

Abstract
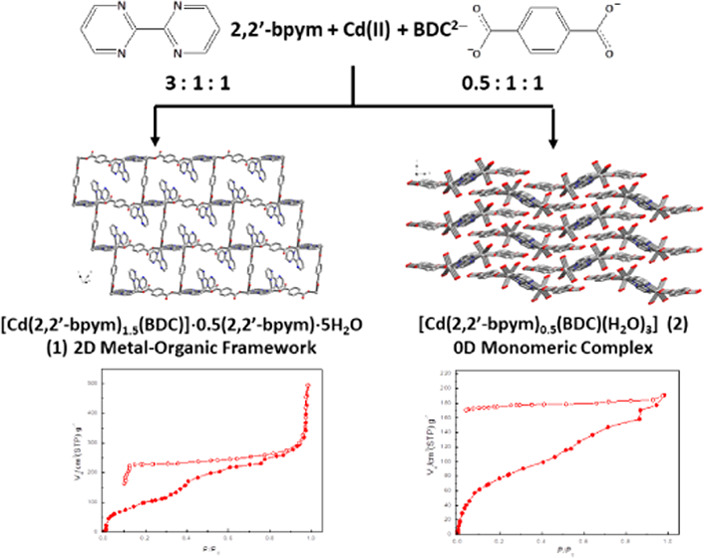
Two ligand ratio-dependent supramolecular networks, [Cd(2,2′-bpym)1.5(BDC)]·0.5(2,2′-bpym)·5H2O (1) and [Cd(2,2′-bpym)0.5(BDC)(H2O)3] (2), (BDC2– = dianion of terephthalic acid and 2,2′-bpym = 2,2′-bipyrimidine) have been synthesized and structurally characterized by the single-crystal X-ray diffraction method. Structural determination reveals that compound 1 is a two-dimensional (2D) layered metal–organic framework (MOF) constructed via the bridges of Cd(II) ions with 2,2′-bpym and BDC2– ligands, and compound 2 is a zero-dimensional (0D) 2,2′-bpym-bridged di-Cd(II) monomeric complex. When the thermally dehydrated powders of 1 (at 100 °C) were immersed into water solution, most of the dehydrated powders of 1 underwent structural transformation back to rehydrated 1, but very little amounts of the dehydrated powders of 1 were decomposed to light-brown crystals of 2 or colorless crystals of a new coordination polymer (CP), [Cd(2,2′-bpym)(BDC)(H2O)]·3H2O (3), with its one-dimensional (1D) zigzag chain-like framework being constructed via the bridges of Cd(II) ions with the BDC2– ligand. Structural analysis reveals that all 3D supramolecular networks of 1–3 are further constructed via strong intermolecular interactions, including hydrogen bonds and π–π stacking interactions. Compounds 1 and 2 both exhibit significant water vapor hysteresis isotherms, and their cyclic water de-/adsorption behavior accompanied with solid-state structural transformation has been verified by de-/rehydration TG analyses and powder X-ray diffraction (PXRD) measurements.
Introduction
Three-dimensional (3D) supramolecular networks built up via the self-assembly of metal complexes, coordination polymers (CPs),1 or metal–organic frameworks (MOFs)1,2 are an important research issue, not only in their structural diversity of supramolecular networks assembled via various types of intermolecular forces3−18 but also in their potentially diverse functional applications.19−26 Bridging ligands have been widely used in the developments of CPs or MOFs and are now at the heart of supramolecular coordination chemistry.27 Rigid multi-carboxylate ligands have been extensively employed to link metal ions and produce high-dimensional frameworks.28−30 For example, 1,4-benzenedicarboxylate (BDC2–) is the key bridging ligand with various kinds of coordination modes in the construction of archetypical MOFs.29,30 In addition, conjugated N-containing heterocycles are important bridging ligands. The coordination chemistry of 2,2′-bipyrimidine ligands (2,2′-bpym), however, has been understudied and underdeveloped compared to its more prominent N-heterocyclic analogues 2,2′-bipyridine31 and 5,5′-bipyrimidine (5,5′-bpym).32 Unlike 5,5′-bpym acting as bridging ligands with μ-type multi-monodentate coordination modes,32 2,2′-bpym can coordinate with metal ions with chelating or bis-chelating coordination modes. Nevertheless, 2,2′-bpym shares the key feature of each of these ligands: a π-conjugated pathway that can mediate electronic or magnetic interactions between metal centers.33 Recently, porous 3D MOFs have been examined for their water capture properties, and they were found to be highly promising materials.34−36 Water adsorption in MOFs can occur following three distinct pathways: chemisorption on open metal sites, physisorption in the form of layers or clusters, or capillary condensation.34,36 However, water capture behaviors applied on 3D supramolecular networks assembled via 0D metal complexes, 1D CPs, or 2D MOFs are seldom, and only few cases have been examined.37−39 In our previous study, we have successfully synthesized a series of 2D or 3D MOFs constructed using Cd(II) with 2,2′-bpym and oxocarbon dianion (CnOn2–, n = 4, 5, 6) ligands, creating interesting structural topology.40,41 On the other hand, the 2,2′-bpym can be used as an ancillary ligand, together with the carboxylate ligand, to meet the requirement of coordination geometries of metal ions in the assembly process. With our continuous effort on the study of 3D supramolecular networks assembled with metal complexes, CPs, or MOFs, the structural characterization of metal ions with the used mixed ligands, BDC2– and 2,2′-bpym, seems to be interesting and is worth further investigating. Focusing on this approach, we report here two ligand ratio-dependent supramolecular networks, [Cd(2,2′-bpym)1.5(BDC)]·0.5(2,2′-bpym)·5H2O (1) and [Cd(2,2′-bpym)0.5(BDC)(H2O)3] (2), with their 3D supramolecular networks being self-assembled by 2D layered-like MOFs and 0D dinuclear Cd(II) monomeric complexes, respectively. Interestingly, when the thermally dehydrated powder samples of 1 (at 100 °C) were immersed into water solution, most of the dehydrated powders of 1 underwent structural transformation back to rehydrated 1, but very little amounts of the dehydrated powders of 1 were decomposed to light-brown crystals of 2 or colorless crystals of a new polymeric zigzag chain-like framework, [Cd(2,2′-bpym)(BDC)(H2O)]·3H2O (3). Compounds 1 and 2 both exhibit significant water vapor hysteresis isotherms, and their reversible water de-/adsorption behavior has been evidenced by cyclic de-/rehydration gravimetric analysis (TGA) and in situ temperature-dependent powder X-ray diffraction (PXRD).
Experimental Section
General Procedures and Physical Measurements
All chemicals were reagent grade and were used as commercially obtained without further purification. Elementary analyses (carbon, hydrogen, and nitrogen) were performed using a Perkin-Elmer 2400 elemental analyzer. The infrared spectra were recorded on a Nicolet Fourier Transform IR, MAGNA-IR 500 spectrometer in the range of 500–4000 cm–1 using the KBr disc technique. Thermogravimetric analyses (TGA) were performed on a computer-controlled Perkin-Elmer 7 Series/UNIX TGA7 analyzer. Single-phased powder samples were loaded into alumina pans and heated with a ramp rate of 5 °C/min from room temperature to 800 °C under a nitrogen flux. The adsorption isotherms of N2 gas (77 K)H2O (298 K) were measured in the gaseous state using BELSORP-max volumetric adsorption equipment from BEL, Osaka, Japan. In the sample cell (∼1.8 cm3) maintained at T ± 0.03 K was placed the adsorbent sample (∼100–150 mg), which has been prepared at 100 °C for 1 and at 180 °C for 2 and 10–2 Pa for about 24 h prior to the measurement of the isotherm. The adsorbate was placed into the sample cell, and then, the change of pressure was monitored, and the degree of adsorption was determined by the decrease in pressure at the equilibrium state. All operations were automatically computer-controlled.
