

Abstract
BACKGROUND:
Prexasertib (LY2606368) is a novel, second generation, selective dual inhibitor of checkpoint kinase proteins 1 (CHK1) and 2 (CHK2). We conducted a Phase 1 trial of prexasertib to estimate the maximum tolerated dose (MTD) and/or recommended Phase 2 dose (RP2D), to define and describe the toxicities, and to characterize the pharmacokinetics (PK) of prexasertib in pediatric patients with recurrent or refractory solid and central nervous system (CNS) tumors.
METHODS:
Prexasertib was administered intravenously (IV) on days 1 and 15 of a 28-day cycle. Four dose levels, 80, 100, 125, and 150 mg/m2, were evaluated using a rolling-six design. PK analysis was performed during cycle 1. Tumor tissue was examined for biomarkers (CHK1, TP53) of prexasertib activity.
RESULTS:
Thirty patients were enrolled; twenty-five were evaluable. Median age was 9.5 years (range: 2–20) and 21 (70%) were male. Twelve patients (40%) had solid tumors and 18 patients (60%) had CNS tumors. There were no cycle 1 or later dose-limiting toxicities. Common cycle 1, drug-related grade 3/4 toxicities (>10% of patients) included neutropenia (100%), leukopenia (68%), thrombocytopenia (24%), lymphopenia (24%), and anemia (12%). There were no objective responses; best overall response was stable disease in 3 patients for 5 cycles (hepatocellular carcinoma), 3 cycles (ependymoma), and 5 cycles (undifferentiated sarcoma). The PK appeared dose proportional across the 80–150 mg/m2 dose range.
CONCLUSIONS:
While the MTD of prexasertib was not defined by this study, 150 mg/m2 administered IV on day 1 and 15 of a 28-day cycle was determined to be the RP2D.
Clinicaltrials.gov Registry: NCT02808650
Keywords: prexasertib, LY2606368, CHK1/2, CHK1 inhibitor, pediatric, Phase 1
INTRODUCTION
In response to DNA damage or replication stress, cells initiate a DNA damage response (DDR) that activates cell cycle checkpoints to halt progress through the cell cycle to allow for DNA repair.1 DNA damage can arise from normal metabolic processes within the cell, e.g. replication stress, as well as from external sources such as environmental insults, e.g. UV radiation and DNA-damaging agents.2 Checkpoint kinase proteins 1 (CHK1) and 2 (CHK2) are conserved serine/threonine kinases that are key effectors of multiple checkpoint responses and are activated in response to genotoxic stress.1 Activated CHK1 plays a key role in the intra-S and G2/M DNA damage checkpoints through slowing DNA replication and limiting mitotic entry, respectively, which alleviates replication stress and supports cell survival.3–5 CHK1 has also been shown to directly affect DNA repair, confirming its role in maintaining genomic integrity.6
Inhibition of CHK1 abrogates the DDR checkpoint, allowing cells that have sustained DNA damage to prematurely enter mitosis and undergo mitotic catastrophe due to incompletely replicated chromosomes.7, 8 Prexasertib (LY2606368) is a novel, second generation, selective, dual inhibitor of CHK1/2 that achieves adequate CNS penetration and target engagement in preclinical models.9 In vitro studies with pediatric cancer cell lines indicate that prexasertib is a potent inhibitor of cell proliferation at low nanomolar concentrations with most cell lines showing evidence of cytotoxicity.10 Additionally, in pediatric tumor xenograft models, prexasertib was well-tolerated and produced objective responses in multiple tumors including neuroblastoma, rhabdomyosarcoma, Ewing sarcoma, and desmoplastic small round cell tumor.10
A Phase I trial in adults with advanced or metastatic solid tumors demonstrated that prexasertib is generally well tolerated with common grade 3 or 4 treatment-related side effects of neutropenia, leukopenia, anemia, thrombocytopenia, and fatigue.11 The recommended Phase 2 dose (RP2D) from this adult study was 105 mg/m2 intravenously (IV) once every 14 days. There were no dose-limiting toxicities at the RP2D. The primary objectives of this pediatric Phase 1 trial were to establish the maximum tolerated dose (MTD) and/or RP2D of prexasertib, and to characterize the toxicities and pharmacokinetics (PK) of prexasertib in children with relapsed or refractory solid and central nervous system (CNS) tumors.
METHODS
Patient Eligibility
Eligible patients were between the ages of 1 and 21 years and had a recurrent or refractory solid or CNS tumor; measurable or evaluable disease; no known curative therapy or therapy proven to prolong survival with an acceptable quality of life; a Lansky (≤16 years) or Karnofsky (> 16 years) performance status ≥ 50%; and had recovered from the acute toxic effects of prior anticancer therapy. In addition, eligible patients had adequate organ function as defined in Appendix 1. Patients were not eligible if they had a history of allergic reactions attributed to compounds of similar chemical or biologic composition to prexasertib, were receiving strong CYP1A2 inhibitors, or required escalating doses of steroids within 7 days prior to enrollment.
The study was conducted in accordance with good clinical practices and the Declaration of Helsinki, and NCI Pediatric Central Institutional Review Board approval was obtained. Informed consent, and assent as appropriate per institutional guidelines, was obtained from patients and their guardians prior to enrollment. The trial was listed on clinicaltrials.gov as NCT02808650.
