

Abstract
The Three Prime Repair EXonuclease I (TREX1) is critical for degrading post‐apoptosis DNA. Mice expressing catalytically inactive TREX1 (TREX1 D18N) develop lupus‐like autoimmunity due to chronic sensing of undegraded TREX1 DNA substrates, production of the inflammatory cytokines, and the inappropriate activation of innate and adaptive immunity. This study aimed to investigate Thelper (Th) dysregulation in the TREX1 D18N model system as a potential mechanism for lupus‐like autoimmunity. Comparison of immune cells in secondary lymphoid organs, spleen and peripheral lymph nodes (LNs) between TREX1 D18N mice and the TREX1 null mice revealed that the TREX1 D18N mice exhibit a Th1 bias. Additionally, the T‐follicular helper cells (Tfh) and the germinal celter (GC) B cells were also elevated in the TREX1 D18N mice. Targeting Bcl6, a lineage‐defining transcription factor for Tfh and GC B cells, with a commercially available Bcl6 inhibitor, FX1, attenuated Tfh, GC, and Th1 responses, and rescued TREX1 D18N mice from autoimmunity. The study presents Tfh and GC B‐cell responses as potential targets in autoimmunity and that Bcl6 inhibitors may offer therapeutic approach in TREX1‐associated or other lupus‐like diseases.
Keywords: autoimmunity, Bcl6, inflammation, Tfh, Th1, TREX1
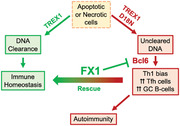
Defects in DNA degrading enzyme such as TREX1 induce immune dysregulation that includes a Th1 bias as well as increase in T‐follicular helper (TfH) and germinal center (GC) B‐cells leading to autoimmune disease. This study offers the potential of using Bcl6 inhibition for restoring immune homeostasis and rescuing from autoimmunity.
Introduction
Billions of cells die each day as part of normal homeostatic processes, and the genetic material of these cells are dismantled in a coordinated fashion. Defects in this process trigger inappropriate immune responses, which may promote autoimmunity, as indicated by mice with defects in apoptotic cell clearance developing systemic lupus erythematosus (SLE or lupus)‐like autoimmune diseases [1, 2, 3]. Among the several DNA exonucleases involved in DNA metabolism, TREX1 is a major 3‐prime to 5‐prime exonuclease that can digest ss‐ and ds DNA [4, 5, 6].
Several mutations contributing to a spectrum of human autoimmune disorders have been identified in this single‐exon containing gene. Large deletions or frame shifts in the TREX1 locus cause Aicardi‐Goutières syndrome (AGS), a severe neuroinflammatory disease, while point mutations impacting catalytic activity cause familial chilblain lupus (FCL), an inflammatory skin disorder [7]. Additionally, certain TREX1 mutations have been identified as common risk alleles for the development of SLE [8]. The TREX1 D18N allele causes FCL in humans [9] and when translated into a murine model, mimics the disease phenotype and immune dysregulation observed in these patients [10].
Cytosolic DNA sensing and innate immune responses are vital for the maintenance of normal DNA metabolism and antiviral responses. The cGAS‐STING pathway contributes to immune‐activation with AIM2 inflammasome reported to be dispensable in TREX1 null mice [11, 12]. While TREX1 null animals represent a good model of the acutely aggressive AGS phenotype, mutations observed in lupus patients are not known to represent a complete deletion of the TREX1 gene. Differences in the autoimmune phenotype and the role of various DNA sensors between global TREX1 null (KO) and TREX1 D18N animals remain to be fully explored.
Recent studies supports that there could either be a T‐follicular helper cells (Tfh cells) transition into Th1 cells or Tfh cells can make more Th1‐like cytokines in autoimmunity and other inflammatory conditions [13, 14, 15, 16]. Tfh cells represent a specialized CD4 Th cell subset, which can be activated by conventional APCs. They are identified by the expression of surface molecules CXCR5 and PD‐1 and the transcription factor Bcl6. It is known that antigen‐activated T cells expressing CXCR5 localize at the T‐cell/B‐cell border, and some of these cells commit to Tfh lineage to provide help to activated B cells in the GC reaction. Overactivation of this process triggers autoimmune responses and production of high‐affinity autoantibodies [17]. Whether such immune dysregulation occurs as a result of uncleared DNA is currently unknown.
The current study aimed to investigate immune dysregulation in the TREX1 D18N mutant mouse model, relative to the more broadly studied TREX1 null model and to explore therapeutic strategies to ameliorate autoimmunity. We present the first evidence of a Th1 bias in the TREX1 D18N mutant mice, which correlates with a heightened Tfh–GC B‐cell response. We successfully implement a therapeutic strategy of Bcl6 inhibition to target the Tfh–GC B‐cell crosstalk and demonstrate the rescue of TREX D18N mutant animals from autoimmunity. These results highlight the Tfh–GC B‐cell response as a potential therapeutic target in autoimmunity and spontaneous lupus‐like inflammatory diseases triggered by defective DNA clearance.
Results
Status of CD4 T‐helper cells and T‐effector memory in TREX1 deficiency
Previous studies with 6‐ to 12‐month‐old TREX1 D18N mice showed high mortality and immune activation [10]. To investigate the early events that may contribute to autoimmunity in the TREX1 mutant mice, we first investigated the status of CD4 Th cells and effector memory cells in the secondary lymphoid organs (spleen and pooled peripheral LNs) of 8‐ to 12‐week‐old TREX1 D18N and TREX1 null (KO) mice (Fig. 1). Both TREX1 D18N and TREX1 KO mice exhibited elevated numbers of activated (CD69+) CD4 Th cells in spleen and LNs. The numbers of CD44+CD62Llo T‐effector memory were also significantly higher in both TREX1 D18N and TREX1 KO mice, indicating an active T‐cell dysregulation at the age of 8–12 weeks. Similarly, the proportions of the activated and effector memory cells within the CD4 T‐cell population were also increased in the TREX1 D18N and TREX1 KO mice (Supporting information Figs. S1A and B, S8A and B).
Figure 1.
