

SUMMARY
Mutations in BRCA1 or BRCA2 (BRCA) is synthetic lethal with poly(ADP-ribose) polymerase inhibitors (PARPi). Lethality is thought to derive from DNA double-stranded breaks (DSBs) necessitating BRCA function in homologous recombination (HR) and/or fork protection (FP). Here, we report instead that toxicity derives from replication gaps. BRCA1- or FANCJ-deficient cells, with common repair defects but distinct PARPi responses, reveal gaps as a distinguishing factor. We further uncouple HR, FP, and fork speed from PARPi response. Instead, gaps characterize BRCA-deficient cells, are diminished upon resistance, restored upon resensitization, and, when exposed, augment PARPi toxicity. Unchallenged BRCA1-deficient cells have elevated poly(ADP-ribose) and chromatin-associated PARP1, but aberrantly low XRCC1 consistent with defects in backup Okazaki fragment processing (OFP). 53BP1 loss resuscitates OFP by restoring XRCC1-LIG3 that suppresses the sensitivity of BRCA1-deficient cells to drugs targeting OFP or generating gaps. We highlight gaps as a determinant of PARPi toxicity changing the paradigm for synthetic lethal interactions.
In brief
Cong et al. report that replication gaps couple with PARPi sensitivity, whereas defects in DNA repair, fork protection, or fork speed can be uncoupled, implicating gaps as the sensitizing lesion. Correspondingly, gap suppression fully aligns with resistance including cells with BRCA1 and 53BP1 deficiency that regain Okazaki fragment processing.
Graphical Abstract
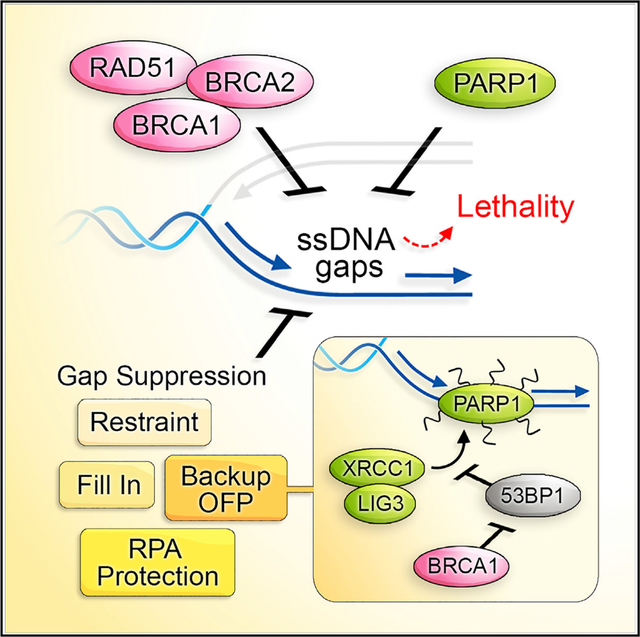
INTRODUCTION
Since the landmark finding that poly(ADP-ribose) polymerase inhibition (PARPi) is synthetic lethal with deficiency in the hereditary breast cancer genes BRCA1 and BRCA2 (BRCA), the primary sensitizing lesion has been attributed to DNA double-stranded breaks (DSBs) (Bryant et al., 2005; Farmer et al., 2005). PARPi has significant anticancer activity in cancers harboring defects in BRCA or other genes required for the repair of DSBs by homologous recombination (HR) (Moynahan et al., 1999). BRCA proteins also function in fork protection (FP), which limits excessive nucleolytic degradation of stalled replication forks. In their absence, stalled forks are expected to collapse into DSBs (Hashimoto et al., 2010; Lomonosov et al., 2003; Schlacher et al., 2011, 2012). Correspondingly, the PARPi olaparib is believed to trap PARP1 on DNA and interfere with DNA replication and promote fork collapse (D’Andrea, 2018; Michelena et al., 2018; Murai et al., 2012; Pommier et al., 2016). Thus, PARPi is considered especially toxic in BRCA-deficient cells because forks collapse into DSBs that are not repaired due to HR defects. The observation that restoration of HR and/or FP is associated with chemoresistance in BRCA-deficient cancer is consistent with this DSB model of therapy response (Bouwman et al., 2010; Bunting et al., 2010; Edwards et al., 2008; Ray Chaudhuri et al., 2016; Sakai et al., 2008).
More recently, the stalled fork model was confronted with the observation that the PARPi olaparib did not slow or block DNA replication, but rather accelerated DNA replication (Maya-Mendoza et al., 2018). This unrestrained replication was consistent with the role of PARP1 in promoting replication-fork reversal, a mechanism by which replication forks slow, pause, and reverse direction when confronted with replication obstacles (Berti et al., 2013; Caldecott et al., 1996; Leppard et al., 2003; Lopes et al., 2006; Maya-Mendoza et al., 2018; Ray Chaudhuri et al., 2012; Ray Chaudhuri and Nussenzweig, 2017; Sogo et al., 2002; Sugimura et al., 2008). To fit the DSB framework of PARPi toxicity, it was proposed that unrestrained replication leads to DSB formation (Maya-Mendoza et al., 2018; Quinet and Vindigni, 2018). Thus, while the proposed mechanism by which PARPi cause DSBs has evolved, the underlying concept that DSBs drive synthetic lethality in BRCA-deficient cancers has remained largely unchallenged.
While current models emphasize DSBs as the sensitizing lesions of PARPi, single-stranded DNA (ssDNA) breaks or gaps are commonly associated with agents inducing replication stress and were historically considered in genotoxic chemotherapy response (Mladenov et al., 2013; Olive, 1998). Likewise, PARPi causes an accumulation of single-strand breaks (SSBs), nicks, and gaps (Lord and Ashworth, 2012; van Wietmarschen and Nussenzweig, 2018). The source of the gaps could range from loss of a diverse set of PARP1 functions such as ssDNA break repair or Okazaki fragment processing (OFP) (Berti et al., 2013; Caldecott et al., 1996; Hanzlikova et al., 2018; Leppard et al., 2003). Functionally, PARP1 recruits proteins to ssDNA by its ability to synthesize protein-conjugated polymers of ADP-ribose or PAR. Incompletely processed Okazaki fragments that escape processing by canonical FEN1 and LIG1 are bound by PARP1, and PAR signaling is activated. PARP1 then recruits X-ray repair cross-complementing protein 1, XRCC1, to complete OFP (Azarm and Smith, 2020; Hanzlikova and Caldecott, 2019; Hanzlikova et al., 2018). Thus, PARPi is predicted to disrupt OFP and generate lagging strand gaps (van Wietmarschen and Nussenzweig, 2018). In conventional models, nicks/gaps or PARPi trapped proteins are proposed to be converted to DSBs during DNA replication and sensitize BRCA-deficient cells lacking DSB repair.
Hseveral lines of evidence suggest that PARPi toxicity could stem from sources distinct from the eventual induction of DSBs. For example, PARPi sensitizes cells deficient or mutated in OFP factors such as flap endonuclease I (FEN1), LIG1, XRCC1, or proliferating cell nuclear antigen (PCNA) (Hanzlikova et al., 2018; Lehmann et al., 1988; Ström et al., 2011; Teo et al., 1983; Thakar et al., 2020), implying that combined replication gaps are insurmountable and lead to cell death. Intriguingly, and further raising lagging strand gaps as a cause of toxicity, FEN1 loss is synthetic lethal in BRCA-Fanconi anemia (FA)-deficient cells (Guo et al., 2020; Mengwasser et al., 2019; Ward et al., 2017). In addition, PARPi is synthetic lethal, with loss of genes having no corresponding defect in HR or FP (Ström et al., 2011; Zimmermann et al., 2018), and HR deficiency is not required for PARPi response in the clinic (Ledermann and Pujade-Lauraine, 2019), indicating that lesions distinct from DSBs could drive cell death. Notably, aside from HR and FP, BRCA proteins function in preventing replication-associated gaps (Hashimoto et al., 2010; Henry-Mowatt et al., 2003; Kolinjivadi et al., 2017a, 2017b; Panzarino et al., 2021; Schlacher et al., 2011; Somyajit et al., 2021; Su et al., 2008; Zellweger et al., 2015). In particular, the central recombination protein RAD51 functions in replication gap suppression (RGS) in a manner that is separated for its role in HR or post-replication gap filling (Kolinjivadi et al., 2017b; Su et al., 2008). Replication gaps could be the basis for underreplication in BRCA-deficient cells that provides opportunities for synthetic lethal interactions, with loss of genes functioning during or in the resolution of replication (Adam et al., 2021; Álvarez-Quilón et al., 2020; Feng and Jasin, 2017; Lai et al., 2017)
We also noted that DSB framework requires concessions because HR and FP vary in their relation to PARPi response in several cell models (Cantor and Calvo, 2017; Kolinjivadi et al., 2017b; Mijic et al., 2017; Schlacher et al., 2011; Wang et al., 2015; Zadorozhny et al., 2017). Here, we considered that immediate induction of widespread replication gaps could drive synthetic lethality. Consistent with this interpretation, we report that BRCA-RAD51-deficient cells accumulate excessive gaps in response to PARPi. Our findings indicate that gaps result in PARPi-induced sensitivity due to OFP defects that exhaust replication protein A (RPA) pools. Correspondingly, BRCA1-deficient cells are vulnerable to the inhibition of OFP and ssDNA binding by RPA that exposes gaps. OFP defects in BRCA1-deficient cells are rescued by the loss of 53BP1, indicating that lagging strand gaps are a critical determinant of PARPi synthetic lethality and should be considered to be a biomarker of PARPi response.
RESULTS
The length of nascent DNA following PARPi is aberrantly enhanced in PARPi-sensitive BRCA1-deficient cells and hyperrestrained in FANCJ-deficient cells that are not PARPi sensitive
To explore the connection between replication dynamics and PARPi response, we analyzed replication fork progression following PARPi treatment. We performed DNA fiber assays that monitor fork dynamics by incorporating nucleoside analogs into newly synthesized DNA strands, which can then be fluorescently labeled. We used human retinal pigmented epithelial (RPE1-human telomerase reverse transcriptase [hTERT], TP53−/−; herein, RPE1 control) cells and confirmed that BRCA1 deficiency (BRCA1 knockout [KO] RPE1-hTERT, TP53−/− cells) caused sensitivity to PARPi and the DNA cross-linking agent cisplatin (Figures 1A and 1B). Next, we verified that cells displayed longer dual-labeled replication tracts (5-iodo-2′-deoxyuridine [IdU] and 5-chloro-2′-deoxyuridine [CldU]) when treated at 10 μM PARPi for 2 h (Maya-Mendoza et al., 2018; Figure 1C). Moreover, we confirmed that BRCA1-deficient cells have greater fork asymmetry that is reduced by PARPi (Maya-Mendoza et al., 2018; Figure S1A).
Figure 1. PARPi-induced fork lengthening and ssDNA gaps are greater in BRCA1-deficient but not in FANCJ-deficient cells.
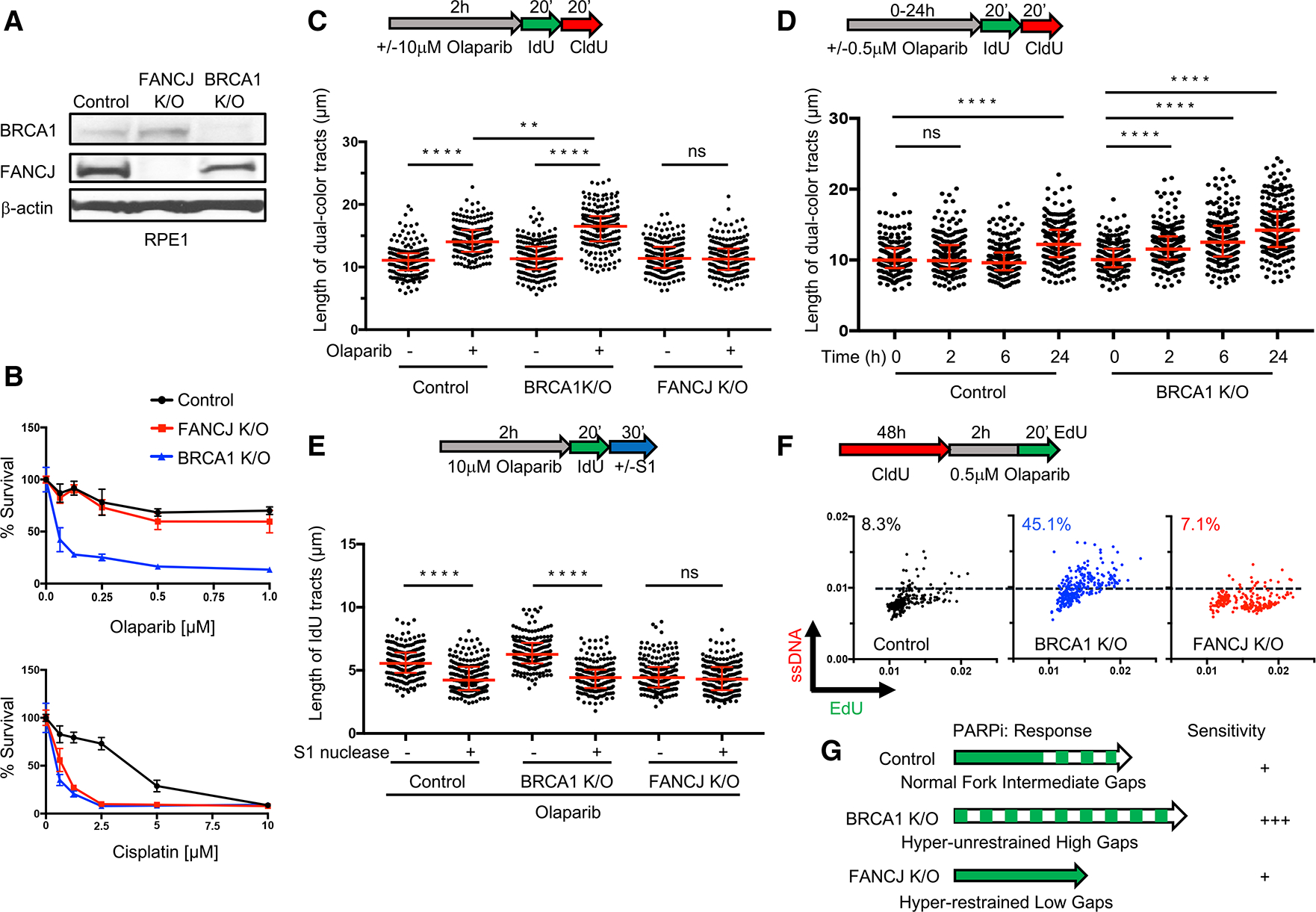
(A) Western blot analysis with the indicated antibodies of lysates from control, FANCJ KO, and BRCA1 KO RPE1 cells.
(B) Cell survival assays for indicated cells under increasing concentrations of olaparib or cisplatin. Data represent the mean percentage ± SD of survival for each dot.
(C) Schematic and quantification of the DNA fiber assays for the length of dual-color tracts in indicated cells following olaparib treatment (10 μM, 2 h).
(D) DNA fiber assays for the length of dual-color tracts in indicated cells following olaparib treatment (0.5 μM) at different periods of time.
(E) DNA fiber assays for IdU tracts with or without S1 nuclease incubation in indicated cells following olaparib treatment (10 μM, 2 h). For (C)–(E), each dot represents 1 fiber; at least 200 fibers are quantified from 2 biological independent experiments (n = 2). Red bars represent the median ± interquartile range. All statistical analysis according to Kruskal-Wallis test, followed by Dunn’s test. **p < 0.01; ****p < 0.0001; ns, not significant.
(F) Schematic and quantification of mean ssDNA intensity for indicated cells following CldU pre-labeling and olaparib release in 0.5 μM, 2 h with EdU added at the last 20 min. At least 200 cells are quantified from n = 2. Dashed lines indicate a cutoff intensity level (at 0.01) for all cell lines. Cells with intensity higher than that are calculated for percentages. EdU and ssDNA are measured by arbitrary units.
(G) Model illustrating gap formation following PARPi treatment in control, BRCA1 KO, and FANCJ KO RPE1 cells.
The total length of nascent DNA tracts following PARPi was not reported to be different between BRCA1-deficient and -proficient cells at 24 h post-10 μM PARPi (Maya-Mendoza et al., 2018). However, we found that BRCA1-deficient cells had significantly longer replication tracks at 2 h post-10 μM PARPi (Figure 1C), suggesting that the initial response to PARPi is distinct when BRCA1 is absent. To decipher whether aberrant lengthening of nascent DNA following in the immediate aftermath of PARPi reflects cell sensitivity, we analyzed DNA fibers at several time points post-0.5 μM PARPi, a dose in which BRCA1 KO cells are more sensitive than controls (Figure 1B). We observed longer DNA tracts in BRCA1 KO cells as compared to control cells at all time points before 24 h (Figure 1D). These findings suggest that fork lengthening within a few hours of PARPi could distinguish sensitive versus resistant lines.