Trial Design
The primary objectives for this study were to estimate the MTD and/or RP2D of prexasertib administered as an intravenous infusion over 60 minutes, every 14 days of a 28-day cycle, to children with recurrent or refractory solid tumors and CNS tumors, to define and describe the toxicities of prexasertib administered on this schedule, and to characterize the PK of prexasertib in children with recurrent or refractory cancer. The secondary objectives were to preliminarily define the antitumor activity of prexasertib within the confines of a Phase 1 study, to examine CHK1/2 expression status in archival tumor tissue from pediatric patients with solid and CNS tumors using immunohistochemistry (IHC), to evaluate tumor tissue for deletion and/or mutation of TP53 as a potential biomarker of prexasertib sensitivity, and to evaluate autophosphorylation of CHK1 and γH2AX in peripheral blood mononuclear cells (PBMCs) as potential pharmacodynamic markers of target engagement by prexasertib.12–15
Prexasertib mesylate monohydrate (LY2606368; Eli Lilly and Company, Indianapolis, IN) was supplied as a lyophilized, light yellow powder in a single-use vial, and was reconstituted with water to make a solution of 2 mg/mL of prexasertib for injection. Prexasertib was administered IV over 60 minutes on days 1 and 15 of a 28-day cycle. Subsequent cycles could begin if the patient had at least stable disease, met eligibility lab parameters, and did not meet criteria for removal from protocol therapy due to toxicity or otherwise. Cycles could be repeated every 28 days for up to 13 cycles or up to a total duration of therapy of approximately 12 months.
The starting dose of prexasertib was 80 mg/m2/dose, approximately 80% of the adult RP2D of 105 mg/m2/dose.11 Dose escalations to 100 mg/m2, 125 mg/m2, and 150 mg/m2 were planned with a possible dose de-escalation if dose-limiting toxicity (DLT) occurred at the starting dose level. A rolling-six design was used for dose escalation.16 No intra-patient dose escalation was allowed. The MTD was defined as the maximum dose at which fewer than one-third of patients experienced DLT during Cycle 1 of therapy. Once the MTD or RP2D was defined, we planned to enroll up to 6 additional patients in a PK expansion cohort to acquire additional PK data in a representative number of patients < 12 years of age.
Toxicities were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 5.0. Non-hematologic DLT was defined as any grade 3 or 4 non-hematologic toxicity that was possibly, probably, or definitely attributable to prexasertib with the exclusion of the following: grade 3 nausea and vomiting < 3 days duration; grade 3 liver enzyme elevation, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), and gamma-glutamyl transferase (GGT), that returned to grade ≤ 1 or baseline prior to the time for the next treatment cycle; grade 3 fever; grade 3 infection; and grade 3 hypophosphatemia, hypokalemia, hypocalcemia or hypomagnesemia responsive to supplementation. In addition, any non-hematologic toxicity that caused a delay of ≥ 14 days between treatment cycles was considered a DLT. Upon meeting eligibility parameters or returning to baseline, patients who experienced a non-hematologic DLT were able to continue on protocol therapy with a one dose level reduction.
Hematologic DLT was defined as grade 4 neutropenia > 7 days, platelet count < 20,000/mm3 on 2 separate days, or requiring a platelet transfusion on 2 separate days, within a 7-day period, or myelosuppression that caused a delay of > 14 days between treatment cycles. In addition, grade 4 neutropenia or grade 3 thrombocytopenia that occurred on Day 15 and did not resolve to absolute neutrophil count (ANC) ≥ 500/mm3 and platelet count ≥ 50,000/mm3 (transfusion independent) by Day 18 was considered dose-limiting. Grade 3 or 4 febrile neutropenia was not considered a DLT. Upon meeting eligibility parameters, patients who experienced dose-limiting thrombocytopenia were able to remain on protocol therapy with a one dose level reduction. Patients who experienced dose-limiting neutropenia with no other DLT received the same dose in the next cycle with myeloid growth factor support. If dose-limiting neutropenia recurred after myeloid growth factor was added the patient was given the next lower dose level for subsequent cycles.
Radiographic response was assessed using the Response Evaluation Criteria in Solid Tumors (RECIST) guideline (version 1.1) for patients with solid tumors and modified Response Assessment in Neuro-Oncology (RANO) for patients with CNS tumors.17, 18 Tumor disease evaluations, including bone marrow evaluation, if applicable, were performed at the end of cycles 1, 3, and 5, and then every 3 cycles thereafter. Partial or complete responses and prolonged stable disease (≥ 6 cycles) were confirmed by central radiologist’s review.
Pharmacologic Studies
In course 1, blood samples were drawn before treatment, and 1 hour (hr) (end of infusion), 1.5 hrs, 2 hrs, 4 hrs, 8 hrs, 24 hrs and 96 (± 24) hrs after beginning the Day 1 infusion. Additional samples were drawn on Day 8 during complete blood count (CBC) evaluation and on Day 15 before and 1 hr (end of infusion) after the infusion. Prexasertib plasma concentrations were determined using a validated liquid chromatography, tandem mass spectrometry (LC-MS/MS) method. Prexasertib PK were estimated by standard non-compartmental analysis using the program Phoenix® WinNonlin® Version 6.4 (Certara Corporation, Princeton, NJ) (see Appendix 2 for PK Methods).
Tumor tissue, either from diagnosis or relapse, was collected, when available, or analyzed for phospho-CHK 1/2 and TP53 expression by IHC (See Appendix 3 for IHC Methods).
RESULTS
Patient Characteristics
Thirty patients enrolled on study, twenty-four in the dose-escalation cohort and 6 in the PK expansion cohort, and all were eligible. Baseline characteristics for all eligible patients are presented in Table 1. The median age was 9.5 years (range: 2–20), and 21 patients (70%) were male. Eighteen patients (60%) had a brain tumor and 12 (40%) had a solid tumor, with the most common diagnoses being high-grade glioma (27%), ependymoma (13%), and rhabdomyosarcoma (13%). Twenty-nine (97%) patients had previously received chemotherapy with a median of 2 (range: 1–8) prior chemotherapy regimens. One patient with diffuse intrinsic pontine glioma was previously treated with radiation alone. Twenty-four (80%) patients had previously received radiation with a median of 1 (range: 1–3) prior radiation courses.
