

ABSTRACT
The glucocorticoid-induced tumor necrosis factor receptor (GITR) agonistic antibody (DTA-1) has been proved to elicit robust immune response in various kinds of tumors. However, only a few of the HCC patients could benefit from it, and the mechanism of DTA-1 resistance remains unknown. Here, we measured GITR expression in different immunocytes in HCC microenvironment, and we observed that tumor-infiltrating regulatory T cells (Ti-Tregs) significantly expressed GITR, which were associated with poor prognosis. Meanwhile, we analyzed the variation of tumor-infiltrating immune components and associated inflammation response after DTA-1 treatment in orthotopic liver cancer model of mice. Surprisingly, DTA-1 treatment reduced the infiltration of Tregs but failed to activate CD8+ T cells and elicit antitumor efficacy. In particular, DTA-1 treatment enforced alternative M2 polarization of macrophage, and macrophage depletion could enhance DTA-1-mediated antitumor efficacy in HCC. Mechanistically, macrophage M2 polarization attributed to the IL-4 elevation induced by Th2 immune activation in the treatment of DTA-1, resulting in DTA-1 resistance. Furthermore, Toll-like receptor 4 (TLR4) agonist could diminish the macrophage (M2) polarization and reverse the M2-mediated DTA-1 resistance, eliciting robust antitumor effect in HCC. Our finding demonstrated that the TLR4 agonist synergized with DTA-1 was a potential strategy for HCC treatment.
KEYWORDS: GITR, regulatory T cells, Th2 response, M2 polarization of macrophage, Toll-like receptor 4
Introduction
Cancer immunotherapy, aiming to rekindle or reinforce natural body defense to eliminate malignant cells, mainly involves around immune checkpoint blockade, including CD28/B7 superfamily (e.g.,CTLA-4,PD-1).1 Multiple cancer types like advanced melanoma, non–small cell lung cancer (NSCLC), and renal cell carcinoma (RCC) have shown efficacious clinical responses to immunotherapy.2,3 Disappointedly, HCC-associated clinical immunotherapy has limited outcomes, and the clinical objective response rate of HCC treated with immunological agents alone was only ~15–20%.4 GITR agonist treatment displayed robust efficacy in CT26, MC38, and B16 melanoma subcutaneous tumor model. However, other studies had shown that although none of the 39 patients had complete response (CR) or partial response (PR) when treated with GWN323 alone (an IgG monoclonal antibody against GITR), combination with pembrolizumab and nivolumab had a favorable efficacy. Given the heterogeneity of microenvironment, combination therapy was a promising strategy in cancer immunotherapy. Suppressive microenvironment promoting primary or acquired resistance to immunotherapy mainly involves around the infiltration of immunosuppressive cells, including Treg cells, tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs). For instance, benefits from PD-L1 antibody treatment could be abolished by TREM-1+ TAM infiltration in HCC.5 However, the roles of these immunosuppressive cells played and the mechanism of their immunosuppression in DTA-1 treatment in HCC need further investigation.
Accumulation of macrophage density is thought to be related with poor prognosis and survival in lung cancer, prostate cancer, and HCC.6 For example, increased recruitment of tumor-infiltrating macrophage via CCR2/CCL2 signaling resulted in immunosuppressive tumor microenvironment and drove tumor growth.6,7 However, distinct status of macrophage has been considered with either protective or pathogenic function in HCC.8 The protective phenotype in HCC has been described for M1 macrophages, which activate tumor-killing mechanism. M2 macrophage has been shown to suppress adaptive immune response and promote tumor growth, invasion, and metastasis. M2 polarization is usually related to microenvironment metabolism/hypoxia/Th2 immunity (secretion of IL-4, IL-13, IL-10, etc.) in tumor microenvironment.9–13 Nevertheless, whether DTA-1 could influence polarization status of macrophages remains to be studied.
Tumor necrosis factor receptor superfamily, including GITR (TNFRSF18), OX40, CD27, and 4–1BB, can enhance T-cell responses and survival through synergizing with TCR signaling.14 GITR is constitutively expressed on activated CD8+ or CD4+ Foxp3+ Treg cells, at low levels on naïve T cells, natural killer (NK) cells, and macrophages.15,16 In mice treated with DTA-1, Ti-Tregs are more likely to be depleted and CD8+ T cells could attain the ability to resist Treg inhibition. However, DTA-1 treatment could also increase Th2 response, which is associated with resolution of inflammation, tissue repair, and regeneration.13 Given that various kinds of tumor-infiltrating immune cells, composed of adaptive T cells and innate immune cells (like Natural killing cells, Dendritic cells, and macrophages), contribute to the modulation of tumor progression, instead of blind clinical application of immune-associated agents, personalized strategies to enhance clinical response of immunotherapy either by biomarker-targeted therapy17 or by reasonable design of prudent combinations for DTA-1 treatment are still desperately in need.