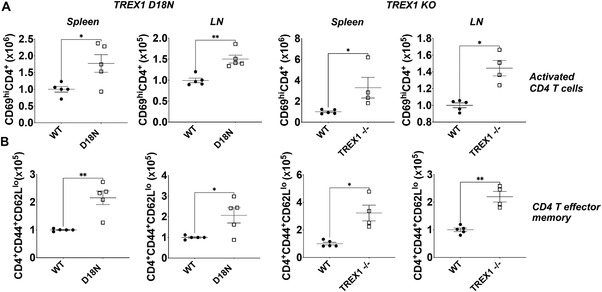
Elevated levels of CD4 T helper cells and T effector memory (Tem) in TREX1 deficiency. Spleen and pooled peripheral lymph nodes (cervical, axillary, brachial, and inguinal lymph nodes) were obtained from female (8–12 weeks old) TREX1 D18N and TREX1 knockout mice and levels of (A) activated CD4+ T cells (CD4+CD69hi) and (B) Tem status (CD4+CD44+CD62Llo) were analyzed by flow cytometry. Absolute number data are presented as mean ± SEM relative to WT mice (normalized absolute numbers) and a total of four to five mice per group over three independent experiments, with the dots representing individual mice. *p < 0.01, **p < 0.001 as calculated by two‐tailed unpaired Student's t‐test and Mann–Whitney test. Also see Supporting information Fig. S1 for data in percentages.
Increased cytokine production by CD4 T cells and Th1 bias in TREX1 deficiency
To further delineate the mechanisms rendering TREX1 mutant mice prone to autoimmunity, we performed intracellular cytokine analysis to investigate Th1 and Th2 phenotypes. CD4+ Th cells in the spleen and LNs of both TREX1 D18N and TREX1 KO mice displayed higher number and proportion of the IFN‐γ producing Th1 cells as compared to WT littermates (Fig. 2A and Supporting information Fig. S2A). CD4 T cells from TREX1 D18N mice also had high levels of cells producing the proinflammatory cytokine TNF‐α, whereas the CD4 T cells from TREX1 KO mice unexpectedly had significantly less cells producing TNF‐α than WT littermates (Fig. 2B and Supporting information Fig. S2B). Differences in TNF‐α production between TREX1 D18N and TREX1 KO cells is particularly interesting and require further investigation. Splenic CD4+ T cells producing the Th2 cytokines IL‐4 and IL‐5 were also higher in the TREX1 D18N and TREX1 KO mice compared to the WT littermates (Fig. 2C and D and Supporting information Fig. S2C and D). Interestingly, CD4 T cells from the LNs of TREX1 D18N or TREX1 KO animals did not show elevated levels of IL‐4 producing CD4 T cells as compared to the WT littermates, although the number and proportion of IL‐5 producing cells were higher in the LNs of TREX1 D18N mice (Fig. 2D and Supporting information Fig. S2D).
Figure 2.
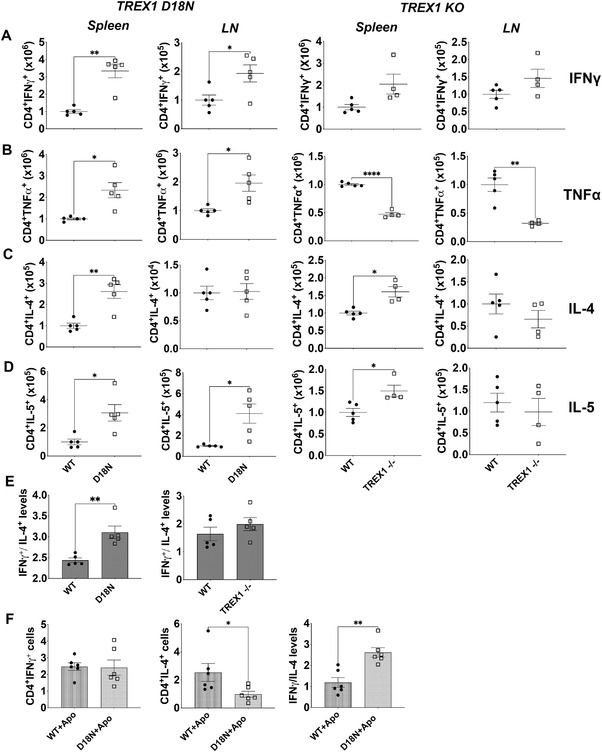
Status of T‐helper cytokines in TREX1 deficiency. Spleen and lymph nodes (LN) from female TREX1 D18N and TREX1 knockout mice were stimulated with PMA/Ionomycin for 5 h and levels of Th1 cytokines IFN‐γ (A), TNF‐α (B), and Th2 cytokines IL‐4 (C) and IL‐5 (D) were analyzed by flow cytometry. (E) The ratio of IFN‐γ to IL‐4 producing CD4 T cells indicated a significant Th1 bias as shown in the spleen of TREX1 D18N mice, but did not reach statistical significance in TREX1 KO animals. (F) In vitro T‐cell stimulation assay using BM‐derived macrophages as described in the Materials and Methods showed that TREX D18N macrophages induced similar levels of %IFN‐γ producing T cells, but much fewer %IL‐4 producing cells. A ratio of IFN‐γ/IL‐4 confirmed the greater Th1 bias induced by the TREX1 D18N macrophages. Data presented as mean ± SEM relative to WT mice (normalized absolute numbers) and a total of four to five mice per group over three independent experiments, with the dots representing individual mice, and three independent experiments for in vitro assays. *p < 0.01, **p < 0.001 as calculated by two‐tailed unpaired Student's t‐test. Also see Supporting information Fig. S2 for data in percentages.
Calculation of the ratio of the absolute numbers of CD4+ T cells producing IFN‐γ over IL‐4 revealed a significantly higher IFN‐γ/IL‐4 ratio in the TREX1 D18N mice, suggestive of a Th1 bias. TREX1 KO mice also showed a similar trend, though it did not reach statistical significance (Fig. 2E). The ratio of the percentages of IFN‐γ and IL‐4 producing cells produced the same results, because the absolute numbers were calculated based on the percentages. To confirm the Th1 bias in the TREX1 D18N mice, we established an in vitro assay by employing BM‐derived macrophages (BMDM), treated with apoptotic cells, to delineate the role of defective TREX1 in inducing a dysregulated T helper response extrinsically in WT T cells. The rationale behind this experiment was that postengulfment, apoptotic cells would represent a source of DNA that would be degraded in WT BMDM and not in TREX1 D18N BMDM, thus, inducing proinflammatory signals, leading to the observed T‐cell polarization. The BMDM were cocultured for 16 h with apoptotic WT thymocytes as described in the Material and Methods. Apoptosis induction was confirmed by flow cytometry analysis by employing Annexin V and 7AAD staining (Supporting information Fig. S5). Subsequently, naïve CD4+ T cells from WT mice were added and the ability of the BMDM to dictate the Th1 and Th2 differentiation was investigated. Both WT and D18N BMDM induced similar IFN‐γ production in the responder WT T cells, whereas in the presence of TREX1 D18N BMDM, IL‐4 production in the responder CD4 T cells was significantly attenuated (Fig. 2F). Calculation of the ratio of IFN‐γ/IL‐4 recapitulated the observed in vivo Th1 bias of the TREX1 D18N mice. It should be taken into account that in this assay we only looked at WT cells as T‐effector cells. Normalization on Th2 shows Th1 elevation. We therefore only see an extrinsic effect. The BMDM experiments suggest that macrophages from the D18N mutant mice, induce less anti‐inflammatory cytokine IL‐4 without affecting the IFN‐γ production, which may contribute to the Th1 bias as observed in vivo. We also investigated the levels of Th17 cells and observed a marginal elevation of IL‐17 producing cells in spleen of TREX1 D18N mice that was close to significance, however, no significant differences were observed in the LNs and in FX1‐treated animals (See Fig. 4 below and Supporting information Fig. S10). There was also a marginal elevation of RORg transcript levels in the spleens without reaching statistical significance (Supporting information Fig. S10C).