We previously found that the aberrant fork lengthening during replication stress was dependent on the FANCJ (BACH1/BRIP1) helicase (Cantor et al., 2001; Peng et al., 2018). In most respects, however, FANCJ phenocopies BRCA1 with functions not only in the suppression of hereditary breast/ovarian cancer and FA but also in promoting HR and FP (Cantor et al., 2004; Levitus et al., 2005; Levran et al., 2005; Litman et al., 2005; Nath et al., 2017; Peng et al., 2006, 2018; Sawyer et al., 2015; Suhasini et al., 2013). Thus, we analyzed the response of FANCJ-deficient (FANCJ KO RPE1-hTERT, TP53−/−) cells to PARPi. In striking contrast to BRCA1 KO, we found that FANCJ KO cells, along with U2OS and 293T FANCJ KO cell lines, were not sensitive to PARPi, while, as expected, they were sensitive to cisplatin, mitomycin C, or the topoisomerase inhibitor camptothecin (CPT) (Figures 1A, 1B, and S1B–S1D). In contrast to BRCA1 KO cells, FANCJ KO cells did not show DNA tract lengthening at 2 h post-10 μM PARPi (Figure 1C), suggesting that FANCJ is required for fork lengthening and sensitivity following PARPi. These findings also highlighted that not all HR- and FP-deficient lines are sensitive to PARPi.
Replication-associated ssDNA gaps are enhanced in BRCA1-deficient cells and hyperrestrained in FANCJ-deficient cells
We hypothesized that fork lengthening in response to PARPi leads to the formation of replication gaps that are exacerbated in BRCA-deficient cells due to their aberrant fork-lengthening phenotype that correlates with gap formation (Lossaint et al., 2013; Panzarino et al., 2021; Peng et al., 2018; Zellweger et al., 2015). To determine whether gaps form in the vicinity of the accelerated replication forks following PARPi treatment, cells were labeled with IdU/CldU and incubated with the S1 nuclease to digest ssDNA regions (Quinet et al., 2016, 2017). If nascent ssDNA regions are within the labeled replication tracts, then S1 nuclease will cut and shorten the visible IdU/CldU replication tracts. Following 2 h PARPi (10 μM), conditions under which lengthening is readily observed in both the control and BRCA1 KO cells (Figure 1C), we observed that S1 nuclease treatment also reduced replication tract lengths (Figure 1E). Following the lower-dose 0.5 μM PARPi, in which tracts lengthened in BRCA1 KO cells, tracts also shortened with S1 nuclease treatment (Figure S1E). By contrast, in FANCJ KO cells, tracts did not lengthen or display S1 nuclease sensitivity even following high-dose 10 μM PARPi (Figure 1E), consistent with our previous finding that unrestrained replication and ssDNA gap formation is dependent on FANCJ and that when fork lengthening is not detected, neither are gaps (Panzarino et al., 2021; Peng et al., 2018). Moreover, these findings suggest that PARPi sensitivity correlates with initial lengthening and/or gap formation.
If gaps indicate PARPi sensitivity and RGS indicates PARPi resistance, then it will be critical to readily detect them. To assess whether genome-wide immunoreactive ssDNA gaps could be observed immediately following PARPi, cells were pre-labeled with nucleoside analogs, and non-denaturing immunofluorescence (IF) was performed (Couch et al., 2013). Following 10 μM PARPi that lengthens replication tracts in both control and BRCA1 KO cells (Figure 1C), we observed a positive signal for ssDNA, as compared to FANCJ KO cells that did not score positive (Figure S1F). Following 0.5 μM PARPi for 2 h that lengthens replication tracts in BRCA1 KO cells but not controls (Figure 1D), it was evident that BRCA1 KO cells had more replicating cells with elevated ssDNA intensity (Figure 1F). These findings suggest that replication-associated ssDNA can be detected by IF. Moreover, PARPi sensitivity could derive from either excessive replication fork speed and/or ssDNA gaps that are elevated in BRCA1 KO cells as compared to control and are avoided in FANCJ KOs that are not PARPi sensitive (Figure 1G).
Fork lengthening alone does not underlie synthetic lethality caused by PARPi
To understand whether lengthening or gaps confer PARPi toxicity, we sought to uncouple them. One way to accelerate replication is to deplete the cell-cycle regulator p21, which when combined with PARPi, further lengthens replication tracts (Maya-Mendoza et al., 2018), a finding we confirmed in both RPE1 and U2OS cells (Figures 2A, 2B, S2A, and S2B). Notably, the lengthening of tracts in p21-depleted RPE1 or U2OS cells did not enhance PARPi sensitivity (Figures 2C and S2C). By contrast, p21 depletion significantly decreased ssDNA formation (Figures 2D and S2D), consistent with previous observations (Galanos et al., 2016). These findings suggest that fork lengthening following PARPi treatment can occur without a significant induction of ssDNA or sensitivity and that gaps rather than speed could also underlie toxicity (Figure 2E).
Figure 2. PARPi-induced fork acceleration and sensitivity can be uncoupled.
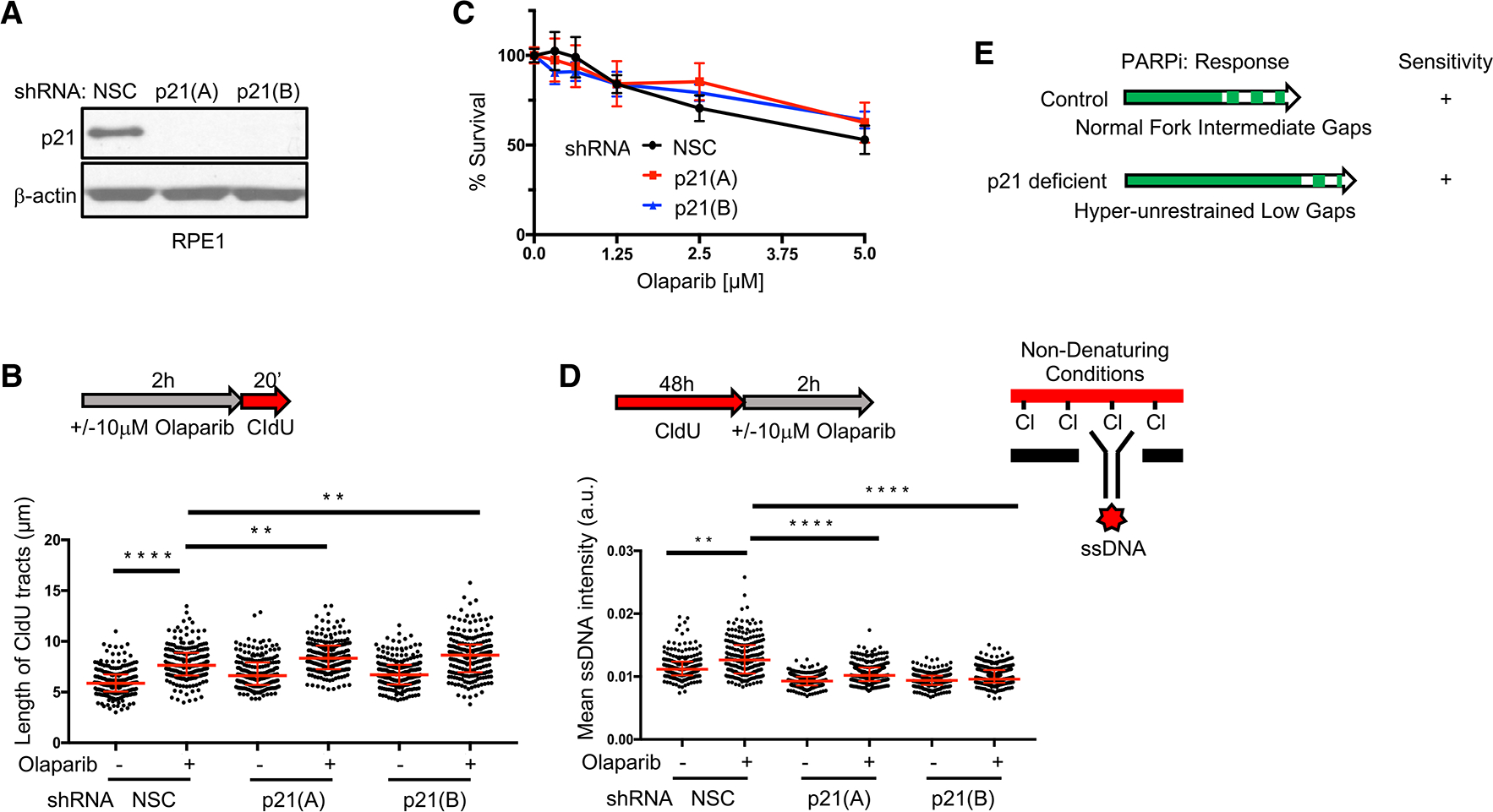
(A) Western blot analysis with the indicated antibodies of lysates from RPE1 cells expressing small hairpin RNA (shRNA) against non-silencing control (NSC), p21(A), and p21(B).
(B) Schematic and quantification of the length of CldU tracts in indicated RPE1 cells following olaparib treatment (10 μM, 2 h). Each dot represents 1 fiber; at least 200 fibers are quantified from 2 biological independent experiments (n = 2).
(C) Cell survival assays for indicated RPE1 cells under increasing concentrations of olaparib. Data represent the mean percentage ± SD of survival for each dot.
(D) Schematic and quantification of mean ssDNA intensity for indicated RPE1 cells following CldU pre-labeling and olaparib release (10 μM, 2 h). At least 200 cells are quantified from n = 2. For (B) and (D), red bars represent the median ± interquartile range. a.u., arbitrary units. Statistical analysis according to Kruskal-Wallis test, followed by Dunn’s test. **p < 0.01, ****p < 0.0001.
(E) Model for fork acceleration without gaps following PARPi treatment in p21-proficient and -deficient backgrounds.
See also Figure S2.
Gap suppression correlates with PARPi resistance and resensitization restores gaps
If a gap toxicity threshold is fundamental to PARPi sensitivity, then RGS should confer resistance. To test this hypothesis, we analyzed the BRCA1-deficient mouse ovarian tumor cell line BR5 and the PARPi-resistant derived cell line BR5-R1 with an unknown mechanism of resistance (Yazinski et al., 2017). We confirmed PARPi and cisplatin sensitivity of BR5 compared to BRCA1-proficient T2 tumor cells (Yazinski et al., 2017; Figures 3A and S3A). At 10 μM PARPi, a concentration at which replication tract lengthening is readily detected in control cells (Figure 1C), replication tract lengths were increased in the BR5 cells and BRCA1-proficient T2 cells, but not in the PARPi resistant BR5-R1 cells (Figure S3B). Moreover, the PARPi-resistant BR5-R1 cells did not show an increase in PARPi-induced gaps compared to the PARPi-sensitive BR5 cells (Figure 3B). Thus, similar to the FANCJ KO cells, BR5-R1 are sensitive to cisplatin (Figure S3A) but resistant to PARPi by a mechanism that includes both fork restraint and RGS.
Figure 3. Gap suppression correlates with PARPi resistance and resensitization restores gaps.
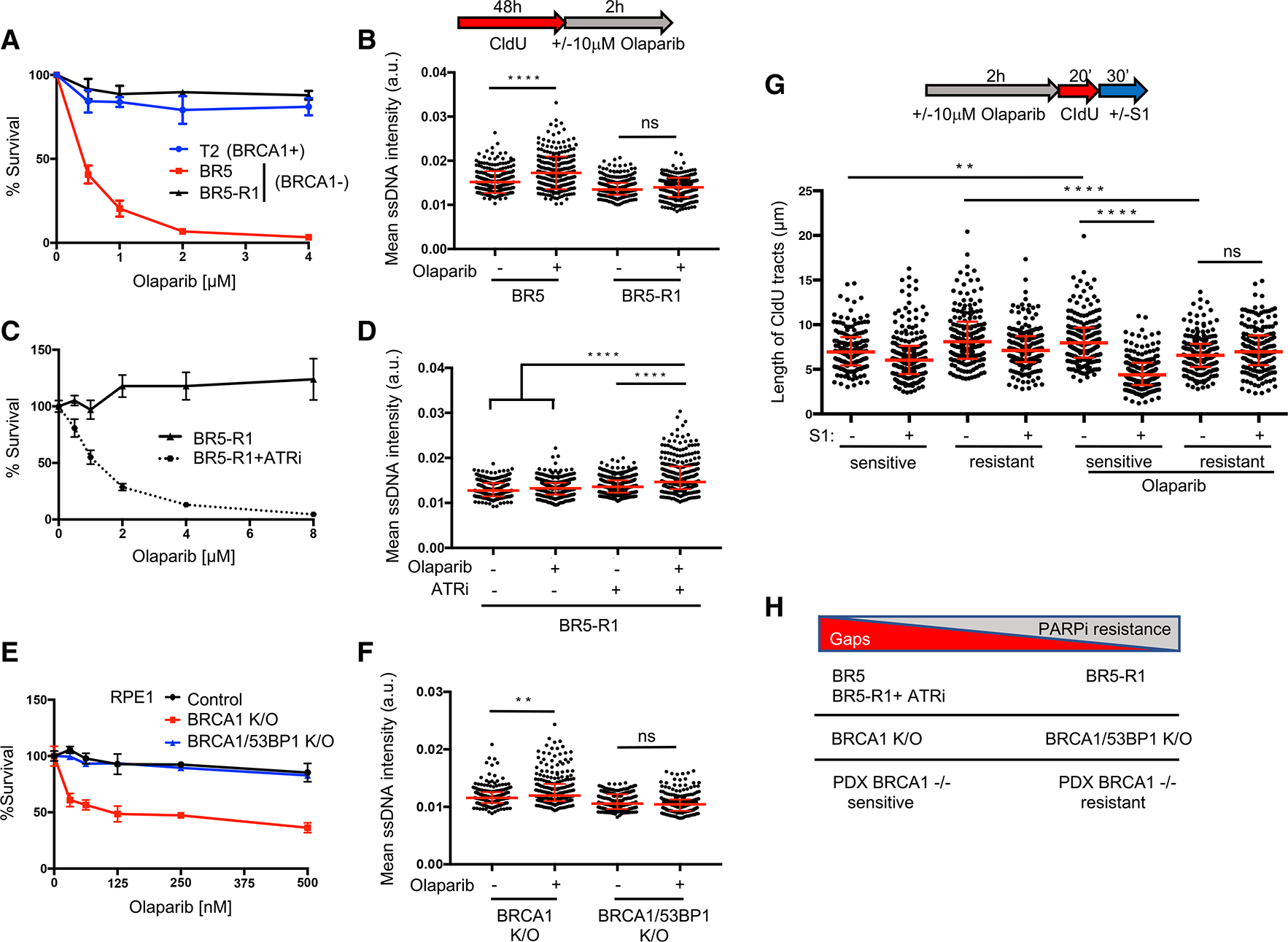
(A) Cell survival assays for T2, BR5, and BR5-R1 (BR5-derived PARPi-resistant cells) cells under increasing concentrations of olaparib.
(B) Schematic and quantification of mean ssDNA intensity for BR5 and BR5-R1 cells following CldU pre-labeling and olaparib release (10 μM, 2 h).
(C) Cell survival assays for BR5-R1 cells under increasing concentrations of olaparib with or without ATR inhibitor (VE-821, 1 μM).
(D) Quantification of mean ssDNA intensity for BR5-R1 cells following CldU pre-labeling and olaparib release (10 μM, 2h), with or without ATR inhibitor (VE-821, 1 μM).
(E) Cell survival assays for indicated RPE1 cells under increasing concentrations of olaparib.
(F) Quantification of mean ssDNA intensity for indicated cell lines following CldU pre-labeling and olaparib release (10 μM, 2 h).
(G) Schematic and quantification of the length of CldU tracts with or without S1 nuclease incubation in indicated PDX samples and olaparib treatment (10 μM, 2 h). Each dot represents 1 fiber; at least 150 fibers are quantified for each sample independently. Red bars represent the median ± interquartile range. Statistical analysis according to Kruskal-Wallis test, followed by Dunn’s test. **p < 0.01, ****p < 0.0001.
(H) Model indicating that gap suppression predicts resistance in response to PARPi in both de novo and engineered backgrounds.
For (A), (C), and (E), data represent the mean percentage ± SD of survival for each dot. For (B), (D), and (F), red bars represent the median ± interquartile range.At least 200 cells are quantified from 2 biological independent experiments.
To further query the relationship between gaps and PARPi toxicity, we analyzed the effect of an ataxia telangiectasia and Rad3-related inhibitor (ATRi), VE-821, shown to restore PARPi sensitivity to PARPi-resistant BR5-R1 cells (Yazinski et al., 2017). We observed that along with PARPi sensitivity, co-incubation with the ATRi generated widespread gap induction (Figures 3C and 3D), raising the possibility that ATRi synergizes with PARPi due to gap induction.
