

Abstract
The ability to assay large numbers of low-frequency mutations is useful in biomedicine, yet, the technical hurdles of sequencing multiple mutations at extremely high depth, with accuracy, limits their detection in clinical practice. Low-frequency mutations can typically be detected by increasing the sequencing depth, however this limits the number of loci that can be probed for simultaneously. Here, we report a technique to accurately track thousands of distinct mutations with minimal reads, termed MAESTRO (minor allele enriched sequencing through recognition oligonucleotides), which employs massively-parallel mutation enrichment to enable duplex sequencing to track up to 10,000 low-frequency mutations, yet requiring up to 100-fold less sequencing. We show that MAESTRO could inform the mutation validation of whole-exome sequencing and whole genome sequencing data from tumor samples, enable chimerism testing, and is suitable for the monitoring of minimal residual disease via liquid biopsies. MAESTRO may improve the breadth, depth, accuracy, and efficiency of sequencing-based mutational testing.
Editorial summary:
Massively-parallel mutation enrichment enables the tracking of up to 10,000 low-frequency mutations, via duplex sequencing, requiring up to 100-fold less sequencing depth.
Mutations in DNA emerge from single cells1, define cell populations2, and establish genetic diversity3. Considering the vast genetic diversity of living organisms and the significance of mutations in disease biology4, there is a growing need to assay many distinct, low-abundance mutations in multiple areas of biomedicine spanning oncology5, obstetrics6, transplantation7,8, infectious disease9, genetics10, microbiomics11, forensics12, and beyond. Yet, the intrinsic tradeoff in breadth-versus-depth of DNA sequencing means that either few mutations can be assayed at high depth, or many mutations at low depth—not both. High depth (i.e. many reads per genomic locus) is required to accurately detect low-abundance mutations, but this severely limits breadth (i.e. number of distinct loci). This explains why, despite massive reductions in sequencing costs, it remains prohibitively expensive to test for large numbers of distinct, low-abundance mutations.
Duplex sequencing is one of the most accurate methods for mutation detection, with 1000-fold fewer errors than standard sequencing, but adds significant cost13. By requiring mutations to be present in replicate reads from both strands of each DNA duplex, many of the errors in sample preparation and sequencing can be overcome to enable reliable detection of low-abundance mutations. Yet, up to 100-fold more reads per locus are required—a challenge that is exacerbated when tracking many low-abundance mutations. Less stringent methods exist that require fewer reads, but compromising specificity to save cost would be deeply problematic for applications that impact patient care. While methods to enrich rare mutations have been developed14–21, none have employed high-accuracy sequencing, nor tracked many rare mutations.
Here, we describe MAESTRO (minor allele enriched sequencing through recognition oligonucleotides), a technique which combines massively-parallel mutation enrichment with duplex sequencing to enable accurate mutation testing with minimal sequencing. In contrast to conventional hybrid-capture duplex sequencing22–24 (herein referred to as ‘Conventional’), which uses long probes to capture mutant and wild type with similar efficiency, MAESTRO uses short probes to enrich for patient-specific mutant alleles and uncovers the same mutant duplexes using up to 100-fold fewer reads. We first establish its performance and then present three exemplary use cases.
Results
MAESTRO uncovers mutant duplexes with ~100-fold fewer reads
We have established an accurate and efficient technique to track large numbers of low-abundance mutations in clinical specimens (Fig. 1A). Our technique, called MAESTRO, utilizes allele-specific hybridization with short probes, leveraging thermodynamic differences in heteroduplex versus homoduplex DNA (Fig. 1C), to enrich barcoded library molecules bearing up to 10,000 prespecified mutations. Minimal sequencing is applied, and mutations are detected on both strands of each DNA duplex (Fig. 1B). MAESTRO also employs a tunable noise filter which excludes error-prone loci (Supplementary Fig. 3, Methods).
Figure 1: MAESTRO enables accurate mutation tracking using minimal sequencing in clinical specimens.
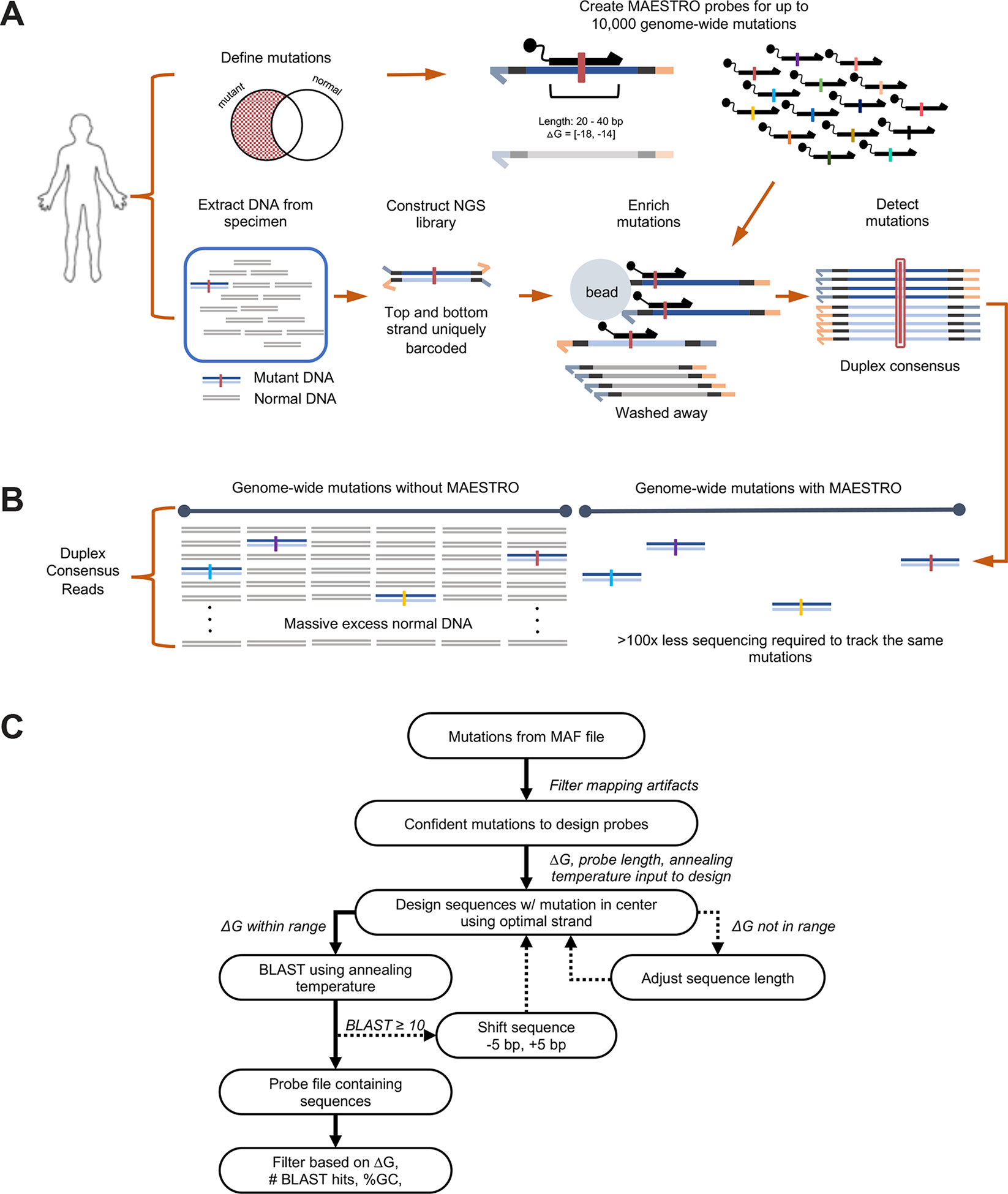
(A) Up to 10,000 MAESTRO probes are designed with stringent length and ΔG for single-nucleotide discrimination of predefined mutations. DNA libraries containing uniquely barcoded top and bottom strands are subject to hybrid capture using allele-specific MAESTRO probes. Only molecules containing tracked mutations are captured and sequenced with duplex consensus for error suppression. (B) Using MAESTRO, the same mutations are discovered using up to 100x less sequencing because uninformative regions are depleted. (C) Probe design overview. See probe design section of Methods.