Elevated T‐follicular helper cell and activated B‐cell responses in TREX1 deficiency
We next sought to delineate the mechanism leading to the observed skewing of the CD4 T‐cell compartment toward the Th1 lineage. Tfh can facilitate autoimmunity by helping autoreactive B cells to undergo maturation and produce autoantibodies [18, 19, 20]. Since common transcriptional features between Th1 and Tfh generating cells has been reported in autoimmunity [21, 22, 23], we hypothesized that the Th1 bias observed in TREX1 D18N animals may coincide with an increased and potentially proautoimmune Tfh response. Indeed, elevated levels of CXCR5+PD1+CD4+ Tfh cells in the TREX1 D18N and TREX1 KO mice were observed consistent with this hypothesis. Correspondingly, using gating strategy based on earlier studies to measure B‐cell responses [24], we also observed elevated levels of GL7+CXCR5+B220+ cells in the secondary lymphoid organs of the both TREX1 KO and TREX1 D18N mice as compared to WT littermates (Fig. 3A and B, Supporting information Figs. S3A‐B and 9A‐B), suggesting an exacerbated GC reaction. Indeed, the TREX1 D18N mice displayed greater GC B cells, measured as GL7hiFas+ cells in the B220 gated cells (Supporting information Fig. S6A), which correlated well with the increased GL7+CXCR5+B220+ cells in the TREX1 mutant mice. Thus, TREX1 D18N mice exhibit a simultaneous dysregulation of Th1 as well as Tfh‐/B‐cell responses, which may drive the autoimmune phenotype.
Figure 3.
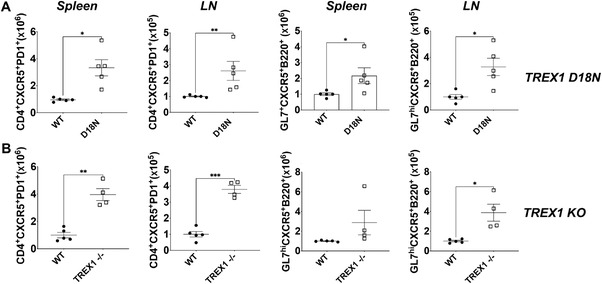
Elevated T‐follicular helper (Tfh) cell response and germinal center reaction in TREX1 deficiency. Levels of Tfh cells (CD4+CXCR5+PD1+) and GL7hiCXCR5+B220+ B‐cells in (A) TREX1 D18N and (B) TREX1 KO female mice analyzed by flow cytometry. Data presented as mean ± SEM relative to WT mice (normalized absolute numbers) and a total of four to five mice per group over three independent experiments, with the dots representing individual mice. *p < 0.01, **p < 0.001, ***p < 0.0001 as calculated by two‐tailed unpaired Student's t‐test. Also see Supporting information Fig. S3 for data in percentages.
Pharmacologic targeting of Bcl6 ameliorates T‐cell dysregulation in TREX1 D18N mice
Bcl6 is an important transcription factor known for its role in the differentiation of Tfh and GC B‐cells [18, 25‐27]. Bcl6 is also highly expressed in B‐cell lymphoma and acts as a survival factor. Targeting of Bcl6 has been tested in experimental studies for the treatment of B‐cell lymphoma [28, 29, 30, 31]. We hypothesized that Bcl6 may be targeted to intervene with the Tfh and GC B cells that may also intervene with the Th1 bias as a potential treatment for TREX1 D18N‐associated autoimmunity. To test this hypothesis, we used the commercially available Bcl6 inhibitor FX1, which was shown to phenocopy Bcl6 deletion in mice by inhibiting GC formation in the anti‐sheep RBC murine model [29] and also suppressed GC‐diffuse large B‐cell lymphomas (DLBCL) [29]. We treated TREX1 D18N mice with FX1 for 8 days followed by detailed investigation on day 10. The levels of activated CD4 T helper cells were significantly reduced in the FX1‐treated TREX1 D18N mice as compared to the vehicle controls down to the levels of WT controls in LNs, with a similar trend in spleens as well (Fig. 4A and Supporting information Fig. S4A). FX1‐treated TREX1 D18N mice also demonstrated a significant attenuation of the Th1 response in the LNs and showed a similar trend in the spleen, as measured by the reduced ability of the CD4 T cells to produce IFN‐γ (Fig. 4B and Supporting information Fig. S4B). The TREX1 D18N mice treated with FX1 exhibited a noticeable trend toward attenuation of Tfh and GC B‐cell responses, with the most prominent effects being observed in splenic GC B cells (Fig. 4C and D and Supporting information Fig. S4C and D). Histological analysis of spleens showed that TREX1 D18N GCs are significantly enlarged relative to WT mice (Fig. 4E). Such histological analysis using H&E staining for investigation of GC changes have also been utilized by others [32] [https://www.arppress.org/v/vspfiles/assets/images/Chapter1T25LymphNodes.pdf; Figs. 1‐6]. As anticipated, the GC size was markedly reduced in TREX1 D18N mice treated with FX1. This finding was further confirmed by immunofluorescence analysis with peanut agglutinin (PNA) staining, which similarly demonstrated a reduction in FX1‐treated TREX1 D18N mice (Fig. 4F). We also performed immunofluorescence analysis with GL7 staining, which displayed a similar trend of a reduction of GC size in FX1‐treated TREX1 D18N mice (Supporting information Fig. S4E). We scored multiple splenic follicles from multiple mice for total PNA and GL7 fluorescence that further confirmed a reduction of GC (Fig. 4G and Supporting information Fig. S4F). We also investigated the transcript levels of Tbet and Gata3 and observed significantly higher levels of Gata3 in the spleens of FX1‐treated TREX1 D18N mice. The ratios of Tbet:Gata3 further confirmed the observed inhibitory effects on the Th1 responses in FX1‐treated mice (Supporting information Fig. S6B).