In this work, we characterized GITR expression pattern of different immune cells in HCC microenvironment. We found that GITR is selectively expressed on tumor-infiltrating Treg. Then, in orthotopic liver tumor models, although significant decreasing tumor-infiltrating Tregs were observed when treating with DTA-1, the mice could not benefit from the therapy. Intriguingly, we found that polarization of alternative macrophage can compromise the well fractured immunosuppression related to decreased infiltration of Treg, which can account for the failure of tumor load reduction. To further explore, we demonstrated that upregulation of Th2 response induced by DTA-1 contributed to M2 polarization via IL-4 elevation. Finally, DTA-1 combined with TLR4 monoclonal antibody can abrogate M2 polarization-mediated immunosuppression. These results suggest that DTA-1 combined with TLR4 agonist is a candidate strategy to supplement anti-HCC immune therapeutic efficacy in HCC.
Materials and method
Clinical samples
After the patients gave their fully informed consent and signed the informed consent, 17 fresh liver cancer samples, peripheral blood samples, and para-carcinoma samples were collected from the first Affiliated Hospital of Zhejiang University School of Medicine after liver cancer resection. These fresh specimens were treated into single cell suspensions for subsequent sorting and flow cytometry. This project has been approved by the Ethical Committee of the First Affiliated Hospital of Zhejiang University School of Medicine, and relevant clinical data are summarized in Table S1.
Sample dissociation and sorting
The tumor and para-cancer tissues were treated with Tumor Dissociation Kit, human (Miltenyi, 130–095-929) according to the manufacturer’s instruction, and a part of the single cell suspensions was reserved for labeling CD68+ macrophages. Subsequently, CD4+ T cells and CD8+ T cells were separated from single cell suspension using REAlease® CD4/CD8 (TIL) MicroBead Kit, human (Miltenyi, 130–121-561) for following label and flow cytometry.
Establishment of mouse tumor model and treatment
The mice involved in this experiment mainly included C57BL/6 J, Rag1-KO, and Foxp3DTR mice, male and 6–8 weeks of age. All the mice were fed in the SPF experimental animal center. After the establishment of the mouse tumor model, drug treatments were described, given in details in the supplementary material.
Cell culture and induced differentiation assay
For T-cell differentiation experiment, naive T cells were sorted from fresh spleen tissue by flow sorter. The induction conditions of iTreg were RMPI 1640 contained TGF-β (10 ng/ mL), IL-2 (20 ng/ml), and DynabeadsTM Mouse T-activator CD3/CD28 (Thermo fisher, 11542D) (at ratio of 2:1). The induction conditions of Th2 were RMPI 1640 contained IL-4 (10 ng/ mL), IL-2 (20 ng/ml), and DynabeadsTM Mouse T-activator CD3/CD28 (at ratio of 2:1). The induction conditions of Th1 were RMPI 1640 contained IL-12 (4 ng/ ml), IL-2 (20 ng/ml), and DynabeadsTM Mouse T-activator CD3/CD28 (at ratio of 2:1). The concentration of GITR-ligand is 5 µg/ml. For the experiment of macrophage differentiation, monocytes derived from mouse bone marrow were extracted and stimulated with GM-CSF (40 ng/ml, Novus Biologicals, Littleton, CO) for 7 days for further experiment in vitro. The macrophages were induced to differentiate under IL-4 (10 ng/ml) conditions with or without GITR-ligand (5 µg/ml).
Elisa assay
The tumor draining lymph node was lysed by RIPA, and the protein concentration was determined and then diluted to 2 mg/ml of total protein concentration. Mouse Interleukin 4, IL-4 ELISA KIT (Cusabio, CSB-E04634m) was used to determine IL-4 concentration according to the manufacturer’s instruction.
Single cell suspension preparation and flow cytometry
The mice were sacrificed on the indicated day, and then the tumor in the left outer lobe of the liver was collected. Mononuclear cells were separated from single-cell suspension by OptiPrep Density Gradient Medium. CD4+ T cells and CD8+ T cells were enriched by Magnetic beads (BD, 551539 and 551516) and then detected by multicolor flow cytometry after antibody incubation. CD8+ T cells were stimulated by Leukocyte Activation Cocktail (550583, BD Pharmingen, San Diego, CA) for 5 h for the analysis of cytotoxic function. Specific staining steps and corresponding antibodies are shown in the supplementary materials.
Immunohistochemistry
Tissue sections with a thickness of 3 μm were used for immunohistochemical assay, and the images were analyzed by two independent pathologists. Specific immunohistochemistry method is provided in the supplementary materials.