Synthesis of [Cd(2,2′-bpym)1.5(BDC)]·0.5(2,2′-bpym)·5H2O (1)
A MeOH/H2O (1:1) solution (3 mL) of Na2BDC (21.0 mg, 0.1 mmol) was added to a MeOH/H2O (1:1) solution (6 mL) of Cd(NO3)2·4H2O (30.8 mg, 0.1 mmol) and 2,2′-bpym (47.4 mg, 0.3 mmol) at room temperature to give a colorless solution. The resulting solution was allowed to stand for two months, crystallizing into colorless plate-like crystals of 1 in the yield of 91.5% (62.48 mg) based on Cd(NO3)2·4H2O. Anal. calcd for C24H26Cd1N8O9 (1): C, 42.24; N, 16.48; H, 3.55. Found: C, 42.21; N, 16.41; H,3.84. Selected IR data (cm–1, KBr pellet): 3504 (m), 3391 (m), 3062 (m), 3044 (m), 1665 (m), 1568 (vs), 1497 (m), 1406 (vs),1376 (s), 1142 (m), 1013, (m), 826(s), 763(s), 687 (m), 655 (s) cm–1.
Synthesis of [Cd(2,2′-bpym)0.5(BDC)(H2O)3] (2)
A MeOH/H2O (1:1) solution (3 mL) of Na2BDC (21.0 mg, 0.1 mmol) was added to a MeOH/H2O (1:1) solution (6 mL) of Cd(NO3)2·4H2O (30.8 mg, 0.1 mmol) and 2,2′-bpym (7.9 mg, 0.05 mmol) at room temperature to give a colorless solution. The resulting solution was allowed to stand for one month, crystallizing into pale-brown block-like crystals of 2 in the yield of 85.68% (35.09 mg) based on Cd(NO3)2·4H2O. Anal. calcd for C12H13Cd1N2O7 (2): C, 35.18; N, 6.84; H, 3.20. Found: C, 35.13; N, 6.87; H, 3.52. Selected IR data (cm–1, KBr pellet): 3386 (m), 3073 (m), 1574 (vs), 1543 (m), 1414 (vs),1306 (s), 1151 (m), 1015, (m), 830(s), 754(s), 669 (s), 518 (s) cm–1.
Synthesis of [Cd(2,2′-bpym)(BDC)(H2O)]·3H2O (3)
Crystal samples of 1 were put into the bottom of a sample tube and heated up to 100 °C for one week. During the heating period, some colorless crystals were attached on the wall of the sample tube, which were collected and structurally identified as free bpym ligands by the single-crystal X-ray diffraction method. The dehydrated powders at the bottom were collected and then immersed into the water solution. After standing for several days, little amounts of colorless block-like crystals of [Cd(2,2′-bpym)(BDC)(H2O)]·3H2O (3) were obtained. Anal. calcd for C16H18Cd1N4O8 (3): C, 37.22; N, 11.03; H, 3.19. Found: C, 37.92; N, 11.06; H, 3.58. Selected IR data (cm–1, KBr pellet): 3367 (m), 1567 (vs), 1500 (m), 1404 (s),1358 (m), 1149 (w), 1014, (w), 826(m), 745(m), 668 (s), 508 (s) cm–1.
Crystallographic Data Collection and Refinement
Single-crystal structural analysis of compounds 1–3 were performed on a Siemens SMART diffractomer with a CCD detector with Mo Kα radiation (λ = 0.71073 Å) at room temperature. A preliminary orientation matrix and unit cell parameters were determined from 3 runs of 15 frames each; each frame corresponds to a 0.3° scan in 10 s, following by spot integration and least squares refinement. For each structure, data were measured using ω scans of 0.3° per frame for 20 s until a complete hemisphere has been collected. Cell parameters were retrived using SMART42 software and refined with SAINT43 on all observed reflections. Data reduction was performed with SAINT35 software and corrected for Lorentz and polarization effects. Absorption corrections were applied with the program SADABS.44 Direct phase determination and subsequent difference Fourier map synthesis yielded the positions of all non-hydrogen atoms, which were subjected to anisotropic refinements. For compounds 1–3, all hydrogen atoms were generated geometrically (C–Hsp2 = 0.93) with the exception of the hydrogen atoms of the coordinated and solvated water molecules, which were located in the difference Fourier map with the corresponding positions and isotropic displacement parameters being refined. The final full-matrix, least squares refinement on F2 was applied for all observed reflections [I > 2σ(I)]. All calculations were performed using the SHELXTL-PC V 5.03 software package.45 Crystallographic data and details of data collections and structure refinements of compounds 1–3 are listed in Table 1.
Table 1. Crystal Data and Structural Refinement for Compounds 1, 2, and 3b.
| 1 | 2 | 3 | |
|---|---|---|---|
| formula | C24H26Cd1N8O9 | C12H13Cd1N2O7 | C16H18Cd1N4O8 |
| fw | 682.93 | 409.64 | 506.74 |
| cryst syst | triclinic | monoclinic | monoclinic |
| space group | P1̅ | P21/c | P21/n |
| a (Å) | 11.1088(6) | 9.5092(4) | 12.6390(7) |
| b (Å) | 11.4336(6) | 20.3463(9) | 10.7412(6) |
| c (Å) | 12.8554(6) | 7.1187(3) | 15.5868(9) |
| α (°) | 65.687(1) | 90.0 | 90.0 |
| β (°) | 67.701(1) | 94.256(1) | 112.986(1) |
| γ (°) | 75.149(1) | 90.0 | 90.0 |
| V (Å3) | 1366.99(12) | 1373.51(10) | 1948.02(19) |
| Z | 2 | 4 | 4 |
| Dcalcd (g cm–3) | 1.659 | 1.981 | 1.728 |
| μ (mm–1) | 0.866 | 1.629 | 1.173 |
| F(000) | 692 | 812 | 1016 |
| max and min trans | 0.8883, 0.6712 | 0.8291, 0.7370 | 0.7992, 0.7424 |
| reflns collected | 18133 | 13588 | 14228 |
| unique reflns (I > 2σ(I)) | 5887 | 2899 | 3888 |
| R1, wR2a (I > 2σ(I)) | 0.0254, 0.0629 | 0.0232, 0.0534 | 0.0337, 0.0914 |
| R1, wR2a (all data) | 0.0277, 0.0641 | 0.0264, 0.0548 | 0.0401, 0.0948 |
| GOF on F2 | 1.056 | 1.054 | 1.055 |
| no. of variable | 379 | 199 | 293 |
R1 = Σ||Fo – Fc||/Σ|Fo|; wR2(F2) = [Σw|Fo2 – Fc2|2/Σw(Fo4)]1/2.
GOF = {Σ[w|Fo2 – Fc2|2]/(n – p)}1/2.
In Situ and Ex Situ X-Ray Powder Diffraction
High-resolution synchrotron powder X-ray diffraction measurements were performed at the TPS 19A and 09A in the National Synchrotron Radiation Research Center. X-ray energies of 16 and 15 keV are used at 19A and 09A, respectively. All powder samples were packed into a 0.2 mm boronsilicate glass capillary and were rotated fast at 500 RPM during data collection. The powder diffraction patterns were recorded using a position-sensitive detector, MYTHEN. All the geometry and sample position offset corrections were calibrated using an NIST standard material, LaB6(660c), using GSAS-II suite46 and GSASIIscriptable modules.47 For the high-temperature experiment, a hot air gas blower was placed 2 mm under the sample with a ramp rate of 0.2°/s.