De novo PARPi resistance in BRCA1- or BRCA2-deficient cancers is linked to BRCA reversion mutations that reinstate reading frames and restore BRCA function (Edwards et al., 2008; Norquist et al., 2011; Sakai et al., 2008). To identify whether fork restraint and RGS were also achieved by genetic reversion, we analyzed the BRCA2 mutant PEO1 ovarian cancer cell line and the derived BRCA2 reversion cell clone C4-2 (Sakai et al., 2009). We found that the C4-2 clone had not only gained PARPi resistance but also displayed fork restraint and reduced gap induction (Figures S3C–S3E). These findings suggest that both BRCA1 and BRCA2 are important for restraining replication and preventing gaps in response to PARPi and that resistance by either de novo or reversion mutation correlates with RGS.
Restored FP alone has been proposed to confer chemoresistance in BRCA-deficient cancer cells (Ray Chaudhuri et al., 2016). In particular, depletion of the chromatin remodeler chromodomain helicase DNA binding protein 4 (CHD4) in BRCA2-deficient cells enhances PARPi resistance and restores FP (Guillemette et al., 2015; Ray Chaudhuri et al., 2016). Notably, along with PARPi resistance, we found that CHD4 depletion suppressed gaps following PARPi treatment (Figures S3F–S3H), suggesting that RGS also contributes to PARPi resistance in this model. Restored FP and RGS could be linked, in that one is required for the other. However, this does not appear to be the case. The (BRCA2 S3291A) mutant that is defective in FP (Schlacher et al., 2011) is by contrast proficient in RGS and confers PARPi resistance to BRCA2-deficient Chinese hamster V-C8 cells similar to BRCA2 wild type (WT) (Figures S3I–S3K). These findings further uncouple FP from therapy response (Schlacher et al., 2011) and reveal in another BRCA2-deficient and -proficient model that RGS correlates with PARPi resistance.
Restored HR alone has also been proposed to confer chemoresistance in BRCA-deficient cells. In particular, HR is restored in BRCA1-deficient cells by loss of DNA end resection factors, such as 53BP1 (Bouwman et al., 2010; Bunting et al., 2010). We confirmed that in BRCA1 KO, deletion of 53BP1 (DKO) enhances PARPi resistance (Noordermeer et al., 2018; Figures 3E and S3L) and, remarkably, observed significant RGS (Figure 3F). Correspondingly, we found that 53BP1 depletion in the BRCA1-deficient cancer cell line, BR5, enhanced PARPi resistance and RGS (Figures S3M and S3N). Thus, 53BP1 loss restores both HR (Bouwman et al., 2010; Bunting et al., 2010) and RGS as found here.
To consider the clinical relevance of gaps further, we analyzed PARPi-sensitive versus PARPi-resistant BRCA1 mutant patient-derived xenograft (PDX) that gained PARPi resistance upon serial passage in mice. Tumor samples that were PARPi sensitive showed significant PARPi-induced lengthening of tracts (Figure 3G). Consistent with this lengthening reflecting replication with gaps, elongated tracts dramatically shortened upon S1-nuclease treatment (Figure 3G). In contrast, PARPi-resistant tumor samples did not show PARPi-induced lengthening (but showed shortening), and tracts were not shortened by S1 nuclease cleavage (Figure 3G), indicating that replication gaps were not present. Collectively, these models demonstrate that replication-restraint defects and gaps characterize BRCA deficiency in human and mouse cancer cell lines as well as human tumors, and that restored fork restraint and RGS are associated with both known and de novo mechanisms of PARPi resistance. Moreover, ATRi serves as a tool to resensitize and restore gaps (Figure 3H).
Gaps predict PARPi sensitivity in HR- and FP-proficient cells
PARPi is thought to lead to an accumulation of DSBs that are not effectively repaired in BRCA-deficient cells (Byrum et al., 2019; McCabe et al., 2006). However, PARPi also sensitizes HR-proficient cells (Ström et al., 2011; Zimmermann et al., 2018), suggesting a distinct phenomenon such as gaps could confer PARPi sensitivity. To assess the role of gaps, we sought to identify a model system in which the BRCA pathway was compromised for RGS, but HR and FP were intact. The FA patient fibroblast line with one mutant RAD51 allele (RAD51-T131P) is sensitive to cisplatin or PARPi, but unexpectedly is HR proficient, leading to the proposal that the FP defect underlies the sensitivity (Kolinjivadi et al., 2017b; Mijic et al., 2017; Wang et al., 2015; Zadorozhny et al., 2017; Figure 4A). Conversion of the mutant RAD51 allele to WT enhanced PARPi resistance as expected and restored RGS (Figures 4B and 4C), consistent with the function of RAD51 in RGS (Hashimoto et al., 2010; Henry-Mowatt et al., 2003; Su et al., 2008; Vallerga et al., 2015; Zellweger et al., 2015), and that either the restored RGS and/or FP conferred the greater resistance. By comparison, deletion of the mutant allele (−/WT) provides only partial resistance and RGS as compared to the parental (Figures S4A and S4B), suggesting that two RAD51 alleles are essential for the restoration of RGS and/or FP. As reported, we found that FP is restored by the depletion of RADX, a negative regulator of RAD51 (Bhat et al., 2018; Figures S4C and S4D). Given that HR and FP are presumed to confer PARPi resistance, it was surprising that restored FP in the FA or BRCA1-deficient cells did not alter PARPi sensitivity. Given that gaps remained, it suggested that gaps do not derive from degraded forks and that gaps could mediate the PARPi sensitivity in this model (Figures 4A, 4D, 4E, and S4C–S4F).
Figure 4. PARPi induces gaps and sensitizes cells regardless of HR and fork protection (FP) proficiency.
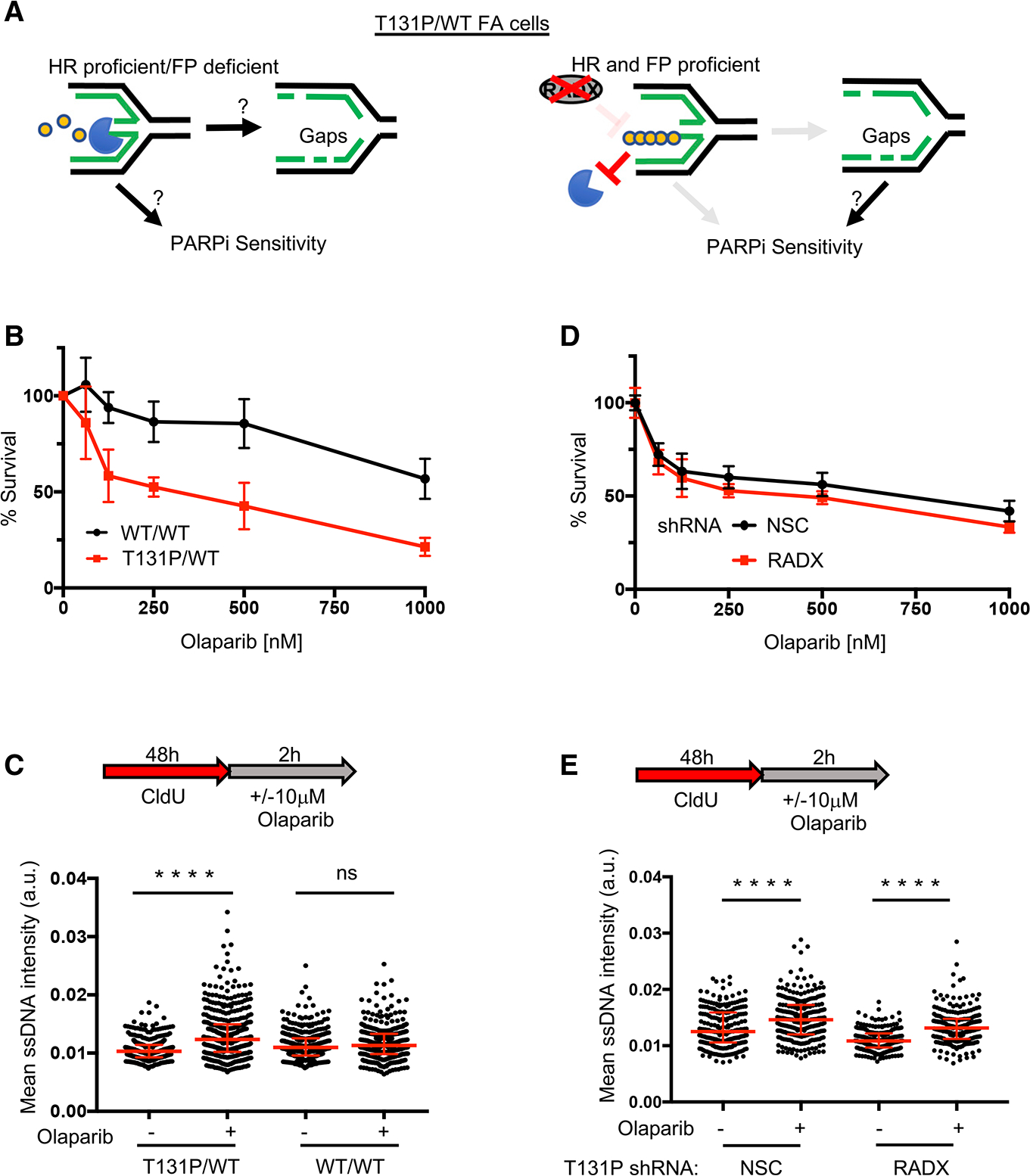
(A) Left: is fork degradation the cause for gaps and PARPi sensitivity in HR-proficient cells? Right: model indicating that PARPi-induced gaps still sensitize cells when FP is restored by depleting RADX.
(B) Cell survival assays for patient fibroblasts (RA2630) RAD51 T131P (T131P/WT) and RAD51 double-allele CRISPR-corrected (WT/WT) cells under increasing concentrations of olaparib.
(C) Schematic and quantification of mean ssDNA intensity for indicated cell lines in (B) following CldU pre-labeling and olaparib release (10 μM, 2 h).
(D) Cell survival assays for indicated cells under increasing concentrations of olaparib. For all cell survival assays, data represent the mean percentage ± SD of survival for each dot.
(E) (E) Quantification of mean ssDNA intensity for indicated cell lines following CldU pre-labeling and olaparib release (10 μM, 2 h). For (C) and (E), red bars represent the median ± interquartile range. At least 200 cells are quantified from 2 biological independent experiments. All statistical analysis according to Kruskal-Wallis test, followed by Dunn’s test. ****p < 0.0001.
Targeting RPA augments synthetic lethality between PARPi and BRCA1 deficiency
The increased replication gaps in BRCA-deficient cells could render the single-stranded binding protein RPA essential for viability. To address this idea, we used single-molecule localization microscopy (STORM) for direct visualization and quantification of ssDNA-bound RPA based on individual replication assemblies (5-ethynyl-2′-deoxyuridine-positive [EdU+], minichromosome maintenance complex component 6-positive [MCM6+], and PCNA+ sites) (Lee et al., 2021). To measure the amounts of incorporated RPA within individual replisomes, we performed unbiased correlation-based image analyses in control, BRCA1-, and FANCJ-depleted osteosarcoma cell line (U2OS) cells following PARPi treatment (Figures 5A, 5B, and S5A). At 10 μM PARPi, we observed that the average RPA fork density was enhanced in the BRCA1-depleted and control cells, as compared to FANCJ-depleted U2OS cells (Figure 5B), which is consistent with BRCA1 being required to suppress gaps and FANCJ being required to make gaps.
Figure 5. PARPi synthetic lethality in BRCA1-deficient cells is linked to RPA exhaustion and can be augmented by targeting RPA.
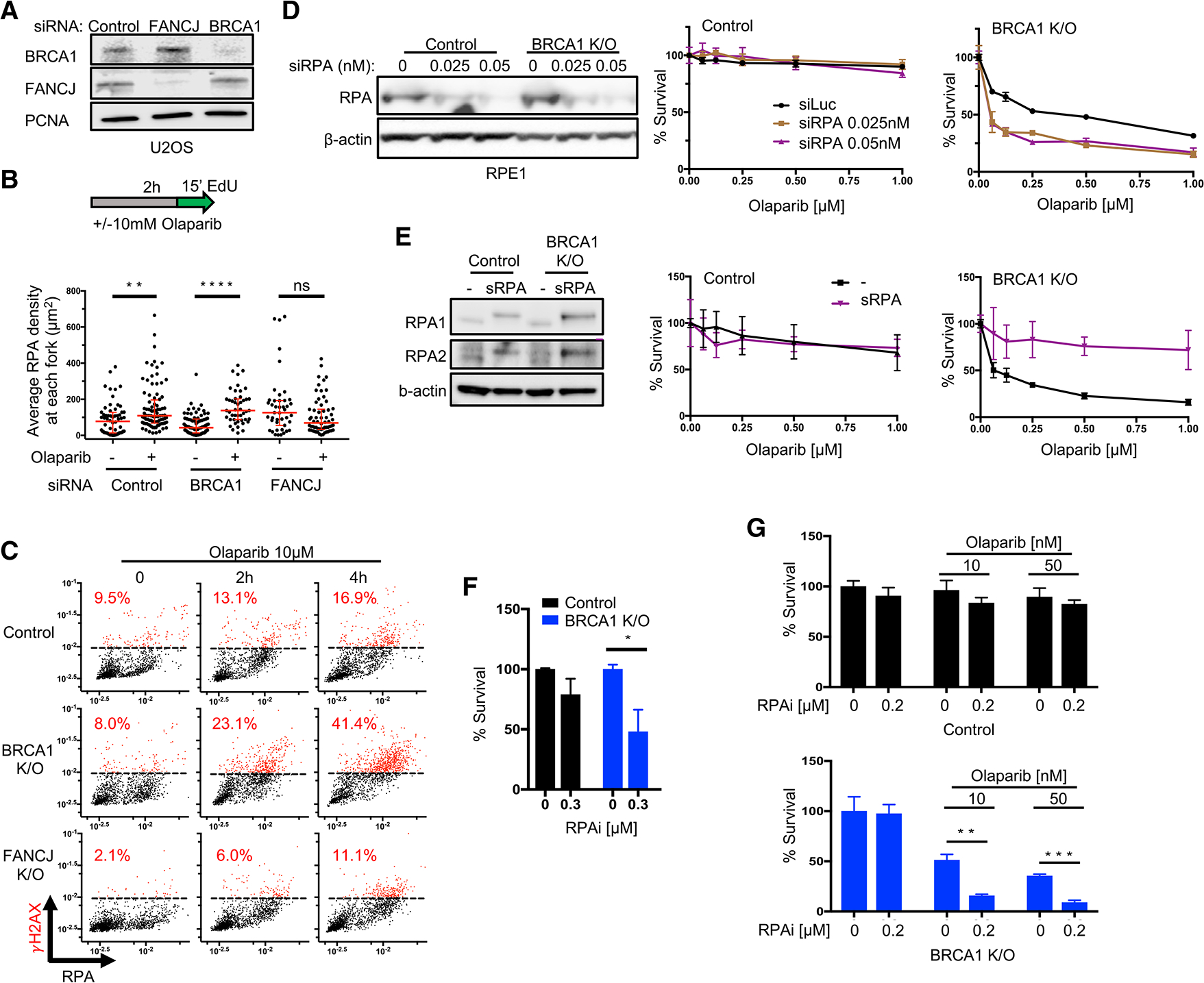
(A) Western blot analysis with the indicated antibodies of lysates from U2OS cells expressing siRNA against non-silencing control, FANCJ, and BRCA1.
(B) Schematic and quantification of average RPA density at each replication fork in STORM analysis for indicated cells and treatment (10 μM, 2 h olaparib). At least 40 single cells are quantified from 2 biological independent experiments (n = 2), respectively, for each group; every dot represents the pair-localized RPA density from 1 nucleus. All red bars represent the median ± interquartile range. Statistical analysis according to Kruskal-Wallis test, followed by Dunn’s test.
(C) Control, BRCA1 KO and FANCJ KO RPE1 cells are treated with olaparib (10 μM) at indicated times and stained for CB-γH2AX/RPA. Dashed lines indicate maximum γH2AX levels in untreated control cells. Cells higher than those are marked in red and calculated for percentages. Each dot represents 1 cell; cells (n = 1,500 ± 300) are collected from n = 2.
(D) Western blot analysis with the indicated antibodies of lysates from control and BRCA1 KO RPE1 cells expressing siRNA against RPA in increasing doses (0, 0.025, and 0.05 nM), and cell survival assays for cells above under increasing concentrations of olaparib.
(E) Western blot analysis for indicated antibodies in control and BRCA1 KO RPE1 cells with negative mock transfection (−) and overexpression of superRPA cDNA (sRPA shows higher bands in immunoblots due to P2A tag), and cell survival assays for cells above under increasing concentrations of olaparib.