TABLE 1.
Characteristics of eligible patients (N=30)
| Characteristic | Number (%) |
|---|---|
| Median age (range), years | 9.5 (2 – 20) |
| Sex | |
| Male | 21 (70) |
| Female | 9 (30) |
| Race | |
| White | 20 (67) |
| Black or African American | 3 (10) |
| Asian | 1 (3) |
| Multiple | 1 (3) |
| Unknown | 5 (17) |
| Ethnicity | |
| Non-Hispanic | 23 (77) |
| Hispanic | 5 (17) |
| Unknown | 2 (7) |
| Diagnosis (N=30) | |
| Brain Tumor (N=18) | |
| Choroid plexus carcinoma | 1 (3) |
| Ependymoma | 4 (13) |
| High-grade glioma | 8 (27) |
| Medulloblastoma | 1 (3) |
| Oligodendroglioma | 1 (3) |
| Pineoblastoma | 1 (3) |
| Primitive neuroectodermal tumor (PNET) | 2 (7) |
| Solid Tumor (N=12) | |
| Adenocarcinoma | 1 (3) |
| Ewing sarcoma | 1 (3) |
| Hepatocellular carcinoma | 1 (3) |
| Neuroendocrine carcinoma | 1 (3) |
| Osteosarcoma | 2 (7) |
| Rhabdomyosarcoma | 4 (13) |
| Undifferentiated sarcoma | 1 (3) |
| Wilms Tumor | 1 (3) |
| Prior Therapy | |
| Median prior chemotherapy regimens (range) (N=29) | 2 (1 – 8) |
| Median prior radiation courses (range) (N=24) | 1 (1 – 3) |
Toxicity
Twenty-five patients were evaluable for DLT determination. Three patients were inevaluable due to progressive disease and 2 patients due to refusal of further protocol therapy during cycle 1. The median number of cycles received by evaluable patients was 1 (range: 1–5). No DLTs were experienced by any patient on study (Table 2). The most common toxicities experienced by patients on study were hematologic. Common grade 3/4 non-DLT cycle 1 hematologic toxicities occurring in > 10% of patients that were at least possibly attributable to prexasertib included neutropenia (100%), leukopenia (68%), thrombocytopenia (24%), lymphopenia (24%), and anemia (12%) (Table 3). Grade 4 neutropenia was experienced by 88% (22/25) of patients; however, no patient required growth factor support during cycle 1. The frequency and type of toxicities experienced were similar across all dose levels. There were only two grade 3/4 non-DLT cycle 1 non-hematologic toxicities that were at least possibly attributable to prexasertib: grade 3 ALT elevation and grade 3 febrile neutropenia. There was not a trend towards increased hematologic toxicity at higher dose levels, as the number and/or frequency of each toxicity was similar among dose levels. The timing of ANC nadir and recovery was predictable following each dose with no notable differences across dose levels (Figure 1). While the MTD of prexasertib was not reached, the dose of 150 mg/m2 administered IV on day 1 and 15 of a 28- day cycle was tolerable, and hence, determined to be the RP2D.
TABLE 2.
Summary of DLTs
| Dose Level | Prexasertib Dose (mg/m2) | Part | No. of Patients Entered | No. of Patients Evaluable | No. of Patients with Cycle 1 DLTs | No. of Patients with Later-Cycle DLTs |
|---|---|---|---|---|---|---|
| 1 | 80 | A | 6 | 6 | 0 | 0 |
| 2 | 100 | A | 6 | 5 | 0 | 0 |
| 3 | 125 | A | 6 | 4 | 0 | 0 |
| 4 | 150 | A | 6 | 5 | 0 | 0 |
| 4 | 150 | PK | 6 | 5 | 0 | 0 |
Abbreviations: A= dose-escalation cohort; DLT= dose-limiting toxicity; No.= number; PK= Pharmacokinetic expansion cohort
TABLE 3.