Results and Discussion
Synthesis and IR spectroscopy of Compounds 1–3
The synthetic representation of compounds 1–3 is shown in Scheme 1, in which 1 and 2 were ligand ratio-dependent products, which can be synthesized independently by controlling the molar ratio of Cd(II) salts and 2,2′-bpym and Na2BDC ligands at 1:3:1 and 1:0.5:1, respectively. Compound 3 is the thermal pyrolysis product when crystal samples of 1 were heated at 100 °C for one week to obtain dehydrated powders and then immersed into water solution for several days at room temperature to generate light-brown crystals of 2 or colorless crystals of 3. The most relevant IR features for 1–3 are those associated with the 2,2′-bpym and BDC2– ligands and almost identical throughout the region from 500 to 4000 cm–1, showing strong characteristic absorption bands in the 1300–1600 cm–1 range attributed to the carboxylate groups of BDC2– ligands.
Scheme 1. Synthetic Representation of Compounds 1–3.

Structural Description of [Cd(2,2′-bpym)1.5(BDC)]·0.5(2,2′-bpym)·5H2O (1)
The asymmetric unit of compound 1 is composed of one Cd(II) ion, one and a half of coordinated 2,2′-bpym, two halves of BDC2– ligands, half of a guest 2,2′-bpym, and five solvated water molecules. The molecular structure of 1 is shown in Figure S1 (Supporting Information), in which Cd(II) ions are seven-coordinate bonded to four nitrogen donors of two crystallographically independent 2,2′-bpym ligands and three oxygen donors of two crystallographically independent BDC2– ligands. Relevant bond lengths and angles around the Cd(II) center are listed in Table S1 (Supporting Information). In 1, two crystallographically independent BDC2– ligands act as bridging ligands with bis-monodentate and bis-chelating coordination modes, connecting the Cd(II) ions to form a one-dimensional (1D) zigzag-like chain. Adjacent chains are mutually linked via the bridges of Cd(II) ions and 2,2′-bpym ligands in the bis-chelating coordination mode to generate an undulating two-dimensional (2D) layered MOF (Figure 1a, left) with puckered honeycomb cavities. From a topological perspective, each Cd(II) center can be viewed as a three-connected node, and the BDC2– (red lines) and 2,2′-bpym (green lines) ligands are linear linkers. Thus, such a 2D network can be regarded as a honeycomb puckered 63-hcb net topology. The Cd(II)···Cd(II) separations through bis-monodentate/bis-chelating BDC2– and bis-chelating 2,2′-bpym bridges are 11.678(8)/11.439(4) and 6.372(5) Å, respectively. Adjacent layers are then arranged in an orderly AAA manner via the π–π interaction between the pyrimidyl rings of the interlayer chelating 2,2′-bpym ligands with the ring centroid distance of 3.377 Å (for related parameters, see in Table S3, Supporting Information) to complete its 3D supramolecular networks (Figure 1b). It is worth noting that all five guest H2O molecules behave both as two hydrogen bond donors and one hydrogen bond acceptors and then are mutually interlinked via O–H···O hydrogen bonding interactions to generate a 1D hydrogen-bonded polymeric chain (shown in Figure 1c, yellow dashed lines) using a puckered hydrogen-bonded (H2O)4 tetramer and (H2O)6 hexamer as basic building units with O···O distances in the ranges of 2.726(2)–2.888(2) Å. Meanwhile, the synergistic interactions through the hydrogen bonding interaction between the guest water molecules and oxygen atoms (O(1) and O(2)) of BDC2– and nitrogen atom (N(4)) of 2,2′-bpym (see Figure 1b) render the O···O distances of 2.749(2), 2.868(2), and 3.023(2) Å, respectively, thus providing another key stabilization energy to hold the aggregation of 1D water chains in the 3D supramolecular network. In other words, the distinctive alternate −(H2O)4–(H2O)6– hydrogen-bonded chain-like structure is grown and stabilized within the 3D supramolecular network and vice versa, which also acts as the base substrate for further stabilization of the as-prepared 2D MOF (shown in Figure 1c). Relevant parameters of O–H···O hydrogen bonds are summarized in Table S2 (Supporting Information). Furthermore, the puckered cavities in the 3D network encapsulate free 2,2′-bpym molecules (Figure 1d, green 2,2′-bpym molecules) stabilized by the combination of two intermolecular interactions, including the O–H···N hydrogen bond between the water and free 2,2′-bpym molecules with an O···N distance of 2.920(2) Å (Table S2, Supporting Information) and two sets of π–π interactions between the pyrimidyl rings of chelating 2,2′-bpym ligands and free 2,2′-bpym molecules with the ring centroid distances of 3.595 and 3.648 Å (for related parameters, see Table S3, Supporting Information) (Figure 2).
Figure 1.

(a) Left: 2D layered MOF of 1; right: 2D topological representation of 1 with BDC2– (red lines) and bpym (green lines) as linear linkers. The guest bpym, five water molecules, and hydrogen atoms are omitted for clarity. (b) 3D supramolecular network of 1 assembled by 2D layered MOFs viewed along the c axis. (c) 1D hydrogen-bonded polymeric chains assembled by five guest water molecules in the 3D supramolecular network using puckered hydrogen-bonded (H2O)4 tetramers and (H2O)6 hexamers as building units. (d) Guest bpym (green color) molecules in the 3D supramolecular network.
Figure 2.
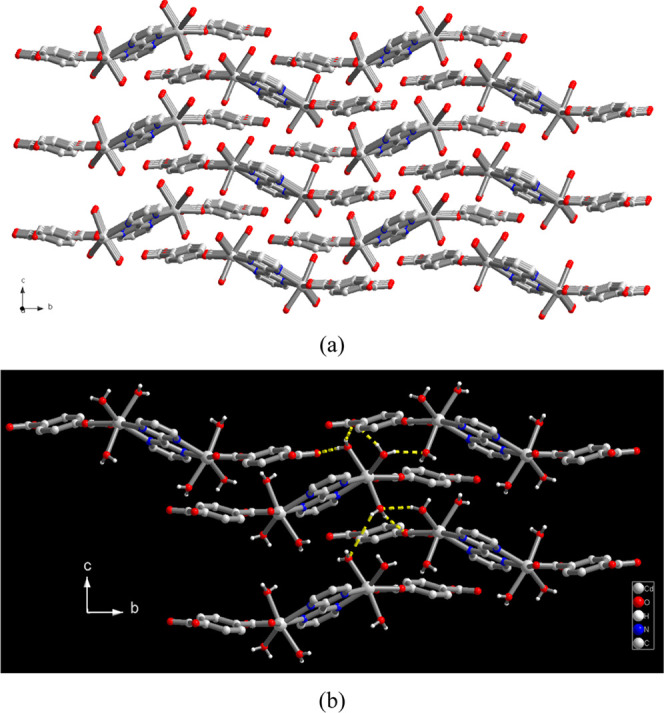
(a) 3D Supramolecular network of 2 and (b) representation of hydrogen bonding interactions among monomeric molecules.