(F) Clonogenic assays for control and BRCA1 KO cells treated with RPA inhibitor (RPAi, NERx-329) under indicated doses. Mean survival percentages of n = 4 with SEM are collected for all of the cells.
(G) Clonogenic assays for indicated cells under increasing olaparib with sublethal dose of RPAi (NERx-329). Mean survival percentages of n = 3 with SEM are collected for all of the cells. Statistical analysis according to t test. All p values are described in Quantification and statistical analysis.
For (D) and (E), data represent the mean percentage ± SD of survival for each dot.
See also Figure S5.
To test the hypothesis that PARPi is synthetic lethal with BRCA1 deficiency due to the generation of gaps that exceed levels that can be protected by RPA, we addressed whether following PARPi, the BRCA1 KO cells more readily exhausted the pool of RPA (Toledo et al., 2013). Following PARPi, the RPE1 cell lines gained chromatin-bound (CB)-RPA that correlated with an increase in γ-H2AX, most notably in BRCA1 KO cells (Figures 5C and S5B). Consistent with an enhanced reliance on RPA to protect ssDNA, we also observed that RPA depletion reduced the survival of BRCA1 KO cells to PARPi as compared to the other RPE1 cells (Figures 5D and S5C). When RPA subunits were instead overexpressed (Toledo et al., 2013), PARPi resistance was uniquely elevated in BRCA1 KO cells (Figure 5E). These results suggest that RPA is a critical buffer of PARPi toxicity in BRCA1-deficient cells. Consistent with this premise, a small molecule (RPAi, compound NERx-329) that blocks RPA-ssDNA interaction (Gavande et al., 2020; Sriramkumar et al., 2020) displayed single-agent activity in the BRCA1 KO cells (Figures 5F and S5D–S5F). Furthermore, at a sublethal dose, the RPAi augmented synthetic lethality between BRCA1 KO and PARPi while having little impact on the control or FANCJ KO RPE1 cells (Figures 5G, S5F, and S5G). These findings further implicate replication gaps in PARPi synthetic lethality and indicate that targeting the RPA protection of gaps magnifies synthetic lethality in BRCA1-deficient cells.
Gaps converted to DSBs could underlie the killing of BRCA-deficient cells such as when gaps either interface with a second round of replication or are digested by nucleases (Toledo et al., 2013). Alternatively, DSBs could derive from other sources such as apoptosis. To address this idea, we identified a dose of PARPi that BRCA1 KO cells uniquely activated apoptosis as measured by induction of PARP1 and caspase-3 cleavage and verified suppression by apoptosis inhibitors, Z-VAD-FMK, or emricasan (Figures S5H and S5I). We observed that the inhibition of apoptosis not only elevated the resistance of BRCA1 KO but also suppressed PARPi-induced DSBs (Figures S5J and S5K), suggesting that apoptosis contributes to the loss of cell viability and DSB formation in BRCA1-deficient cells.
BRCA1-deficient cells have OFP defects
PARPi is synthetic lethal with loss of genes functioning in OFP and PARP1 functions in OFP (Hanzlikova et al., 2018; Ward et al., 2017). To address the possibility that PARPi is synthetic lethal with BRCA1 deficiency due to combined OFP defects, we sought to analyze endogenous PAR polymerization, which when elevated in unperturbed cells, suggests a lagging strand problem (Hanzlikova et al., 2018). We verified FEN1 depletion in conjunction with the PAR catabolism by PAR glycohydrolase inhibitor (PARGi) (Hanzlikova et al., 2018) elevated PAR levels as also observed following treatment with methyl methanesulfonate (MMS), a drug that induces ssDNA gaps (Hashimoto et al., 2010; Lundin et al., 2005; Sgagias et al., 2004; Figures S6A and S6B). While following these perturbations, PAR was similarly elevated in the distinct RPE1 cell lines (Figures S6C and S6D), and we found that they were distinct in unchallenged conditions. Specifically, as compared to control cells or FANCJ KO cells that displayed remarkably low PAR levels, BRCA1 KO cells had higher PAR levels (Figure 6A), suggesting that the source of PAR was not from DNA base damage but from an OFP defect.
Figure 6. PARPi-sensitive cells display high PAR reversed upon PARPi resistance.
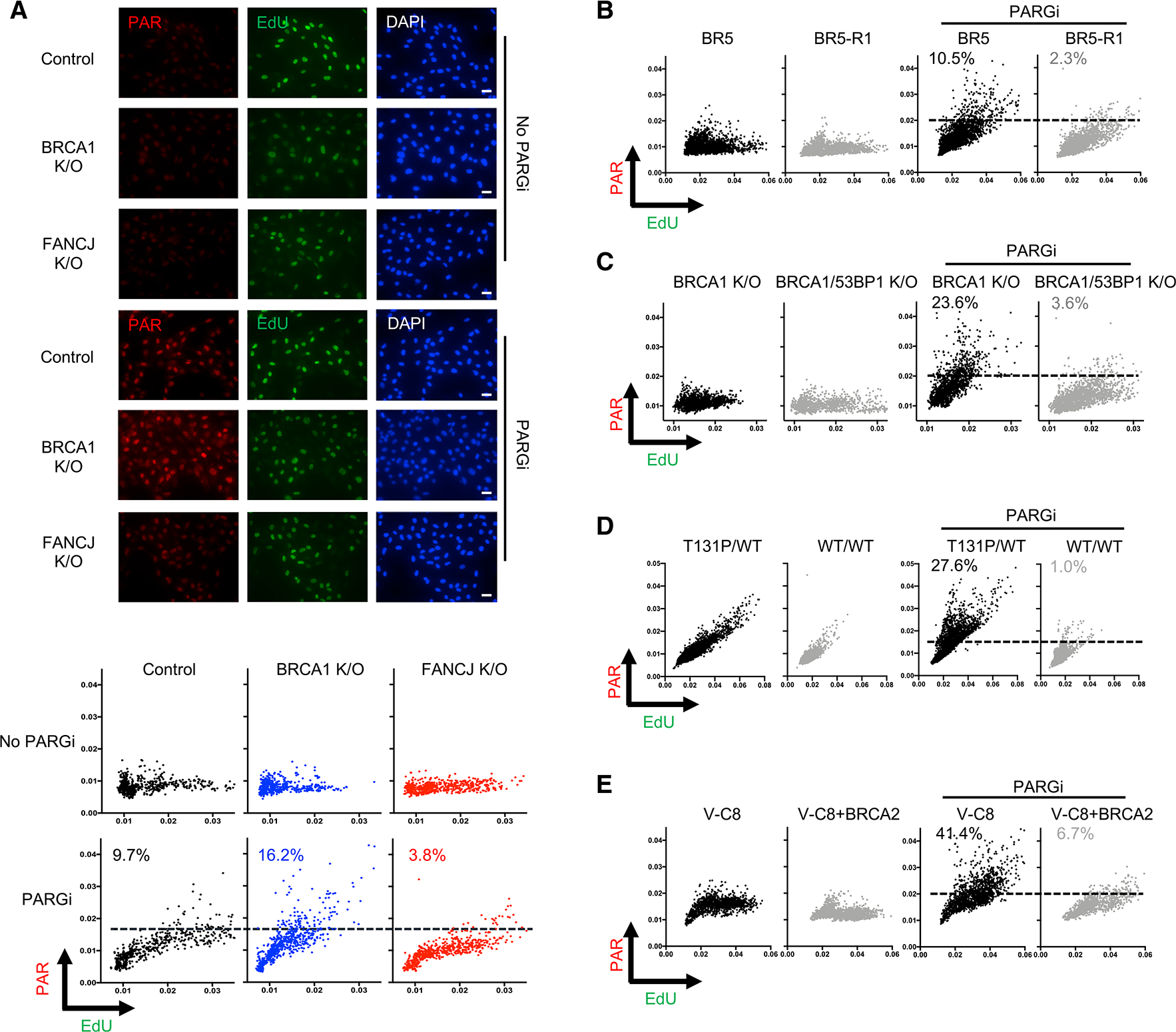
(A) Top: representative images of immunofluorescence for control, BRCA1 KO, and FANCJ KO RPE1 cells stained for poly(ADP-ribose) (PAR) and EdU. Cells were incubated without (top) or with (bottom) PARG inhibitor (10 μM) for PAR and EdU for 20 min to detect DNA synthesis. Scale bars, 50 μm. Bottom: scatterplot of indicated cells for mean PAR and EdU intensity per nucleus after incubation with EdU with or without PARG inhibitor (10 μM, 20 min, using DMSO as control). Dashed lines indicate maximum PAR level in untreated control cells. Each dot represents 1 cell; at least 500 cells are collected for each from 3 biological independent experiments (n = 3).
(B) Scatterplot of mean PAR and EdU intensity for BR5 and BR5-R1 cells. Cells are incubated with EdU with or without PARG inhibitor (10 μM, 20 min) for indicated groups. At least 2,000 cells are collected from n = 3. Dashed lines indicate maximum PAR level in untreated BR5-R1 cells.
(C) Scatterplot of PAR and EdU for BRCA1 and BRCA1/53BP1 KO cells. Cells are incubated the same as above for indicated groups. At least 1,200 cells are collected from n = 3. Dashed lines indicate maximum PAR level in untreated BRCA1 KO cells.
(D) Scatterplot of PAR and EdU for patient fibroblasts (RA2630) RAD51 T131P (T131P/WT) and RAD51 double-allele CRISPR-corrected (WT/WT) cells. Cells are treated the same as above. At least 2,000 cells are collected from n = 3. Dashed lines indicate maximum PAR level in untreated WT/WT cells.
(E) Scatterplot of PAR and EdU for V-C8 and V-C8+BRCA2 cells. Cells are treated the same as above. At least 1,200 cells are collected from n = 2. Dashed lines indicate maximum PAR level in untreated V-C8+BRCA2 cells. For all of the above, cells higher than those lines are calculated for percentages, respectively.
See also Figure S6.
To further assess whether OFP defects were the source of S phase PAR in BRCA1 KO cells, we used emetine, an inhibitor of DNA replication that prevents the formation of OFs (Burhans et al., 1991). As found previously, emetine completely suppressed the appearance of S phase PAR in control and FEN1-depleted cells (Hanzlikova et al., 2018), as well as in BRCA1 KO cells (Figures S6E and S6F), consistent with PAR requiring lagging strand synthesis. In summary, these findings strongly implicate unligated OFs are the source of gaps and cause of S phase PARylation in BRCA1 KO cells.
Next, we examined whether PAR mirrored gap induction in the PARPi-sensitive and -resistant models. PAR was significantly lower in the de novo PARPi-resistant BR5-R1 cells as compared to PARPi-sensitive BR5 cells (Figure 6B). Moreover, PARPi-resistant BRCA1 and 53BP1 DKO cells displayed significantly lower PAR levels than PARPi-sensitive BRCA1 KO cells (Figure 6C). Furthermore, PARPi-resistant corrected FA cells (WT/WT) had dramatically reduced PAR levels compared to the parental cells (T131P/WT) (Figure 6D). In PARPi-sensitive V-C8 cells, PAR was also suppressed, with BRCA2 complementation (Figure 6E) akin to previous findings (Gottipati et al., 2010). These results suggest that OFP defects are corrected in cells that gain PARPi resistance.
53BP1 deletion restores lagging strand synthesis in BRCA1 KO cells via alternative OFP
To decipher the OFP defect in BRCA1 KO cells, we considered that FEN1 depletion induced similar PAR levels with or without BRCA1 (Figures S6A–S6D), consistent with intact canonical OFP in BRCA1 KO cells. Thus, we analyzed the chromatin accumulation of PARP1 that functions in backup OFP. As compared to controls, we observed that CB-PARP1 levels were increased in BRCA1 KO cells (Figure 7A), consistent with the higher PAR. In fact, the PARP1 “trapping” in untreated BRCA1 KO cells was similar to control cells treated with PARPi (Figure 7A), indicating that PARP1 was aberrantly CB in BRCA1 KO cells. Lagging strand defects generate gaps that trigger PARP1 activation that attempts to rectify the problem by recruiting ssDNA binding protein XRCC1 (Caldecott et al., 1996; El-Khamisy et al., 2003; Hanzlikova et al., 2017; Masson et al., 1998; Schreiber et al., 2002). However, unlike PARP1, XRCC1 was aberrantly low in BRCA1 KO cells, with its reduction primarily in EdU+ cells (Figures 7B and 7C). BRCA1 KO cells also had a reduced EdU intensity in EdU+ cells and elevated 53BP1 nuclear foci (Figures 7D, 7E, and S7A), suggesting that replication was less robust and/or underreplicated (Lukas et al., 2011; Michelena et al., 2021; Pellegrino et al., 2017; Saredi et al., 2016). These data suggest that in BRCA1 KO cells, PARP1 fails to effectively recruit XRCC1 to engage backup lagging strand synthesis (Hanzlikova and Caldecott, 2019).
Figure 7. OFP defects in BRCA1-deficient cells are suppressed by 53BP1 depletion.
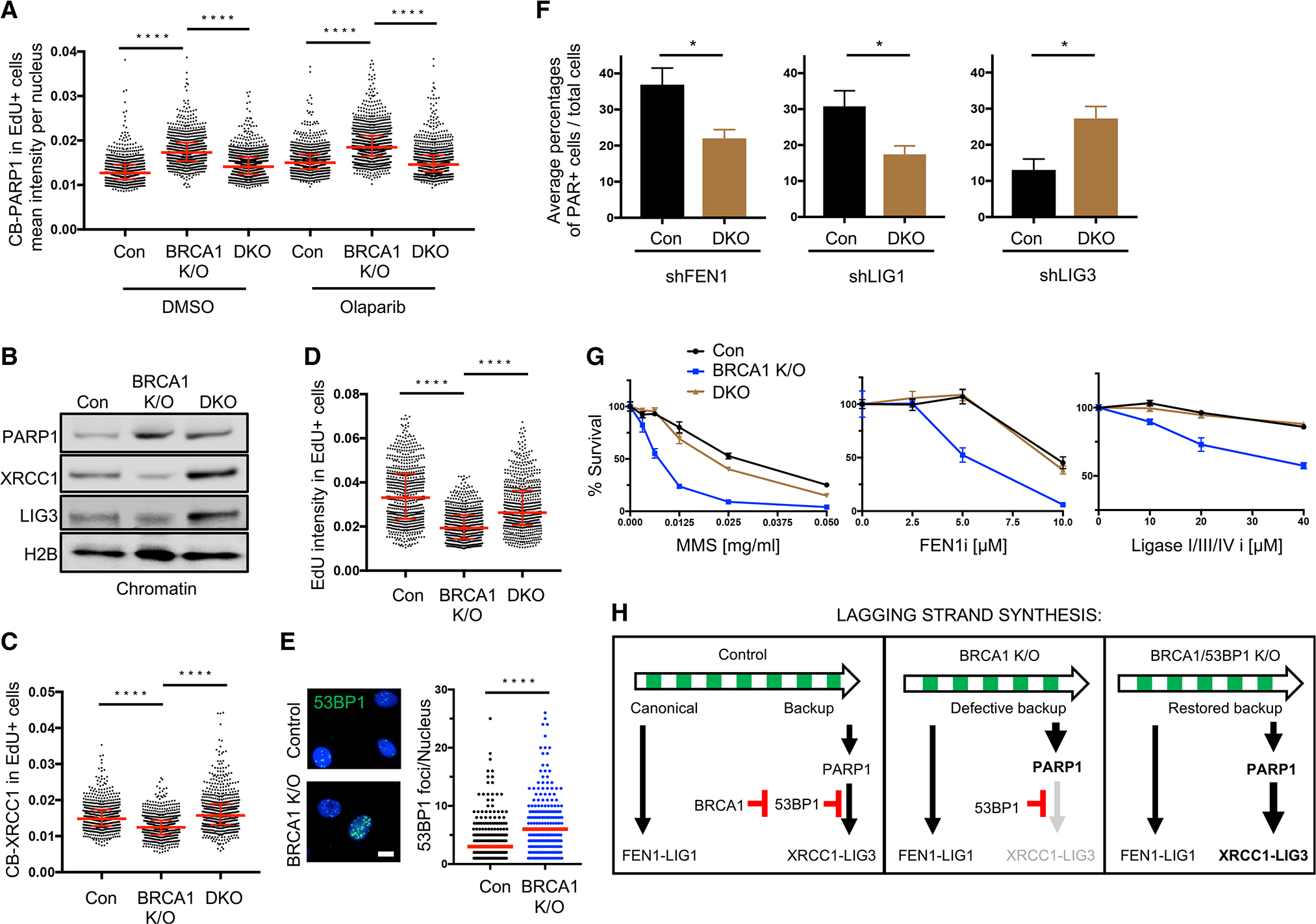
(A) Quantification of chromatin-bound PARP1 (CB-PARP1) for control, BRCA1 KO, and BRCA1/53BP1 KO (labeled as Con, DKO in figure, same as follows) RPE1 cells with or without treatment (0.5 μM, 2 h olaparib), with EdU added to the final 20 min. For quantification, EdU+ cells were gated according to positive EdU incorporation.