We first sought to maximize fold-enrichment while minimizing loss of mutations. We created a 1/1k dilution of sheared genomic DNA from two human cell lines, identified exclusive single nucleotide polymorphisms (SNPs) as proxies for clonal mutations, and generated duplex sequencing libraries that were split for hybrid capture. Using qPCR, we confirmed that adapter ligation efficiencies are consistent with prior reports (Supplementary Table 1), and that MAESTRO capture efficiency is only slightly lower than Conventional capture (35% vs. 57% respectively, Supplementary Table 2). After sequencing, we compared raw variant allele fraction (raw VAF) and recall of mutant duplexes (Supplementary Table 3, Supplementary Fig. 2B) using MAESTRO versus Conventional (120 bp probes, 65°C annealing). By adjusting probe length and hybridization parameters, we established conditions (ΔG −18 to −14 kcal/mol, T=50°C, Fig. 1C, Supplementary Fig. 1, Supplementary Fig. 2A) that yielded strong fold-enrichment of mutant vs. wild type alleles (median 948.3-fold, range 8.1 to 3.4E4) while uncovering the majority of mutant duplexes (Fig. 2A,B, Supplementary Table 3). Indeed, the median raw VAF with MAESTRO was 0.97 (range 5.03E-3 to 1), in contrast to 6.98E-4 (range 3.00E-5 to 3.87E-3) with Conventional. The fraction of recoverable mutations (or, enrichment ‘success rate’) was 72.5%. Interestingly, we did not observe equal and opposite magnitude raw VAF changes when swapping strands of C and G reference base probes (Supplementary Fig. 2C). We believe this may be due to differences in probe characteristics (i.e. delta G, length) for each base category but further investigation is needed. MAESTRO cannot uncover more mutations than physically present in a sample; yet, by detecting each with up to 100x fewer reads, it can recover more total unique mutations, particularly when it would not otherwise be possible (e.g. due to cost) to sequence a sample to saturation.
Figure 2: MAESTRO uncovers most mutant duplexes using significantly fewer reads.
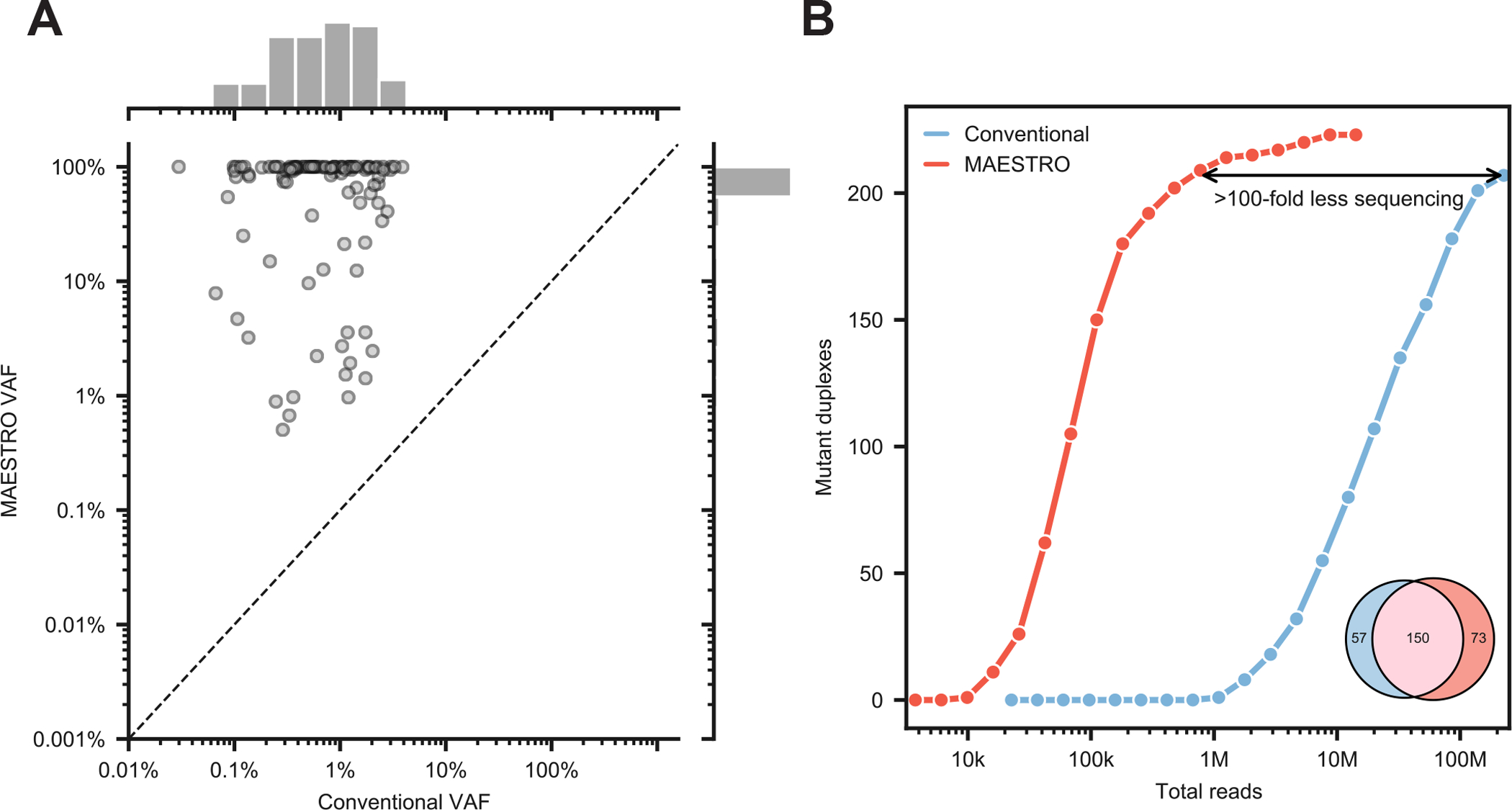
(A) Comparison of variant allele frequency with Conventional and MAESTRO with 438 probe panel at 1/1k dilution. (B) Downsampling of Conventional and MAESTRO. As an inset, mutant duplex overlap is shown; of the 57 mutant duplexes exclusive to Conventional, 42 were detected by MAESTRO but excluded by the noise filter. The initial sample was barcoded with UMIs (unique molecular indices) which allowed for tracking individual duplex molecules through different experimental conditions.
We next tuned the MAESTRO noise filter. This filter was designed to protect against the possibility that errors could arise independently on both strands of library molecules and, given enrichment bias, ‘collide’ to form a duplex (Supplementary Fig. 3A,B). It works based on the assumptions that (i) errors should be impartial to read family, and (ii) error-prone loci should therefore exhibit a disproportionate number of double- (DSC) to single- (SSC) strand consensus read families bearing mutations (Supplementary Fig. 3B). Indeed, we found that sites with DSC/SSC ratios below 0.15 had poor reproducibility in replicate captures of a non-mutant library (our negative control) (Supplementary Fig. 3C). We also found that our filter protected against errors introduced by excessive PCR (Supplementary Fig. 3D), and further confirmed that MAESTRO probes—which contain the mutant base—do not create false mutant duplexes (Supplementary Fig. 4). Filtering by DSC/SSC ratio was found to be robust to changes in sequencing depth with similar concordance observed at 10% of the original sequencing depth (Supplementary Fig. 5).
Considering the profound enrichment, we then asked how many fewer reads would be required to detect the same mutant duplexes as Conventional. We found that MAESTRO could uncover the majority (n=150/207) using ~100-fold less sequencing (Fig. 2B), while providing comparable specificity (Supplementary Fig. 6C). Of the 57 mutant duplexes exclusive to Conventional, 42 were detected by MAESTRO but excluded by the noise filter. Though some of these excluded mutations may be real, the current filter threshold prioritizes specificity over sensitivity but could be tuned for varied application. Our results suggest that MAESTRO can uncover the majority of mutant duplexes using significantly less sequencing.
MAESTRO could enable mutation verification
Expansive methods such as whole-exome and whole-genome sequencing stand to unravel the genetic basis of human diseases. However, it remains challenging to resolve low-level mutations (e.g. < 10% VAF) given insufficient depth to read each DNA molecule enough times to suppress errors. Currently, mutations discovered in sequencing studies may be orthogonally validated via technologies such as digital droplet PCR or multiplex amplicon sequencing. However, these are not highly scalable approaches and are usually restricted to a handful of mutations suspected of having potential clinical significance. We reasoned that MAESTRO could enable accurate verification of large numbers of mutations discovered from whole-exome and -genome sequencing. The net result would be that lower abundance mutations could be reliably discovered and verified from comprehensive sequencing studies.
To explore this, we performed whole-exome sequencing of tumor biopsies (of varied tumor purity; median 63%, range 26 – 100%) and matched normal DNA from 16 patients. We identified a median of 40 mutations per patient (median 40, range 13–130) and created both a MAESTRO and Conventional panel comprising all mutations for which we could design probes. Requiring the true mutations to be detected on both strands of each duplex, we found similar fractions of validated mutations between MAESTRO and Conventional, with slightly lower fractions for MAESTRO likely due to probe dropout (Fig. 3A). Yet, the fraction of validated mutations was much higher for those which had been identified at >0.10 VAF from tumor whole-exome sequencing (median 0.75, range 0.21–0.90 for MAESTRO; median 0.98, range 0.40–1.0 for Conventional), in comparison to those which had been identified at <0.10 VAF (median 0.29, range 0.07–0.82 for MAESTRO; median 0.35, range 0.04–1.0 for Conventional, Fig. 3A). Indeed, the mutations which were found to be “not validated” tended to have the lowest VAFs from tumor whole-exome sequencing (median 0.04, range 0.01–0.83, Fig. 3B). Expectedly, we observed higher fractions of MAESTRO-validated mutations in fresh-frozen (median 0.65, range 0.62–0.77) as compared to formalin-fixed (median 0.58, range 0.10–0.76) tumor biopsies. Our results suggest that MAESTRO could inform mutation validation and optimization of variant detection pipelines from WES and WGS data.