Structural Description of [Cd(2,2′-bpym)0.5(BDC)(H2O)3] (2)
The asymmetric unit of compound 2 is composed of one Cd(II) ion, half of a 2,2′-bpym ligand, one BDC2– ligand, and three coordinated water molecules. The molecular structure of 2 is a dinuclear Cd(II) monomeric complex bridged by a 2,2′-bpym ligand in the bis-chelating coordination mode (shown in Figure S2, Supporting Information), where each Cd(II) ion is seven-coordinate bonded to two nitrogen donors of 2,2′-bpym ligands, two oxygen donors of one BDC2– ligand, and three water molecules. Relevant bond lengths and angles around the Cd(II) center are listed in Table S4 (Supporting Information). Adjacent dinuclear monomers are then assembled together via six sets of intermolecular O–H···O hydrogen bonding interactions between the coordinated H2O molecules and oxygen atoms of the BDC2– ligand to generate a hydrogen-bonded 3D supramolecular network with O···O distances in the ranges of 2.648(2)–2.865(2) Å (shown in Figure 3b, yellow dashed lines). It is worth noting that among three coordinated H2O molecules, two (O(5) and O(6)) behave as two hydrogen bond donors, while the third one (O7) behaves not only as two hydrogen bond donors but also one hydrogen bond acceptor. Such a difference in the intermolecular interactions among the three coordinated water molecules results in an obvious two-step weight loss during the thermal dehydration processes, which will be discussed in the next section of thermal stability by TG analysis. Meanwhile, the uncoordinated oxygen atom (O(3)) in the BDC2– ligand forms a unique trifurcated hydrogen bonds with three coordinated water molecules. Relevant parameters of O–H···O hydrogen bonds are summarized in Table S5 (Supporting Information).
Figure 3.

(a) 1D Zigzag-like polymeric chain of 3, (b) 2D hydrogen-bonded (yellow dashed lines) and π–π (green dashed lines) assembled layered framework in 3, and (c) 3D parallel and interpenetrating supramolecular network of 3.
Structural Description of [Cd(2,2′-bpym)(BDC)(H2O)]·3H2O (3)
The asymmetric unit of compound 3 is composed of one Cd(II) ion, one 2,2′-bpym, two halves of BDC2– ligands, and one coordinated water and three solvated water molecules. The molecular structure of 3 is shown in Figure S3 (Supporting Information), in which Cd(II) ions are six-coordinate bonded to two nitrogen donors of the 2,2′-bpym ligand, three oxygen donors of two crystallographically independent BDC2– ligands, and one water molecule. Relevant bond lengths and angles around the Cd(II) center are listed in Table S6 (Supporting Information). In 3, two crystallographically independent BDC2– ligands both act as bridging ligands with bis-monodentate and bis-chelating coordination modes, connecting the Cd(II) ions to form a one-dimensional (1D) zigzag-like chain (Figure 3a). The Cd(II)···Cd(II) separations through bis-monodentate and bis-chelating BDC2– bridges are 11.304(5) and 11.256(2) Å, respectively. Adjacent chains are arranged in an orderly AAA manner via the hybrid interchain interaction of bifurcated O–H···N hydrogen bonds between the coordinated water molecule (O(5)) and chelating 2,2′-bpym ligands (N(2) and N(3)) with the O···N distances of 3.056(4) and 3.072(4) Å and two sets of π–π interactions between the pyrimidyl rings of chelating 2,2′-bpym ligands with the ring centroid distance of 3.700 Å to form a hydrogen-bonded and π–π-stacked 2D layered framework (shown in Figure 3b). Relevant parameters of O–H···N hydrogen bonds and π–π interactions are summarized in Table S7 and Table S8, respectively, for 3 (Supporting Information). It is noteworthy that adjacent hydrogen-bonded and π–π-stacked layers are further assembled in a combined parallel and mutually interpenetrated manner (Figure 3c, left) via interlayer π–π interactions between the benzene rings of the BDC2– ligand and two pyrimidyl rings of the chelating 2,2′-bpym ligands in a sandwich-type πpyrimidyl–πbenzene–πpyrimidyl fashion with the ring centroid distance of 3.691 Å (see Table S8, Supporting Information), to afford its 3D supramolecular network with porous 1D channels (Figure 3c, right). Three guest water molecules are intercalated into the 1D porous channels (Figure 3c, right, yellow ones) via the intermolecular O–H···O hydrogen bonding interactions among water molecules with O···O distances in the ranges of 2.739(2)–2.880(2) Å, and the synergistic interactions via the O–H···O hydrogen bonding interaction between the guest water molecules (O(6) and O(7)) and oxygen atoms (O(1)) in the BDC2– ligand render the O···O distances of 2.850(2) and 2.962(2) Å, respectively, thus providing another key stabilization energy to hold the aggregation of the guest water molecules in the porous channels of the 3D supramolecular network. Relevant parameters of O–H···O hydrogen bonds are summarized in Table S7 (Supporting Information).
It is noteworthy that compounds 2 or 3 can be obtained via the thermal pyrolysis reaction of 1 at 100 °C. Based on the structural characteristics of 1–3, the potential mechanism of the structural transformation may be attributed to the bond breaking of Cd–N bonds of chelating 2,2′-bpym or bis-chelating 2,2′-bpym in 1 to generate 2 or 3, respectively, as shown in Scheme 2. Pathway A reveals that the thermal pyrolysis reaction of 1 undergoes the bond breaking of Cd–N bonds of chelating 2,2′-bpym ligands and one side Cd–O bond of BDC2– ligands to produce monomeric [Cd2(2,2′-bpym)(BDC)2] fragments and then rehydration to generate compound 2 by immersion into water solution. On the contrary, pathway B reveals that the thermal pyrolysis reaction of 1 undergoes the bond breaking of Cd–N bonds of bis-chelating 2,2′-bpym ligands to produce 1D polymeric chain-like [Cd(2,2′-bpym)(BDC)] fragments and then re-hydration to generate compound 3 by immersion into water solution.
Scheme 2. Possible Mechanism of the Pyrolysis Reaction of Compound 1 to Compound 2 (Pathway A) and Compound 3 (Pathway B).

Thermal Stability and Structural Variation of 1 and 2
The thermal stability and related temperature-dependent structural variation of compounds 1 and 2 were investigated and characterized by TGA and insitu PXRD measurements. TG analysis of 1 (Figure S4a, Supporting Information) reveals that during the heating process, a one-step weight loss of 12.6%, occurred in the temperature range of 37.7–85.2 °C, which is closely equal to the loss of five guest H2O molecules (calcd 13.2%), and then, thermal stability is observed up to 109.9 °C. On further heating, sample 1 decomposed. TG analysis of 2 (Figure S5a, Supporting Information) reveals that during the heating process, a two-step weight loss occurred with a weight loss of 9.2% for the first step and 4.3% for the second step, in the temperature ranges of 87.9–115.2 and 147.0–179.0 °C, respectively, which are closely equal to the losses of two (calcd 8.8%) and one (calcd 4.4%) coordinated H2O molecules, respectively, and then, thermal stability is observed up to 282.6 °C. On further heating, sample 2 decomposed. Based on structural analysis described in the previous section, the two-step weight-loss behavior shown in Figure S5a may be attributed to different numbers of hydrogen bonds for the three coordinated water molecules; two (O(5) and O(6)) of them behave as two hydrogen bond donors, while the third one (O(7)) acts both as two hydrogen bond donors and one hydrogen bond acceptor.
To investigate the temperature-dependent structural variation during the thermal dehydration procedure, in situ high-resolution PXRD experiments for 1 and 2 were performed and analyzed as shown in Figures S4b and S5b, respectively (Supporting Information). The powder XRD patterns of 1 at room temperature and the selected temperatures revealed that the structure of polycrystalline 1 at room temperature closely matches the simulation one obtained from its single-crystal structure, and its crystallinity was maintained up to 60 °C. A phase transformation happened at around 75°C and completed at 100 °C, corresponding to its dehydrated samples. The powder XRD patterns of 2 at room temperature and the selected temperatures reveal that the structure of polycrystalline 2 closely matches the simulation one obtained from its single-crystal structure. During 90–100 °C, the diffraction pattern change was observed. With the temperature up to 120 °C, an obvious phase transformation occurred, and then, another phase transformation occurred from 150 to 175 °C. The structural variations based on the temperature-dependent PXRD patterns are consistent with the results from TGA.