Hematologic and non-hematologic non-DLTsa,b observed during cycle 1 in evaluable patients across all dose levels (N=25)
| Dose Level and Toxicity Grade, No. (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| All Dose Levels (N=25) | Dose Level 1 (N=6) | Dose Level 2 (N=5) | Dose Level 3 (N=4) | Dose Level 4 (N=10) | ||||||
| Toxicity Type | All | ≥ Grade 3 | All | ≥ Grade 3 | All | ≥ Grade 3 | All | ≥ Grade 3 | All | ≥ Grade 3 |
| Neutrophil count decreased | 25 (100) | 25 (100) | 6 (100) | 6 (100) | 5 (100) | 5 (100) | 4 (100) | 4 (100) | 10 (100) | 10 (100) |
| White blood cell decreased | 24 (96) | 17 (68) | 5 (83) | 4 (67) | 5 (100) | 3 (60) | 4 (100) | 3 (75) | 10 (100) | 7 (70) |
| Platelet count decreased | 21 (84) | 6 (24) | 4 (67) | 1 (17) | 5 (100) | 2 (40) | 4 (100) | 2 (50) | 8 (80) | 1 (10) |
| Anemia | 19 (76) | 3 (12) | 5 (83) | 0 (0) | 3 (60) | 1 (20) | 3 (75) | 1 (25) | 8 (80) | 1 (10) |
| Lymphocyte count decreased | 12 (48) | 6 (24) | 3 (50) | 2 (33) | 2 (40) | 0 (0) | 2 (50) | 2 (50) | 5 (50) | 2 (20) |
| Electrocardiogram QT corrected interval prolonged | 9 (36) | 0 (0) | 2 (33) | 0 (0) | 1 (20) | 0 (0) | 2 (50) | 0 (0) | 4 (40) | 0 (0) |
| Fatigue | 6 (24) | 0 (0) | 0 (0) | 0 (0) | 2 (40) | 0 (0) | 2 (50) | 0 (0) | 2 (20) | 0 (0) |
| Vomiting | 6 (24) | 0 (0) | 1 (17) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 0 (0) | 4 (40) | 0 (0) |
| Alanine aminotransferase increased | 5 (20) | 1 (4) | 2 (33) | 0 (0) | 2 (40) | 1 (20) | 0 (0) | 0 (0) | 1 (10) | 0 (0) |
| Hypokalemia | 5 (20) | 0 (0) | 0 (0) | 0 (0) | 2 (40) | 0 (0) | 1 (25) | 0 (0) | 2 (20) | 0 (0) |
| Nausea | 5 (20) | 0 (0) | 1 (17) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 0 (0) | 3 (30) | 0 (0) |
| Aspartate aminotransferase increased | 4 (16) | 0 (0) | 0 (0) | 0 (0) | 3 (60) | 0 (0) | 0 (0) | 0 (0) | 1 (10) | 0 (0) |
| Diarrhea | 4 (16) | 0 (0) | 2 (33) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 0 (0) | 1 (10) | 0 (0) |
| Hypophosphatemia | 4 (16) | 0 (0) | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (30) | 0 (0) |
| Anorexia | 3 (12) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (10) | 0 (0) |
| Headache | 3 (12) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (25) | 0 (0) | 2 (20) | 0 (0) |
| Infusion-related reaction | 3 (12) | 0 (0) | 1 (17) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 0 (0) | 1 (10) | 0 (0) |
| Myalgia | 3 (12) | 0 (0) | 0 (0) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 0 (0) | 2 (20) | 0 (0) |
Abbreviation: DLT= dose-limiting toxicity; No.= number; WBC= white blood cell
Toxicities occurring in >10% of evaluable patients
Includes toxicities attributed as possibly, probably, or definitely related to prexasertib
Figure 1. Effect of Prexasertib on Absolute Neutrophil Count (ANC) during Cycle 1 Across All Dose Levels.
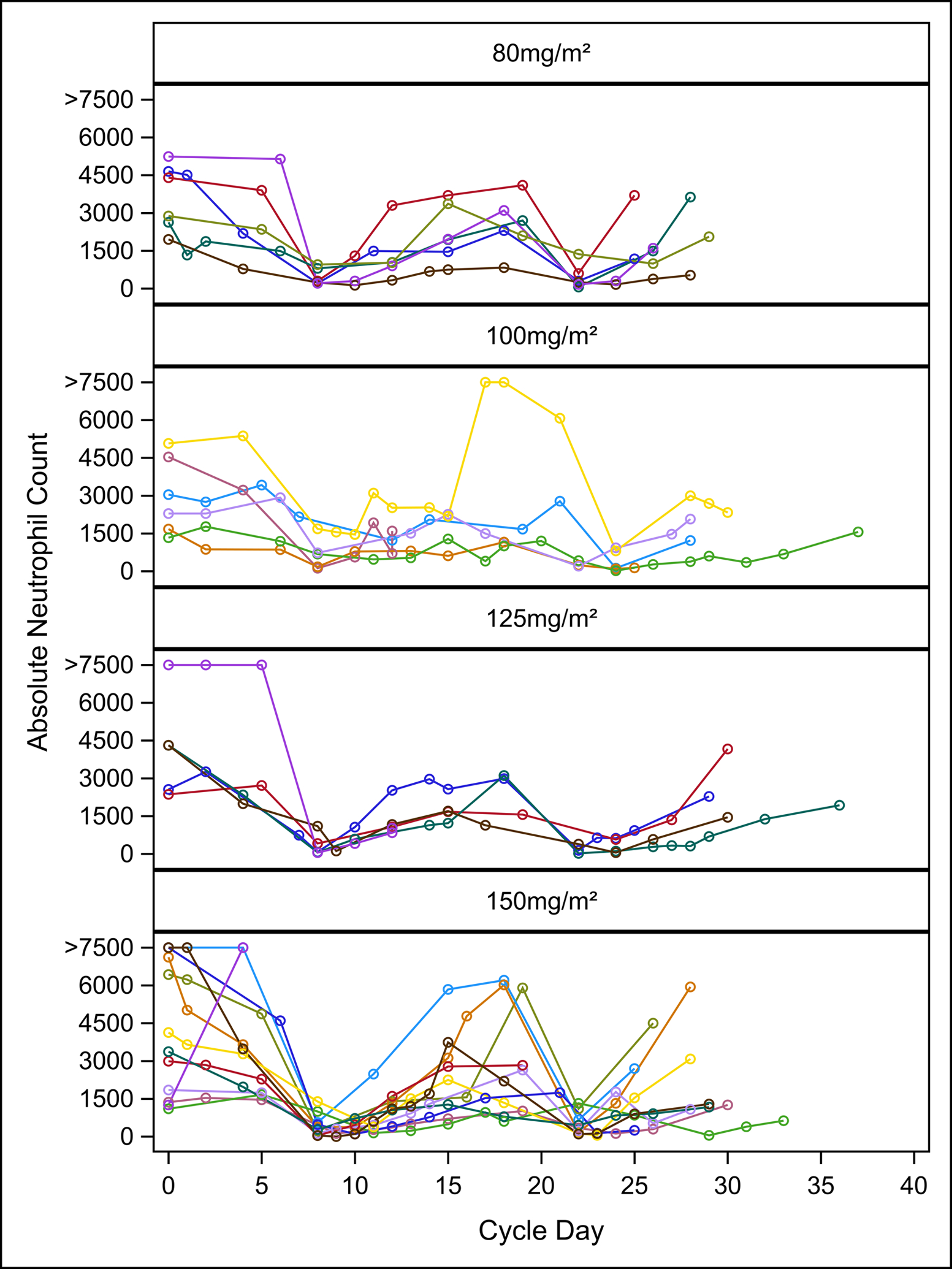
Different line colors represent individual patients.