Figure 3: MAESTRO fingerprint validation of whole exome tumor samples.
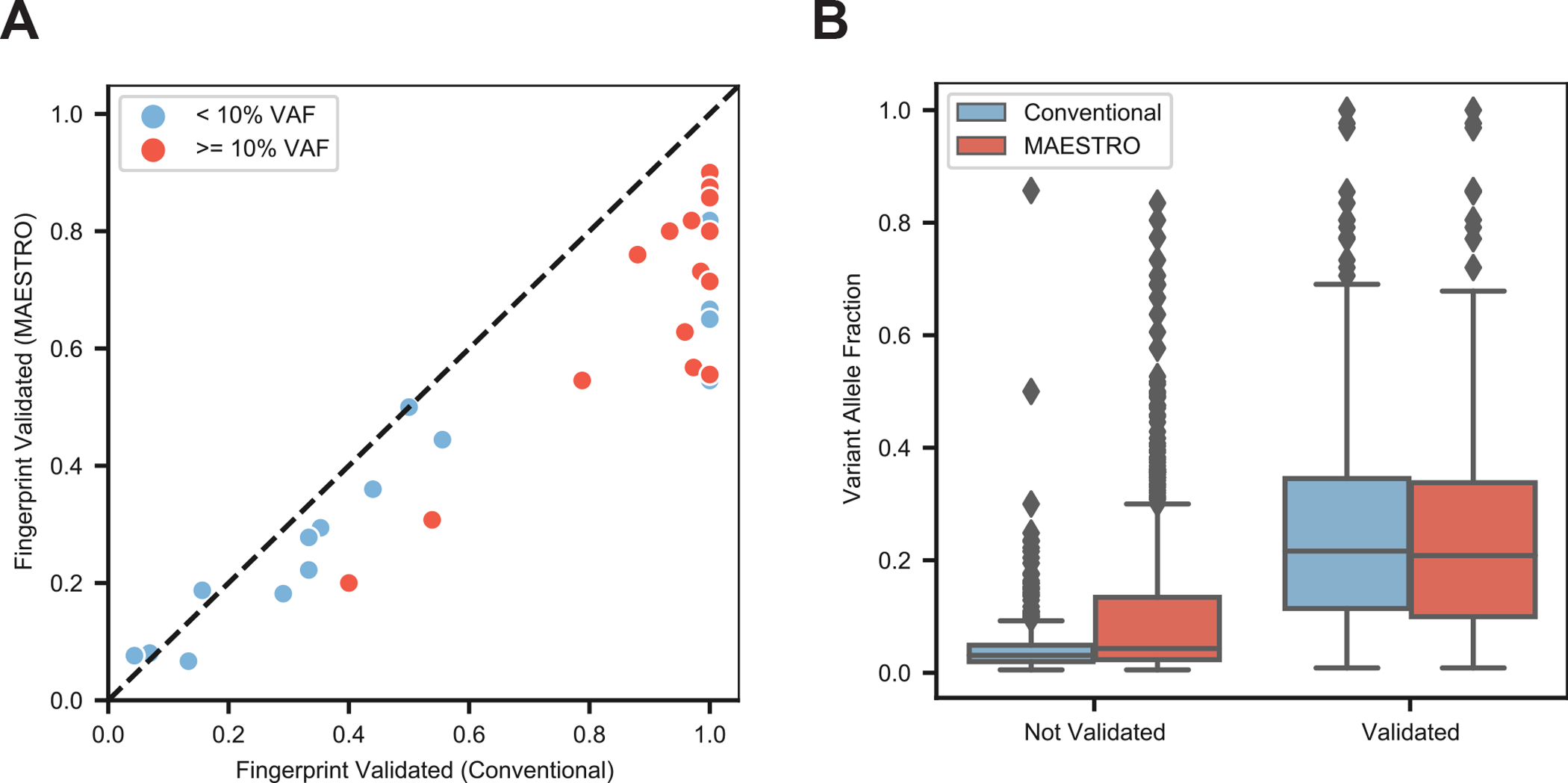
(A) Performance of n=16 tumor fingerprints using both Conventional and MAESTRO. Mutations were called from the n=16 tumor biopsies and both Conventional and MAESTRO probe sets (fingerprints) were created for all possible mutations from each tumor. The tumor biopsy libraries were captured with the Conventional and MAESTRO fingerprints and duplexes were sequenced. Fingerprints were split into two groups based on whether or not their original tumor VAF was < 10%. A mutation was considered validated if it was observed in the sequenced duplexes of the Conventional or MAESTRO sample. (B) Comparing variant allele fraction across all mutations from all Conventional (n=16) and MAESTRO (n=16) panels. Center line represents the median and bounds of the box indicate the first and third quartiles.
MAESTRO could enable chimerism testing
Chimerism testing involves detecting admixtures of cells that arise from different zygotes25. For patients who undergo stem cell transplantation, such as in the treatment of hematologic malignancies, assessment of donor chimerism has been shown to inform likelihood of recurrence and graft-versus-host disease26,27. Existing methods such as short tandem repeat analysis and qPCR testing can detect chimerism levels ranging from 1–5% to 0.05–0.1%, respectively28. We sought to determine whether higher sensitivity could be achieved by tracking more SNPs and from limited blood volume, e.g. a 1uL fingerprick contains ~10,000 white blood cells.
We first compared MAESTRO to Conventional for tracking 438 SNPs in 18 × replicate 1/100k dilutions and 17 × replicate negative control samples. We used sheared genomic DNA from the same two cell lines described in the previous section8,23,29–33 to reflect donor vs. recipient DNA and isolated 20 ng for each replicate. MAESTRO uncovered 81% (n=47/58) and 80% (n=4/5) of the donor-exclusive SNP duplexes detected with Conventional across all 1/100k and negative control samples, respectively, using much less sequencing (Supplementary Fig. 6A). Most that were exclusive to Conventional in the 1/100k samples (n=6/11) were detected by MAESTRO but excluded by the noise filter. MAESTRO also uncovered an additional 52 and 16 donor-exclusive SNP duplexes across all 1/100k and negative control samples, respectively, but most were near fragment ends, which proved less likely to be captured by Conventional in these experiments (Supplementary Fig. 6B). If we consider these differences, the concordance is nearly perfect (Supplementary Fig. 6C). However, for the rest of the study we do not remove the molecules that were less likely to be captured with Conventional. Importantly, MAESTRO detected significantly more donor-exclusive SNP duplexes in the 1/100k samples than the negative controls (Fig. 4A, p=1.16E-5, Welch’s t-test). We also confirmed that without duplex error suppression, we would have been unable to resolve chimerism at these limiting dilutions (Supplementary Fig. 6D).
Figure 4: MAESTRO can detect signal above noise at 1/100k dilution.
