochim. Biophys. Acta 2008, 1784 (1), 76–93. 10.1016/j.bbapap.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoGrasso P.; Kamenecka T. Inhibitors of c-jun-N-terminal kinase (JNK). Mini Rev. Med. Chem. 2008, 8 (8), 755–66. 10.2174/138955708784912120. [DOI] [PubMed] [Google Scholar]
- Siddiqui M. A.; Reddy P. A. Small molecule JNK (c-Jun N-terminal kinase) inhibitors. J. Med. Chem. 2010, 53 (8), 3005–12. 10.1021/jm9003279. [DOI] [PubMed] [Google Scholar]
- Dou X.; Huang H.; Jiang L.; Zhu G.; Jin H.; Jiao N.; Zhang L.; Liu Z.; Zhang L. Rational modification, synthesis and biological evaluation of 3,4-dihydroquinoxalin-2(1H)-one derivatives as potent and selective c-Jun N-terminal kinase 3 (JNK3) inhibitors. Eur. J. Med. Chem. 2020, 201, 112445. 10.1016/j.ejmech.2020.112445. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Park H.; Bauer L.; Ryu J. C.; Yoon S. O. Thiophene-Pyrazolourea Derivatives as Potent, Orally Bioavailable, and Isoform-Selective JNK3 Inhibitors. ACS Med. Chem. Lett. 2021, 12 (1), 24–29. 10.1021/acsmedchemlett.0c00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan C. H.; Light S. H.; Dunne S. F.; Anderson W. F. Ligand screening using fluorescence thermal shift analysis (FTS). Methods Mol. Biol. 2014, 1140, 263–89. 10.1007/978-1-4939-0354-2_20. [DOI] [PubMed] [Google Scholar]
- Bergsdorf C.; Fiez-Vandal C.; Sykes D. A.; Bernet P.; Aussenac S.; Charlton S. J.; Schopfer U.; Ottl J.; Duckely M. An Alternative Thiol-Reactive Dye to Analyze Ligand Interactions with the Chemokine Receptor CXCR2 Using a New Thermal Shift Assay Format. J. Biomol Screen 2016, 21 (3), 243–51. 10.1177/1087057115619597. [DOI] [PubMed] [Google Scholar]
- Krishna S. N.; Luan C. H.; Mishra R. K.; Xu L.; Scheidt K. A.; Anderson W. F.; Bergan R. C. A fluorescence-based thermal shift assay identifies inhibitors of mitogen activated protein kinase kinase 4. PLoS One 2013, 8 (12), e81504 10.1371/journal.pone.0081504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupesh K. R.; Smith A.; Boehmer P. E. Ligand induced stabilization of the melting temperature of the HSV-1 single-strand DNA binding protein using the thermal shift assay. Biochem. Biophys. Res. Commun. 2014, 454 (4), 604–608. 10.1016/j.bbrc.2014.10.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson U. B.; Hallberg B. M.; Detitta G. T.; Dekker N.; Nordlund P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 2006, 357 (2), 289–98. 10.1016/j.ab.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Vu H.; Pham N. B.; Quinn R. J. Direct screening of natural product extracts using mass spectrometry. Journal of Biomolecular Screening 2008, 13 (4), 265–275. 10.1177/1087057108315739. [DOI] [PubMed] [Google Scholar]
- Yang B.; Feng Y. J.; Vu H.; McCormick B.; Rowley J.; Pedro L.; Crowther G. J.; Van Voorhis W. C.; Forster P. I.; Quinn R. J. Bioaffinity mass spectrometry screening. Journal of Biomolecular Screening 2016, 21 (2), 194–200. 10.1177/1087057115622605. [DOI] [PubMed] [Google Scholar]
- Sternicki L. M.; Nonomiya J.; Liu M.; Mulvihill M. M.; Quinn R. J. Native Mass Spectrometry for the Study of PROTAC GNE-987-Containing Ternary Complexes. ChemMedChem. 2021, 16 (14), 2206–2210. 10.1002/cmdc.202100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Han J.; Feng Y.; Guymer G.; Forster P. I.; Quinn R. J. Antimicrobial Benzyltetrahydroisoquinoline-Derived Alkaloids from the Leaves of Doryphora aromatica. J. Nat. Prod. 2021, 84, 676. 10.1021/acs.jnatprod.0c01093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnaas A. R.; Grice D.; Han J. Y.; Feng Y. J.; Capua A. D.; Mak T.; Laureanti J. A.; Buchko G. W.; Myler P. J.; Cook G.; Quinn R. J.; Liu M. M. Discovery of a Natural Product That Binds to the Mycobacterium tuberculosis Protein Rv1466 Using Native Mass Spectrometry. Molecules 2020, 25 (10), 2384. 10.3390/molecules25102384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler D. R.; Liu M.; McAuley J. L.; Lowery S. A.; Illing P. T.; Gully B. S.; Purcell A. W.; Chandrashekaran I. R.; Perlman S.; Purcell D. F. J.; Quinn R. J.; Rossjohn J. A natural product compound inhibits coronaviral replication in vitro by binding to the conserved Nsp9 SARS-CoV-2 protein. J. Biol. Chem. 2021, 297, 101362. 10.1016/j.jbc.2021.101362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifaioglu A. S.; Atas H.; Martin M. J.; Cetin-Atalay R.; Atalay V.; Dogan T. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief Bioinform 2019, 20 (5), 1878–1912. 10.1093/bib/bby061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza Neto L. R.; Moreira-Filho J. T.; Neves B. J.; Maidana R.; Guimaraes A. C. R.; Furnham N.; Andrade C. H.; Silva F. P. Jr. In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front Chem. 2020, 8, 93. 10.3389/fchem.2020.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Bhardwaj V. K.; Sharma J.; Das P.; Purohit R. Discovery and in silico evaluation of aminoarylbenzosuberene molecules as novel checkpoint kinase 1 inhibitor determinants. Genomics 2021, 113 (1 Pt 2), 707–715. 10.1016/j.ygeno.2020.10.001. [DOI] [PubMed] [Google Scholar]
- Bhardwaj V. K.; Singh R.; Sharma J.; Das P.; Purohit R. Structural based study to identify new potential inhibitors for dual specificity tyrosine-phosphorylation- regulated kinase. Comput. Methods Programs Biomed 2020, 194, 105494. 10.1016/j.cmpb.2020.105494. [DOI] [PubMed] [Google Scholar]
- Ferreira L. G.; Dos Santos R. N.; Oliva G.; Andricopulo A. D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20 (7), 13384–421. 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B. L.; Sasaki D. T.; Murray B. W.; O’Leary E. C.; Sakata S. T.; Xu W.; Leisten J. C.; Motiwala A.; Pierce S.; Satoh Y.; Bhagwat S. S.; Manning A. M.; Anderson D. W. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (24), 13681–6. 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T.; Barber G. N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-kappa B Activation through TBK1. J. Virol 2014, 88 (10), 5328–5341. 10.1128/JVI.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelder J.; Grootenhuis P. D.; Bayada D. M.; Delbressine L. P.; Ploemen J. P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16 (10), 1514–9. 10.1023/A:1015040217741. [DOI] [PubMed] [Google Scholar]
- Ertl P.; Rohde B.; Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43 (20), 3714–7. 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- Scapin G.; Patel S. B.; Lisnock J.; Becker J. W.; LoGrasso P. V. The structure of JNK3 in complex with small molecule inhibitors: structural basis for potency and selectivity. Chem. Biol. 2003, 10 (8), 705–12. 10.1016/S1074-5521(03)00159-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.