Response
Among the 25 patients evaluable for response, there were no objective responses. Three patients had a best overall response (BOR) of stable disease (SD): SD for 5 cycles in a patient with hepatocellular carcinoma, SD for 3 cycles in a patient with anaplastic ependymoma, and SD for 5 cycles in a patient with undifferentiated sarcoma. These patients were treated at dose levels 1, 2, and 3, respectively. All other patients had a BOR of progressive disease.
Pharmacokinetic and Pharmacodynamic Studies
The PK of prexasertib were studied in 29 patients during cycle 1. The plasma concentration versus time profile for patients treated at the RP2D (150 mg/m2) is illustrated in Figure 2A. Prexasertib PK were determined after the first dose by non-compartmental analysis and parameter estimates are summarized in Table 4. Following administration of the intravenous infusion, multi-exponential decline in plasma concentration was observed with a mean terminal elimination half-life of 8.9 hours. Based on the approximately dose-proportional increase in AUC0−∞ as dose was increased from 80 – 150 mg/m2 (Figure 2B) and BSA-normalized clearance that was independent of dose (Figure 2C), the prexasertib PK appear to be linear after a single dose. The plasma clearance was 45.6 ± 13.1 L/hr/m2 for males (N=20) and 47.1 ± 9.7 L/hr/m2) for females (N=9). The BSA-adjusted plasma clearance in children <12 years (N=16) was 49.4 ± 10.0 L/hr/m2 compared to 42.2 ± 13.3 L/hr/m2 for those ≥12 years (N=13).
Figure 2. Prexasertib Pharmacokinetics in Pediatric Patients with Recurrent or Refractory Solid and CNS Tumors.
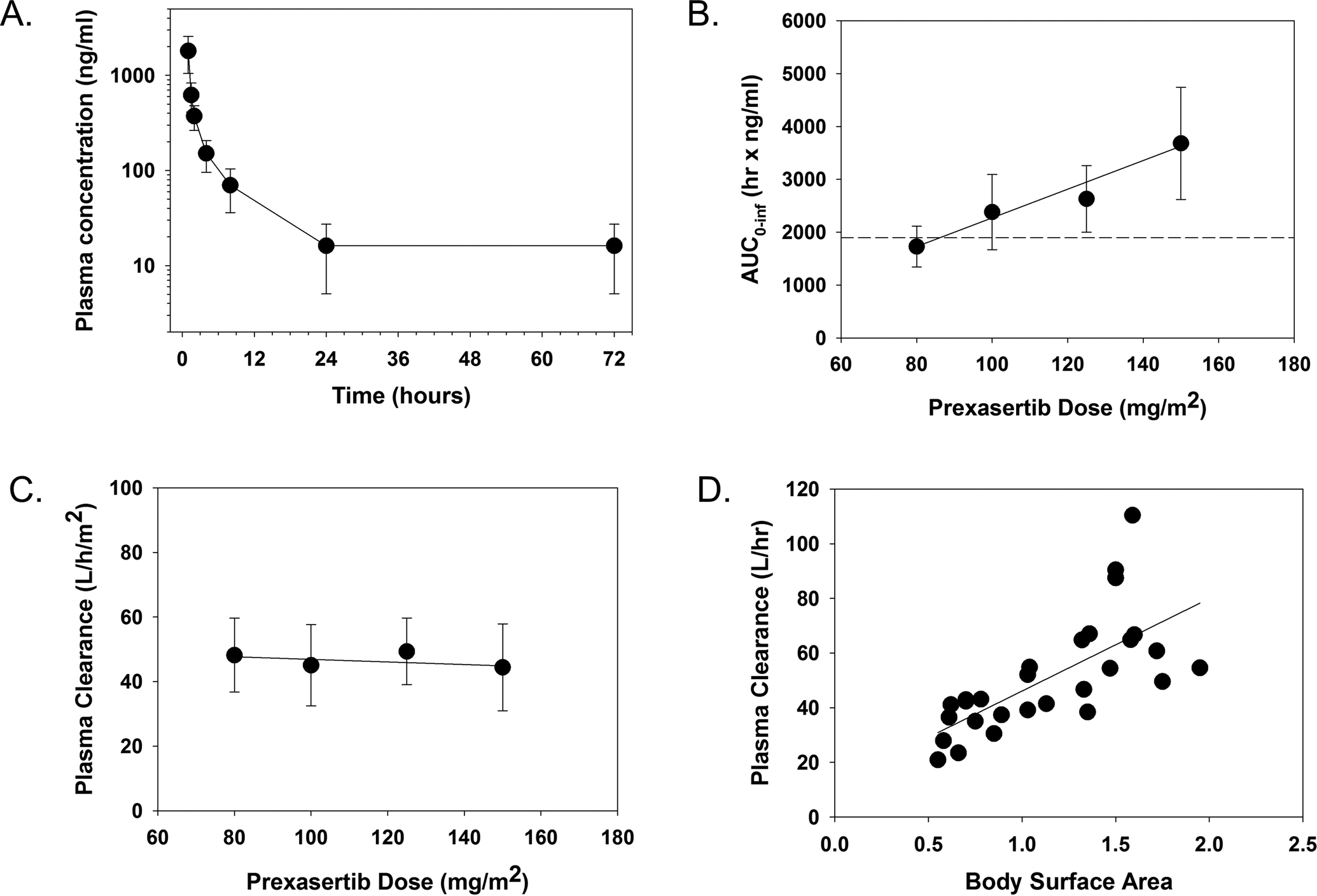
The mean plasma concentration as a function of time for patients treated with 150 mg/m2 of prexasertib (A), mean AUC0−∞ for each dose level with horizontal line on the plot representing median AUC0–72h value (1896 ng*hr/ml) predicted for efficacy based on the Calu-6 preclinical PK/PD model11 (B), mean plasma clearance for each dose level (C), and plasma clearance as a function of body surface area (D). Error bars represent the standard deviation for each observation.