Figure 4.
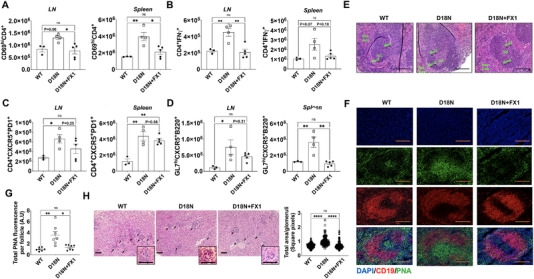
Bcl6 inhibition rescues from autoimmunity in TREX1 D18N mutant mice. Mice were treated as described in Materials and Methods and analyzed by flow cytometry for (A) Activated (CD69+) CD4; (B) IFN‐γ producing CD4 T cells; (C) CD4+CXCR5+PD1+ T follicular helper cells; and (D) GL7hiCXCR5+B220+ B cells; (E) Representative H&E stained sections of the spleens. Spleens from WT, D18N mutant DMSO control, and D18N mutant FX1‐treated animals were stained with H&E and screened for germinal centers (GC), Mantle zone (ManZ), Marginal zone (MarZ), and Red pulp, Scale bars 50 μm. (F) Immunofluorescence for PNA/CD19/DAPI, Scale bars 50 μm and (G) Quantification of total PNA fluorescence per splenic follicle by ImageJ. (H) Representative H & E stained sections of the kidneys and quantification of total area per glomeruli by ImageJ, Scale bars 50 μm (Insert shows glomeruli). Arrows point to individual glomeruli. Data are shown as mean ± SEM and show a total of three to five mice per group over two independent experiments and shown as relative to D18N animals. *p < 0.01, **p < 0.001, ***p < 0.0001 as calculated by multiple comparison by one‐way ANOVA. Also see Supporting information Fig. S4 for data in percentages and GL‐7 staining for GC B cells.
FX1‐mediated targeting of Bcl6 rescues TREX1 D18N mice from autoimmunity
To determine the effects of the Tfh‐GC inhibition by FX1 on autoimmunity in TREX1 D18N mutant mice, we performed immunofluorescence analysis of the immune complex deposition in the kidneys. Although the TREX1 D18N mutant mice do not develop glomerulonephritis‐related loss of renal function at this age, they demonstrate strong immune‐complex deposition in glomeruli and tubulointerstitium [10]. Analysis of immune‐complex deposition in the kidneys revealed that there was an apparent reduction of IgG deposits in FX1‐treated mice in comparison to vehicle control in the glomerular and extra‐glomerular regions in the kidneys (Supporting information Fig. S7A). We observed a significant reduction in the glomerular hypertrophy upon FX1 treatment as compared to the vehicle‐treated TREX1 D18N mice (Supporting information Fig. S7A). Morphometric analysis of the size of glomeruli using the H & E stained sections confirmed that compared to the WT mice, the glomeruli in the TREX1 D18N mutant mice are significantly enlarged and that FX1 treatment of TREX1 D18N mice restored the size of the glomeruli in similar to WT mice (Fig. 4H). However, no other pathological changes were obvious in the kidneys. We also investigated the levels of Blood Urea Nitrogen in these mice to measure renal function and observed that the levels were not affected (Supporting information Fig. S7D). The TREX1 D18N mice, thus, does not exhibit overt renal pathology, but display glomerular hypertrophy, which was inhibited by FX1 treatment (Fig. 4H and Supporting information Fig. S7A). The data are suggestive of not only immunological changes systemically, but also restoration of organ‐specific autoimmunity as indicated by glomerular integrity upon FX1 treatment (Fig. 4H). We also did not observe any obvious adverse effects at the dose of FX1 utilized in the TREX1 D18N mice.
Next, we used a Western blot assay to evaluate the autoantibodies in plasma samples of WT, TREX1 D18N mutant mice treated or untreated with FX1 by measuring reactivity to murine B‐cell lymphoma WEHI‐231 cell extract. Compared to the WT littermates, the plasma of 100% of TREX1 D18N mice showed higher reactivity to WEHI‐231 cell extract. On the other hand, the plasma reactivity in 60% of FX1‐treated animals was similar to the WT mice and lower than the control TREX1 D18N mice, indicating a rescue from autoimmunity (Supporting information Fig. S7B). We then investigated the plasma samples for anti‐dsDNA antibody levels, as a surrogate marker for autoantibody production and observed reduced levels in the FX1‐treated mice. Although there is a noticeable decline in the anti‐dsDNA antibody levels, the difference however did not reach statistical significance (Supporting information Fig. S7C). We further compared the plasma samples from the FX1‐treated mice with WT and vehicle‐treated samples using a novel autoantibody array. Heat map and clustering analysis of the antibody array confirms differential autoantibody patterns in FX1‐treated and untreated TREX1 D18N mice. However, it also indicates the large variability in the autoantibody profiles of WT as well as TREX1 D18N mutant mice (Supporting information Fig. S7E). Nevertheless, three out five FX1‐treated mice showed a strong reduction in the levels of autoantibodies especially against the cluster of autoantigens containing dsDNA, ssDNA, genomic DNA, ssRNA, Histone H4 along with several others that include matrix‐associated proteins collagen I and II, proteoglycan, chondroitin sulfate, heparin sulfate, sphingomyelin, and laminin in the bottom of the heat map. Overall, these data demonstrate that Bcl6 inhibition in TREX1‐deficient mice induced a trend for the reduction in the autoantibody response, which could contribute to a rescue from the autoimmune phenotype observed in the TREX1 D18N mice. Furthermore, FX1‐treated mice exhibited a significant decline in helper T cell dysregulation, indicating a kinetic disconnect of the Th1/Tfh immune response with the circulating autoantibody levels. These data indicate that future long‐term studies using Bcl6 inhibition for the treatment of lupus‐like autoimmune diseases are warranted.
Discussion
Current treatment options available for autoimmune diseases, such as lupus nephritis, involves steroids and immunosuppressants that have serious side‐effects [33, 34, 35, 36, 37]. TREX1 is a key cytoplasmic exonuclease which protects the cell from self‐DNA‐induced immune activation [4, 5, 6]. The TREX1 D18N mutation found in human patients was translated into a murine model, and these mice exhibit spontaneous lupus‐like features [10]. Both TREX1 D18N and TREX1 KO mice exhibited an active disease state at 8 to 12 weeks of age as indicated by elevated levels of CD4 T helper cells and elevated T effector memory status (Fig. 1).