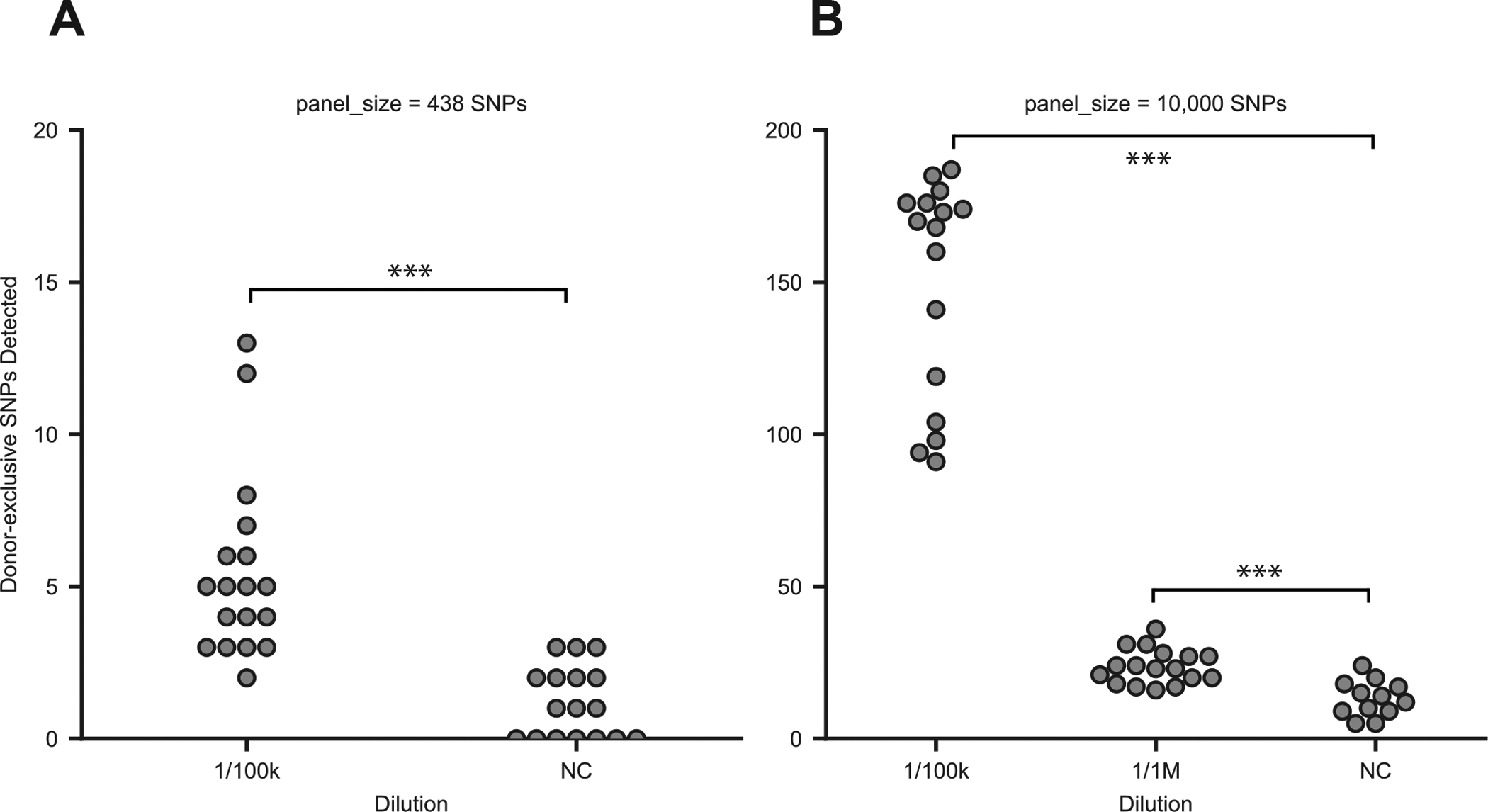
(A) Donor-exclusive SNPs detected in MAESTRO using a 438 probe panel across 18 × biological replicates of a 1/100k dilution and 17 × biological replicates of a negative control (p=1.16E-5, two-sided Welch’s t-test). (B) Donor-exclusive SNPs detected in MAESTRO using a 10,000 probe panel across 16 × biological replicates of a 1/100k dilution, 17 × biological replicates of 1/1M, and 12 × negative controls. The Welch’s t-test (two-sided) was used to determine whether significantly more donor-exclusive SNPs were uncovered in each dilution compared to the negative controls (1/100k vs. NC p=7.23E-11; 1/1M vs NC p=7.47E-5).
While MAESTRO provided comparable sensitivity and specificity using significantly less sequencing, the number of donor-exclusive SNPs detected at 1/100k dilution, of 438 tracked, was not much greater than the negative controls. We thus hypothesized that tracking even more, e.g. 10,000, could improve our signal-to-noise ratio. Yet, this could only be done feasibly with MAESTRO, as Conventional would require >10 billion reads (~$20,000 on the Illumina HiSeqX) to saturate duplex recovery, in contrast to about ~100 million reads (~$200) with MAESTRO.
Applying MAESTRO to track 10,000 donor-exclusive SNPs in 16 × replicate 1/100k dilutions, 17 × 1/1M dilutions and 12 × negative controls, we observed a large increase in number detected in the 1/100k samples (median mutations=169, range 91 to 187), which was significantly higher than the negative controls (median 13 mutations, range 5 to 24, p=7.23E-11, Fig. 4B). Higher counts were also observed in the 1/1M dilutions (median 23, range 16 to 36, p=7.47E-5), although further refinements are likely needed to enable reliable detection at 1/1M. Our results suggest that tracking thousands of genome-wide, donor-specific SNPs provides a profound boost in our signal-to-noise ratio, which is likely to be important for detecting very low levels of chimerism.
As for the signal in the negative controls, we reason that these could either be (a) true mutations that arose spontaneously with each cell division, (b) cross-contamination when cell lines were cultured, or (c) technical artifacts that have yet to be overcome in duplex sequencing. While we cannot yet discern the source, the counts were consistent with what was expected for scanning tens of millions of bases for potential mutation (10,000 mutations × few thousand haploid genomes of DNA) given the reported error rate of ~1×10−6 in duplex sequencing13,23,24. By retesting specific loci, we also verified that the majority would have been detected with Conventional (Supplementary Fig. 7), suggesting that most are not artifacts of the MAESTRO protocol.
MAESTRO could enable high-sensitivity liquid biopsy testing
Liquid biopsy represents an application for which accurate tracking of many distinct mutations could empower clinical decisions34. For instance, applying liquid biopsies to detect minimal residual disease (MRD) after cancer treatment5,35–38 has the potential to inform whether surgery is needed after neoadjuvant therapy39–41, whether adjuvant therapy is needed after surgery42, and ultimately, whether it is safe to stop treatment43. One promising way to improve sensitivity of liquid biopsies is to track many patient-specific tumor mutations in cell-free DNA (cfDNA), recognizing that not all mutations may be present in an individual blood tube when tumor DNA in the bloodstream is sparse (i.e. less than a genome equivalent of tumor DNA per tube)23,39,40,44,45. Yet, this has been challenging, because to rely upon any subset for MRD detection requires extremely accurate sequencing of many rare mutations. We reasoned that MAESTRO could enable sensitive monitoring of tumor mutation fingerprints from cell-free DNA (cfDNA).
To explore whether MAESTRO could enhance MRD detection, we created dilutions targeting between 1/100 to 1/300k tumor fraction (i.e. the fraction of tumor-derived DNA versus total DNA) using previously characterized cfDNA from a cancer patient spiked into cfDNA from a healthy donor (Fig. 5A). Replicate 20 ng libraries at each dilution were amplified and split between MAESTRO and Conventional capture, tracking 978 genome-wide SNVs exclusive to the patient’s tumor WGS. Of the 978 mutations tracked, 94.7% were validated with MAESTRO when applied to the patient’s tumor DNA (Fig. 5A inset), meaning that the remaining 5.3% were either artifacts from tumor sequencing or were missed by MAESTRO. MAESTRO uncovered a comparable number of mutations as Conventional at tumor fractions down to 1/100k and required ~50-fold less sequencing (Supplementary Fig. 8). To consider a sample “MRD positive” we used our previously defined threshold of requiring >1 mutation to detect MRD for both MAESTRO and Conventional23,37,44. Applying this threshold, none of the eight healthy donor cfDNA (negative control) samples tested positive for MRD. Leveraging probes with imperfect enrichment, we show MAESTRO retains the ability to estimate tumor fraction (r2 = 0.92, Fig. 5B, Methods). Our results show that MAESTRO’s limit-of-detection is comparable to that of Conventional in cfDNA despite ~50-fold less sequencing. MAESTRO is well-positioned to affordably track thousands of genome-wide mutations in cfDNA, well beyond what is feasible with Conventional.
Figure 5: MAESTRO improves detection of MRD in pre-operative setting.