TABLE 4.
Summary of prexasertib pharmacokinetics in pediatric patients with recurrent or refractory solid and CNS tumorsa
| Dose Level (mg/m2) | 80 | 100 | 125 | 150 | All Patients |
|---|---|---|---|---|---|
| No. of Patients | 6 | 6 | 5 | 12 | 29 |
| Cmax (ng/ml) | 858 ± 311 | 984 ± 289 | 1227± 212 | 1697 ± 822 | ———— |
| Tmax (hrs) | 1.4 ± 0.9 | 1.1 ± 0.2 | 1.1 ± 0.1 | 1.2 ± 0.5 | 1.2 ± 0.5 |
| T1/2 (hrs) | 7.6 ± 2.3 | 10.0 ± 3.9 | 8.5 ± 4.5 | 9.2 ± 3.5 | 8.9 ± 3.4 |
| AUC0–24h (ng•hr/ml) | 1624 ± 358 | 2011 ± 416 | 2462 ± 545 | 3359 ± 898 | ———— |
| AUC0−∞ (ng•hr/ml) | 1726 ± 387 | 2381 ± 712 | 2638 ± 632 | 3681 ± 1059 | ———— |
| Vss (L/m2) | 245 ± 67 | 332 ± 85 | 292 ± 169 | 257 ± 98 | 276 ± 102 |
| CLp (L/hr/m2) | 48.2 ± 11.5 | 45.1 ± 12.6 | 49.3 ± 10.3 | 44.4 ± 13.5 | 46.1 ± 12.0 |
Abbreviations: AUC= area under the concentration versus time curve; CLp= plasma clearance; Cmax= peak plasma concentration; CNS= central nervous system; hr= hour; No.= number; PK= pharmacokinetic; Tmax= amount of time that a drug is present at the maximum concentration in serum; T1/2= half-life; Vss= volume of distribution at steady state
Shown as median (range)
Tissue for immunohistochemical analysis was available for 19 patients. Most tumor tissue showed immunoreactivity for both Trp53 and Chk1S345 with areas of intensely labelled cells throughout the field (Supplemental Table S1 and Supplemental Figure S1). Despite robust immunohistochemical labelling for both Trp53 and Chk1S345 in cells throughout more than half of the tumor tissue, this labelling did not correlate with clinical response.
Samples for peripheral blood pharmacodynamic (PD) marker studies of target engagement were obtained from 23 patients. However, insufficient quantity and quality of protein was available from the limited blood samples to detect either markers of DNA damage (phospho-H2AX) or reduced phosphorylation of Chk1.
DISCUSSION
In this first-in-pediatrics trial evaluating prexasertib in patients with recurrent or refractory solid and CNS tumors, we did not reach an MTD. The RP2D was determined to be 150 mg/m2 administered IV on day 1 and 15 of a 28-day cycle. There were no DLTs during cycle 1 or in later cycles. Prexasertib was well tolerated with grade 3/4 drug-related toxicity being exclusively hematologic in nature, most notably neutropenia, which was seen in all patients, and leukopenia. The neutropenia was clinically inconsequential in nearly all cases as there were no DLTs due to delay in starting a cycle or receiving the day 15 dose, and only one episode of febrile neutropenia. There were no objective responses and BOR was stable disease in 3 patients. The prexasertib exposure appeared to be proportional to dose.
The single agent RP2D of prexasertib administered in adult patients on an every 14-day schedule was determined to be 105 mg/m2.11, 19, 20 In a Phase 1b expansion that treated over 100 patients with squamous cell carcinoma at this dose and schedule, 89% of patients had treatment-emergent grade 3/4 neutropenia, which is similar to the 100% seen on our study.19 Rates of grade 3/4 anemia (14%) and thrombocytopenia (16%) on this adult study were also comparable to ours, 12% and 24%, respectively. In contrast, the frequency of grade 3/4 leukopenia (68% vs. 26%) and lymphopenia (24% vs. not reported) were higher on our pediatric study. The frequency of non-hematologic, drug-related toxicity was similarly low on both trials.
Prexasertib PK were measured in this trial to ascertain whether target plasma concentrations and exposure values were achieved at the RP2D and to determine if the pediatric PK were similar to adults. The mean Cmax, half-life, AUC0–24h, and plasma clearance at the RP2D (150 mg/m2) were 1697 ng/ml, 9.2 hours, 3359 ng•hr/ml, and 44.4 L/hr/m2, respectively. The median plasma concentration measured 24 hours after the first dose (C-24h) was 13.4 ng/ml with a range of 7.6–43.6 ng/ml. Thus, prexasertib plasma concentrations were maintained above the IC50 (14.1 ng/ml) for pCHK1 inhibition for approximately 24 hours and the drug exposure measured as the AUC0–24h was greater than the median AUC0–72h value (1896 ng•hr/ml) predicted for efficacy based on the Calu-6 preclinical PK/PD model.11 There are limited published adult PK data for prexasertib administered as a single agent in Phase I clinical trials, and no adult data at the pediatric RP2D.11, 20 Thus, we compared the pediatric PK data measured at 100 mg/m2/dose with the adult PK data measured at 105 mg/m2/dose. As illustrated in the PK profiles shown in those reports, similar drug disposition was observed in the pediatric and adult studies. The mean AUC0–24h value of 2011 ng•hr/ml found in our study was within the range of mean AUC0–24h values (1130 and 1690 ng•hr/ml, respectively) found in the studies by Hong et al. and Iwasa et al., after administration of 105 mg/m2. Additionally, the mean CLp value of 45.1 L/hr/m2 found in our study was within the range of mean CLp values (78.2 and 45.4 L/hr/m2, respectively), when adjusted for average adult BSA of 1.7 m2.