Dysregulated cytokine production is central to autoimmunity, with alterations in the balance between Th1 and Th2 signaling strongly implicated in the loss of self‐tolerance [38, 39, 40]. Although both Th1 and Th2 cytokine production was elevated in the secondary lymphoid organs of TREX1‐deficient mice (Fig. 2), analysis of the Th1/Th2 ratio revealed an Th1 bias in TREX1 D18N mice (Fig. 2E), though not as robustly in the TREX1 KO mice. TNF‐α production was also elevated in the TREX1 D18N mutant mice but was surprisingly downregulated in TREX1 KO mice, an observation warranting further investigation. These data demonstrate that TREX1 D18N animals mimic the Th1 bias commonly observed in SLE patients [41, 42, 43].
Tfh dysregulation can facilitate proautoimmune GC responses observed in SLE patients [15, 20, 44]. Cells generating both Th1 and Tfh cells have been postulated to have features of both subsets, with possible interconversion [13, 14, 15, 16]. The transcription factor Bcl6 participates in the differentiation and maintenance of both Tfh and GC B‐cells [25, 26, 27]. Targeting Bcl6, therefore, represents an attractive strategy for ameliorating autoimmune disease involving Tfh‐GC B‐cell dysregulation and abnormal B‐cell activation. The Bcl6 inhibitor FX1 exhibits therapeutic efficacy in targeting B‐cell lymphoma, and was shown to reduce Tfh and GC B‐responses in sheep RBC immunization model [29], making it a promising candidate for attenuation of the TREX1 D18N autoimmune phenotype.
As evident from Fig. 4A and B, activated CD4+ T cells and IFN‐γ producing cells in LNs were significantly reduced in FX1‐treated mice with spleen samples displaying a trend toward reduction. This may either be due to a reduction in the IFN‐γ producing Tfh cells or a direct inhibition of activated T cells, because of a direct effect of FX1 on CD69, a target of Bcl6 [29]. FX1‐treated TREX1 D18N mice exhibited a trend toward reduction of Tfh levels and GC B cells in the secondary lymphoid organs, with the levels in spleen approaching the WT levels (Fig. 4C and D). This was further confirmed with histological analysis with H&E staining as well as immunofluorescence, revealing effects on germinal centers with FX1 treatments (Fig. 4E‐G). A variety of autoantibodies have been reported with antigenic locations in nuclear, cytoplasmic, or mitochondrial, and extracellular in SLE [45]. Three mechanisms have been postulated as being at the forefront of autoantibody production: (1) cell death which leads to the release of intracellular components; (2) neoantigens that are produced as a result of modification of self‐antigens to trigger B‐cell autoreactive responses; and (3) responses toward foreign antigens [45]. Treatment with FX1 induced a discernible effect on the circulating autoantibody levels in the WEHI‐231 B‐cell lymphoma Western blot assay, anti‐dsDNA antibody assay, as well as the multiplex autoantibody profiling (Supporting information Fig. S4).
Interestingly, these effects coincided with the reduction of a generalized immune complex deposition in the kidneys of FX1‐ treated mice, and with significant indications of preserved glomerular integrity and rescue from autoimmunity (Fig. 4H). Mild reduction of the autoimmune readouts were expected, as FX1 treatment was only given short‐term. Long‐term treatment was not possible due to the intraperitoneal route of drug administration, and better delivery systems need to be developed. Notably, FX1 was recently shown to alter Tfh‐GC responses to sheep RBC injection and lymphoid hyperplasia in mice as well as a short‐term nonhuman primate study [46, 47]. However, if approaches such as these were to be employed for treatment options in autoimmune diseases, studies would need to be conducted to establish their safety and interference with the host‐immunity to infectious diseases or response to immunizations. Investigations into an effect of such therapies on myeloid, T cell, B cell, or mesangial compartments are particularly interesting and are in prospects.
Nevertheless, these data demonstrate that Bcl6‐targeting with FX1 may not just inhibit the Tfh/GC B‐cell axis, but more critically translate into a reduced Th1 response in the secondary lymphoid organs and ultimately to the rescue of TREX1 D18N mutant mice from end‐organ autoimmunity. These findings further our understanding of autoimmune pathogenesis caused by failed DNA clearance and may have relevance for the treatment of autoimmunity in human TREX1 deficiency and other lupus‐like diseases.
Materials and methods
Animals
TREX1D18N/D18N (TREX1 D18N) mutant mice were generated on a 129S6/SvEvTac background using an allelic replacement strategy as described before [10]. The mice were backcrossed 10 generations with C57BL/6J (B6) mice. Transmission of the TREX1 D18N allele was confirmed by tail‐DNA sequencing. All experiments were performed in accordance with the protocols approved by the Institutional Animal Care and Use Committee at the University of Virginia and Wake Forest Baptist Medical Center. TREX‐null (KO) mice revived from cryopreserved embryos were obtained from KOMP Repository, UC Davis. Eight to twelve weeks old female mice were used in this study.
Flow cytometry and intracellular cytokine analysis
Flow cytometry analysis was performed as described previously [48, 49]. Antibodies used for analysis are listed in Supporting information Table S1. Data were acquired on a FACSCalibur (BD Biosciences) and analyzed with FlowJo software (BD Biosciences) using the gating strategies shown in Supporting information Figs. 1 and 2. Data are presented as absolute number relative to WT mice and normalized to treatment controls for in vitro analysis.
Generation of apoptotic cells and BM‐derived myeloid cells (BMDM) for in vitro experiments
Thymii from WT female B6 mice were crushed in RPMI complete media and the cell suspension was filtered using a 70‐micron cell strainer. A total of 5 × 106 cells/mL were incubated in a 10 cm bacterial dish with 5 μM dexamethasone for 6 h in a 37°C and 5% CO2 incubator. The cells were then washed 3× with 15 mL DPBS before using for the experiment. Cells were stained with FITC‐labeled Annexin‐V and 7AAD for flow cytometry to confirm apoptotic cell generation. BMDM have been used to study T‐cell stimulation [50] and were generated as before [51]. Briefly, single‐cell suspension of BM, flushed from the femurs and tibias of mice, was cultured in bacteriological petri plates in 10 mL RPMI complete media (RPMI containing 2 mM l‐glutamine, 100 units/mL penicillin G, 100 μg/mL streptomycin (all from Life Technologies), and 10% fetal bovine serum (Sigma) supplemented with 15% L929 conditioned media [52] initially and 50% media replacement every 3 days. The L929 conditioned media was prepared by culturing the murine L929 cells to confluency in complete DMEM for 7 days, followed by filtering through 0.22 μm and stored at −20°C in aliquots.