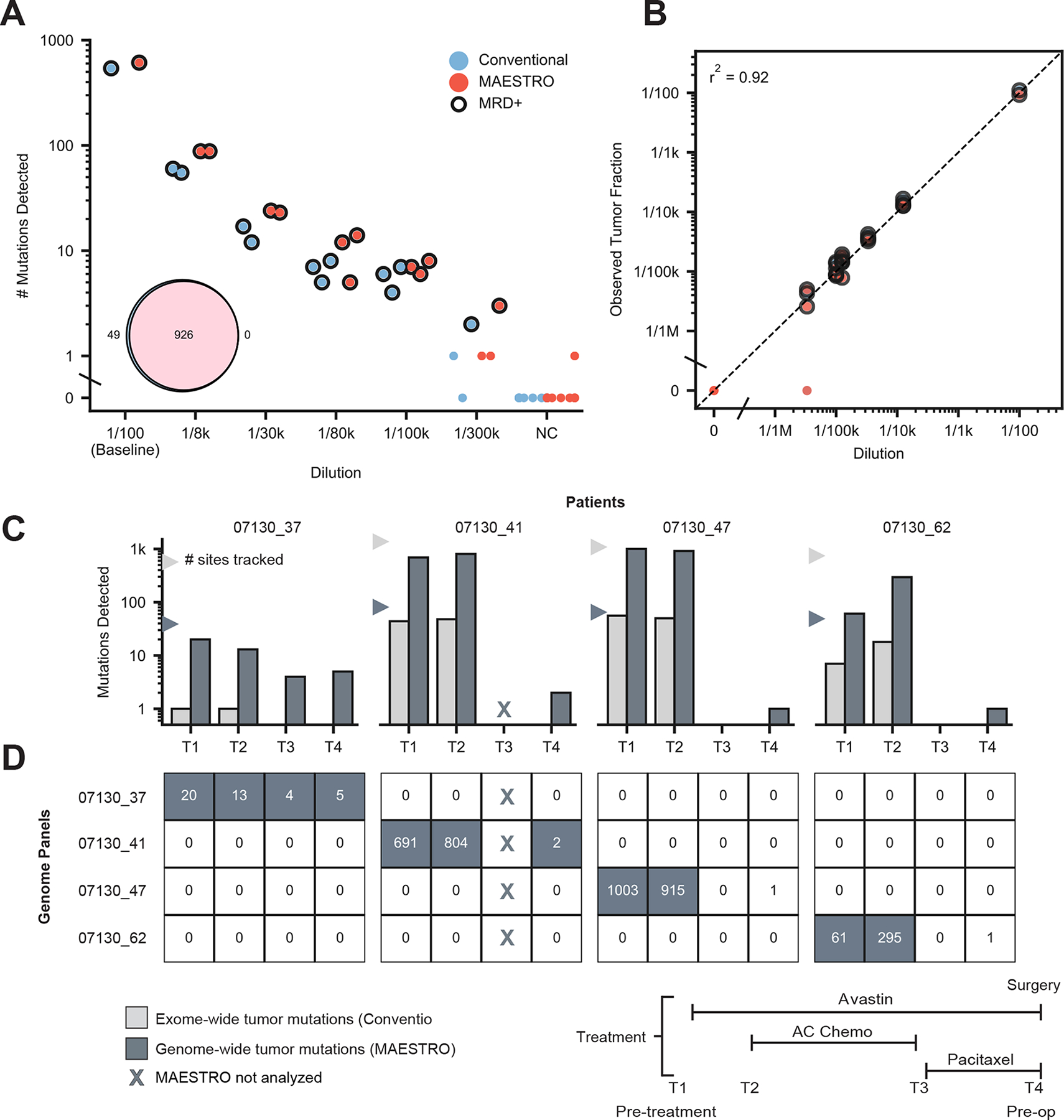
(A) Number of mutations detected at various tumor fraction dilutions between 1/100 and 1/300k using the cfDNA from a cancer patient and healthy donor. A 978 SNV genome-wide fingerprint exclusive to the cancer patient’s tumor was used for all replicates. Eight unrelated healthy donors were used as negative controls (NC). Closed circles indicate MRD(+). Inset shows number of mutations validated using Conventional and MAESTRO when each panel was applied to patient’s tumor DNA. (B) Observed tumor fraction versus expected dilution for Conventional and MAESTRO probes. (C) Genome-wide tumor mutations detected with MAESTRO compared to exome-wide tumor mutations detected with a personalized MRD test built on our Conventional assay23. Fingerprint sizes for the two conditions are shown with triangles. Mutations from all patients were combined into a single panel for MAESTRO and the same panel was applied to all samples. (D) The heatmap shows mutation counts detected using MAESTRO with patient-specific mutations on the diagonal. Filled boxes indicate MRD(+); Unfilled boxes indicate MRD(−). When each panel was applied to the other three patients’ timepoints, there was no evidence of false positives (off-diagonal), highlighting MAESTRO’s specificity.
We next sought to determine whether tracking thousands of genome-wide tumor mutations could enhance MRD detection after cancer therapy. This is significant because (i) detecting MRD remains a significant unmet medical need, and (ii) while MRD detection correlates with the number of tumor mutations tracked in cfDNA23,39,45, existing techniques have had limited breadth or depth. For instance, cancer gene panels typically cover just a few mutations per patient24; patient-specific assays track tens to hundreds39,44; and whole-genome sequencing remains far too costly to apply beyond minimal depth46. For patients with some common, aggressive forms of breast cancer, standard care involves preoperative systemic chemotherapy for its utility in guiding subsequent response-based treatment47,48. We analyzed patients with breast cancer enrolled in a clinical trial (16 patients) of preoperative therapy (Supplementary Fig. 9A), reasoning we could (i) determine the detectability of tumor-derived cfDNA at diagnosis, (ii) describe how cfDNA trends with clinical response over the course of treatment, and (iii) determine whether preoperative MRD testing could predict the presence of residual cancer in the surgical specimen.
Reasoning that genome-wide mutation tracking would be most useful in samples with low tumor fraction, we first tracked all exome-wide tumor mutations using a personalized cfDNA test built on our Conventional assay23. We found that most patients had detectable circulating tumor DNA at diagnosis (median tumor fraction 0.00858, range 0 to 0.21, Supplementary Fig. 9B) and that a decrease in tumor fraction in cfDNA between the first two time points (T1, T2) trended with clinical response (Supplementary Fig. 9C) which is consistent with prior reports39–41. Yet, MRD was detected preoperatively (T4) in only one of eight patients with residual disease at the time of surgery and in only one of five who experienced future distant recurrence. We chose the remaining four patients to explore whether genome-wide mutation tracking could enhance MRD detection.
For these four patients who had tested MRD-negative preoperatively but experienced future distant recurrence, we performed PCR-free whole-genome sequencing of their tumor biopsy specimens and blood normal DNA. We identified a median of 5575.5 (range 3385 to 8783) somatic mutations per patient and, using stringent criteria for probe design, we created one MAESTRO test comprising 55–58% of exonic mutations and 30–38% of intronic mutations from all patients (Supplementary Fig. 10). We applied the MAESTRO test to tumor and normal DNA and found 52% (range 41–56%) of probed mutations to be verified (Supplementary Fig. 11). We then applied the assay to all available cfDNA samples from all four patients, such that we assess all mutations in all patients, using the unmatched samples as controls for one another. By also applying MAESTRO tests to matched germline DNA from each patient, we further limited the potential impact of variants arising from clonal hematopoiesis.
We found that tracking all tumor mutations with MAESTRO uncovered more mutations per patient in cfDNA compared to Conventional (Fig. 5C) and detected no false mutations in any unmatched samples (Fig. 5D). Previous studies have shown that using > 1 mutation for MRD detection helps to protect against error23,37,44. We uncovered multiple tumor mutations preoperatively for two of the four patients, while observing profound signal enhancement in the earlier time points from all patients. These results demonstrate that a threshold of >1 mutation could provide reasonable specificity for MRD detection but further testing is needed to determine clinical relevance. These proof-of-principle results suggest that MAESTRO could enhance MRD detection by enabling all genome-wide tumor mutations to be accurately tracked in cfDNA.
Discussion
We have demonstrated a practical approach to extend the breadth, depth, accuracy, and efficiency of mutation tracking in clinical specimens. Our technique breaks the breadth-vs-depth ‘glass ceiling’ of DNA sequencing, enabling thousands of low-abundance mutations to be accurately tracked using minimal sequencing. As a hybrid capture and sequencing methodology, MAESTRO is easy to adopt and highly adaptable across a range of sample types. This is likely to empower many types of biomedical research and diagnostic tests that demand accurate and efficient tracking of many rare mutations.
MAESTRO is the first method, to our knowledge, to simultaneously enrich and detect thousands of mutations with high-accuracy sequencing. In a dilution series involving sheared genomic DNA, we demonstrated a median ~1000-fold enrichment from 0.1% VAF to nearly pure mutant DNA, which enabled us to detect most mutant duplexes using ~100-fold less sequencing. We showed that MAESTRO could track up to 10,000 distinct, low-abundance (< 0.1% VAF) mutations to enable both mutation validation and chimerism testing. Finally, we show signal enhancement in liquid biopsies beyond what is feasible with existing methods.
MAESTRO addresses a fundamental challenge in the mutation enrichment field by using molecular barcodes to discern true mutations from low-level errors that may also be enriched. While tracking more mutations increases our detection sensitivity at limiting dilutions, it will never be possible to detect below sequencing error rates. Accordingly, we opted to employ the most accurate sequencing method, duplex sequencing. Further, our DSC/SSC ratio filter is a unique advance that measures intrinsic noise within each sample, but two current limitations are (i) that it needs to be tuned, and (ii) that error-prone loci are discarded, which impacts sensitivity when these regions contain real mutations. We imagine a tunable noise filter could be leveraged to achieve the right balance of sensitivity and specificity across various applications.