Reversibility of Water De-/Adsorption in 1 and 2
The cyclic TG measurements by thermal de-/rehydration treatment have been carried out under water vapor to verify the reversibility of water de-/adsorption behaviors in 1 and 2 during the thermal heating/cooling procedures. Compound 1 displays a 13.2% weight decrease, corresponding to losses of five guest H2O molecules, to produce the dehydrated form after thermal treatment up to 90 °C (Figure 4). When the dehydrated samples were cooled down to room temperature and exposed to water vapor, H2O molecules can be readsorbed to produce the rehydrated form with a weight increase of 6.4%, closely equal to 2.4 water molecules. Such heating/cooling procedures have been repeated for five cycles (Figure 4), to confirm the partial reversibility of the water de-/adsorption.
Figure 4.

TG measurements of 1 during the cyclic de-/rehydration procedures were repeated five times from RT to 95 °C. Solid red line: the variation of weight loss with time; blue dash–dot line: the variation of temperature with time.
The thermal stability study by TG analysis reveals that the weight losses of the first and second stages in compound 2 correspond to the releases of two and one coordinated water molecules, respectively. It is worth noting that the de-/adsorption of two coordinated water molecules for the first weight-loss stage in 2 is proven to be reversible, which is demonstrated using cyclic TG measurements under water vapor via the repeated de-/rehydration procedures as a function of time and temperature (Figure 5a). The dehydrated 2 after the dehydration process by the thermal treatment (up to 90 °C) shows 9.1% weight loss, corresponding to 2.0 water molecules. As the samples were cooled down to RT, the water molecules can then be readsorbed by exposing the dehydrated 2 to water vapor, forming rehydrated 2 with a weight increase of 8.7%, corresponding to approximately 2.0 water molecules. Such heating and cooling procedures were repeated five cycles with almost the same weight increase/weight decrease percentages to demonstrate the stable reversibility during the thermal re/dehydration procedures in the first stage. These results evidence that compound 2 has a reversible water de-/adsorption behavior between the dehydrated and rehydrated forms in the first stage driven by thermal re-/dehydration treatments. However, in contrast to the complete reversibility of two coordinated water molecules in the first stage, the de-/adsorption with the removal of three coordinated water molecules in the second stage is more likely to be partial reversible (Figure 5b). When all three coordinated water molecules (13.1%) were completely removed by the thermal treatment (up to 180 °C), only partial weight increase (5.6%) with approximately one water molecule under water vapor was recovered during the cooling process. Such heating and cooling procedures were also repeated for five cycles (Figure 5b) to demonstrate the stable but only partially reversible water ad-/desorption behavior during the thermal re/dehydration procedures. This result reveals that after removal of three coordinated water molecules in the second stage, the molecular structure and the coordination environment of Cd(II) ions in the completely dehydrated form of 2 in the second stage might be changed and cannot be recovered to the original structure.
Figure 5.

TG measurements of 2 during the cyclic de-/rehydration processes (a) from RT to 90 °C and (b) from RT to 185 °C were repeated five times. Solid red line: the variation of weight loss with time; blue dash–dot line: the variation of temperature with time.
The N2 gas isotherms of 1 and 2 at 78 K both display typical type-II adsorption profiles (see Figures S6a and S6b, respectively, Supporting Information) with a very low N2 gas uptake, suggesting only surface adsorption. To explore the water adsorption ability of these two supramolecular networks, water vapor sorption isotherms of pretreated dehydrated 1 and 2 were measured at 298 K. The water vapor adsorption isotherm of dehydrated 1 (Figure 6) exhibits a steady and gradual increase of adsorbed water vapor at 0 < relative P/P0 < 0.90, with a maximum amount of 296.1 cm3 g–1 at relative P/P0 = 0.94, approximately equal to 7.8 water molecules, and then an abrupt increase of adsorbed water vapor at 0.94 < relative P/P0 < 0.98, with a maximum amount of 498.8 cm3 g–1 at relative P/P0 = 0.98, approximately equal to 13.1 water molecules, indicating that the pretreated dehydrated 1 can adsorb more than five guest water molecules in 1. It is worth noting that the desorption curve did not follow the adsorption curve any longer, exhibiting a notable hysteresis loop with the amount of 224.2 cm3 g–1 (approximately equal to 5.9 water molecules) at lower relative P/P0 = 0.12. This result demonstrates that the porous dehydrated 1 exhibits high water sorption capability. A similar water vapor ad-/desorption behavior can be found in 2. The water vapor sorption isotherm of dehydrated 2 with three-coordinated water weight loss in the second stage (Figure 7) also exhibits a steady and gradual increase of adsorbed water vapor at 0 < relative P/P0 < 0.98, with a maximum amount of 191.1 cm3 g–1 at relative P/P0 = 0.98, approximately equal to 3.0 water molecules, indicating three coordinated water molecules being readsorbed in pretreated dehydrated 2. Similar to 1, the desorption curve also did not follow the adsorption curve any longer, which exhibited a notable hysteresis loop with the value of 170.7 cm3 g–1 (approximately equal to 2.7 water molecules) at lower relative P/P0 = 0.04. The water ad-/desorption isotherm of dehydrated 2 is consistent with results from the cyclic TGA and indicates that the Cd(II) center of the dehydrated forms provides the vacant sites for the bond reformation of water molecules with the dehydrated samples being exposed to water vapor. The water sorption isotherms with notable hysteresis loops observed in 1 and 2 are significant and may be exploited in potential applications of water-harvesting materials.
Figure 6.

Water vapor ad-/desorption isotherms of dehydrated 1 at 298 K.
Figure 7.

Water vapor ad-/desorption isotherms of dehydrated 2 at 298 K.
Water De-/Adsorption of 1 and 2 Accompanied with Structural Transformation
The correlation between the reversible guest water de-/adsorption behaviors and the dynamic structural transformation in de-/rehydrated 1 and 2 is an important issue. The interesting point is that the structure changes are induced by packing water molecules and coordinated water molecules in 1 and 2, respectively.
In sample 1, as shown in Figure 8, the dehydrated species can adsorb water molecules to reconstruct the rehydrated species when the dehydrated species at 100 °C are cooled down to room temperature and then exposed to water (i.e., the dehydrated samples are placed in a glass capillary and immersed into the beaker). The PXRD patterns of the rehydrated species immersed into the water solution (Figure 8d) mostly match well those of freshly synthesized samples (Figure 8b), indicating that the structure of rehydrated species may be nearly close to the original structure 1. This result reveals that 1 may undergo reversible dynamic structural transformations between the dehydrated species and rehydrated species; it has also been identified that the guest water molecules are readsorbed by cycle de-/adabsorption TGA and water vapor isotherms (shown in Figures 4 and 6). Because the structure of dehydrated species at a high temperature cannot be directly solved through PXRD data, the structural transformation mechanism cannot be directly deduced. However, the cell indexing and Le Bail refinement of the dehydrated form of 1 were performed successfully. The dehydrated form retains triclinic P1̅ symmetry, and the cell volume is 1239.6 Å3 (a = 12.0332 Å, b = 11.7545 Å, c = 10.6020 Å, α = 111.626°, β = 105.373°, γ = 63.622°) which is reduced by 10% compared to that of the rehydrated species. However, the closely similar PXRD patterns between the rehydrated species and the fresh original 1 provide an indirect but significant proof to certify the reversibility of guest water molecules during the de-/rehydration procedures. Consequently, the reversible structural transformation mechanisms may be attributed to the substantial hydrogen bonding interactions among the guest water molecules.