Bcl6 inhibitor treatments
Bcl6 inhibitor FX1 (Sigma) was dissolved in DMSO (Sigma) at 25 mg/mL concentration and stored as aliquots at −20° C. Aliquots were thawed and diluted to 300 μL in USP‐grade sterile saline before intraperitoneal injections in TREX1 D18N mice at 40 mg/kg body weight. An equal volume of DMSO similarly diluted in saline was used as placebo controls. Treatments involved daily injection for 8 days and the mice were euthanized on day 10.
Immunofluorescence and microscopy
Kidneys frozen in liquid nitrogen were embedded in Optimal Cutting Temperature compound (Ted Pella Inc.). Tissue was cryosectioned (5 μm) and permeabilized with cold acetone for 20 min at room temperature. The slides were washed with PBS and blocked with 2% horse serum prepared in 3% BSA/PBS for 20 min at room temperature. The sections were stained for 30 min with 1:200 diluted horse anti‐mouse IgG‐Fluorescein (Vector Laboratories) followed by extensive washing in PBS containing 0.1% Tween 20 and mounting using ProLong Diamond Antifade (Thermo Fisher Scientific) containing DAPI counterstain. Images were acquired using Axiovert 200 microscopy system with Apotome imaging and Axiovision software (Carl Zeiss Microscopy, LLC). For GC studies, spleen sections were rehydrated for 30 min with PBS and blocked for 1 h with 10% BSA/PBS at room temperature. The sections were stained for 1 h with 1:100 diluted PNA‐Fluorescein (Vector Laboratories) or anti‐GL7‐Alexafluor 488 and washed three times with PBS, followed by incubation with 1:50 diluted PE‐anti‐mouse CD19 antibody (Thermo Fisher Scientific) at room temperature for 1 h. The slides were washed and mounted using ProLong Diamond Antifade (Thermo Fisher Scientific) containing DAPI. Germinal centers were mapped using the selection tool and quantified using ImageJ (NIH). H & E stained spleen sections were also imaged for GC analyses.
Anti‐dsDNA assay
Serum samples collected at necropsy were analyzed for anti‐dsDNA antibodies by ELISA as described earlier [49, 53]. Briefly, pGEM‐3zf plasmid DNA (Promega) was digested with S1‐nuclease (Thermo Fisher Scientific) and biotinylated using Photoprobe Biotin (Vector Labs) following the manufacturer's instructions. Biotinylated dsDNA (1 ng/mL) was used for the assay.
SDS‐PAGE and immunoblot analysis
B‐cell lymphoma WEHI‐231 cell extract prepared by repeated freeze thawing was quantified for protein content with bicinchoninic acid protein assay kit (Pierce Chemical). Total 150 μg of protein extract was loaded onto a preparative 10% SDS‐PAGE gel and transferred to Immun‐Blot PVDF Membrane (Bio‐Rad). After blocking the membrane in 3% BSA for 1 h, thin strips were cut and probed with 1:50 dilution of plasma samples overnight at 4°C. The strips were washed with PBS containing 0.1% Tween 20 and were incubated in 1:5000 IRDye 680RD anti‐Mouse IgG (H+L) secondary antibody (LI‐COR Biosciences) for 1 h at room temperature. The strips were aligned and imaged using Odyssey Fc Imaging System (LI‐COR Biosciences).
Multiplex autoantibody profiling
Blood was collected at euthanasia by retro‐orbital bleeding using heparinized capillary tubes (Fisher Scientific). Plasma obtained by centrifugation was used for the multiplex autoantibody profiling at UT Southwestern Medical Center Genomics and Microarray core facility (https://microarray.swmed.edu/products/product/autoantigen‐microarray‐service‐igg‐igm/).
Statistical analysis
All statistical analyses were performed using Prism 8 software (GraphPad). Representative data from three or more independent experiments are shown as mean ± SEM. For comparison of more than two groups, one‐way ANOVA or Kruskal–Wallis test was used. For statistical analysis testing of two groups, two‐tailed unpaired Student's t‐test or Mann‐Whitney U test was used.
Conflict of interest
RS is an inventor for US patents 6897041 and 9840545 licensed to Slate Bio Inc., holds equity and is a consultant for Slate Bio Inc., unrelated to this work. The research was conducted in the absence of any commercial or financial relationships that could pose a potential conflict of interest.
Author contribution
RS, RV, and FWP conceived the project. RS, JMG, and FWP provided overall supervision. RV conducted the majority of the experiments. RV, VS, MD, SRS, and RS collected and analyzed data, and wrote the manuscript. NY helped with the autoantibody multiplex array. SEH, RV, and SM managed the mouse colony.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202149324.
Abbreviations
- AGS
Aicardi‐Goutières syndrome
- BMDM
BM‐derived macrophages
- FCL
familial chilblain lupus
- PNA
peanut agglutinin
- SLE
systemic lupus erythematosus
- Tfh cells
T‐follicular helper cells
Supporting information
Supporting information
Acknowledgments
This research was supported by R01AI116725 (FWP, subcontract to RS) from the National Institute of Allergy and Infectious Diseases. Additional support was from the National Institute of Diabetes and Kidney Diseases R01DK105833 (multi‐PI:RS and SMF), R01DK104963 (PI: RS), R21DK112105 (multi‐PI: RS, M. Rosner, and K. Lynch), UVA School of Medicine LaunchPad Diabetes Fund (RS), T32AI007401 (SRS), the Alliance for Lupus Research (RS), the Cowgill and Artom Memorial Fellowship (SRS), and the Comprehensive Cancer Center of Wake Forest University National Cancer Institute (Cancer Center Support Grant P30CA012197). The content is solely the responsibility of the authors and does not represent the official views of the funding agencies. We also thank the University of Virginia (UVA) Research Histology Core for processing tissue samples and UT Southwestern Medical Center Genomics and Microarray Core for Autoantibody assay.
Data Availability Statement
The data that support the findings of this study are present in the supplementary materials. Further data is available from the corresponding author upon reasonable request.