Quantitative RT-PCR
After total RNA extraction and reverse transcription in complementary DNA, real-time quantitative PCR was performed using the SYBR qPCR Master Mix (Vazyme, Q711-02) according to the manufacturer’s instruction. The threshold cycle value was used to analyze the gene expression among the different groups. Independent experiment was repeated at least three times. Primers are shown in the Supporting Table S2.
Statistical approach
Statistical analysis was performed using GraphPad Prism 8 software (La Jolla, CA). Flow cytometry data were analyzed by FlowJo.10 (TreeStar, Ashland, OR). Student two-tailed t test was used for comparison of quantitative data from different groups. P < .05 was considered statistically significant (*P < .05; **P < .01; ***P < .001).
Results
Ti-Treg shows significant high expression of GITR, which was associated with poor prognosis
To determine which components of immune cell would be prioritized to be targeted by anti-GITR agonist in HCC, we researched GITR expression in different immune cell through a public database (http://tisch.comp-genomics.org/home/), and we found Treg showed the highest expression of TNFRSF18 in single-cell sequencing dataset (Figure 1a). To further investigate, we collected 17 HCC samples from patients and further analyzed GITR expression in CD4+ T cells, CD8+ T cells, and macrophage by flow cytometry assay. We found that GITR was mainly expressed on CD4+ T cells and hardly expressed on CD8+ T cells and macrophages in HCC microenvironment (Figure 1b, S1A). Intriguingly, Treg selectively express GITR when compared to naïve CD4+ T cell and conventional CD4+ T cell (Figure 1c). Moreover, GITR expression on Treg showed region specificity. When compared with Treg from para-carcinoma liver, tumor-infiltrating Treg showed higher expression of GITR (Figure 1d). Coupled with that, Treg abundance was higher in tumor microenvironment when compared with the liver (S1B), suggesting that these Ti-Tregs are more likely to be regulated.
Figure 1.
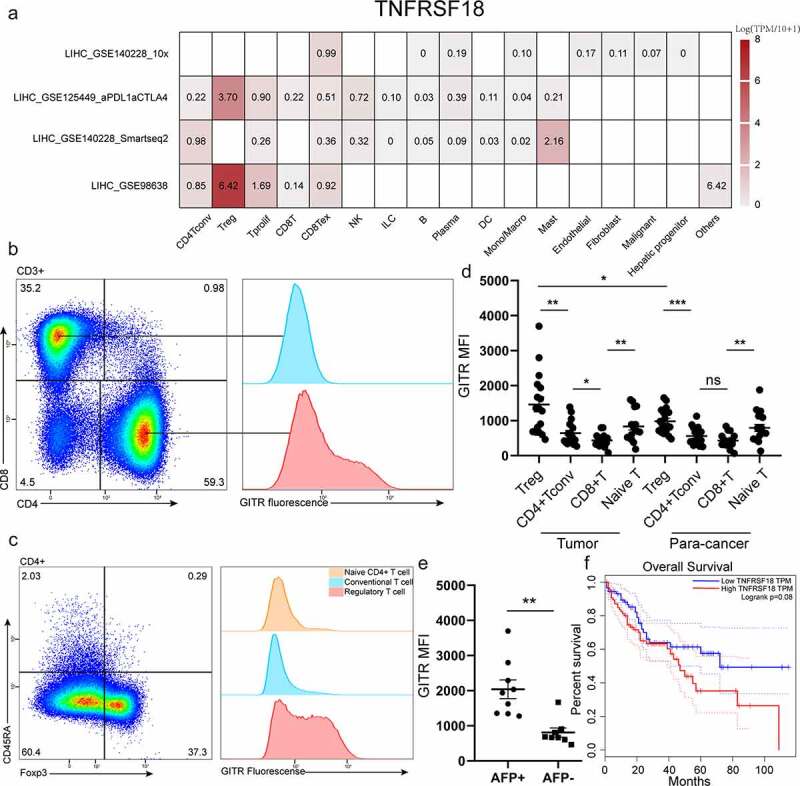
Ti-Treg shows significant high expression of GITR, which was associated with poor prognosis. (a) TNFRSF18 expression pattern in different immune cells in four single cell sequences of LIHC (liver hepatocellular carcinoma). (b) CD4+ T cells and CD8+ T cells in fresh clinical HCC sample were enriched and then labeled with CD3, CD4, CD8a, CD45RA, and GITR for analyzing GITR expression on different subpopulation by flow cytometry. (c)The different expression patterns of GITR on Naïve CD4+ T cells (labeled as CD3+ CD4+ CD45RA+), conventional CD4+ T cells (labeled as CD3+ CD4+ Foxp3− CD45RA−), and Treg (labeled as CD3+ CD4+ Foxp3+) in tumor are shown. (d) Comparison of GITR MFI of various subgroups (CD3+ CD4+ Foxp3+ Treg cell, CD3+ CD4+ Foxp3− CD45RA−Tconv cell, CD3+ CD8+ T cell, CD3+ CD45RA+ Naïve T cell) in tumor and para-cancer tissue. (e) The different expression of GITR of Ti-Treg between AFP-positive group (AFP > 20 ng/ml) and AFP-negative group (AFP ≤ 20 ng/ml). Data are shown as mean ± SEM. *P < .05; **P < .01; ***P < .001. (f) Correlation analysis of TNFRSF18 gene expression level and survival prognosis of HCC patients. High TNFRSF18 group (n=91) (red line) was defined as the top 25% in TNFRSF18 gene expression level, and low TNFRSF18 group (n=90) (blue line) was defined as the end 25% in TNFRSF18 gene expression level.