While an MTD was not reached in our study, the dose of 150 mg/m2 was well tolerated and provided twice the exposure than the RP2D from either adult study. In the absence of strong PK or PD data to suggest otherwise, the typical approach taken in pediatric phase 1 trials is to use the estimated highest tolerable dose as the RP2D. Our study was limited by the lack of protein quantity and quality in plasma samples which precluded investigation for a peripheral blood biomarker; however, previous clinical trials testing single agent prexasertib in adult patients have failed to identify a clear biomarker of response.11 In the absence of further PD data, we determined 150 mg/m2 to be the pediatric RP2D. While responses were observed in the adult single agent prexasertib studies at 105 mg/m2, a dose which produced similar PK as the 100 mg/m2 dose in our study, it is not accurate to make assumptions about the pediatric RP2D based on activity observed in adult cancers, as adult and pediatric tumors have different molecular etiologies. In addition, the small numbers and heterogeneous patient population treated in our study, which included CNS tumors, treated across multiple dose levels, limits the conclusions that can be drawn about activity. Furthermore, lack of activity of a targeted kinase inhibitor when dosed as a single agent is not predictive of potential activity when given in combination with another targeted or cytotoxic agent. Additional refinement of prexasertib dose would need to be undertaken in a future combination trial.
Tumor tissue from 19 patients was analyzed for CHK 1/2 and TP53 expression by IHC. In normal tissues, wild-type P53 is rapidly degraded and mutation of the gene is believed to result in increased protein stability, allowing for its detection by immunohistochemistry in many tumor tissues.21 While most tissue sections had considerable Trp53 and Chk1S345 immunoreactivity, there were no significant correlations with clinical response to prexasertib. This is consistent with preclinical studies suggesting p53 status does not influence prexasertib efficacy as a monotherapy.12, 22 This may also be due to the limited clinical response in patients to single-agent prexasertib and, therefore, the lack of true positive responders to prexasertib exposure. One possible explanation for the lack of response in patients treated on our study is the presence of either acquired or de novo resistance to prexasertib through mechanisms such as upregulation of Wee1.23 The limited tumor tissue available as preserved tumor tissue from only 19 out of 29 patients precluded our ability to look for somatic mutations in genes such as CDK1/CyclinB1 and the DDR pathway that have been demonstrated to confer resistance to CHK1 inhibition.24 Additionally, we are not aware of the mutational status of DDR genes in this patient cohort as germline sequencing was not within the scope of this study.
In conclusion, the RP2D of prexasertib in pediatric patients with recurrent or refractory solid and CNS tumors is 150 mg/m2 administered IV on day 1 and 15 of a 28-day cycle. Prexasertib was well tolerated with the only grade 3/4 regimen-related toxicity being hematologic, predominantly neutropenia. However, there were no DLTs at any dose level and no delays in starting subsequent cycles. The best overall response on this study was stable disease. Preclinical models of pediatric tumors including neuroblastoma, osteosarcoma, Ewing sarcoma, and alveolar rhabdomyosarcoma demonstrate improved antitumor activity when prexasertib is given concurrently with several different DNA damaging agents including irinotecan, cyclosphosphamide, and doxorubicin.10 Gemcitabine, a potent inducer of replication stress and DNA damage, induces S phase arrest and activation of the CHK1-dependent DNA damage pathway.25, 26 MYC overexpression or MYCN amplification causes unscheduled replication origin firing and replication stress which also activates CHK1. CHK1 inhibitors like prexasertib induce apoptosis during S phase of the cell cycle, providing additional mechanistic rationale for combining this class of agents with gemcitabine in patients with neuroblastoma, rhabdomyosarcoma, and medulloblastoma where MYC overexpression or amplification frequently occurs.27, 28 Given the potential for overlapping hematologic toxicity of prexasertib and cytotoxic chemotherapy, additional dose finding studies will be needed to determine the optimal dose and schedule.
Supplementary Material
Supplemental Figure S1. Immunohistochemical labelling of positive cells in tumor tissue. A) Positive cells showing immunoreactivity for Trp53; B) Positive cells showing immunoreactivity for Chk S345; C) Cells showing no immunoreactivity for Trp53; D) Cells showing no immunoreactivity for Chk S345. Scale bars are 200 uM.
ACKNOWLEDGEMENTS
We would like to acknowledge Bradley Hanberry, PhD and the Children’s Healthcare of Atlanta and Emory University’s Children’s Clinical and Translational Discovery Core for their assistance with the collection, processing, and storage of specimens for our correlative studies. We would like to acknowledge Eli Lilly and Company for supplying prexasertib and support of pharmacokinetic analysis.
Research funding was provided by the National Cancer Institute (NCI) of the NIH under award number UM1CA228823 and the Cookies for Kids’ Cancer Foundation. Dr. Reid was supported in part by Grant Number P30 CA015083 from the NCI.