While our approach requires whole-genome sequencing and individualized probe design, the cost of each continues to decline, and biotinylation of oligonucleotides in-house can further help to limit costs (see Methods). Bespoke genome-wide liquid biopsies reflect one potential application for MAESTRO. In cases like this, we expect upfront costs could be amortized over many serial tests, while being offset by large savings in sequencing required per test. We’ve shown that MAESTRO can be used to estimate the dilution (e.g. tumor fraction) of a sample, even though mutation enrichment may lose the ability to quantify the VAF of each mutation. It is possible that mutations could be missed by MAESTRO due to poor probe performance or exclusion by the noise filter. We are working to address this by incorporating internal controls to calibrate enrichment performance on a locus-by-locus basis, as well as probes against fixed sequences to more systematically estimate the total molecular diversity of the library and to confirm whether we have sequenced to saturation. We also focused on enrichment of point mutations, but expect that MAESTRO could also be useful for tracking other types of alterations such as insertions and deletions or structural variants. Notably, the reduction in reads required by MAESTRO can be up to 100-fold but varies with the fold-enrichment of the assayed mutations.
In all, MAESTRO provides a powerful approach to (i) convert low-abundance mutations into high-abundance mutations, and (ii) enable their detection with high-accuracy sequencing using significantly fewer reads. This means that it is no longer necessary to trade breadth for depth, or accuracy for efficiency, when tracking many low-abundance mutations in clinical samples.
Methods
Patients and Samples
All patients provided written informed consent to allow the collection of blood and/or tumor tissue and analysis of clinical and genetic data for research purposes. Adult patients with HER2-negative breast cancer and a tumor size >1.5 cm were prospectively enrolled into Dana-Farber Cancer Institute IRB-approved treatment protocol 07130 (NCT00546156). Primary endpoint of the trial was pathologic complete response (pathCR) to preoperative therapy. Data were collected at the Dana-Farber Cancer Institute and Massachusetts General Hospital in Boston, Massachusetts. All patients completed the following course of neoadjuvant Phase II therapy: Bevacizumab × 1 dose; Doxorubicin/Cyclophosphamide × 4 cycles plus Bevacizumab; Paclitaxel × 4 cycles plus Bevacizumab. Patients had plasma isolated from 20 cc blood in EDTA tubes at baseline and prior to each treatment and tissue sampling performed at diagnosis. A Residual Cancer Burden (RCB) score was calculated after surgery. We selected for analysis all enrolled patients with ER-negative, PR-negative, HER2-negative (triple-negative) breast cancer (TNBC). For those patients with sufficient tumor tissue, exome-sequencing identified mutations that we captured using our Conventional assay23. From within this cohort we identified four TNBC patients who had tested MRD-negative using the exome-wide panel but who experienced metastatic recurrence. For these patients, we applied MAESTRO to analyze genome-wide tumor mutations. HapMap DNA from NA12878 and NA19238 was purchased from Corielle. This research was conducted in accordance with the provisions of the Declaration of Helsinki and the U.S. Common Rule.
Defining Mutations to Track
For the HapMap panels, VCF files were taken from the Genome in a Bottle Consortium49 (NA12878) and 1000 Genomes project50 (NA19238). Sites specific to NA12878 were subsampled to create MAF files and were subsequently run through probe design to create the 438 and 10,000 SNV (single nucleotide variant) fingerprints.
Tumor DNA was extracted from fresh-frozen tumor samples. All patients’ tumor DNA underwent whole-exome sequencing to identify trackable mutations for conventional capture. Of the four patients selected for MAESTRO, tumor DNA underwent PCR-free whole-genome sequencing. Illumina output from whole-genome sequencing was processed by the Broad Picard pipeline and aligned to hg19 using BWA. We ran the GATK best practices workflow on the Terra platform to detect somatic SNVs and indels in our deep whole-genome sequencing data using tumor/normal calling (see Terra workflow). We subset the somatic mutation calls to only SNVs and passed the candidate SNVs for tracking to our probe design pipeline. By sequencing each patient’s tumor and normal to adequate depth we can avoid tracking variants arising from clonal hematopoiesis.
Probe Design
Mutations in MAF (mutation annotation format) were first checked for specificity in the reference genome to filter out potential mapping artifacts. The resulting filtered MAF was then used as input into probe design. Conventional probe design was performed on the filtered MAF as previously described23. For MAESTRO probe design, along with the mutation file, initial probe length (default = 30 bp), annealing temperature (default = 50°C), and ΔG range (default = −18 to −14 kcal/mol) were used as input. For ΔG and melting temperature calculations, we use the annealing temperature, [Na+] = 50 mM, [Mg2+] = 0 mM, and [DNA] = 250 nM. An initial sequence was designed for the given length with the mutation at its center. If the sequence was within the specified ΔG range, it proceeded through the subsequent design steps, otherwise the sequence length was adjusted until it fell within the range. A modified BLAST was performed where the melting temperature for each hit was calculated and if it was less than the annealing temperature, it was removed. If there were 10 or greater pass-filter BLAST hits, we attempted to redesign the sequence using a sliding window. This resulted in the mutation being offset from the center of the sequence, but still provided good enrichment. The sequence with the minimum BLAST hits was then chosen. All sequences were output in a tab-delimited file, and the results were filtered based on length, GC content, ΔG, and the number of BLAST hits before ending up with the final panel design (Supplementary Fig. 1). All probe sequences can be found in Probe Sequences File.
In-house Biotinylation of Probe Panel
Patient-specific oligo pools ordered from Twist Bioscience contained universal forward and reverse primer binding sites. Amplification of the oligo pool was performed using an internally biotin-modified forward primer containing a dU base directly 5’ to the biotinylated dT and an unmodified reverse primer containing a BciVI recognition sequence at its 3’ end. The PCR product was purified using Zymo’s DNA Clean & Concentrator-25 columns. Two micrograms of biotinylated, double-stranded product were sequentially subject to the following 100 μL one-tube enzymatic reaction: 40 units BciVI for 60 minutes at 37°C; 10 units Lambda Exonuclease for 30 minutes at 37°C followed by 20 minutes at 80°C; 7 units USER Enzyme for 30 minutes at 37°C (NEB). Zymo’s Oligo Clean & Concentrator columns were used to purify short, single-stranded, biotinylated probes for hybrid capture.
Creating Spike-in Dilution Series with Sheared gDNA
Healthy gDNA from two HapMap cell lines, NA12878 and NA19238, were sheared to 150 bp fragments using a Covaris E220/LE220 Ultrasonicator. Sheared DNA was quantified using the Quant-iT Picogreen dsDNA assay kit on a Hamilton STAR-line liquid handler. Dilutions were created by spiking sheared gDNA from NA12878 (“mutant”) into NA19238 (“normal”) at 0, 1:1k, 1:10k, 1:100k, and 1:1M. All libraries were constructed with 20 ng sheared gDNA using the Kapa Hyper Prep Kit with custom dual-index duplex UMI adapters (IDT). These UMI adapters allowed tracking of the top and bottom strand of each unique starting molecule despite rounds of amplification.
In cases where there was insufficient library remaining for a subsequent capture, 200 ng of library was subject to additional rounds of PCR to generate workable mass (>1 μg) for hybrid capture using KAPA’s library amplification primer mix. In cases where technical replicates of the same library were needed, libraries were reindexed using a new set of P5/P7 indices (IDT).
Creating Spike-in Dilution Series with cell-free DNA
Processing of patient blood samples followed the same protocol as previously described51. Germline DNA (gDNA) was extracted from either buffy coat or whole blood using the QIAsymphony DSP DNA Mini kit and sheared. Cell-free DNA (cfDNA) was extracted from plasma using the QIAsymphony DSP Circulating DNA Kit. 20 ng libraries were made for patients’ cfDNA timepoints and sheared gDNA.
Healthy donor cfDNA and gDNA were obtained from fresh plasma and whole blood, respectively, at Research Blood Components and processed in the same manner as patient samples. For the cfDNA dilution series, tumor fraction dilutions were created by spiking cancer patient’s cell-free DNA (starting tumor fraction ~1/100) into healthy donor cfDNA at 0, 1:8k, 1:30k, 1:80k, 1:100k, and 1:300k tumor fractions. Replicate 20ng libraries were made for the dilution series as described in the above section.
MAESTRO capture
Hybrid capture using biotinylated, short probe panels was performed using xGen Hybridization and Wash Kit with xGen Universal Blockers (IDT) using a protocol adapted from Schmitt, et al22. Each hybrid capture contained 1 μg of library and 0.75 pmol/μL of MAESTRO probes (IDT or Twist Bioscience), using wells in the middle of the 96-well plate to prevent temperature fluctuations. The hybridization program began at 95°C for 30 seconds. This was followed by a stepwise decrease in temperature from 65°C to 50°C, dropping 1°C every 48 minutes. Finally, the plate was held at 50°C for at least four hours, making the total time in hybridization 16 hours. Heated wash buffer was kept at 50°C (lid temp 55°C) and heated wash steps were performed at 50°C. After the first round of hybrid capture, 16 cycles of PCR were applied. The product was subject to a second round of hybrid capture using half volumes of Cot-1 DNA, xGen Universal Blockers, and probes. This was followed by another 16 cycles of PCR. Apart from these differences, MAESTRO double capture was performed using the same protocol as outlined in Parsons, et al23. Final captured product was quantified and pooled for sequencing on an Illumina HiSeq 2500 (101 bp paired-end reads) or a HiSeqX (151 bp paired-end reads) with a target raw depth of 10,000 × per site.