Next, we analyzed the relationship between GITR expression and clinical information of patients. Clinical features of patients and tumors are described in Table S1. We found that Treg cells showed higher MFI of GITR in alpha fetoprotein (AFP) positive group patients compared with alpha fetoprotein (AFP) negative group patients (Figure 1e), and GITR expression was independent of age and sex (table S1). High expression of GITR is positively associated with survival prognosis of HCC patients in GEPIA (http://gepia.cancer-pku.cn/, dataset: TCGA-LIHC) (Figure 1f). Collectively, these results indicate that tumor-infiltrating Treg cells were the main target of GITR agonist.
DTA-1 treatment elicited robust B16/F10 tumor regression but failed to impede HCC growth
In preclinical studies, DTA-1 have been reported to cause robust tumor inhibition of various kinds of tumor and increase survival prognosis.18,19 To further investigate the role DTA-1 played in anti-HCC response, hepa1-6 in situ in mouse model was established and treated with DTA-1. Disappointedly, DTA-1 treatment failed to curb HCC growth (Figure 2a,b). To verify the effective function of DTA-1 and compare sensitivity of different tumor to DTA-1, we simultaneously built B16/F10 and hepa1-6 subcutaneous tumor model, and DTA-1 treatment caused significant B16/F10 tumor regression, even partially cured (Figure 2c, d). However, the tumor growth of subcutaneous Hepa1-6 tumor still shown no difference after DTA-1 treatment (Figure 2e, f). Taken together, these data demonstrated that HCC tumor is selectively resistant to DTA-1 treatment.
Figure 2.
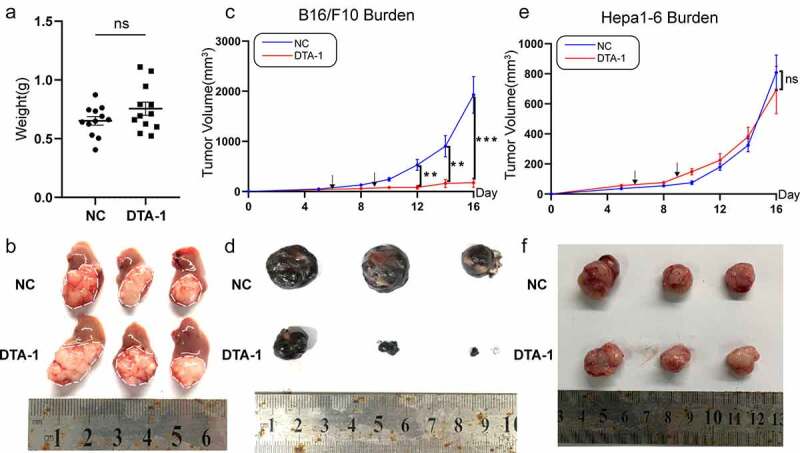
DTA-1 treatment elicited robust B16/F10 tumor regression, but failed to impede HCC growth. (a-b) The hepa1-6 cells (5X105) were injected within left lobe of liver of C57BL/6 in situ, followed by treatment of DTA-1 (300ug) on day 6 and day 9 after inoculation. Tumor weight was examined on Day 13 (n=12 for each group), and tumor appearance was compared with other group. (c) Average tumor growth of subcutaneously injected B16/F10 cells was examined (n=6 for each group), and the treatment of DTA-1 was administered on day 6 and day 9. (d) B16/F10 tumor appearance was compared with other group (day 17). (e) Average tumor growth of subcutaneously Hepa1-6 cells was examined (n = 6) for each group. The treatment of DTA-1 was administered the same as before. (f) Hepa1-6 tumor appearance was compared with other group (day 17). Data are shown as mean ± SEM. *P < .05; **P < .01; ***P < .001.