References
- 1. Lu, Q. and Lemke, G. , Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2001. 293: 306–311. [DOI] [PubMed] [Google Scholar]
- 2. Cohen, P. L. , Caricchio, R. , Abraham, V. , Camenisch, T. D. , Jennette, J. C. , Roubey, R. A. , Earp, H. S. et al., Delayed apoptotic cell clearance and lupus‐like autoimmunity in mice lacking the c‐mer membrane tyrosine kinase. J. Exp. Med. 2002. 196: 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hanayama, R. , Tanaka, M. , Miyasaka, K. , Aozasa, K. , Koike, M. , Uchiyama, Y. and Nagata, S. , Autoimmune disease and impaired uptake of apoptotic cells in MFG‐E8‐deficient mice. Science 2004. 304: 1147–1150. [DOI] [PubMed] [Google Scholar]
- 4. Perrino, F. W. , Miller, H. and Ealey, K. A. , Identification of a 3'→5'‐exonuclease that removes cytosine arabinoside monophosphate from 3' termini of DNA. J. Biol. Chem. 1994. 269: 16357–16363. [PubMed] [Google Scholar]
- 5. Mazur, D. J. and Perrino, F. W. , Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3'→5' exonucleases. J. Biol. Chem. 1999. 274: 19655–19660. [DOI] [PubMed] [Google Scholar]
- 6. Mazur, D. J. and Perrino, F. W. , Excision of 3' termini by the Trex1 and TREX2 3'→5' exonucleases. Characterization of the recombinant proteins. J. Biol. Chem. 2001. 276: 17022–17029. [DOI] [PubMed] [Google Scholar]
- 7. Rice, G. I. , Rodero, M. P. and Crow, Y. J. , Human disease phenotypes associated with mutations in TREX1. J. Clin. Immunol. 2015. 35: 235–243. [DOI] [PubMed] [Google Scholar]
- 8. Lee‐Kirsch, M. A. , Gong, M. , Chowdhury, D. , Senenko, L. , Engel, K. , Lee, Y. A. , de Silva, U. et al., Mutations in the gene encoding the 3'‐5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 2007. 39: 1065–1067. [DOI] [PubMed] [Google Scholar]
- 9. Lee‐Kirsch, M. A. , Chowdhury, D. , Harvey, S. , Gong, M. , Senenko, L. , Engel, K. , Pfeiffer, C. et al., A mutation in TREX1 that impairs susceptibility to granzyme A‐mediated cell death underlies familial chilblain lupus. J. Mol. Med. (Berl) 2007. 85: 531–537. [DOI] [PubMed] [Google Scholar]
- 10. Grieves, J. L. , Fye, J. M. , Harvey, S. , Grayson, J. M. , Hollis, T. and Perrino, F. W. , Exonuclease TREX1 degrades double‐stranded DNA to prevent spontaneous lupus‐like inflammatory disease. Proc. Natl. Acad. Sci. U. S. A. 2015. 112: 5117–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao, D. , Li, T. , Li, X. D. , Chen, X. , Li, Q. Z. , Wight‐Carter, M. and Chen, Z. J. , Activation of cyclic GMP‐AMP synthase by self‐DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. U. S. A. 2015. 112: E5699–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gray, E. E. , Winship, D. , Snyder, J. M. , Child, S. J. , Geballe, A. P. and Stetson, D. B. , The AIM2‐like receptors are dispensable for the interferon response to intracellular DNA. Immunity 2016. 45: 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cucak, H. , Yrlid, U. , Reizis, B. , Kalinke, U. and Johansson‐Lindbom, B. , Type I interferon signaling in dendritic cells stimulates the development of lymph‐node‐resident T follicular helper cells. Immunity 2009. 31: 491–501. [DOI] [PubMed] [Google Scholar]
- 14. Mesquita, D. , Jr., Cruvinel, W. M. , Resende, L. S. , Mesquita, F. V. , Silva, N. P. , Camara, N. O. and Andrade, L. E. , Follicular helper T cell in immunity and autoimmunity. Braz. J. Med. Biol. Res. 2016. 49: e5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gensous, N. , Schmitt, N. , Richez, C. , Ueno, H. and Blanco, P. , T follicular helper cells, interleukin‐21 and systemic lupus erythematosus. Rheumatology (Oxford) 2017. 56: 516–523. [DOI] [PubMed] [Google Scholar]
- 16. Bocharnikov, A. V. , Keegan, J. , Wacleche, V. S. , Cao, Y. , Fonseka, C. Y. , Wang, G. , Muise, E. et al., PD‐1hi CXCR5‐ T peripheral helper cells promote B cells responses in lupus via MAF and IL‐21. JCI Insight 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mountz, J. D. , Hsu, H. C. and Ballesteros‐Tato, A. , Dysregulation of T follicular helper cells in lupus. J. Immunol. 2019. 202: 1649–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crotty, S. , T follicular helper cell differentiation, function, and roles in disease. Immunity 2014. 41: 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ueno, H. , T follicular helper cells in human autoimmunity. Curr. Opin. Immunol. 2016. 43: 24–31. [DOI] [PubMed] [Google Scholar]
- 20. Kim, S. J. , Lee, K. and Diamond, B. , Follicular helper T cells in systemic lupus erythematosus. Front. Immunol. 2018. 9: 1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakayamada, S. , Kanno, Y. , Takahashi, H. , Jankovic, D. , Lu, K. T. , Johnson, T. A. , Sun, H. W. et al., Early Th1 cell differentiation is marked by a Tfh cell‐like transition. Immunity 2011. 35: 919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma, C. S. , Deenick, E. K. , Batten, M. and Tangye, S. G. , The origins, function, and regulation of T follicular helper cells. J. Exp. Med. 2012. 209: 1241–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmitt, N. , Liu, Y. , Bentebibel, S. E. and Ueno, H. , Molecular mechanisms regulating T helper 1 versus T follicular helper cell differentiation in humans. Cell Rep. 2016. 16: 1082–1095. [DOI] [PubMed] [Google Scholar]
- 24. Kerfoot, S. M. , Yaari, G. , Patel, J. R. , Johnson, K. L. , Gonzalez, D. G. , Kleinstein, S. H. and Haberman, A. M. , Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 2011. 34: 947–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi, Y. S. , Kageyama, R. , Eto, D. , Escobar, T. C. , Johnston, R. J. , Monticelli, L. , Lao, C. and Crotty, S. , ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 2011. 34: 932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hatzi, K. , Nance, J. P. , Kroenke, M. A. , Bothwell, M. , Haddad, E. K. , Melnick, A. and Crotty, S. , BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J. Exp. Med. 2015. 212: 539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang, J. A. , Tubo, N. J. , Gearhart, M. D. , Bardwell, V. J. and Jenkins, M. K. , Cutting edge: Bcl6‐interacting corepressor contributes to germinal center T follicular helper cell formation and B cell helper function. J. Immunol. 2015. 194: 5604–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cerchietti, L. and Melnick, A. , Targeting BCL6 in diffuse large B‐cell lymphoma: what does this mean for the future treatment? Expert. Rev. Hematol. 2013. 6: 343–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cardenas, M. G. , Yu, W. , Beguelin, W. , Teater, M. R. , Geng, H. , Goldstein, R. L. , Oswald, E. et al., Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J. Clin. Invest. 2016. 126: 3351–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cardenas, M. G. , Oswald, E. , Yu, W. , Xue, F. , MacKerell, A. D., Jr. and Melnick, A. M. , The expanding role of the BCL6 oncoprotein as a cancer therapeutic target. Clin. Cancer Res. 2017. 23: 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leeman‐Neill, R. J. and Bhagat, G. , BCL6 as a therapeutic target for lymphoma. Expert Opin. Ther. Targets 2018. 22: 143–152. [DOI] [PubMed] [Google Scholar]