Figure 8.
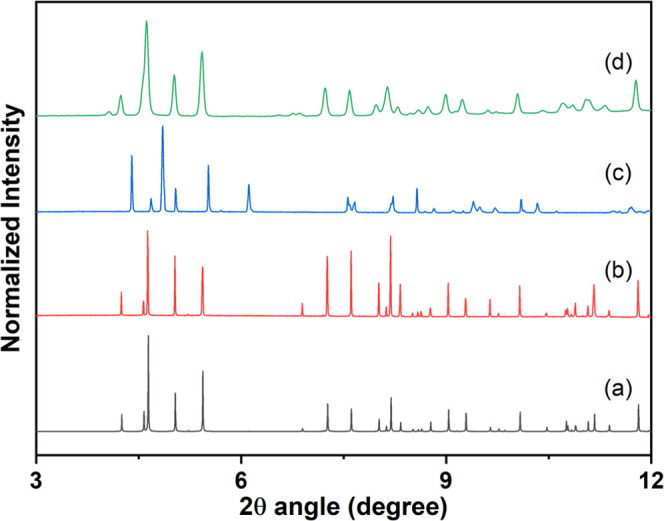
Ex situ X-ray powder diffraction measurements of 1: (a) Simulation from single-crystal X-ray diffraction data; (b) fresh powder samples at RT; (c) dehydrated powder samples at 100 °C; and (d) rehydrated powder samples obtained by immersion of the dehydrated powder samples into water solution.
In sample 2, as shown in Figure 9, when the dehydrated form at 120 °C, for the first stage with two-water molecule weight losses, is cooled down to room temperature and then exposed to water vapor, the dehydrated form readsorbs water molecules to reconstruct the rehydrated form. The closely similar powder patterns between the rehydrated species and the fresh original 2 provide an indirect but significant proof to certify the reversibility of Cd(II)–O(water) bond breaking and bond reforming during the cyclic de-/rehydration procedures. Consequently, the reversible structural transformation mechanism may be attributed to the substantial hydrogen bonding interactions between two coordinated water molecules and BDC2– ligands. Reasonable unit cell parameters of the dehydrated form were indexed successfully; the crystal system retains monoclinic P21/c with a = 9.5006 Å, b = 17.9234 Å, c = 7.6206 Å, and β = 90.825°. Obviously, the b axis is shortened by 2.5 Å, and the c axis is increased by 0.5 Å, where the β angle is reduced by 3.4°. The volume of the dehydrated form of 2 is 1297.5 Å3, which is reduced 5% from that (1373.5 Å3) of the original crystal structure 2. Unfortunately, the structure of dehydrated species cannot be simply determined through the Rietveld refinement process, which gives a hint here that an obvious structural transformation occurred during the dehydration process. Based on the structural analysis, the distance of hydrogen-bonded O(4)···O(6) in 2 is 2.649 Å, which is quite close to the amount of b axis reduction (2.5 Å), which gives a plausible ratiocination that when the coordinated water molecules (O(5) and O(6)) are removed, the uncoordinated oxygen atom (O(4)) of the BDC2– ligand replaces the positions of water molecules. Thus, the water molecules coordinated to Cd(II) centers are relatively easily broke via a neighboring BDC2–-assisted process under a gentle thermal treatment. The probable and logical pathway of reversible structural conversion between dehydrated and rehydrated species could help deduce that the vacant coordinate sites of Cd(II) ions in 2 are produced by the removal of the water molecules (O(5) and O(6)), initiating the opportunity for the approach of the adjacent BDC2– ligands, which are oriented toward the coordinated water molecules of adjacent units through hydrogen bonding interactions. These contacts shorten in the track of structural transformation and change into bonds with Cd(II) centers after removal of the coordinated water molecules, generating the dehydrated form, which can readsorb water molecules to produce the rehydrated form after being exposed to water vapor.
Figure 9.
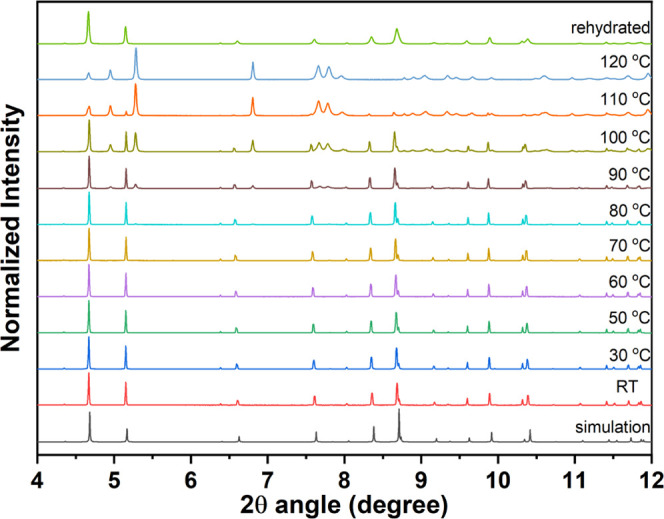
Temperature-dependent X-ray powder diffraction measurements and ex situ water adsorption powder X-ray diffraction measurements of 2.
Conclusions
In summary, two ligand ratio-dependent Cd(II) compounds, [Cd(2,2′-bpym)1.5(BDC)]·0.5(2,2′-bpym)·5H2O (1) and [Cd(2,2′-bpym)0.5(BDC)(H2O)3 (2)] and a thermal pyrolysis compound [Cd(2,2′-bpym)(BDC)(H2O)]·3H2O (3) have been successfully structurally characterized. All of their 3D supramolecular networks are unique and quite interesting, which are constructed using 2D layered MOFs, 0D monomers, and 1D polymeric chains for 1, 2, and 3, respectively. In 1–3, 2,2′-bpym and BDC2– both behave as multifunctional ligands for the construction of their 3D supramolecular networks, including coordination ligands with various coordination modes, hydrogen-bonding acceptors, and π–π stacking constructors. During thermal re-/dehydration processes, compound 1 possesses a reversible water de-/adsorption behavior accompanied with a reversible thermally induced de-/rehydrated structural transformation, while in 2, reversible and partially reversible water de-/adsorption behaviors are observed in the first- and second-step stages, respectively, accompanied with a reversible thermally induced de-/rehydrated structural transformation in the first-step stage. Notably, the water vapor ad-/desorption isotherms of dehydrated 1 and 2 both exhibit high water capture uptakes with unique and significant hysteresis loops. We thus believe that these two 3D supramolecular compounds may have the potential for development in the field of water-harvesting materials. Further study on the water uptakes under different humidities for atmospheric water harvesting (AWH) applications36 will be carried out in the future.
Acknowledgments
The authors wish to thank the Ministry of Science and Technology, Taiwan, for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00652.
Selected bond lengths (Å) and angle (°) around the Cd(II) ion for 1–3; parameters of O–H···N and O–H···O hydrogen bonds for 1–3; π–π interactions (face-to-face) between pyrimidyl rings of bpym ligands and benzene ring of BDC2– ligands in 1 and 3; coordination environments about the Cd ion in 1–3; thermogravimetric measurements of 1 and 2; temperature-dependent powder X-ray diffraction patterns of 1 and 2; and N2 ad-/desorption isotherms of dehydrated species 1 and 2 (PDF)
Crystallographic data for [Cd(2,2′-bpym)1.5(BDC)]·0.5(2,2′-bpym)·5H2O (1) (CIF)
Crystallographic data for [Cd(2,2′-bpym)0.5(BDC)(H2O)3] (2) (CIF)
Crystallographic data for [Cd(2,2′-bpym)(BDC)(H2O)]·3H2O (3) (CIF)
Accession Codes
CCDC2131947–2131949 for compounds 1–3, respectively, contain the supplementary crystallographic data for this paper. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: (internet.) +44-1223/336-033; E-mail: deposit@ccdc.cam.ac.uk].