DTA-1 treatment reduced infiltration of Treg but failed to activate CD8+ T cell in HCC immune microenvironment.
Previous study had shown that reduced tumor infiltrating Treg and increased proportion of CD8+ T cell with less exhausted phenotype caused by DTA-1 treatment contributed to the robust immune response in MC38 immune microenvironment.20 Thus, we analyze the proportion of infiltrating CD4+ T and CD8+ T cell in HCC immune microenvironment. Consistent with previous studies, we found decreased proportion of CD4+ T cells in total CD45+ CD3+ T cell after DTA-1 treatment but with no significantly proportional change within CD8+ T cells (Figure 3a). The main decreased subpopulation was CD45+ CD3+ CD4+ Foxp3+ Treg (Figure 3b-d), the same results as in the lymph node (S1C). We also showed that GITR-ligand could compromise expression of Foxp3 under induced Treg (iTreg) polarizing condition in vitro (S1D). We also found high expression of GITR on Ti-Treg in orthotopic Hepa1-6 tumor mouse model, and when treated with DTA-1, GITR expression on Foxp3+ T cells drastically decreased (S1E). However, the expressions of suppressive ligand such as PD-L1, CTLA4 showed no significant difference after treatment (Figure 3e). In addition, DTA-1 treatment maintained sufficient stability of peripheral Treg, which may avoid an overactive peripheral immune response (S1F). In spite of decreased infiltration of Ti-Treg, the cytotoxic function of CD8+ T cell showed little advance and only showed a slight elevation of TNF-α production, and the proportion of PD-1+ in CD8+ T decreased. The proportion of PD-1+TIM3+ and secretion of Granzyme B in CD8+ T cells show no difference (Figure 3f). Evident reduction of infiltrating of repressive Foxp3+ Treg cell failed to generate the activation of CD8+ T cells and mighty anti-tumor effect, thus, the potential resistance mechanism of DTA-1 treatment needs further investigation.
Figure 3.
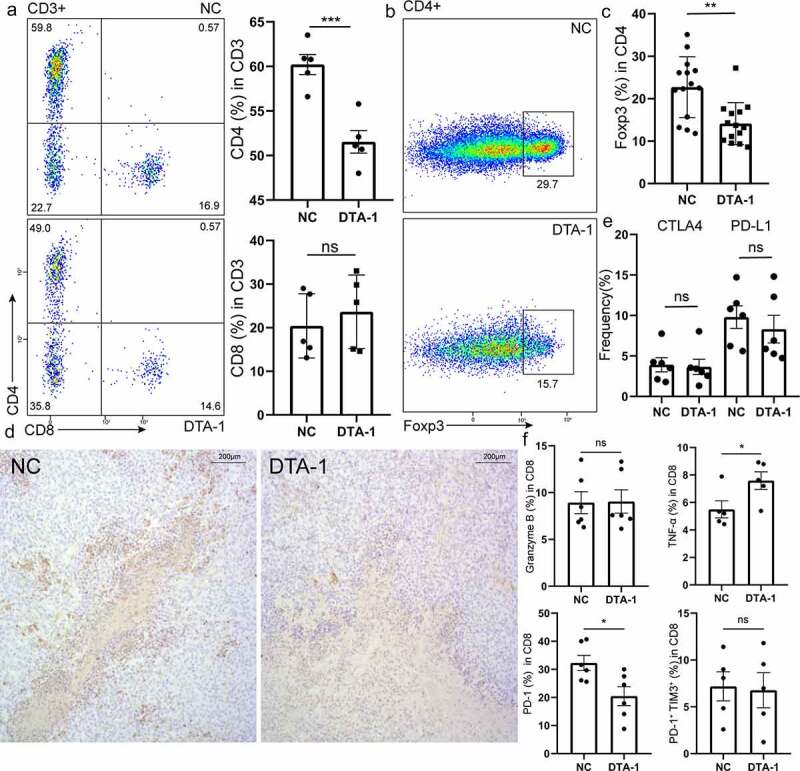
DTA-1 treatment reduced infiltration of Treg but failed to activate CD8+ T cell in HCC immune microenvironment. (a) Analysis of the variety of percentage of infiltrating CD4+ T cells and CD8+ T cells in CD3+ T cells within tumor lesion from hepa-1-6 bearing mice by flow cytometry after DTA-1 treatment (n = 5). (b-c) Tumor lesions were harvested for analyzing percentage of CD4+ Foxp3+ Tregs population. Representative flow images are presented. Data are shown as mean ± SEM. (d) Representative immunohistochemical images stained with Foxp3 for each group. Magnification, x100. (e) The suppressive ligands of CD45+ CD3+ CD4+ Foxp3+ Treg are examined for each group. Data are shown as mean ± SEM (n = 6). (f) CD8+ T cells were labelled with CD3, CD8a, granzyme B, TNF-α, PD-1, and TIM-3 for analyzing cytotoxic function and exhausted status (PD-1+ Tim3+) by flow cytometry. Data are shown as mean ± SEM (n=5 or 6). *P < .05; **P < .01; ***P < .001.