- 32. Zandvoort, A. and Timens, W. , The dual function of the splenic marginal zone: essential for initiation of anti‐TI‐2 responses but also vital in the general first‐line defense against blood‐borne antigens. Clin. Exp. Immunol. 2002. 130: 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ethgen, O. , de Lemos Esteves, F. , Bruyere, O. and Reginster, J. Y. , What do we know about the safety of corticosteroids in rheumatoid arthritis? Curr. Med. Res. Opin. 2013. 29: 1147–1160. [DOI] [PubMed] [Google Scholar]
- 34. Rainsford, K. D. , Anti‐inflammatory drugs in the 21st century. Subcell. Biochem. 2007. 42: 3–27. [DOI] [PubMed] [Google Scholar]
- 35. Curtis, J. R. , Westfall, A. O. , Allison, J. , Bijlsma, J. W. , Freeman, A. , George, V. , Kovac, S. H. et al., Population‐based assessment of adverse events associated with long‐term glucocorticoid use. Arthritis Rheum. 2006. 55: 420–426. [DOI] [PubMed] [Google Scholar]
- 36. Murray, M. D. and Brater, D. C. , Renal toxicity of the nonsteroidal anti‐inflammatory drugs. Annu. Rev. Pharmacol. Toxicol. 1993. 33: 435–465. [DOI] [PubMed] [Google Scholar]
- 37. Fujita, T. , Kutsumi, H. , Sanuki, T. , Hayakumo, T. and Azuma, T. , Adherence to the preventive strategies for nonsteroidal anti‐inflammatory drug‐ or low‐dose aspirin‐induced gastrointestinal injuries. J. Gastroenterol. 2013. 48: 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guan, Q. , Gao, X. , Wang, J. , Sun, Y. and Shekhar, S. , Cytokines in autoimmune disease. Mediators Inflamm. 2017. 2017: 5089815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moudgil, K. D. and Choubey, D. , Cytokines in autoimmunity: role in induction, regulation, and treatment. J. Interferon Cytokine Res. 2011. 31: 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shachar, I. and Karin, N. , The dual roles of inflammatory cytokines and chemokines in the regulation of autoimmune diseases and their clinical implications. J. Leukoc. Biol. 2013. 93: 51–61. [DOI] [PubMed] [Google Scholar]
- 41. Akahoshi, M. , Nakashima, H. , Tanaka, Y. , Kohsaka, T. , Nagano, S. , Ohgami, E. , Arinobu, Y. et al., Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus. Arthritis Rheum. 1999. 42: 1644–1648. [DOI] [PubMed] [Google Scholar]
- 42. Talaat, R. M. , Mohamed, S. F. , Bassyouni, I. H. and Raouf, A. A. , Th1/Th2/Th17/Treg cytokine imbalance in systemic lupus erythematosus (SLE) patients: Correlation with disease activity. Cytokine 2015. 72: 146–153. [DOI] [PubMed] [Google Scholar]
- 43. Jabbari, A. , Suarez‐Farinas, M. , Fuentes‐Duculan, J. , Gonzalez, J. , Cueto, I. , Franks, A. G., Jr. and Krueger, J. G. , Dominant Th1 and minimal Th17 skewing in discoid lupus revealed by transcriptomic comparison with psoriasis. J. Invest. Dermatol. 2014. 134: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gensous, N. , Charrier, M. , Duluc, D. , Contin‐Bordes, C. , Truchetet, M. E. , Lazaro, E. , Duffau, P. et al., T follicular helper cells in autoimmune disorders. Front. Immunol. 2018. 9: 1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suurmond, J. and Diamond, B. , Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J. Clin. Invest. 2015. 125: 2194–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cai, Y. , Watkins, M. A. , Xue, F. , Ai, Y. , Cheng, H. , Midkiff, C. C. , Wang, X. et al., BCL6 BTB‐specific inhibition via FX1 treatment reduces Tfh cells and reverses lymphoid follicle hyperplasia in Indian rhesus macaque (Macaca mulatta). J. Med. Primatol. 2020. 49: 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cai, Y. , Poli, A. N. R. , Vadrevu, S. , Gyampoh, K. , Hart, C. , Ross, B. , Fair, M. et al., BCL6 BTB‐specific inhibitor reversely represses T‐cell activation, Tfh cells differentiation, and germinal center reaction in vivo. Eur. J. Immunol. 2021. 51: 2441–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kinsey, G. R. , Sharma, R. , Huang, L. , Li, L. , Vergis, A. L. , Ye, H. , Ju, S. T. et al., Regulatory T cells suppress innate immunity in kidney ischemia‐reperfusion injury. J. Am. Soc. Nephrol. 2009. 20: 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stremska, M. E. , Jose, S. , Sabapathy, V. , Huang, L. , Bajwa, A. , Kinsey, G. R. , Sharma, P. R. et al., IL233, A novel IL‐2 and IL‐33 hybrid cytokine, ameliorates renal injury. J. Am. Soc. Nephrol. 2017. 28: 2681–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hume, D. A. , Macrophages as APC and the dendritic cell myth. J. Immunol. 2008. 181: 5829–5835. [DOI] [PubMed] [Google Scholar]
- 51. Kadl, A. , Meher, A. K. , Sharma, P. R. , Lee, M. Y. , Doran, A. C. , Johnstone, S. R. , Elliott, M. R. et al., Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010. 107: 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Englen, M. D. , Valdez, Y. E. , Lehnert, N. M. and Lehnert, B. E. , Granulocyte/macrophage colony‐stimulating factor is expressed and secreted in cultures of murine L929 cells. J. Immunol. Methods 1995. 184: 281–283. [DOI] [PubMed] [Google Scholar]
- 53. Ge, Y. , Jiang, C. , Sung, S. S. , Bagavant, H. , Dai, C. , Wang, H. , Kannapell, C. C. et al., Cgnz1 allele confers kidney resistance to damage preventing progression of immune complex‐mediated acute lupus glomerulonephritis. J. Exp. Med. 2013. 210: 2387–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
The data that support the findings of this study are present in the supplementary materials. Further data is available from the corresponding author upon reasonable request.