The authors declare no competing financial interest.
Supplementary Material
References
- Batten S. R.; Champness N. R.; Chen X. M.; Garcia-Martinez J.; Kitagawa S.; Öhrström L.; O’keeffe M.; Suh M. P.; Reedijk J. Terminology of metal–organic frameworks and coordination polymers. Pure Appl. Chem. 2013, 85, 1715–1724. 10.1351/PAC-REC-12-11-20. [DOI] [Google Scholar]
- Batten S. R.; Champness N. R.; Chen X. M.; Garcia-Martinez J.; Kitagawa S.; Öhrström L.; O’keeffe M.; Suh M. P.; Reedijk J. Coordination polymers, metal–organic frameworks and the need for terminology guidelines. CrystEngComm. 2012, 14, 3001–3004. 10.1039/c2ce06488j. [DOI] [Google Scholar]
- Takamizawa S.; Akatsuka T.; Ueda T. Gas-conforming transformability of an ionic single-crystal host consisting of discrete charged components. Angew. Chem., Int. Ed. 2008, 47, 1689–1692. 10.1002/anie.200702950. [DOI] [PubMed] [Google Scholar]
- Takamizawa S.; Kohbara M.; Akatsuka T.; Miyake R. Gas-adsorbing ability of tris-ethylenediamine metal complexes (M = Co(III), Cr(III), Rh(III), Ir(III)) as transformable ionic single crystal hosts. New J. Chem. 2008, 32, 1782–1787. 10.1039/b806467a. [DOI] [Google Scholar]
- Pan Q. H.; Li J. Y.; Chen Q.; Han Y. D.; Chang Z.; Song W.-C.; Bu X.-H. [Co(en)3]1/3[In(ox)2]·3.5H2O: A zeolitic metal-organic framework templated by Co(en)3Cl3. Microporous Mesoporous Mater. 2010, 132, 453–457. 10.1016/j.micromeso.2010.03.026. [DOI] [Google Scholar]
- Pan Q. H.; Chen Q.; Song W.-C.; Hu T.-L.; Bu X.-H. Template-directed synthesis of three new open-framework metal(II) oxalates using Co(III) complex as template. CrystEngComm 2010, 12, 4198–4204. 10.1039/c002658a. [DOI] [Google Scholar]
- Bertani R.; Sgarbossa P.; Venzo A.; Lelj F.; Amati M.; Resnati G.; Pilati T.; Metrangolo P.; Terraneo G. Halogen bonding in metal–organic–supramolecular networks. Coord. Chem. Rev. 2010, 254, 677–695. 10.1016/j.ccr.2009.09.035. [DOI] [Google Scholar]
- Ingleson M. J.; Bacsa J.; Rosseninsky M. J. Homochiral H-bonded proline based metal organic frameworks. Chem. Commun. 2007, 3036–3038. 10.1039/b706557d. [DOI] [PubMed] [Google Scholar]
- Wang C. C.; Yang C. C.; Yeh C. T.; Ceng K. Y.; Chang P. C.; Ho M. L.; Lee G. H.; Shih W. J.; Sheu H. S. Reversible Solid-State Structural Transformation of a 1D–2D Coordination Polymer by Thermal De/Rehydration Processes. Inorg. Chem. 2011, 50, 597–603. 10.1021/ic1018345. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R. C–H···O and other weak hydrogen bonds. From crystal engineering to virtual screening. Chem. Commun. 2005, 2995–3001. 10.1039/b504372g. [DOI] [PubMed] [Google Scholar]
- García-Báez E. V.; Martínez-Martínez F. J.; Höpfl H.; Padilla-Martínez I. I. π-Stacking Interactions and C–H···X (X = O, Aryl) Hydrogen Bonding as Directing Features of the Supramolecular Self-Association in 3-Carboxy and 3-Amido Coumarin Derivatives. Cryst. Growth Des. 2003, 3, 35–45. 10.1021/cg0255826. [DOI] [Google Scholar]
- Jabiak C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc., Dalton Trans. 2000, 3885–3896. 10.1039/B003010O. [DOI] [Google Scholar]
- Claessens C. G.; Stoddart J. F. π–π Interactions in Self-assembly. J. Phys. Org. Chem. 1997, 10, 254–272. . [DOI] [Google Scholar]
- Guo H.; Guo X.; Batten S. R.; Song J.; Song S.; Dang S.; Zhang G.; Tang J.; Zhang H. Hydrothermal Synthesis, Structures, and Luminescent Properties of Seven d10 Metal–Organic Frameworks Based on 9,9-Dipropylfluorene-2,7-Dicarboxylic Acid (H2DFDA). Cryst. Growth Des. 2009, 9, 1394–1401. 10.1021/cg8006469. [DOI] [Google Scholar]
- Reger D. L.; Horger J. J.; Smith M. D.; Long G. J.; Grandjean F. Homochiral, Helical Supramolecular Metal–Organic Frameworks Organized by Strong π···π Stacking Interactions: Single-Crystal to Single-Crystal Transformations in Closely Packed Solids. Inorg. Chem. 2011, 50, 686–704. 10.1021/ic102256t. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Chang Z.; Song W. C.; Song H.; Song H. B.; Hu T. L.; Bu X. H. A Controllable Gate Effect in Cobalt(II) Organic Frameworks by Reversible Structure Transformations. Angew. Chem., Int. Ed. 2013, 52, 11550–11553. 10.1002/anie.201306304. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Feng R.; Xu J.; Jia Y. Y.; Wang T. T.; Chang Z.; Bu X. H. Kinetic and Thermodynamic Control of Structure Transformations in a Family of Cobalt(II)–Organic Frameworks. ACS Appl. Mater. Interfaces 2017, 9, 35141–35149. 10.1021/acsami.7b12925. [DOI] [PubMed] [Google Scholar]
- Li A. L.; Gao Q.; Xu J.; Bu X. H. Proton-conductive metal-organic frameworks: Recent advances and perspectives. Coord. Chem. Rev. 2017, 344, 54–82. 10.1016/j.ccr.2017.03.027. [DOI] [Google Scholar]
- Li B.; Chrzanowski M.; Zhang Y.; Ma S. Applications of metal–organic frameworks featuring multi-functional sites. Coord. Chem. Rev. 2016, 307, 106–129. 10.1016/j.ccr.2015.05.005. [DOI] [Google Scholar]
- Zhang X.; Wang W.; Hu Z.; Wang G.; Uvdal K. Coordination polymers for energy transfer: Preparations, properties, sensing applications, and perspectives. Coord. Chem. Rev. 2015, 284, 206–235. 10.1016/j.ccr.2014.10.006. [DOI] [Google Scholar]
- Bradshaw D.; Claridge J. B.; Cussen E. J.; Prior T. J.; Rosseinsky M. J. Design, Chirality, and Flexibility in Nanoporous Molecule-Based Materials. Acc. Chem. Res. 2005, 38, 273–282. 10.1021/ar0401606. [DOI] [PubMed] [Google Scholar]
- Silva P.; Vilela S.M.F.; Tome J.P.C.; Paz F.A.A. Multifunctional metal–organic frameworks: From academia to industrial applications. Chem. Soc. Rev. 2015, 44, 6774–6803. 10.1039/C5CS00307E. [DOI] [PubMed] [Google Scholar]
- Li S.; Huo F. Metal–organic framework composites: From fundamentals to applications. Nanoscale 2015, 7, 7482–7501. 10.1039/C5NR00518C. [DOI] [PubMed] [Google Scholar]