(B) Western blot analysis for indicated antibodies in chromatin extraction for control, BRCA1 KO, and BRCA1/53BP1 KO RPE1 cells.
(C) Quantification of CB-XRCC1 for untreated control, BRCA1 KO, and BRCA1/53BP1 KO RPE1 cells, with EdU incubated for 20 min. For quantification, EdU+ cells were gated according to positive EdU incorporation.
(D) Quantification of mean EdU intensity from EdU+ cells. EdU was incubated for 20 min in indicated cells. For (A), (C) and (D), red bars represent the median ± interquartile range. At least 600 cells are quantified from 3 biological independent experiments (n = 3). All statistical analysis according to Kruskal-Wallis test, followed by Dunn’s test. ****p < 0.0001.
(E) Immunofluorescence showing 53BP1 chromatin foci in untreated control and BRCA1 K/O RPE1 cells. Cells ≥1 foci per nucleus were measured. At least 150 cells were quantified from n = 3. Red bars represent the median number. Statistical analysis according to Mann-Whitney test. ****p < 0.0001. Scale bar, 10 μm.
(F) (F) Average percentages of PAR+ over total for indicated cells after PARGi (10 μM, 20 min) and EdU incubation. For quantification, PAR+ cells were gated by maximum PAR level in control NSC cells for each experiment. Average values of n = 4 with SEM are calculated. *p < 0.05.
(G) Cell survival assays for indicated cells under increasing concentrations of MMS, FEN1 inhibitor (FEN1i), and ligase I/III/IV inhibitor (Ligase I/III/IV i). Data represent the mean percentage ± SD of survival for each dot.
(H) Model summarizing the function of BRCA1 and 53BP1 in regulating lagging strand synthesis, and their interactions with the backup OFP pathway.
See also Figure S7.
Could 53BP1 interfere with the function of PARP1 in lagging strand synthesis? In accordance with the reduced PAR (Figure 6C), we observed that 53BP1 deletion in the BRCA1 KO cells reduced PARP1 chromatin association (Figures 7A and 7B). Notably, XRCC1, LIG3 (Figures 7B and 7C), and replication were instead elevated (Figure 7D), suggesting that backup OFP was in place (Arakawa and Iliakis, 2015; Hanzlikova and Caldecott, 2019; Kumamoto et al., 2021). Similarly, 53BP1 depletion suppressed PAR and elevated XRCC1 and LIG3 in BRCA1-deficient BR5 cells, indicating that restored alternative OFP was not specific to the RPE1 cells (Figures S7B and S7C). Consistent with a switch in the mechanism of OFP, control cells had a greater PAR induction than DKO cells following depletion of the canonical OFP factors FEN1 or LIG1, whereas DKO had a greater PAR induction following depletion of the alternative OFP factor LIG3 (Figures 7F, S7D, and S7E). We also observed that 53BP1 deletion increased the resistance of BRCA1 KO cells to inhibitors of FEN1 (FEN1i) or ligases (ligase I/III/IV i) as well as to the gap-inducing drug MMS (Hashimoto et al., 2010; Lundin et al., 2005; Sgagias et al., 2004). These findings suggest that as compared to the DKO cells and control cells that have intact XRCC1-LIG3 backup, the BRCA1 KO cells are more vulnerable to the titration of a pan-ligase inhibitor or loss of canonical OFP factor FEN1 (Figure 7G). Our data suggest that BRCA1 deficiency activates PARP1, but fails to stimulate XRCC1-LIG3 as a backup pathway for OF ligation unless 53BP1 is deleted (Figure 7H).
DISCUSSION
As clinical interventions for PARPi resistance are lacking, understanding the sensitizing lesion is of critical importance. The DSB framework was logical given the synthetic lethality between PARPi- and BRCA-deficient cells that are defective in HR and FP, functions that fix and limit DSBs. However, this DSB model does not fully align with cell models and clinical outcomes. Furthermore, suppression and repair of replication-associated ssDNA gaps is also a function of the BRCA and PARP1 pathways (Hanzlikova et al., 2018; Kolinjivadi et al., 2017a; Panzarino et al., 2021). Thus, we considered whether ssDNA gaps deriving from the combined loss of PARP1 and BRCA instead caused synthetic lethality. Consistent with the gap model, we observe that RGS underlies acquired PARPi resistance and resensitization restores gaps. We also reveal that replication gaps in BRCA1-deficient cells result from defects in OFP and that 53BP1 deletion reverses this defect by engaging a backup lagging strand ligation pathway mediated by PARP1-XRCC1-LIG3 (Arakawa and Iliakis, 2015; Hanzlikova et al., 2018; Kumamoto et al., 2021). Accordingly, DKO cells gain XRCC1 and LIG3 as well as resistance to inhibitors of PARP1, FEN1, pan-ligases, or the methylating agent MMS. Significantly, LIG3 depletion in DKO cells restores PAR as well as PARPi sensitivity, an outcome linked to widespread gap induction, but not DSBs (Dias et al., 2021). Clinical relevance is also supported as gaps accumulate more excessively in BRCA-deficient immortalized cells, cancer cell lines, and patient tumors as compared with BRCA-proficient controls. Notably, BRCA1-deficient cells also show sensitivity to RPA inhibitors that augments synthetic lethality with PARPi. We, therefore, propose that PARPi kills BRCA1-deficient cells due to compounded lagging strand gaps that exhaust RPA pools and that gap accumulation is the root cause of therapy sensitivity and “BRCAness” (Panzarino et al., 2021).
Our data also highlight that PARPi sensitivity does not stem from a speed threshold, but rather a gap threshold that exceeds RPA pools. As reported, we find that PARPi lengthens replication tracts (Maya-Mendoza et al., 2018) that are significantly longer in BRCA-deficient cells. However, replication tracts are discontinuous. In contrast, hyper-lengthened tracts are continuous in p21-depleted cells that are not PARPi sensitive, implicating gaps as the toxic factor. Lengthening without gaps could be mediated by translesion synthesis (TLS). p21 is a TLS inhibitor (Avkin et al., 2006), and enhanced TLS in BRCA2-deficient cells confers resistance (Guillemette et al., 2015; Nayak et al., 2020). Moreover, TLS suppresses gaps induced by chemotherapy or oncogenes (Nayak et al., 2020) and counters gaps in yeast (Wong et al., 2020). When not avoided by TLS and exposed by drugs, gaps and RPA exhaustion could contribute to outcomes of replication stress (Feng and Jasin, 2017; Lai et al., 2017; Schoonen et al., 2017; Toledo et al., 2013). Correspondingly, modulating the gap threshold via RPA depletion or inhibition enhanced the sensitivity of BRCA1-deficient cells to PARPi, whereas RPA overexpression limited this sensitivity. Again, RPA loss could expose gaps that convert to DSBs; however, our findings suggest that a significant number of PARPi-induced DSBs stem from apoptosis and not directly from PARPi, raising the possibility that DSBs are not the primary killing event. It remains unclear whether DSBs not inhibited by apoptosis stem directly from PARPi and are cell lethal or rather result from the partial inhibition of apoptosis or other technical issues. A full kinetic analysis will also be critical to decipher the relationship between these events, but suggest that genotoxin-induced DSBs derive in large part from apoptosis (Faivre et al., 2003; Panzarino et al., 2021; Sané and Bertrand, 1998).
Our gap model is in line with recent reports revealing that gaps forming in BRCA-deficient cells are independent of DSBs (Somyajit et al., 2021). While it is possible that eventual PARPi-induced fork degradation, collapse, and/or DSBs contribute to the toxicity in BRCA-deficient cells, we present a series of separation-of-function models that indicate a greater prediction between the immediate induction of gaps and PARPi toxicity than loss of either HR or FP (Table S1). An important comparison revealing gaps as a discriminating factor is between BRCA1 and FANCJ. Both are mutated in hereditary breast/ovarian cancer and FA, with their loss disrupting both HR and FP (Litman et al., 2005; Peng et al., 2006, 2018), but unlike BRCA1 loss, FANCJ loss does not sensitize to PARPi or generate gaps or PAR in unchallenged cells. Either FANCJ is not required for processing PARPi-induced DSBs or DSBs are not the sensitizing lesion. Logically, if DSBs were the sensitizing lesion, then HR-proficient cells should repair DSBs and show PARPi resistance. However, the HR-proficient FA cell line RAD51-T131P is sensitive, albeit not hypersensitive, possibly because of its slow growth or proficiency in HR and/or post-replication gap repair that is distinct from RGS (Su et al., 2008; Wang et al., 2015). However, the sensitivity does not change even when FP is restored by RADX depletion, indicating that HR and FP are not sufficient to confer PARPi resistance.
The concept that synthetic lethality derives from a distinct lesion is further supported by the range of synthetic lethal combinations unrelated to DSB repair (Hanzlikova et al., 2018; McDermott et al., 2019; Pillay et al., 2019; Ward et al., 2017; Zimmermann et al., 2018). Moreover, the synthetic lethality between PARPi and XRCC1, FEN1 and BRCA deficiency, or a PCNA OFP mutant and BRCA1 deficiency also implicates gaps due to OFP defects as the sensitizing lesion (Guo et al., 2020; Mengwasser et al., 2019; Ström et al., 2011; Thakar et al., 2020; van Wietmarschen and Nussenzweig, 2018). Even within the classical paradigm of HR deficiency conferring PARPi response, synthetic lethality includes widespread gap induction (Hewitt et al., 2021; Verma et al., 2021) that at the very least indicates that gaps are a biomarker of response. Interestingly, not only does the BRCA-RAD51 pathway function in RGS but it also functions in post-replication gap filling (Hashimoto et al., 2010; Kolinjivadi et al., 2017a; Lossaint et al., 2013; Panzarino et al., 2021; Somyajit et al., 2021; Su et al., 2008). Thus, distinct BRCA functions could facilitate the removal of PARP1, process unligated OFs, or fill gaps in post-replication.
Gaps from OFP defects could lead to the appearance of unrestrained replication. Similar to loss of OFP such as FEN1, LIG1, PCNA, or PARP1, loss of BRCA-RAD51 pathway proteins is associated with unrestrained replication (Hanzlikova et al., 2018; Hashimoto et al., 2010; Henry-Mowatt et al., 2003; Kolinjivadi et al., 2017b; Lachaud et al., 2016; Lossaint et al., 2013; Luke-Glaser et al., 2010; Maya-Mendoza et al., 2018; Panzarino et al., 2021; Saxena et al., 2019; Su et al., 2008; Sugimura et al., 2008; Thakar et al., 2020; Xu et al., 2019). Mechanistically, it is possible that lagging strand gaps interfere with nascent strand annealing reactions that slow and remodel replication forks. Correspondingly, in FANCJ-deficient cells, there is an absence of lagging strand gaps possibly due to parental DNA forming G4s (Lee et al., 2021; Odermatt et al., 2020), and replication is hyperrestrained.
Our studies also have important implications for how 53BP1 deletion rescues BRCA1 deficiency from PARPi sensitivity as well as embryonic lethality (Bouwman et al., 2010; Bunting et al., 2010; Cao et al., 2009). 53BP1 functions in both unperturbed and perturbed replication (Lukas et al., 2011; Schmid et al., 2018). Dual deficiency in replication and BRCA1 expression could enhance 53BP1 in chromatin because replication dilutes its recruiting histone mark that in turn favors BRCA1 binding (Michelena et al., 2021; Pellegrino et al., 2017; Saredi et al., 2016). In BRCA1 KO cells, 53BP1 could interfere with XRCC1 recruitment or its attempts to process the lagging strand. Notably, loss of 53BP1 leads to gap formation (Schmid et al., 2018), providing a platform for PARP1-XRCC1 binding. Alternatively, given that 53BP1 exists in a complex with DNA polymerase α (Polα) (Mirman et al., 2018), 53BP1 deletion could in part rescue replication in BRCA1 KO cells by liberating a sequestered pool of Polα. If this scenario is correct, then inhibitors of Polα would resensitize PARPi-resistant BRCA1/53BP1-deficient cells. Conceivably, lagging strand gaps in BRCA-RAD51-deficient cellscause the vulnerability to cytotoxic nucleotides (Fugger et al., 2021), trigger sister chromatid exchange reactions (Henderson et al., 1985), and/or induce chromatid-type aberrations (Nagaraju and Scully, 2007).
In summary, our findings establish a model to help further develop targeted treatment with PARPi, such that combined therapies maximize exposed lagging strand gaps. It will be crucial to consider whether gaps are the genetic vulnerability resensitizing PARPi-resistant cells (Amé et al., 2009; Gogola et al., 2018; Smith et al., 2015) or whether they underlie combinations that synergize with PARPi (Horton et al., 2014; McDermott et al., 2019; Michelena et al., 2018; Muvarak et al., 2016; Smith et al., 2015). The rationale for limiting PARPi to HR-defective cancers is in question because of significant clinical benefit across ovarian cancer patients regardless of BRCA status (Ledermann and Pujade-Lauraine, 2019; Pilié et al., 2019). Moreover, a key future question will be whether, in a range of synthetic lethality models, gaps confer toxicity, RGS confers chemoresistance, and targeting RGS pathways will maximize the efficacy of genotoxic chemotherapy. These findings highlight the importance of considering ssDNA gaps as a critical biomarker and determinant of PARPi synthetic lethality.
Limitations of the study
Based on RGS being closely coupled with therapy response, whereas HR and FP in several cases do not correlate, our findings support a ssDNA gap framework for PARPi synthetic lethality. However, our findings do not differentiate whether toxicity reflects the number, size, and/or persistence of ssDNA gaps. Short-term gaps promote an adaptive response to tolerate genotoxic chemotherapy (Quinet et al., 2020). Ideally, ssDNA sequencing or direct detection tools will facilitate the defining of a replication gap threshold underlying PARPi toxicity. Moreover, although HR-proficient cells display PARPi sensitivity, suggesting that DSBs are not required, the conversion of gaps to DSBs cannot be ruled out as a contributing factor. Furthermore, our study does not provide visual insight with microscopy toward the relationship between replication gaps, DSBs, and/or stalled forks that in yeast were found to be physically distinct entities (Wong et al., 2020).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sharon Cantor (sharon.cantor@umassmed.edu)
Materials availability
Materials associated with this study are available upon request from the lead contact.
Data and code availability
Unprocessed blots, gels, and microscopy images are available on Mendeley Data at https://doi.org/10.17632/g96fktpb58.1. Raw Data for STORM analysis will be made available by the corresponding author upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and gene editing
Human RPE1-hTERT cell lines were grown in DMEM+GlutaMAX-I (GIBCO, 10569) supplemented with 10% FBS and 1% Pen Strep (100 U/ml). U2OS, 293T, PEO1, C4-2 and V-C8 derived cell lines were grown in DMEM (GIBCO, 11965) supplemented with 10% fetal bovine serum (FBS) and 1% Pen Strep (100 U/ml). T2, BR5, BR5-R1 cell lines were cultured in DMEM (CORNING cellgro, 15-017-CV) with 10% FBS, penicillin and streptomycin (100 U/ml each), and 1% L-glutamine (Yazinski et al., 2017). The resistant cell line BR5-R1 was maintained in 1 μM olaparib. FA Patient fibroblasts (RA2630 T131P, CRISPR corrected clone 3–39 ΔMUT/WT or −/WT, and double-allele CRISPR corrected clone WT/WT) were cultured in DMEM (GIBCO, 11965) supplemented with 10% FBS, 1% Pen Strep (100 U/ml), 1% GlutaMAX-I, 1% MEM NEAA and 1% Sodium pyruvate (Wang et al., 2015). UWB1 and complemented cells were maintained in MEGM Bullet Kit (Lonza, CC-3150): RPMI-1640 (CORNING cellgro, 10-040-CV) 1:1 with 3% FBS and 1% Pen Strep. All the cell lines were cultured at 37°C, 5% CO2. The generation of RPE1-hTERT TP53−/− BRCA1 K/O and BRCA1/53BP1 double K/O Cas9 cells were described elsewhere (Noordermeer et al., 2018). FANCJ gene knockout in RPE1-hTERT TP53−/− Cas9 cells was introduced by using two synthesized gRNAs (gRNA #1: GGGTCGAGGAAAGGTAACGG, gRNA #2: GGCAATCACCACACCCTTCA) and the protocol from IDT technology. For generating FANCJ gene knockout in RPE1-hTERT TP53−/− Cas9 cells, 10 μL tracrRNA (100 μM) and 10 μL 20 nt crRNA (100 μM) were annealed in 80 μL nuclease free duplex buffer (IDT#11-05-01-03) to form a 10 μM gRNA solution. The crRNAs were designed using CRISPR design tools of Benchling. In brief, 100.000 cells were seeded out the day before transfection in 12-well dish in 1 mL medium. 30 minutes prior to transfection the medium was replaced with 750 μL of medium. The 3 μL of 10 μM for each gRNA was added to optiMEM (Life Technologies) to final volume of 125 μL and incubated briefly at RT. Then 6 μL Lipofectamine RNAiMAX (Invitrogen) was added and supplemented to a total volume of 250 μL with optiMEM. Mixture was incubated at RT for 20 min before adding to the cells. The next day the medium was changed, and 2 days after transfection the cells were serial diluted in 96 well plates to obtain single clones. 2 weeks later, the cell clones were passages for new 96 wells, and screened for full loss of both BRIP1 alleles using PCR approach (PCR primer set flanking 5′ end (PCR#78: TTCCATTG-GATGCCGAAGT, PCR#79: CGCTCAAAGGAGGTAAGGATAG), and one primer set flanking the full-length gene (PCR#78 + PCR#43, CCACAACACGTCGGGATTAT). Successful KO clones were validated by WB. U2OS FANCJ K/O and 293T FANCJ K/O cells were generated and maintained as previously described (Peng et al., 2018).