CONFLICT OF INTEREST
TC receives research support from Celgene for an unrelated Phase 1 clinical trial and has served as a consultant for EUSA Pharma. CW has received research support from Eli Lilly for an unrelated Phase 1 clinical trial, and is currently employed by Exelixis, Inc.
Abbreviation Key:
- ALT
alanine aminotransferase
- ANC
absolute neutrophil count
- AST
aspartate aminotransferase
- BOR
Best overall response
- CHK1
checkpoint kinase protein 1
- CHK2
checkpoint kinase protein 2
- CNS
central nervous system
- DDR
DNA damage response
- DLT
dose-limiting toxicity
- hr
hour
- IHC
immunohistochemistry
- IV
intravenously
- MTD
maximum tolerated dose
- PBMC
peripheral blood mononuclear cell
- PD
pharmacodynamic
- PK
pharmacokinetics
- RP2D
recommended Phase 2 dose
- SD
stable disease
Footnotes
Presentation of Study: This work was presented in part at the 2018 EORTC-NCI-AACR Molecular Targets and Cancer Therapeutics Symposium in Dublin, Ireland.
Data Availability Statement:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Kastan MB. DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture. Mol Cancer Res. 2008;6: 517–524. [DOI] [PubMed] [Google Scholar]
- 2.Ferrao PT, Bukczynska EP, Johnstone RW, McArthur GA. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene. 2012;31: 1661–1672. [DOI] [PubMed] [Google Scholar]
- 3.Feijoo C, Hall-Jackson C, Wu R, et al. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154: 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Q, Guntuku S, Cui XS, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14: 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 5.Kotsantis P, Petermann E, Boulton SJ. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018;8: 537–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16: 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer. 2008;98: 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niida H, Tsuge S, Katsuno Y, Konishi A, Takeda N, Nakanishi M. Depletion of Chk1 leads to premature activation of Cdc2-cyclin B and mitotic catastrophe. J Biol Chem. 2005;280: 39246–39252. [DOI] [PubMed] [Google Scholar]
- 9.Campagne O, Davis A, Maharaj AR, et al. CNS penetration and pharmacodynamics of the CHK1 inhibitor prexasertib in a mouse Group 3 medulloblastoma model. Eur J Pharm Sci. 2020;142: 105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowery CD, Dowless M, Renschler M, et al. Broad Spectrum Activity of the Checkpoint Kinase 1 Inhibitor Prexasertib as a Single Agent or Chemopotentiator Across a Range of Preclinical Pediatric Tumor Models. Clin Cancer Res. 2019;25: 2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong D, Infante J, Janku F, et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol. 2016;34: 1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.King C, Diaz HB, McNeely S, et al. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol Cancer Ther. 2015;14: 2004–2013. [DOI] [PubMed] [Google Scholar]
- 13.Cleary JM, Aguirre AJ, Shapiro GI, D’Andrea AD. Biomarker-Guided Development of DNA Repair Inhibitors. Mol Cell. 2020;78: 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bryant C, Rawlinson R, Massey AJ. Chk1 inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer. 2014;14: 570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015;5: 1137–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skolnik JM, Barrett JS, Jayaraman B, Patel D, Adamson PC. Shortening the timeline of pediatric phase I trials: the rolling six design. J Clin Oncol. 2008;26: 190–195. [DOI] [PubMed] [Google Scholar]
- 17.Chukwueke UN, Wen PY. Use of the Response Assessment in Neuro-Oncology (RANO) criteria in clinical trials and clinical practice. CNS Oncol. 2019;8: CNS28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45: 228–247. [DOI] [PubMed] [Google Scholar]
- 19.Hong DS, Moore K, Patel M, et al. Evaluation of Prexasertib, a Checkpoint Kinase 1 Inhibitor, in a Phase Ib Study of Patients with Squamous Cell Carcinoma. Clin Cancer Res. 2018;24: 3263–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwasa S, Yamamoto N, Shitara K, et al. Dose-finding study of the checkpoint kinase 1 inhibitor, prexasertib, in Japanese patients with advanced solid tumors. Cancer Sci. 2018;109: 3216–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iggo R, Gatter K, Bartek J, Lane D, Harris AL. Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet. 1990;335: 675–679. [DOI] [PubMed] [Google Scholar]
- 22.Lowery CD, VanWye AB, Dowless M, et al. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin Cancer Res. 2017;23: 4354–4363. [DOI] [PubMed] [Google Scholar]
- 23.Zhao X, Kim IK, Kallakury B, et al. Acquired small cell lung cancer resistance to Chk1 inhibitors involves Wee1 up-regulation. Mol Oncol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nair J, Huang TT, Murai J, et al. Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer. Oncogene. 2020;39: 5520–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karnitz LM, Flatten KS, Wagner JM, et al. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol Pharmacol. 2005;68: 1636–1644. [DOI] [PubMed] [Google Scholar]
- 26.Thompson R, Eastman A. The cancer therapeutic potential of Chk1 inhibitors: how mechanistic studies impact on clinical trial design. Br J Clin Pharmacol. 2013;76: 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole KA, Huggins J, Laquaglia M, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci U S A. 2011;108: 3336–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohn EA, Ruth ND, Brown MK, Livingstone M, Eastman A. Abrogation of the S phase DNA damage checkpoint results in S phase progression or premature mitosis depending on the concentration of 7-hydroxystaurosporine and the kinetics of Cdc25C activation. J Biol Chem. 2002;277: 26553–26564. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Immunohistochemical labelling of positive cells in tumor tissue. A) Positive cells showing immunoreactivity for Trp53; B) Positive cells showing immunoreactivity for Chk S345; C) Cells showing no immunoreactivity for Trp53; D) Cells showing no immunoreactivity for Chk S345. Scale bars are 200 uM.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.