- Janiak C.; Vieth J. K. MOFs, MILs and more: Concepts, properties and applications for porous coordination networks (PCNs). New J. Chem. 2010, 34, 2366–2388. 10.1039/c0nj00275e. [DOI] [Google Scholar]
- He Y.; Li B.; O’Keeffe M.; Chen B. Multifunctional metal–organic frameworks constructed from meta-benzenedicarboxylate units. Chem. Soc. Rev. 2014, 43, 5618–5656. 10.1039/C4CS00041B. [DOI] [PubMed] [Google Scholar]
- Lin Z. J.; Lu J.; Hong M.; Cao R. Metal–organic frameworks based on flexible ligands (FL-MOFs): Structures and applications. Chem. Soc. Rev. 2014, 43, 5867–5895. 10.1039/C3CS60483G. [DOI] [PubMed] [Google Scholar]
- Steel P. J. Aromatic Nitrogen Heterocycles as Bridging Ligands; a Survey. Coord. Chem. Rev. 1990, 106, 227–265. 10.1016/0010-8545(60)80005-7. [DOI] [Google Scholar]
- Habib H. A.; Sanchiz J.; Janiak C. Mixed-ligand coordination polymers from 1,2-bis(1,2,4-triazol-4-yl)ethane and benzene-1,3,5-tricarboxylate: Trinuclear nickel or zinc secondary building units for three-dimensional networks with crystal-to-crystal transformation upon dehydration. Dalton Trans. 2008, 1734–1744. 10.1039/b715812b. [DOI] [PubMed] [Google Scholar]
- Eddaoudi M.; Kim J.; Rosi N.; Vodak D.; Wachter J.; O’Keeffe M.; Yaghi O. M. Systematic Design of Pore Size and Functionality in Isoreticular MOFs and Their Application in Methane Storage. Science 2002, 295, 469–472. 10.1126/science.1067208. [DOI] [PubMed] [Google Scholar]
- Sun M. Y.; Wang Y.; Bai F. Y.; Xing Y. H. Construction of manganese-based metal organic frameworks derived from aromatic dicarboxylic acids and application for the adsorption of iodine. Main Group Chem. 2022, 1–14. 10.3233/MGC-210178. [DOI] [Google Scholar]; Pre-press
- Kaes C.; Katz A.; Hosseini M. W. Bipyridine: The Most Widely Used Ligand. A Review of Molecules Comprising at Least Two 2,2′-Bipyridine Units. Chem. Rev. 2000, 100, 3553–3590. 10.1021/cr990376z. [DOI] [PubMed] [Google Scholar]
- Xu M. H.; Wang C. F.; Liu Y. T.; Cui Q. L. Co(II) coordination polymer: Treatment activity on the chronic obstructive pulmonary disease by reducing the inflammatory cytokines releasing. Main Group Chem. 2021, 20, 231–239. 10.3233/MGC-210005. [DOI] [Google Scholar]
- Beaulieu-Houle G.; White N. G.; MacLachlan M. J. Coordination Polymers from Functionalized Bipyrimidine Ligands and Silver(I) Salts. Cryst. Growth Des. 2018, 18, 2210–2216. 10.1021/acs.cgd.7b01657. [DOI] [Google Scholar]
- Bon V.; Senkovska I.; Weiss M. S.; Kaskel S. Tailoring of network dimensionality and porosity adjustment in Zr-and Hf-based MOFs. CrystEngComm 2013, 15, 9572–9577. 10.1039/c3ce41121d. [DOI] [Google Scholar]
- Fukushima T.; Horike S.; Inubushi Y.; Nakagawa K.; Kubota Y.; Takata M.; Kitagawa S. Solid Solutions of Soft Porous Coordination Polymers: Fine-Tuning of Gas Adsorption Properties. Angew. Chem., Int. Ed. 2010, 49, 4820–4824. 10.1002/anie.201000989. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Lu H.; Zhao F.; Yu G. Atmospheric Water Harvesting: A review of Material and Structural Designs. ACS Materials Lett. 2020, 2, 671–684. 10.1021/acsmaterialslett.0c00130. [DOI] [Google Scholar]
- Wang C. C.; Ke S. Y.; Chen K. T.; Sun N. K.; Liu W. F.; Ho M. L.; Lu B. J.; Hsieh Y. T.; Lee G. H.; Huang S. Y.; Yang E. C.; et al. Sponge-Like Water De-/Ad-Sorption versus Solid-State Structural Transformation and Colour-Changing Behavior of an Entangled 3D Composite Supramolecuar Architecture, [Ni4(dpe)4(btc)2(Hbtc)(H2O)9]·3H2O. Polymers 2018, 10, 1014 10.3390/polym10091014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S. Y.; Chang Y. F.; Wang H. Y.; Yang C. C.; Ni C. W.; Lin G. Y.; Chen T. T.; Ho M. L.; Lee G. H.; Chuang Y. C.; Wang C. C. Self-Assembly of Four Coordination Polymers (CPs) in A 3D Entangled Architecture Showing Reversible Dynamic Solid-State Structural Transformation and Color-Changing Behaviour by Thermal De-/Re-hydration. Cryst. Growth Des. 2014, 14, 4011–4018. 10.1021/cg500620k. [DOI] [Google Scholar]
- Wang C. C.; Tsai J. H.; Ke S. Y.; Lee G. H.; Chuang Y. C.; Yang E. C. Structural Characterization, Water Adsorption, and Magnetic Properties of Two Composite Mn(II)–Squarate–dpe Supramolecular Architectures. Cryst. Growth Des. 2020, 20, 5395–5405. 10.1021/acs.cgd.0c00608. [DOI] [Google Scholar]
- Wang C. C.; Kuo C. T.; Chou P. T.; Lee G. H. Rhodizonate Metal Complexes with a 2D Chair-like M6 Metal Organic Framework: [M(C6O6)(bpym)(H2O)]·nH2O. Angew Chem., Int. Ed. 2004, 43, 4507–4510. 10.1002/anie.200460278. [DOI] [PubMed] [Google Scholar]
- Wang C. C.; Kuo C. T.; Yang J. C.; Lee G. H.; Shih W. J.; Sheu H. S. Assemblies of Two New Metal-Organic Frameworks (MOFs) Constructed from the CdII with 2,2′-Bipyrimidine and Cyclic Oxocarbon dianions CnOn2– (n = 4, 5). Cryst. Growth Des. 2007, 7, 1476–1482. 10.1021/cg070189r. [DOI] [Google Scholar]
- SMART V 4.043 Software for CCD Detector System; Siemens Analytical Instruments Division: Madison, WI, 1995. [Google Scholar]
- SAINT V 4.035 Software for CCD Detector System; Siemens Analytical Instruments Division: Madison, WI, 1995. [Google Scholar]
- Sheldrick G. M.Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1993. [Google Scholar]
- SHELXTL 5.03 (PC-Version), Program Liberary for Structure Solution and Molecular Graphics; Siemens Analytical Instruments Division: Madison, WI, 1995. [Google Scholar]
- Toby B. H.; Von Dreele R. B. GSAS-II: the genesis of a modern open-source all-purpose crystallography software package″. J. Appl. Crystallogr. 2013, 46, 544–549. 10.1107/S0021889813003531. [DOI] [Google Scholar]
- O’Donnell J. H.; Von Dreele R. B.; Chan M. K. Y.; Toby B. H. A scripting interface for GSAS-II. J. Appl. Crystallogr. 2018, 51, 1244–1250. 10.1107/S1600576718008075. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.