Chemicals
The following drugs were used in the course of this study: PARP inhibitor olaparib (AZD-2281, SelleckChem), cisplatin (Sigma-Aldrich), camptothecin (Sigma-Aldrich), ATR inhibitor (VE-821, SelleckChem), PARG inhibitor (Tocris, PDD 0017273; 5952), Emetine (MCE, HY-B1479B), Methyl methansulfonate (MMS, Sigma-Aldrich 129925), RPA inhibitor (NERx Biosciences, NERx-329), FEN1 inhibitor (LNT1, Tocris 6510), Ligase I/III/IV inhibitor (L189, MedChemExpress HY-15588), Z-VAD-FMK (SelleckChem S7023), Emricasan (SelleckChem S7775). Reagents including 5-chloro-2′-deoxyuridine (CldU) and 5-Iodo-2′-deoxyuridine (IdU) were obtained from Sigma-Aldrich. Concentration and duration of treatment are indicated in the corresponding figures and sections.
METHOD DETAILS
Immunoblotting and antibodies
Cells were harvested, lysed and processed for western blot analysis as described previously using 150mM NETN lysis buffer (20mM Tris (pH 8.0), 150mM NaCl, 1mM EDTA, 0.5% NP-40, 1mM phenylmethylsulfonyl fluoride, 10mg/ml leupeptin, 10mg/ml aprotinin). Chromatin fractionation was performed by subjecting cells to extraction with 0.1% Triton X-100 (Thakar et al., 2020) followed by 10min sonication (medium intensity for 30 s on/30 s off by Diagenode bioruptor) in RIPA buffer (Cold Spring Harbor Protocol). Proteins were separated using SDS–PAGE and electro-transferred to nitrocellulose membranes. Membranes were blocked in 5% not fat dry milk (NFDM) phosphate-buffered saline (PBS)/Tween 20 and incubated with primary antibodies for overnight at 4°C. Antibodies for western blot analysis included anti-β-actin (Sigma), anti-FANCJ (E67), anti-BRCA1 (Cell Signaling Technology), anti-p21 (Cell Signaling Technology), anti-CHD4 (Abcam), anti-BRCA2 (Abcam), anti-53BP1 (Novus Biological), anti-RADX (CXorf57, Abcam), anti-RPA70/RPA1 (Cell Signaling Technology), anti-RPA32/RPA2 (Abcam), anti-FEN1 (Abcam), anti-PARP1 (Abcam), anti-XRCC1 (Abcam), anti-LIG1 (Santa Cruz), anti-LIG3 (GeneTex) and anti-H2B (Cell Signaling Technology), anti-Cleaved Caspase-3 (Cell Signaling Technology) and anti-PARP (Cleaved PARP1, Cell Signaling Technology). Membranes were washed, incubated with corresponding horseradish peroxidase-linked secondary antibodies (Amersham, GE Healthcare) for 1h at room temperature (RT) and detected by chemiluminescence imaging system (Bio-Rad).
Plasmids and RNA interference
cDNAs for human RPA subunits (superRPA) were generated by Toledo lab (Toledo et al., 2013). Plasmid transfections were performed with X-tremeGENE HP DNA Transfection Reagent (Roche 6366244001). U2OS cells were reverse transfected using RNAi-MAX transfection reagent (Life Technologies 13778150) and siRNA targeting FANCJ/BRIP1 (QIAGEN SI03110723), BRCA1 (QIAGEN SI00299495), or scrambled negative control (ORIGENE SR30004) in 6-well plates for 48 h before super-resolution microscopy analysis. RPE1 cells were reverse transfected using Lipofectamine RNAiMAX (Invitrogen P/N 56532) with siRNA targeting RPA1(Dharmacon D-015749-04-0005). Stably transduced cells were generated by infection with pLKO.1 vectors containing shRNAs against non-silencing control (NSC) or one of the shRNAs against corresponding genes:
p21(CDKN1A) includes (A) 5′-TAAGGCAGAAGATGTAGAGCG-3′, (B) 5′-AAAGTCGAAGTTCCATCGCTC-3′;
53BP1 (TP53BP1) includes (A) 5′-AAACCAGTAAGACCAAGTATC-3′, (B) 5′-AATCAATACTAATCACACTGG-3′;
CHD4 includes 5′-AATTCATAGGATGTCAGCAGC-3′;
RADX(CXorf57) includes 5′-ATTTCCGTGGAATACTTTCAG-3′;
FEN1 includes 5′-TACTCTCACAGTAGTCACTGC-3′;
LIG1 includes 5′-TTCACGGACTCGAATAAACCG-3′;
LIG3 includes 5′-AATGTAGTCCTTAAAGTGGGC-3′.
The information was obtained from Dharmacon website (https://horizondiscovery.com), and the shRNAs were obtained from the University of Massachusetts Medical School (UMMS) shRNA core facility. Cells were selected by puromycin for 3–5 days before experiments were carried out.
Immunofluorescence and microscopy
Single-stranded DNA (ssDNA)
Cells were grown on coverslips in 10 μM CldU for 48 h before released into the indicated treatment in figures without CldU. After treatment, cells were washed with PBS and pre-extracted by 0.5% Triton X-100 made in phosphate-buffered saline (PBS) on ice. Cells were then fixed using 4% formaldehyde for 15 min at room temperature (RT), and then permeabilized by 0.5% Triton X-100 in PBS again. Permeabilized cells were then incubated with primary antibodies against CldU (Abcam 6326) at 37°C for 1h. Cells were washed and incubated with secondary antibodies (Alexa Fluor 594) for 1 h at RT. EdU labeling was performed using Click-iT EdU Alexa Fluor 488 Imaging Kit (Invitrogen, C10337) according to the manufacturer’s instructions. After washing, coverslips were mounted onto glass slides using VECTASHIELD mounting medium containing DAPI (Vector Laboratories).
Poly(ADP-ribose)
Cells cultured on coverslips were fixed with 4% formaldehyde in PBS for 10 min at RT and subsequently permeabilized by a 5 min incubation in ice-cold methanol/acetone solution (1:1). After blocking the cells with 10% fetal calf serum for 30 min (alternatively add 3% BSA), coverslips were incubated with the primary antibody (anti-PAR Polyclonal antibody, Trevigen 4336-BPC-100) at 37°C for 1h. Followed by PBS washing, cells were then incubated with the appropriate fluorescently labeled secondary antibody for 1 h at RT. EdU labeling was performed as above. Coverslips were then washed, stained with DAPI (1 μg/ml in PBS, 30 min) and mounted using VECTASHIELD as above.
Chromatin bound proteins
Cells on coverslips were plated on ice for 0.5–1 min before pre-extracted by ice-cold PBS+0.5% Triton for 5 min. Then, cells were fixed by 3% paraformaldehyde/2% sucrose for 10 minutes at RT. Cells were washed twice with PBS-T (0.01% Tween) and incubated with primary antibodies (RPA70/RPA1 Ab, Cell Signaling Technology #2267; anti-phospho-Histone-H2A.X/γ-H2AX, Millipore 05-636 clone JBW301; anti-PARP1, Abcam ab227244; anti-XRCC1, Abcam ab134056; anti-53BP1, Novus Biological, NB100-304) in filtered DMEM + 10% FBS (alternatively add 3% BSA) at 37°C for 1h. After 3x PBS-T washing, coverslips were incubated with appropriate secondary antibodies (in DMEM + 10% FBS) and DAPI. Finally, after washing with PBS-T (x3), coverslips were mounted with Prolong (Invitrogen, P36930). For all assays above, images were collected by fluorescence microscopy (Axioplan 2 imaging and Axio Observer, Zeiss) at a constant exposure time in each experiment. Representative images were processed by ImageJ software. Mean intensity of immunofluorescence for each nucleus were measured with Cell Profiler software version 3.1.5 from Broad Institute.
DNA fiber assay and S1 nuclease analysis
These assays were performed as previously described (Peng et al., 2018). Cells were labeled by sequential incorporation of two different nucleoside analogs, IdU and CldU, into nascent DNA strands for the indicated time and conditions. After nucleoside analogs were incorporated in vivo, the cells were collected, washed, spotted, and lysed on positively charged microscope slides by 7.5 mL spreading buffer for 8 min at room temperature. For experiments with the ssDNA-specific endonuclease S1, after the CldU pulse, cells were treated with CSK100 buffer for 10 min at room temperature, then incubated with S1 nuclease buffer with or without 20 U/mL S1 nuclease (Invitrogen, 18001-016) for 30 min at 37°C. The cells were then scraped in PBS + 0.1% BSA and centrifuged at 7,000 rpm for 5 min at 4 C. Cell pellets were resuspended at 1,500 cells/mL and lysed with lysis solution on slides. Individual DNA fibers were released and spread by tilting the slides at 45 degrees. After air-drying, fibers were fixed by 3:1 methanol/acetic acid at room temperature for 3 min. After air-drying again, fibers were rehydrated in PBS, denatured with 2.5 M HCl for 30 min, washed with PBS, and blocked with blocking buffer (PBS + 0.1% Triton + 3%BSA) for 1 hr. Next, slides were incubated for 2.5 hr with primary antibodies for (IdU, Becton Dickinson 347580; CldU, Abcam 6326) diluted in blocking buffer, washed several times in PBS, and then incubated with secondary antibodies (IdU, goat anti-mouse, Alexa Fluor 488; CldU, goat anti-rat, Alexa Fluor 594) in blocking buffer for 1 hr. After washing and air-drying, slides were mounted with Prolong (Invitrogen, P36930). Finally, visualization of green and/or red signals (measure at least 100 fibers for each experiment) by fluorescence microscopy (Axioplan 2 imaging, Zeiss) provided information about the active replication directionality at the single-molecule level.
Viability assays
For cell survival assays, cells were seeded onto 96-well plates (300–500 cells per well, performed in biological triplicates for each experiment group) and incubated overnight. For patient fibroblasts, cells are seeded onto 6-well plates (5000 cells per well). Clonogenic assays as mentioned were seeded onto 6-cm dishes (500–800 cells per dish) and performed at least in three biological independent experiments for each group. The next day, cells were treated with increasing doses of drugs as indicated in corresponding figures and maintained in complete media for 5 to 7 days for 96-well, 10–12 days for clonogenic assay. Camptothecin (CPT) and methyl methanesulfonate (MMS) were treated for 1 hour and then replaced by fresh complete media. Percentage survival was measured either photometrically using a CellTiter-Glo 2.0 viability assay (Promega) or CCK-8 (Dojindo) in a microplate reader (Beckman Coulter DTX 880 Multimode Detector) or by manual cell counting after methanol/0.5% crystal violet staining.
STORM analysis
For super resolution imaging experiments, cells were trypsinized and seeded on glass coverslips in six-well plates in low density. siRNA transfection, drug treatment, and EdU incorporation were performed directly on cells on coverslips. We used an optimized pre-extraction and fixation protocol for our immunofluorescence experiments in order to clearly visualize chromatin-bound nuclear fraction of cells and to minimize nonspecific antibody labeling from the cytoplasm and non-chromatin bound proteins that could significantly increase noise for image analysis. Cells were permeabilized with 0.5% Triton X-100 in ice-cold CSK buffer (10 mM HEPES, 300 mM Sucrose, 100 mM NaCl, 3 mM MgCl2, and 0.5% Triton X-100, pH = 7.4) in RT for 10 minutes and fixed with 4% paraformaldehyde (Electron Microscopy Sciences 15714) in RT for 30 minutes. Following fixation, cells were washed twice with PBS and blocked with blocking buffer (2% glycine, 2% BSA, 0.2% gelatin, and 50 mM NH4Cl in PBS). For nascent DNA detection, cells were pulse-labeled with 10 μM EdU (ThermoFisher A10044), a thymidine analog, 15 minutes before permeabilization and fixation so that it would be incorporated into nascent DNA during replication in S-phase cells. After fixation, EdU was tagged with Alexa Fluor 647 picolyl azide through click reaction (Click-iT chemistry, ThermoFisher, C10640). The cells were blocked with blocking buffer at least overnight at 4°C. Before imaging, the samples were stained with primary antibodies against rb-RPA70 (Abcam ab79398), rbMCM6 (conjugated to AF568, Abcam ab211916), and ms-PCNA (Santa cruz sc-56) in blocking buffer for 1h at RT, then secondary antibodies (goat anti-mouse AF488, Invitrogen A11029; goat anti-rabbit AF 750, Invitrogen A21039) in blocking buffer for 30minutes at RT. Super resolution imaging and other related processes were described before (Yin and Rothenberg, 2016).
PDX studies
Triple-negative breast cancer patient-derived xenograft (PDX), PNX017, was from a patient with a hemizygous germline BRCA1 mutation (1105_1106insTC); the wild-type BRCA1 allele was lost in the tumor, following a Loss of Heterozygosity model. PNX017 was derived at Fox Chase Cancer Center under IRB and IACUC approved protocols. PDX tumors were grown in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice. Resistant tumors were obtained from mice after sensitive tumors progressed on serial treatments of PARP inhibitors (rucaparib, 150mg/kg, 2x daily, 5 days, 2 days off, 5 days). The tumors were harvested at approximately 500 mm3 and dissociated in 0.2% collagenase, 0.33 mg/ml dispase solution for 3h at 37°C. The dissociated cells were maintained at 37°C in RPMI1640 + 10% FBS and used for DNA fiber assays within 24h of tumor extraction. DNA fiber assay and S1 nuclease analysis were performed as described above.