Conventional Capture
The following described protocol was outlined previously in Parsons, et al23. Hybrid capture using a panel consisting of patient-specific (i.e. germline informed), biotinylated 120 nt probes was performed using the xGen Hybridization and Wash Kit with xGen Universal Blockers (IDT). For each Conventional capture reaction, libraries were pooled up to 6-plex with 500 ng input each and 0.56 to 0.75 pmol/μL of probe panel was applied (IDT). The hybridization program began at 95°C for 30 seconds. This was followed by 65°C for 16 hours. Heated wash buffer was kept at 65°C (lid temp 70°C) and heated wash steps were performed at 65°C. After the first round of hybrid capture, 16 cycles of PCR were applied. The product was subject to a second round of hybrid capture using half volumes of Cot-1 DNA, xGen Universal Blockers, and probes. This was followed by another 8 cycles of PCR. Final captured product was quantified and pooled for sequencing on an Illumina HiSeq 2500 (101 bp paired-end reads) or a HiSeqX (151 bp paired-end reads) with a target raw depth of 1,000,000 × per site.
Quantification of Library Conversion Efficiency by ddPCR
To quantify library conversion efficiency, a ddPCR assay was designed to target the flanking adapter regions. Only fragments with successful double ligation were exponentially amplified within the QX200 ddPCR EvaGreen Supermix (Bio-Rad). Varying DNA inputs into LC (3ng, 10ng, 20ng, 50ng) were tested for their varying conversion efficiencies and adjusted to an unligated control. The results are shown in Supplementary Table 1. The sequences of the primers used are as follows:
Primer 1: CACTCTTTCCCTACACGACG
Primer 2: AGTTCAGACGTGTGCTCTTC
Quantification of Probe Capture Efficiency by ddPCR
To quantify probe capture efficiency, a ddPCR assay was designed to target a homozygous mutation site chosen from the 438 SNV HapMap fingerprint (see ddPCR assay design below). Conventional and MAESTRO hybrid capture was performed on pure tumor Hapmap gDNA libraries, with all waste streams collected from washes. The total number of mutant molecules into hybrid capture and lost during hybrid capture were quantified by using the designed ddPCR assay23,44. Probe capture efficiencies were determined using the equation below. The results are shown in Supplementary Table 2. To determine probe capture efficiency, we targeted the homozygous mutation site (g.chr2:29940529A>T) with the following primers and Taqman probe:
Primer 1 (targeting a consensus sequence on our duplex UMI adapters): CACTCTTTCCCTACACGACG
Primer 2: ATGTCCAGGTCATAGCTCC
- Taqman probe: /5HEX/-CAACAAACATGCCATCTCCTTCTCCTGA-/ZEN//3IaBkFQ/
Sequencing and Data Analysis
Sequencing and pre-processing of BAM files followed a similar protocol as previously described23,44 with the following changes. Before grouping reads by UMI, read groups were added to samples from the same library and samples were merged into a single BAM. This ensured identical molecules found in different samples were given the same family ID from Fgbio’s GroupReadsByUmi (Fulcrum Genomics). The resulting BAM was then pushed through GroupReadsByUmi and split afterwards by the added read group tag. The split BAMs were then passed through the consensus calling workflow. Consensus BAM files were indel realigned using GATK 4 before calling mutations using custom scripts. Noise filtering based on DSC/SSC ratio (total mutant DSCs / total mutant SSCs) was performed on all MAESTRO samples. For mutation calling in clinical samples, we used both the matched tumor and normal. We required that each mutation be seen in the tumor and not in the normal in order for the mutation to be considered. Processing of BAM files was automated using a Snakemake52 workflow (Supplementary Fig. 12).
Miredas Minimal Residual Disease Analysis Scripts
A suite of scripts (Miredas) was used for calling mutations and creating metrics files. In the Snakemake workflow, MiredasCollectErrorMetric uses the duplex BAM file to describe the number of errors and calculates errors per base sequenced. MiredasDetectFingerprint uses the duplex BAM file to call mutations and MiredasDetectFingerprintSsc uses the single-stranded BAM file to call mutations. This single-stranded output of MiredasDetectFingerprintSsc is used along with the duplex MiredasDetectFingerprint output to create DSC/SSC ratios.
VAF/Recall
Raw VAF was calculated using the single strand consensus BAMs as consensus bases are more reliable compared to raw sequenced bases and help correct for PCR bias. We opted to use the single strand consensus BAMs rather than the duplex BAMs as we wanted to retain the majority of sequenced reads - with duplex sequencing, more than 50% of reads can be lost due to support only being observed on one strand. For each site, a pileup was created from the single strand consensus BAM and read bases were compared to the called bases in the MAF file. Each base was categorized as reference (REF), alternate (ALT), or OTHER and the consensus family size (number of reads contributing to the consensus) was added to the site’s read counts. Raw VAF could then be calculated by comparing the number of ALT reads to the total reads (REF + ALT + OTHER) for each site. This raw VAF measurement is important for determining the efficiency of sequencing the ALT base, but may not be an accurate readout of true variant allele fraction due to PCR bias. To address this, we have included duplex (DSC) VAF in Supplementary Table 3, where duplex (DSC) VAF is calculated using the consensus duplex fragments rather than family size as used in raw VAF.
To assess recall, the duplex consensus BAM files were used. Our consensus calling workflow gives source molecules the same family ID, so two samples from the same library have many overlapping molecules. Recall was calculated by looking at the overlap of duplex families between two samples (oftentimes a Conventional sample and a MAESTRO sample). See Supplementary Fig. 2B for an example.
Noise Filter
Four replicate negative controls were created from the same source library via reindexing as described in Creating Spike-in Dilution Series with Sheared gDNA. The replicates were captured using the 10,000 SNV MAESTRO panel. For each targeted site with ALT molecules present in any of the replicates, a DSC/SSC ratio was calculated by summing all ALT supporting duplexes and dividing by the total ALT supporting single strand consensus molecules. Targets with ALT duplexes present in more than one replicate were considered “shared” whereas targets with ALT duplexes present in a single replicate were marked as “exclusive”. A single DSC/SSC ratio was chosen that maximized the number of targets shared while minimizing the number of exclusive targets.
Probe Spike-in Experiment
Capture was performed using the 10,000 SNV MAESTRO panel on two replicate negative control samples (no spike-in) and compared to a replicate negative control with 10X the standard concentration of the 10,000 SNV panel (10X spike-in), and two additional negative controls with 1,000X the standard concentration of ten MAESTRO probes added prior to both post-capture PCRs (1,000X spike-in). These ten MAESTRO probes were selected randomly from the 10,000 SNV panel and synthesized by IDT. These spike-in conditions simulated the worst-case scenario that excess probe can create new mutant molecules by extending from real molecules, specifically during post-capture PCR (see Supplementary Fig. 4A for a schematic of this hypothesis).
Tumor Fraction Estimation
Methods for calculating tumor fraction were previously described23,44 but some changes were made for use with MAESTRO. In a Conventional sample, the full wildtype and mutant diversity is available and can inform tumor fraction. This is important as our tumor fraction methods currently rely on first calculating allele fraction (ALT depth / total depth) for all sites. In MAESTRO samples, we often have full mutant diversity, but wildtype molecules have been depleted. Because enrichment is not perfect, for each panel we have some targets that retain the full diversity of wildtype. We currently leverage this imperfect enrichment to estimate what the total potential depth of the sample is (how many cells likely contributed to the cfDNA library). This estimated depth is applied to all targets which allows us to calculate allele fraction (without considering copy number alterations) and subsequently tumor fraction. Fig. 5B shows this strategy and how it compares to actual tumor fractions. We are aware that these methods are not perfect in their current state, but believe that advances in quality control (ie testing for a handful of germline SNPs to measure unique duplexes per loci) could further improve tumor fraction estimation from enriched samples.
Supplementary Material
Acknowledgments
First and foremost, the authors would like to acknowledge the patients and their families for their contributions to this study. The authors would also like to thank the generous support from the Gerstner Family Foundation. This project was supported in part by the Bridge Project (JCL, VAA), a partnership between the Koch Institute for Integrative Cancer Research at MIT and the Dana-Farber/Harvard Cancer Center (DF/HCC). The authors also acknowledge support from National Institutes of Health grants R33 CA217652 (GMM, VAA) and R01 CA221874 (GMM, VAA).