Macrophage M2 polarization conferred resistance to DTA-1 treatment in HCC
Given that liver macrophages are highly plastic and susceptible to adapt their phenotype on the basis of signals from hepatic microenvironment (e.g., danger signals, cytokines from other immune cells)21 and more proportion of macrophage in CD45+ was observed in orthotopic Hepa1-6 tumor when compared with subcutaneous B16/F10 tumor (S1G), we next explore the role macrophages played in DTA-1 therapy. After macrophage depletion (Verified in S2A) by intravenous injection of liposome 1d before building hepa1-6 in situ model, great tumor regression was found in DTA-1 combined treatment group, yet tumor burden increased when macrophage was single depletion or given DTA-1 therapy separately (Figure 4a, 4b). Thus, we next analyze various phenotypes and function of macrophage in HCC microenvironment after DTA-1 treatment, and it is worth noting that enforced alternative activation of macrophage (M2 polarization) was found after the therapy, which characterized as improved expression of CD206 and IL-10 and decreased expression of iNos and TNF-α (Figure 4c, 4d). Together, our results indicated that GITR agonist can skew macrophage polarization from M1 to M2 in some way.
Figure 4.
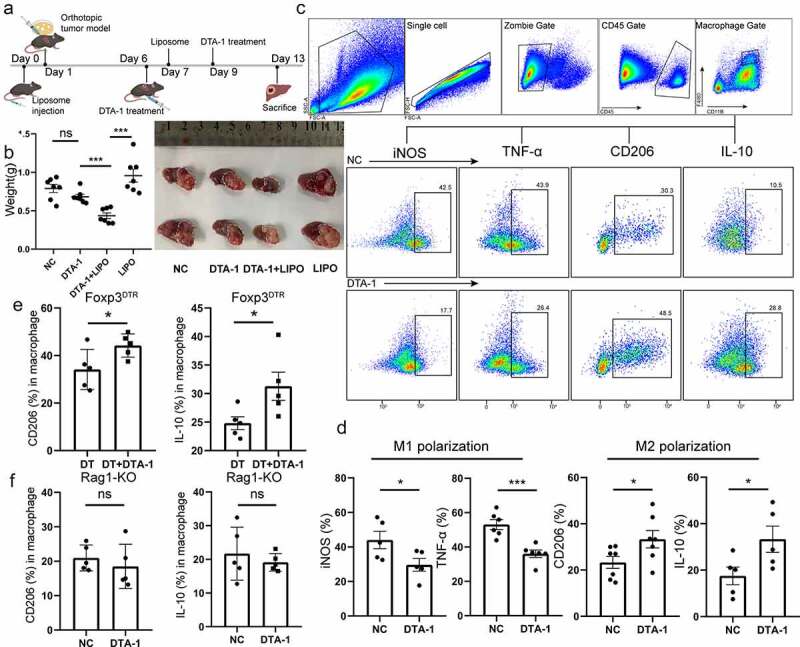
Macrophage conferred resistance to DTA-1 treatment in HCC, and DTA-1 treatment affected M2 polarization indirectly. (a) Schematic illustration of DTA-1 treatment for combination with liposome. The heap1-6 cells were inoculated orthotopically within C57BL/6 mice on day 1, and mice were pretreated with neutral clodronate liposomes (200ul) through tail vein injection on day 0 and 7, followed by treatment of DTA-1 (300ug) on day 6 and 9. (b) All tumors were harvested, and the tumor appearance and weight were compared with each group. Data are shown as mean ± SEM (n = 7). (c-d) Schematic diagram of logical door frame selection of macrophages was listed. The phenotype of macrophages was examined in mice with or without DTA-1 treatment, including M1 polarization (marked as expression of iNos, TNF-α) and M2 polarization (marked as expression of CD206 and IL-10). Data are shown as mean ± SEM (n = 5-7). (e) The M2 polarization of macrophage was examined in mice with or without conditional knockout of Treg cells. Conditional knockout of Foxp3+ Treg was established by regular intraperitoneal injection of DT (2 µg) in tumor-bearing Foxp3DTR mice. Data are shown as mean ± SEM (n=5). (f) The M2 polarization of macrophage was examined in Rag1-KO mice with or without DTA-1 treatment (n=5). Data are shown as mean ± SEM. *P < .05; **P < .01; ***P < .001.