DNA pulsed field capillary electrophoresis
BRCA1 K/O RPE1 cells were assayed for DSBs by pulsed field capillary electrophoresis with the Agilent Femto Pulse systems as previously described (Panzarino et al., 2021). Briefly, 200,000 cells were plated per 10 cm plate and allowed to adhere overnight. Subsequently, the media was aspirated and treated with appropriate volume of complete medium containing 50 μM olaparib in order to match the molar ratio of drug to cells found in the 96-well CellTiter-Glo assay; cells treated with apoptotic inhibitor were pretreated for 2h with 50 μM Emricasan (SelleckChem, S7775) and maintained for the duration of the experiment, and untreated cells received DMSO as control. At 96h, all attached and floated cells were collected, then washed, and high molecular weight genomic DNA (gDNA) was isolated with the FiberPrep Genomic DNA Extraction Kit (Genomic Vision, EXT-001) according to the manufacturer’s instructions; the final digestion step was supplemented with an additional 5 μL of beta-agarase to ensure full digestion of the agarose plug (New England Biolabs, M0392). Genomic DNA was analyzed on the Femto Pulse in the 3h mode for gDNA and large fragments according to the manufacturer’s instructions. The retention time of gDNA and DNA fragments was experimentally determined (intact gDNA observed as signal above 8,000 s of capillary retention time, and DNA fragmentation observed in the 3000 s to 8000 s window of capillary retention time). The percentages of signal from fragments and genomic DNA were measured by ProSize Data Analysis Software and plotted by Graph Pad Prism.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical differences in DNA fiber assays and immunofluorescence intensity were determined by nonparametric Kruskal-Wallis test followed by Dunn’s test for multiple comparations in non-Gaussian populations. Two group comparations were determined using two-tailed Mann-Whitney test. Statistical differences in viability assays with small sample sizes were determined by unpaired t test. Statistical analysis was performed using GraphPad Prism (Version 7.0). In all cases, ns: not significant (p > 0.05), *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-BRCA1 | Cell Signaling Technology | Cat# 9010; RRID: AB_2228244 |
| Rabbit polyclonal anti-FANCJ (E67) | Cantor Laboratory | N/A |
| Mouse monoclonal anti-β-actin, clone AC-15 | Sigma-Aldrich | Cat# A5441; RRID: AB_476744 |
| Mouse monoclonal anti-BrdU (B44) | BD Biosciences | Cat# 347580; RRID: AB_400326 |
| Rat monoclonal anti-BrdU, clone BU1 / 75 (ICR1) | Abcam | Cat# ab6326; RRID: AB_305426 |
| Rabbit monoclonal anti-p21 Waf1/Cip1 (12D1) | Cell Signaling Technology | Cat# 2947; RRID: AB_823586 |
| Rabbit polyclonal anti-53BP1 | Novus Biological | Cat# NB100-304; RRID: AB_10003037 |
| Mouse monoclonal anti-CHD4 [3F2/4] | Abcam | Cat# ab70469; RRID: AB_2229454 |
| Rabbit polyclonal anti-BRCA2 | Abcam | Cat# ab123491; RRID: AB_10972163 |
| Rabbit polyclonal anti-CXorf57 (RADX) | Abcam | Cat# ab228707 |
| Mouse monoclonal anti-PCNA [PC10] | Abcam | Cat# ab29; RRID: AB_303394 |
| Rabbit monoclonal anti-RPA70 [EPR3472] | Abcam | Cat# ab79398; RRID: AB_1603759 |
| Rabbit monoclonal anti-MCM6 [EPR17686] (Alexa Fluor® 568) | Abcam | Cat# ab211916 |
| Mouse monoclonal anti-PCNA (PC10) | Santa Cruz Biotechnology | Cat# sc-56, RRID: AB_628110 |
| Rabbit polyclonal anti-RPA70/RPA1 | Cell Signaling Technology | Cat# 2267; RRID: AB_2180506 |
| Mouse monoclonal anti-γH2AX (Ser139), clone JBW301 | Millipore | Cat# 05-636; RRID: AB_309864 |
| Mouse monoclonal anti-RPA32/RPA2 [9H8] | Abcam | Cat# ab2175; RRID: AB_302873 |
| Mouse monoclonal anti-poly(ADP-ribose) | Trevigen | Cat# 4335-MC-100; RRID: AB_2572318 |
| Rabbit monoclonal anti-FEN1[EPR4460(2)] | Abcam | Cat# ab109132; RRID: AB_10866125 |
| Rabbit polyclonal anti-PARP1 | Abcam | Cat# ab227244 |
| Rabbit monoclonal anti-XRCC1 [EPR4389(2)] |
Abcam | Cat# ab134056 |
| Mouse monoclonal anti-DNA ligase III [1F3] | GeneTex | Cat# GTX70143; RRID: AB_372134 |
| Rabbit polyclonal anti-Histone H2B (V119) | Cell Signaling Technology | Cat# 8135; RRID: AB_10891053 |
| Mouse monoclonal anti-DNA Ligase I (C-5) | Santa Cruz Biotechnology | Cat# sc-271678; RRID: AB_10708845 |
| Rabbit polyclonal anti-Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661; RRID: AB_2341188 |
| Rabbit polyclonal anti-PARP (cleaved PARP1) | Cell Signaling Technology | Cat# 9542; RRID: AB_2160739 |
| ECL anti-rabbit IgG, HRP-linked whole antibody (from donkey) | GE Healthcare | Cat# NA934V; RRID: AB_772206 |
| Rb anti-Ms IgG secondary antibody, HRP conjugate | Thermo Fisher Scientific | Cat# 61-6520; RRID: AB_2533933 |
| Alexa Fluor 488 goat anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat# A-11001; RRID: AB_2534069 |
| Alexa Fluor 594 goat anti-Rat IgG (H+L) | Thermo Fisher Scientific | Cat# A-11007; RRID: AB_141374 |
| Alexa Fluor 568 goat anti-Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# A-11011; RRID: AB_143157 |
| Alexa Fluor 488 Goat Anti-Mouse IgG (H+L) | Invitrogen | Cat# A-11029; RRID: AB_138404 |
| Alexa Fluor 750 Goat Anti-Rabbit IgG (H+L) | Invitrogen | Cat# A-21039; RRID: AB_10375716 |
| Alexa Fluor 555 goat anti-Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# A-21428; RRID: AB_141784 |
| Rhodamine Red-X-conjugated AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 115-295-062; RRID: AB_2338761 |
|
| ||
| Biological samples | ||
|
| ||
| Patient-derived xenograft (PDX) PNX017 | This paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| IdU (5-Iodo-2′-deoxyuridine) | Sigma-Aldrich | Cat# I7125 |
| CldU (5-chloro-2′-deoxyuridine) | Sigma-Aldrich | Cat# C6891 |
| Olaparib | SelleckChem | Cat# AZD2281 |
| Cisplatin | Sigma-Aldrich | Cat# P4394 |
| Camptothecin (CPT) | Sigma-Aldrich | Cat# C9911 |
| Mitomycin C (MMC) | Sigma-Aldrich | Cat# M4287 |
| S1 nuclease | Thermo Fisher Scientific | Cat# 18001-016 |
| VE-821 (ATR inhibitor) | SelleckChem | Cat# S8007 |
| Hydroxyurea | Sigma-Aldrich | Cat# H8627 |
| RPA inhibitor | NERx Biosciences | Cat# NERx-329 |
| PDD 00017273 (PARG inhibitor) | Tocris | Cat# 5952 |
| Emetine | MedChemExpress | Cat# HY-B1479B |
| Methyl methansulfonate (MMS) | Sigma-Aldrich | Cat# 129925 |
| LNT1 (FEN1 inhibitor) | Tocris | Cat# 6510 |
| L189 (Ligase I/II/IV inhibitor) | MedChemExpress | Cat# HY-15588 |
| Z-VAD-FMK (pan-caspase inhibitor) | SelleckChem | Cat# S7023 |
| Emricasan (pan-caspase inhibitor) | SelleckChem | Cat# S7775 |
| DMSO (Dimethyl sulfoxide) | Sigma-Aldrich | Cat# D5879 |
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| Crystal violet | Sigma-Aldrich | Cat# C0775 |
| Formaldehyde | Fisher Chemical | Cat# F79-500 |
| EdU (5-ethynyl-2′-deoxyuridine) | Thermo Fisher Scientific | Cat# A10044 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15714 |
| Paraformaldehyde | Sigma-Aldrich | Cat# P6148 |
| Acetone | Fisher Chemical | Cat# A18 |
| Methanol | Fisher Chemical | Cat# A412 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | Thermo Fisher Scientific | Cat# D1306 |
| ProLong Gold Antifade reagent | Thermo Fisher Scientific | Cat# P36930 |
| VECTASHIELD mounting media | Vector Laboratories | Cat# H-1200 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Click-iT EdU Cell Proliferation Kit for Imaging, Alexa Fluor 488 | Invitrogen | Cat# C10337 |
| CellTiter-Glo 2.0 Assay | Promega | Cat# G9242 |
| Click-iT Plus EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | Cat# C10640 |
| Cell Counting Kit-8 (CCK-8) | Dojindo | Cat# CK04 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| Human: RPE1-hTERT TP53−/− | Durocher Laboratory | https://pubmed.ncbi.nlm.nih.gov/30022168/ |
| Human: RPE1-hTERT TP53−/−;BRCA1−/− | Durocher Laboratory | https://pubmed.ncbi.nlm.nih.gov/30022168/ |
| Human: RPE1-hTERT TP53−/−;BRCA1−/−;TP53BP1−/− |
Durocher Laboratory | https://pubmed.ncbi.nlm.nih.gov/30022168/ |
| Human: RPE1-hTERT TP53−/−;FANCJ−/− | This paper | N/A |
| Human: U2OS | Cantor Laboratory | https://pubmed.ncbi.nlm.nih.gov/30232006/ |
| Human: U2OS FANCJ−/− | Cantor Laboratory | https://pubmed.ncbi.nlm.nih.gov/30232006/ |
| Human: 293T | Cantor Laboratory | https://pubmed.ncbi.nlm.nih.gov/30232006/ |
| Human: 293T FANCJ−/− | Cantor Laboratory | https://pubmed.ncbi.nlm.nih.gov/30232006/ |
| Mouse: T2 | Zou Laboratory | https://pubmed.ncbi.nlm.nih.gov/28242626/ |
| Mouse: BR5 | Zou Laboratory | https://pubmed.ncbi.nlm.nih.gov/28242626/ |
| Mouse: BR5-R1 | Zou Laboratory | https://pubmed.ncbi.nlm.nih.gov/28242626/ |
| Human: PEO1 | Taniguchi Laboratory | https://pubmed.ncbi.nlm.nih.gov/19654294/ |
| Human: C4-2 | Taniguchi Laboratory | N/A |
| Hamster: V-C8 | Jasin Laboratory | https://pubmed.ncbi.nlm.nih.gov/21565612/ |
| Hamster: V-C8+BRCA2 | Jasin Laboratory | https://pubmed.ncbi.nlm.nih.gov/21565612/ |
| Hamster: V-C8+BRCA2 S3291A | Jasin Laboratory | https://pubmed.ncbi.nlm.nih.gov/21565612/ |
| Human: FA Patient fibroblasts RA2630 T131P |
Smogorzewska Laboratory | https://pubmed.ncbi.nlm.nih.gov/26253028/ |
| Human: FA Patient fibroblasts CRISPR corrected clone 3–39 ΔMUT/WT or -/WT | Smogorzewska Laboratory | https://pubmed.ncbi.nlm.nih.gov/26253028/ |
| Human: FA Patient fibroblasts CRISPR double-allele corrected clone | Smogorzewska Laboratory | Available by request |
| Human: UWB1 | Turchi Laboratory | N/A |
| Human: UWB1+BRCA1 | Turchi Laboratory | N/A |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| Mouse: NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ | Johnson Laboratory | https://pubmed.ncbi.nlm.nih.gov/30257212/ |
|
| ||
| Oligonucleotides | ||
|
| ||
| FANCJ CRISPR K/O gRNA | This paper | Table S2 |
| shRNA sequences | This paper | Table S2 |
| siRNA scrambled negative control | ORIGENE | Cat# SR30004 |
| siRNA FANCJ/BRIP1 | QIAGEN | Cat# SI03110723 |
| siRNA BRCA1 | QIAGEN | Cat# SI00299495 |
| siRNA RPA1 | Dharmacon | Cat# D-015749-04-0005 |
|
| ||
| Recombinant DNA | ||
|
| ||
| Plasmid: AcGFP-RPA3-P2A-RPA1-P2A-RPA2 fusion construct | (Toledo et al., 2013) | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism 7.0 | GraphPad | https://www.graphpad.com/scientificsoftware/prism/ |
| CellProfiler-3.1.5 | Broad Institute | https://cellprofiler.org/releases |
| Fiji (ImageJ) | NIH | https://imagej.nih.gov/ij/docs/guide/146-2.html |
| ZEN blue 2.5 pro | Zeiss | N/A |
|
| ||
| Deposited data | ||
|
| ||
| Raw and uncropped data | This paper; Mendeley | https://doi.org/10.17632/g96fktpb58.1 |
|
| ||
| Other | ||
|
| ||
| Lipofectamine RNAiMAX transfection reagent | Invitrogen | Cat# 13778150 and P/N 56532 |
| X-tremeGENE HP DNA Transfection Reagent | Roche | Cat# 6366244001 |
| Genomic DNA Extraction Kit | Genomic Vision | Cat# EXT-001 |
| β-agarase I | New England Biolabs | Cat# M0392 |
Highlights.
PARP inhibitor (PARPi) sensitivity aligns with extent of replication gap formation
Targeting gaps resensitizes and augments PARPi synthetic lethality
Gaps in BRCA1-deficient cells are due to Okazaki fragment processing (OFP) defects
OFP defects in BRCA1-deficient cells are rescued by 53BP1 loss
ACKNOWLEDGMENTS
We thank the Cantor laboratory for helpful discussions. We thank Dr. Daniel Durocher for RPE1 cell lines, including control, BRCA1 KO, and BRCA1/53BP1 KO; Lee Zou for the T2, BR5, and BR5-R1 cells; Agata Smogorzewska for the FA patient and CRISPR-corrected cells; Maria Jasin for the V-C8 and derived cells; and Luis Toledo for the superRPA construct. This work was supported by R01 CA254037, R01 CA225018-02, the Fanconi Anemia Research Fund, charitable contributions from Mr. and Mrs. Edward T. Vitone, Jr and the Lipp Family Foundation (to S.B.C.), and R01 CA214799 and OC130212 (to N.J.). We thank Drs. Igor Astutarov and Vladimir Khazak for establishing and sharing PNX0204.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.06.011.