Competing Interests:
The authors declare the following competing interests:
A.D. Choudhury reports advisory board roles with Clovis, Dendreon, and Bayer and research funding from Bayer. S.M. Tolaney reports research funding to the institution from AstraZeneca, Eli Lilly, Merck, Novartis, Nektar, Pfizer, Genentech, Immunomedics, Exelixis, Bristol-Myers Squibb, Eisai, Nanostring, Cyclacel, Sanofi, Odonate, and Seattle Genetics. S.M. Tolaney also reports honorarium for consulting/advisory board participation from AstraZeneca, Eli Lilly, Merck, Novartis, Nektar, Pfizer, Genentech, Immunomedics, Bristol-Myers Squibb, Eisai, Nanostring, Sanofi, Odonate, Seattle Genetics, Puma, Anthenex, OncoPep, Abbvie, G1 Therapeutics, Silverback Therapeutics, and Celldex. I.E. Krop reports research funding to the institute from Genentech, Pfizer, Daichii-Sankyo. I.E. Krop also reports honorarium for consulting/advisory board participation from Genentech, Daichii-Sankyo, Macrogenics, Context Therapeutics, Taiho Oncology, Merck, Novartis, Bristol-Myers Squibb. H.A. Parsons reports a paid consultant role for Foundation Medicine. T.R. Golub is a paid advisor to GlaxoSmithKline, and is a co-founder and equity holder of Sherlock Biosciences and FORMA Therapeutics. V.A. Adalsteinsson reports a patent application filed with Broad Institute and is a member of the scientific advisory boards of Bertis Inc and AGCT GmbH, which were not involved in this study. The remaining authors report no conflicts of interest.
Footnotes
Code Availability
Software code can be found at the following GitHub link: https://github.com/broadinstitute/MAESTRO-probe_designer
Data Availability
The data supporting the results in this study are available within the paper and its Supplementary Information. Sequencing data have been deposited into the controlled access database, Data Use Oversight System (DUOS; http://duos.broadinstitute.org), under the following accession number: DUOS-000135.
References
- 1.Luquette LJ, Bohrson CL, Sherman MA & Park PJ Identification of somatic mutations in single cell DNA-seq using a spatial model of allelic imbalance. Nat. Commun 10, 3908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ludwig LS et al. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176, 1325–1339. e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zahn LM Mapping genotype to phenotype. Science vol. 362 555.4–556 (2018). [Google Scholar]
- 4.D’Gama AM & Walsh CA Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci 21, 1504–1514 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Garcia-Murillas I et al. Assessment of Molecular Relapse Detection in Early-Stage Breast Cancer. JAMA Oncol (2019) doi: 10.1001/jamaoncol.2019.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canick JA, Palomaki GE, Kloza EM, Lambert-Messerlian GM & Haddow JE The impact of maternal plasma DNA fetal fraction on next generation sequencing tests for common fetal aneuploidies. Prenat. Diagn 33, 667–674 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Bejar R et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J. Clin. Oncol 32, 2691–2698 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snyder TM, Khush KK, Valantine HA & Quake SR Universal noninvasive detection of solid organ transplant rejection. Proc. Natl. Acad. Sci. U. S. A 108, 6229–6234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blauwkamp TA et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat Microbiol 4, 663–674 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Boyd SD et al. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci. Transl. Med 1, 12ra23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilbert JA et al. Current understanding of the human microbiome. Nature Medicine vol. 24 392–400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lowe A, Murray C, Whitaker J, Tully G & Gill P The propensity of individuals to deposit DNA and secondary transfer of low level DNA from individuals to inert surfaces. Forensic Sci. Int 129, 25–34 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Schmitt MW et al. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A 109, 14508–14513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song C et al. Elimination of unaltered DNA in mixed clinical samples via nuclease-assisted minor-allele enrichment. Nucleic Acids Res. 44, e146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J & Mike Makrigiorgos G COLD-PCR: a new platform for highly improved mutation detection in cancer and genetic testing. Biochemical Society Transactions vol. 37 427–432 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Wu LR, Chen SX, Wu Y, Patel AA & Zhang DY Multiplexed enrichment of rare DNA variants via sequence-selective and temperature-robust amplification. Nat Biomed Eng 1, 714–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeffreys AJ & May CA DNA enrichment by allele-specific hybridization (DEASH): a novel method for haplotyping and for detecting low-frequency base substitutional variants and recombinant DNA molecules. Genome Res. 13, 2316–2324 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaudet M, Fara A-G, Beritognolo I & Sabatti M Allele-Specific PCR in SNP Genotyping. Methods in Molecular Biology 415–424 (2009) doi: 10.1007/978-1-60327-411-1_26. [DOI] [PubMed] [Google Scholar]
- 19.Vargas DY, Marras SAE, Tyagi S & Kramer FR Suppression of Wild-Type Amplification by Selectivity Enhancing Agents in PCR Assays that Utilize SuperSelective Primers for the Detection of Rare Somatic Mutations. J. Mol. Diagn 20, 415–427 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Li J et al. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nature Medicine vol. 14 579–584 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Li J, Milbury CA, Li C & Makrigiorgos GM Two-round coamplification at lower denaturation temperature-PCR (COLD-PCR)-based sanger sequencing identifies a novel spectrum of low-level mutations in lung adenocarcinoma. Hum. Mutat 30, 1583–1590 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmitt MW et al. Sequencing small genomic targets with high efficiency and extreme accuracy. Nat. Methods 12, 423–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons HA et al. Sensitive detection of minimal residual disease in patients treated for early-stage breast cancer. Clin. Cancer Res (2020) doi: 10.1158/1078-0432.CCR-19-3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman AM et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol 34, 547–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Machin G Non-identical monozygotic twins, intermediate twin types, zygosity testing, and the non-random nature of monozygotic twinning: a review. Am. J. Med. Genet. C Semin. Med. Genet 151C, 110–127 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Shimoni A & Nagler A Non-myeloablative stem cell transplantation (NST): chimerism testing as guidance for immune-therapeutic manipulations. Leukemia 15, 1967–1975 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Breuer S et al. Early recipient chimerism testing in the T- and NK-cell lineages for risk assessment of graft rejection in pediatric patients undergoing allogeneic stem cell transplantation. Leukemia vol. 26 509–519 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Tyler J, Kumer L, Fisher C, Casey H & Shike H Personalized Chimerism Test that Uses Selection of Short Tandem Repeat or Quantitative PCR Depending on Patient’s Chimerism Status. J. Mol. Diagn 21, 483–490 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Newman AM et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med 20, 548–554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H, Park C, Na W, Park KH & Shin S Precision cell-free DNA extraction for liquid biopsy by integrated microfluidics. npj Precision Oncology 4, 3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mauger F et al. Comparison of commercially available whole-genome sequencing kits for variant detection in circulating cell-free DNA. Sci. Rep 10, 6190 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu D et al. Multiplex Cell-Free DNA Reference Materials for Quality Control of Next-Generation Sequencing-Based In Vitro Diagnostic Tests of Colorectal Cancer Tolerance. Journal of Cancer vol. 9 3812–3823 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsao DS et al. A novel high-throughput molecular counting method with single base-pair resolution enables accurate single-gene NIPT. Sci. Rep 9, 14382 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pantel K & Alix-Panabières C Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat. Rev. Clin. Oncol 16, 409–424 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Tie J et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med 8, 346ra92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaudhuri AA et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 7, 1394–1403 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coombes RC et al. Personalized Detection of Circulating Tumor DNA Antedates Breast Cancer Metastatic Recurrence. Clin. Cancer Res 25, 4255–4263 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Wan JCM et al. High-sensitivity monitoring of ctDNA by patient-specific sequencing panels and integration of variant reads. bioRxiv 759399 (2019) doi: 10.1101/759399. [DOI] [Google Scholar]
- 39.McDonald BR et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci. Transl. Med 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butler TM et al. Circulating tumor DNA dynamics using patient-customized assays are associated with outcome in neoadjuvantly treated breast cancer. Cold Spring Harb Mol Case Stud 5, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magbanua MJM et al. Circulating tumor DNA in neoadjuvant treated breast cancer reflects response and survival. Oncology (2020) doi: 10.1101/2020.02.03.20019760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moding EJ et al. Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small-cell lung cancer. Nature Cancer vol. 1 176–183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Etienne G et al. Long-Term Follow-Up of the French Stop Imatinib (STIM1) Study in Patients With Chronic Myeloid Leukemia. J. Clin. Oncol 35, 298–305 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Abbosh C et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 545, 446–451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan JCM et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci. Transl. Med 12, (2020). [DOI] [PubMed] [Google Scholar]
- 46.Zviran A et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med 26, 1114–1124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masuda N et al. Adjuvant Capecitabine for Breast Cancer after Preoperative Chemotherapy. N. Engl. J. Med 376, 2147–2159 (2017). [DOI] [PubMed] [Google Scholar]
- 48.von Minckwitz G et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med 380, 617–628 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Zook JM et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol 37, 561–566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adalsteinsson VA et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun 8, 1324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Köster J & Rahmann S Snakemake--a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the results in this study are available within the paper and its Supplementary Information. Sequencing data have been deposited into the controlled access database, Data Use Oversight System (DUOS; http://duos.broadinstitute.org), under the following accession number: DUOS-000135.