Research has shown that macrophages express GITR at low level.16,22 Consisted with antecedent studies, we also verified that CD45+ CD68+ macrophage showed low expression of GITR (S1A). In addition, CD206 expression showed no difference in macrophage when cultured under M2 polarization condition with or without GITR-ligand in vitro (S2B), suggesting that GITR agonist would not intervene macrophage polarization directly. Thus, we sought to determine the way DTA-1 influenced macrophage polarization. We first conditionally cleared Foxp3+ Treg in Foxp3DTR mice by intraperitoneal injection of diphtheriatoxin before DTA-1 treatment, and we found that M2 polarization bias of macrophage still existed (Figure 4e), and the enhanced Th2 response still remain (S2C); CD8+ T cells showed no significant difference in PD-1 expression (S2D). However, when we repeated the above tests in Rag1-KO mice, DTA-1 treatment could not skew M2 polarization (Figure 4f). These results strongly suggested that GITR agonists indirectly favor M2 macrophage phenotype through co-stimulation of T cells, which is independent on tumor-infiltrating Treg.
DTA-1 treatment triggered Th2 response in TME lead to increasing M2 polarization
Previous discovery had reported that GITR co-stimulation can enhance Th2 responses19,23 to investigate the potential mechanism of T-cell-mediated increasing M2 polarization after treatment, and we sought to investigate whether DTA-1-induced resistance in HCC model was associated with Th2 immunity. Indeed, flow cytometry assay showed accelerating IL-4 secretion in CD4+ T cell was detected by treatment with the DTA-1 antibody (Figure 5a). By using Elisa array, the levels of IL-4 were enhanced in Hilar lymph nodes after treatment (Figure 5b). When total CD4+ T cells isolated from spleen give GITR-ligand in vitro, the Elisa assay showed that the supernatant contained more IL-4 (Figure 5c). Similarly, we observed that the transcript levels of mRNA encoding interleukins 4, 13, and 10 (Il4, Il13, and Il10, respectively) in tumor-draining lymph nodes (Hilar lymph nodes) were all significantly upregulated (Figure 5d), which are associated with suppressive immune response. In vitro assay also showed that GITR-ligand significantly favor expression of GATA3 under Th2-polarizing condition (Figure 5e), but shown no influence on Th1 differentiation (S2E). Collectively, given that GITR-ligand would not influence polarization of macrophage (S2B), these data can explain that enhanced M2 polarization was the results of DTA-1-mediated Th2 response.
Figure 5.
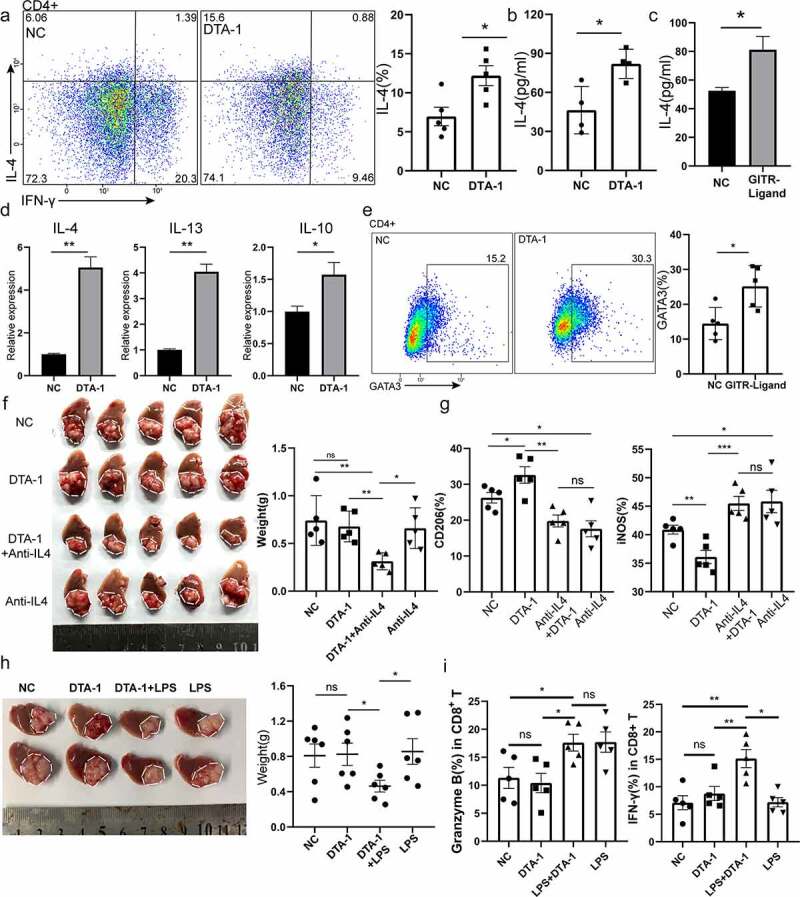
DTA-1 treatment associated Th2 response in TME account for increasing M2 polarization. (a) Analysis of the percentage of Th2 subpopulation in CD4+ T cells by flow cytometry, and percentage of CD3+ CD4+ IL-4+ Th2 population were presented as mean ± SEM (n=5 for each group). (b) ELISA assay was applied to detect the concentration of IL-4 in tumor draining Hilar lymph nodes. (c) CD4+ T cells sorted from spleen were treated with GITR-ligand (5ug/ml) for 24 h, and concentration of IL-4 was measured in supernatant. (d) Tumor-draining lymph nodes were harvested on day 10 from a model of orthotopic Hepa1-6 received DTA-1 treatment on day 6 and 9 or not. Expression of IL4, IL13, IL-10 transcripts in Hilar lymph nodes of Hepa1-6 tumor bearing mice. Data were normalized to the expression of GAPDH and represented as mean ± SEM. (e) In vitro assay for Th2 differentiation. Naïve CD4+ T cells (labeled as CD4+ CD44− CD62L+) sorting from spleen treated with IL-4 condition (10 ng/ml) under the condition of GITR-ligand (5 µg/ml) or not.