REFERENCES
- Adam S, Rossi SE, Moatti N, Zompit MDM, Ng TF, Álvarez-Quilón A, Desjardins J, Bhaskaran V, Martino G, Setiaputra D, et al. (2021). CIP2A is a prime synthetic-lethal target for BRCA-mutated cancers. bioRxiv. 10.1101/2021.02.08.430060. [DOI] [PubMed] [Google Scholar]
- Álvarez-Quilón A, Wojtaszek JL, Mathieu MC, Patel T, Appel CD, Hustedt N, Rossi SE, Wallace BD, Setiaputra D, Adam S, et al. (2020). Endogenous DNA 3′ Blocks Are Vulnerabilities for BRCA1 and BRCA2 Deficiency and Are Reversed by the APE2 Nuclease. Mol. Cell 78, 1152–1165.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amé JC, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, and Schreiber V (2009). Radiation-induced mitotic catastrophe in PARG-deficient cells. J. Cell Sci 122, 1990–2002. [DOI] [PubMed] [Google Scholar]
- Arakawa H, and Iliakis G (2015). Alternative Okazaki Fragment Ligation Pathway by DNA Ligase III. Genes (Basel) 6, 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avkin S, Sevilya Z, Toube L, Geacintov N, Chaney SG, Oren M, and Livneh Z (2006). p53 and p21 regulate error-prone DNA repair to yield a lower mutation load. Mol. Cell 22, 407–413. [DOI] [PubMed] [Google Scholar]
- Azarm K, and Smith S (2020). Nuclear PARPs and genome integrity. Genes Dev. 34, 285–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. (2013). Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol 20, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Krishnamoorthy A, Dungrawala H, Garcin EB, Modesti M, and Cortez D (2018). RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep. 24, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. (2010). 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol 17, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burhans WC, Vassilev LT, Wu J, Sogo JM, Nallaseth FS, and DePamphilis ML (1991). Emetine allows identification of origins of mammalian DNA replication by imbalanced DNA synthesis, not through conservative nucleosome segregation. EMBO J. 10, 4351–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrum AK, Vindigni A, and Mosammaparast N (2019). Defining and Modulating ‘BRCAness’. Trends Cell Biol. 29, 740–751. [DOI] [PubMed] [Google Scholar]
- Caldecott KW, Aoufouchi S, Johnson P, and Shall S (1996). XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res. 24, 4387–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor SB, and Calvo JA (2017). Fork Protection and Therapy Resistance in Hereditary Breast Cancer. Cold Spring Harb. Symp. Quant. Biol 82, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, and Livingston DM (2001). BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105, 149–160. [DOI] [PubMed] [Google Scholar]
- Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, and Livingston DM (2004). The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc. Natl. Acad. Sci. USA 101, 2357–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. (2009). A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 35, 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Bétous R, Carroll CM, Jung SY, Qin J, Cimprich KA, and Cortez D (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Andrea AD (2018). Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst.) 71, 172–176. [DOI] [PubMed] [Google Scholar]
- Dias MP, Tripathi V, van der Heijden I, Cong K, Manolika E-M, Galanos P, Bhin J, Gogola E, Annunziato S, Lieftink C, et al. (2021). Loss of Nuclear DNA ligase III Can Revert PARP Inhibitor Resistance in BRCA1-deficient Cells by Increasing DNA Replication Stress. bioRxiv. 10.1101/2021.03.24.436323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, and Ashworth A (2008). Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451, 1111–1115. [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Masutani M, Suzuki H, and Caldecott KW (2003). A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 31, 5526–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre S, Chan D, Salinas R, Woynarowska B, and Woynarowski JM (2003). DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells. Biochem. Pharmacol 66, 225–237. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Feng W, and Jasin M (2017). BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat. Commun 8, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugger K, Bajrami I, Silva Dos Santos M, Young SJ, Kunzelmann S, Kelly G, Hewitt G, Patel H, Goldstone R, Carell T, et al. (2021). Targeting the nucleotide salvage factor DNPH1 sensitizes BRCA-deficient cells to PARP inhibitors. Science 372, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanos P, Vougas K, Walter D, Polyzos A, Maya-Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti FM, Gagos S, Tzetis M, et al. (2016). Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol 18, 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavande NS, VanderVere-Carozza PS, Pawelczak KS, Vernon TL, Jordan MR, and Turchi JJ (2020). Structure-Guided Optimization of Replication Protein A (RPA)-DNA Interaction Inhibitors. ACS Med. Chem. Lett 11, 1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, James DI, Guerrero Llobet S, Vis DJ, Annunziato S, et al. (2018). Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 33, 1078–1093.e12. [DOI] [PubMed] [Google Scholar]
- Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, Issaeva N, Sleeth K, Sharma RA, and Helleday T (2010). Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 70, 5389–5398. [DOI] [PubMed] [Google Scholar]
- Guillemette S, Serra RW, Peng M, Hayes JA, Konstantinopoulos PA, Green MR, and Cantor SB (2015). Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 29, 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo E, Ishii Y, Mueller J, Srivatsan A, Gahman T, Putnam CD, Wang JYJ, and Kolodner RD (2020). FEN1 endonuclease as a therapeutic target for human cancers with defects in homologous recombination. Proc. Natl. Acad. Sci. USA 117, 19415–19424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzlikova H, and Caldecott KW (2019). Perspectives on PARPs in S Phase. Trends Genet. 35, 412–422. [DOI] [PubMed] [Google Scholar]
- Hanzlikova H, Gittens W, Krejcikova K, Zeng Z, and Caldecott KW (2017). Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 45, 2546–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, and Caldecott KW (2018). The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 71, 319–331.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, and Costanzo V (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol 17, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LM, Arlett CF, Harcourt SA, Lehmann AR, and Broughton BC (1985). Cells from an immunodeficient patient (46BR) with a defect in DNA ligation are hypomutable but hypersensitive to the induction of sister chromatid exchanges. Proc. Natl. Acad. Sci. USA 82, 2044–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry-Mowatt J, Jackson D, Masson JY, Johnson PA, Clements PM, Benson FE, Thompson LH, Takeda S, West SC, and Caldecott KW (2003). XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol. Cell 11, 1109–1117. [DOI] [PubMed] [Google Scholar]
- Hewitt G, Borel V, Segura-Bayona S, Takaki T, Ruis P, Bellelli R, Lehmann LC, Sommerova L, Vancevska A, Tomas-Loba A, et al. (2021). Defective ALC1 nucleosome remodeling confers PARPi sensitization and synthetic lethality with HRD. Mol. Cell 81, 767–783.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JK, Stefanick DF, Prasad R, Gassman NR, Kedar PS, and Wilson SH (2014). Base excision repair defects invoke hypersensitivity to PARP inhibition. Mol. Cancer Res 12, 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, de Antoni A, Técher H, Baldi G, and Costanzo V (2017a). Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 591, 1083–1100. [DOI] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Técher H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017b). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 67, 867–881.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto S, Nishiyama A, Chiba Y, Miyashita R, Konishi C, Azuma Y, and Nakanishi M (2021). HPF1-dependent PARP activation promotes LIG3-XRCC1-mediated backup pathway of Okazaki fragment ligation. Nucleic Acids Res. 49, 5003–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaud C, Moreno A, Marchesi F, Toth R, Blow JJ, and Rouse J (2016). Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 351, 846–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai X, Broderick R, Bergoglio V, Zimmer J, Badie S, Niedzwiedz W, Hoffmann JS, and Tarsounas M (2017). MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun 8, 15983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledermann JA, and Pujade-Lauraine E (2019). Olaparib as maintenance treatment for patients with platinum-sensitive relapsed ovarian cancer. Ther. Adv. Med. Oncol 11, 1758835919849753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WTC, Yin Y, Morten MJ, Tonzi P, Gwo PP, Odermatt DC, Modesti M, Cantor SB, Gari K, Huang TT, and Rothenberg E (2021). Single-molecule imaging reveals replication fork coupled formation of G-quadruplex structures hinders local replication stress signaling. Nat. Commun 12, 2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR, Willis AE, Broughton BC, James MR, Steingrimsdottir H, Harcourt SA, Arlett CF, and Lindahl T (1988). Relation between the human fibroblast strain 46BR and cell lines representative of Bloom’s syndrome. Cancer Res. 48, 6343–6347. [PubMed] [Google Scholar]
- Leppard JB, Dong Z, Mackey ZB, and Tomkinson AE (2003). Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Mol. Cell. Biol 23, 5919–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, et al. (2005). The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat. Genet 37, 934–935. [DOI] [PubMed] [Google Scholar]
- Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, et al. (2005). The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat. Genet 37, 931–933. [DOI] [PubMed] [Google Scholar]
- Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, and Cantor SB (2005). BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8, 255–265. [DOI] [PubMed] [Google Scholar]
- Lomonosov M, Anand S, Sangrithi M, Davies R, and Venkitaraman AR (2003). Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 17, 3017–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Foiani M, and Sogo JM (2006). Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 21, 15–27. [DOI] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2012). The DNA damage response and cancer therapy. Nature 481, 287–294. [DOI] [PubMed] [Google Scholar]
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Décaillet C, Gari K, and Constantinou A (2013). FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol. Cell 51, 678–690. [DOI] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grøfte M, Chan KL, Hickson ID, Bartek J, and Lukas J (2011). 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol 13, 243–253. [DOI] [PubMed] [Google Scholar]
- Luke-Glaser S, Luke B, Grossi S, and Constantinou A (2010). FANCM regulates DNA chain elongation and is stabilized by S-phase checkpoint signalling. EMBO J. 29, 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman AS, and Helleday T (2005). Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 33, 3799–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, and de Murcia G (1998). XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol 18, 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, and Bartek J (2018). High speed of fork progression induces DNA replication stress and genomic instability. Nature 559, 279–284. [DOI] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, et al. (2006). Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 66, 8109–8115. [DOI] [PubMed] [Google Scholar]
- McDermott N, Buechelmaier ES, and Powell SN (2019). Capitalizing on Cancer Replication Stress by Preventing PAR Chain Turnover: A New Type of Synthetic Lethality. Cancer Cell 35, 344–346. [DOI] [PubMed] [Google Scholar]
- Mengwasser KE, Adeyemi RO, Leng Y, Choi MY, Clairmont C, D’Andrea AD, and Elledge SJ (2019). Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell 73, 885–899.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelena J, Lezaja A, Teloni F, Schmid T, Imhof R, and Altmeyer M (2018). Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat. Commun 9, 2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelena J, Pellegrino S, Spegg V, and Altmeyer M (2021). Replicated chromatin curtails 53BP1 recruitment in BRCA1-proficient and -deficient cells. Life Sci. Alliance 4, e202101023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, and Lopes M (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun 8, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, and de Lange T (2018). 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 560, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladenov E, Magin S, Soni A, and Iliakis G (2013). DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front. Oncol 3, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, and Jasin M (1999). Brca1 controls homology-directed DNA repair. Mol. Cell 4, 511–518. [DOI] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, and Pommier Y (2012). Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muvarak NE, Chowdhury K, Xia L, Robert C, Choi EY, Cai Y, Bellani M, Zou Y, Singh ZN, Duong VH, et al. (2016). Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents - A Potential Therapy for Cancer. Cancer Cell 30, 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju G, and Scully R (2007). Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair (Amst.) 6, 1018–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath S, Somyajit K, Mishra A, Scully R, and Nagaraju G (2017). FANCJ helicase controls the balance between short- and long-tract gene conversions between sister chromatids. Nucleic Acids Res. 45, 8886–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak S, Calvo JA, Cong K, Peng M, Berthiaume E, Jackson J, Zaino AM, Vindigni A, Hadden MK, and Cantor SB (2020). Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv 6, eaaz7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Álvarez-Quilón A, Moatti N, Zimmermann M, et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, and Swisher EM (2011). Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol 29, 3008–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odermatt DC, Lee WTC, Wild S, Jozwiakowski SK, Rothenberg E, and Gari K (2020). Cancer-associated mutations in the iron-sulfur domain of FANCJ affect G-quadruplex metabolism. PLoS Genet. 16, e1008740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive PL (1998). The role of DNA single- and double-strand breaks in cell killing by ionizing radiation. Radiat. Res 150 (5, Suppl), S42–S51. [PubMed] [Google Scholar]
- Panzarino NJ, Krais JJ, Cong K, Peng M, Mosqueda M, Nayak SU, Bond SM, Calvo JA, Doshi MB, Bere M, et al. (2021). Replication Gaps Underlie BRCA Deficiency and Therapy Response. Cancer Res. 81, 1388–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino S, Michelena J, Teloni F, Imhof R, and Altmeyer M (2017). Replication-Coupled Dilution of H4K20me2 Guides 53BP1 to Pre-replicative Chromatin. Cell Rep. 19, 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Litman R, Jin Z, Fong G, and Cantor SB (2006). BACH1 is a DNA repair protein supporting BRCA1 damage response. Oncogene 25, 2245–2253. [DOI] [PubMed] [Google Scholar]
- Peng M, Cong K, Panzarino NJ, Nayak S, Calvo J, Deng B, Zhu LJ, Morocz M, Hegedus L, Haracska L, and Cantor SB (2018). Opposing Roles of FANCJ and HLTF Protect Forks and Restrain Replication during Stress. Cell Rep. 24, 3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilié PG, Gay CM, Byers LA, O’Connor MJ, and Yap TA (2019). PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res 25, 3759–3771. [DOI] [PubMed] [Google Scholar]
- Pillay N, Tighe A, Nelson L, Littler S, Coulson-Gilmer C, Bah N, Golder A, Bakker B, Spierings DCJ, James DI, et al. (2019). DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 35, 519–533.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, O’Connor MJ, and de Bono J (2016). Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med 8, 362ps17. [DOI] [PubMed] [Google Scholar]
- Quinet A, and Vindigni A (2018). Superfast DNA replication causes damage in cancer cells. Nature 559, 186–187. [DOI] [PubMed] [Google Scholar]
- Quinet A, Martins DJ, Vessoni AT, Biard D, Sarasin A, Stary A, and Menck CF (2016). Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Res 44, 5717–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A (2017). DNA Fiber Analysis: Mind the Gap! Methods Enzymol. 591, 55–82. [DOI] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Jackson J, Svikovic S, Lemacon D, Carvajal-Maldonado D, Gonzalez-Acosta D, Vessoni AT, Cybulla E, Wood M, et al. (2020). PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol. Cell 77, 461–474.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, and Nussenzweig A (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol 18, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, and Lopes M (2012). Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol 19, 417–423. [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. (2008). Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451, 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, Wurz K, Higgins J, Villegas E, and Taniguchi T (2009). Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 69, 6381–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sané AT, and Bertrand R (1998). Distinct steps in DNA fragmentation pathway during camptothecin-induced apoptosis involved caspase-, benzyloxycarbonyl- and N-tosyl-L-phenylalanylchloromethyl ketone-sensitive activities. Cancer Res. 58, 3066–3072. [PubMed] [Google Scholar]
- Saredi G, Huang H, Hammond CM, Alabert C, Bekker-Jensen S, Forne I, Reverón-Gómez N, Foster BM, Mlejnkova L, Bartke T, et al. (2016). H4K20me0 marks post-replicative chromatin and recruits the TONSL–MMS22L DNA repair complex. Nature 534, 714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Tian L, Kähkönen M, Schwartzentruber J, Kircher M, Majewski J, Dyment DA, Innes AM, Boycott KM, Moreau LA, et al. ; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium (2015). Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 5, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Dixit S, Somyajit K, and Nagaraju G (2019). ATR Signaling Uncouples the Role of RAD51 Paralogs in Homologous Recombination and Replication Stress Response. Cell Rep. 29, 551–559.e4. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid JA, Berti M, Walser F, Raso MC, Schmid F, Krietsch J, Stoy H, Zwicky K, Ursich S, Freire R, et al. (2018). Histone Ubiquitination by the DNA Damage Response Is Required for Efficient DNA Replication in Unperturbed S Phase. Mol. Cell 71, 897–910.e8. [DOI] [PubMed] [Google Scholar]
- Schoonen PM, Talens F, Stok C, Gogola E, Heijink AM, Bouwman P, Foijer F, Tarsounas M, Blatter S, Jonkers J, et al. (2017). Progression through mitosis promotes PARP inhibitor-induced cytotoxicity in homologous recombination-deficient cancer cells. Nat. Commun 8, 15981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber V, Amé JC, Dollé P, Schultz I, Rinaldi B, Fraulob V, Ménissier-de Murcia J, and de Murcia G (2002). Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem 277, 23028–23036. [DOI] [PubMed] [Google Scholar]
- Sgagias MK, Wagner KU, Hamik B, Stoeger S, Spieker R, Huber LJ, Chodosh LA, and Cowan KH (2004). Brca1-deficient murine mammary epithelial cells have increased sensitivity to CDDP and MMS. Cell Cycle 3, 1451–1456. [DOI] [PubMed] [Google Scholar]
- Smith MA, Reynolds CP, Kang MH, Kolb EA, Gorlick R, Carol H, Lock RB, Keir ST, Maris JM, Billups CA, et al. (2015). Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin. Cancer Res 21, 819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, and Foiani M (2002). Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297, 599–602. [DOI] [PubMed] [Google Scholar]
- Somyajit K, Spies J, Coscia F, Kirik U, Rask MB, Lee JH, Neelsen KJ, Mund A, Jensen LJ, Paull TT, et al. (2021). Homology-directed repair protects the replicating genome from metabolic assaults. Dev. Cell 56, 461–477.e7. [DOI] [PubMed] [Google Scholar]
- Sriramkumar S, Matthews TD, Ghobashi AH, Miller SA, VanderVere-Carozza PS, Pawelczak KS, Nephew KP, Turchi JJ, and O’Hagan HM (2020). Platinum-Induced Ubiquitination of Phosphorylated H2AX by RING1A Is Mediated by Replication Protein A in Ovarian Cancer. Mol. Cancer Res 18, 1699–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ström CE, Johansson F, Uhlén M, Szigyarto CA, Erixon K, and Helleday T (2011). Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 39, 3166–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Bernal JA, and Venkitaraman AR (2008). Cell-cycle coordination between DNA replication and recombination revealed by a vertebrate N-end rule degron-Rad51. Nat. Struct. Mol. Biol 15, 1049–1058. [DOI] [PubMed] [Google Scholar]
- Sugimura K, Takebayashi S, Taguchi H, Takeda S, and Okumura K (2008). PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol 183, 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhasini AN, Sommers JA, Muniandy PA, Coulombe Y, Cantor SB, Masson J-Y, Seidman MM, and Brosh RM Jr. (2013). Fanconi anemia group J helicase and MRE11 nuclease interact to facilitate the DNA damage response. Mol. Cell. Biol 33, 2212–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo IA, Broughton BC, Day RS, James MR, Karran P, Mayne LV, and Lehmann AR (1983). A biochemical defect in the repair of alkylated DNA in cells from an immunodeficient patient (46BR). Carcinogenesis 4, 559–564. [DOI] [PubMed] [Google Scholar]
- Thakar T, Leung W, Nicolae CM, Clements KE, Shen B, Bielinsky AK, and Moldovan GL (2020). Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat. Commun 11, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, and Lukas J (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. [DOI] [PubMed] [Google Scholar]
- Vallerga MB, Mansilla SF, Federico MB, Bertolin AP, and Gottifredi V (2015). Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc. Natl. Acad. Sci. USA 112, E6624–E6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wietmarschen N, and Nussenzweig A (2018). Mechanism for Synthetic Lethality in BRCA-Deficient Cancers: No Longer Lagging Behind. Mol. Cell 71, 877–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma P, Zhou Y, Cao Z, Deraska PV, Deb M, Arai E, Li W, Shao Y, Puentes L, Li Y, et al. (2021). ALC1 links chromatin accessibility to PARP inhibitor response in homologous recombination-deficient cells. Nat. Cell Biol 23, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, et al. (2015). A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol. Cell 59, 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward TA, McHugh PJ, and Durant ST (2017). Small molecule inhibitors uncover synthetic genetic interactions of human flap endonuclease 1 (FEN1) with DNA damage response genes. PLoS ONE 12, e0179278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RP, Garcia-Rodriguez N, Zilio N, Hanulova M, and Ulrich HD (2020). Processing of DNA Polymerase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol. Cell 77, 3–16.e4. [DOI] [PubMed] [Google Scholar]
- Xu X, Chen E, Mo L, Zhang L, Shao F, Miao K, Liu J, Su SM, Valecha M, In Chan U, et al. (2019). BRCA1 represses DNA replication initiation through antagonizing estrogen signaling and maintains genome stability in parallel with WEE1-MCM2 signaling during pregnancy. Hum. Mol. Genet 28, 842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, et al. (2017). ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 31, 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, and Rothenberg E (2016). Probing the Spatial Organization of Molecular Complexes Using Triple-Pair-Correlation. Sci. Rep 6, 30819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadorozhny K, Sannino V, Beláň O, Mlčoušková J, Spírek M, Costanzo V, and Krejčí L (2017). Fanconi-Anemia-Associated Mutations Destabilize RAD51 Filaments and Impair Replication Fork Protection. Cell Rep. 21, 333–340. [DOI] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaitė Ž, Muir M, Fluteau A, Aregger M, McEwan A, et al. (2018). CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed blots, gels, and microscopy images are available on Mendeley Data at https://doi.org/10.17632/g96fktpb58.1. Raw Data for STORM analysis will be made available by the corresponding author upon request.