(f) Combination of DTA-1 and anti-IL4 mAb to treat hepa1-6 bearing mice. Tumor appearance and weight were compared with each group (n=5). (g) The polarization status of macrophage was examined in each group (NC, DTA-1, DTA-1+ Anti-IL4 mAb, anti-IL4 mAb), including expression of CD206 and iNos (n=5). (h) Combination of DTA-1 and TLR4 agonist to treat hepa1-6 bearing mice. Tumor appearance and weight were compared with each group. (i) CD8+ T cells were labelled with CD3, CD8a, granzyme B, IFN-γ for analyzing cytotoxic function (n=5). Data are shown as mean ± SEM. *P < .05; **P < .01; ***P < .001.
Anti-IL4 mAb and LPS treatment synergy with DTA-1 lead to great anti-Hepa1-6 effect
To further prove the link between Th2, TAM2, and tumor growth in vivo, DTA-1 was administered after neutralizing IL-4 in orthotopic Hepa1-6 model. We found obvious decreased tumor burden in anti-IL4 mAb and DTA-1 treatment group (Figure 5f). Meanwhile, when combined with anti-IL4 mAb, we found that DTA-1-induced M2 polarization was abolished and marked as decreased expression of CD206 on macrophage and enhanced iNOS expression (Figure 5g). Next, we intend to design a well-directed drug combination to remove DTA-1-induced HCC resistance. Toll-like receptor 4 agonist could reprogram M2 polarization to M1 polarization and induce pro-inflammatory response.24 When combined with TLR4 agonist LPS, tumor burden showed great decrease compared to single treatment (Figure 5h). Subsequently, we showed that combination of LPS and DTA-1 could reprogramme M2-M1 polarization when compared with NC group or DTA-1 group (S2F). Moreover, the decreased infiltration of Foxp3+ Treg cells was still detected once receiving DTA-1 treatment (S2G). To further detect the function of CD8+ T cells, we found that the combination group had shown the highest secretion of IFN-γ and Granzyme B (Figure 5i). These results could further validate our previous resistance mechanism, and our research may provide a potential approach to GITR-targeted immunotherapy.
Discussion
Revealing the expression profile of specific immune checkpoint on immune cells in tumor microenvironment is the cornerstone of specific and individual immunotherapy. For instance, studies have found that patients with high expression of PD-1 in CD8+ T cells in tumor-immune microenvironment can attain better clinical efficacy when treated with PD-1 monoclonal antibody.25,26 The PD-1 expression balance between effector and regulatory T cells also predicts the clinical efficacy of PD-1 blockade therapies.27 Therefore, revealing GITR expression profile in HCC microenvironment becomes a prerequisite for DTA-1 therapy. Here, we found that different patients presented different types of GITR expression profiles, and this might explain why different individuals have different responses to DTA-1 treatment. Moreover, our study and other research22,28 showed that GITR is mainly highly expressed in tumor-infiltrating Treg, while low expression is found in CD8+ T cells, macrophages, naive T cells, etc. Combined with our results, Ti-Treg were thought as a preferable potential target for DTA-1 treatment. However, varying levels of GITR MFI in Ti-Treg was observed in different patients; patients with AFP positive showed higher GITR MFI. This conclusion suggests that whether DTA-1 treatment should be given priority to patients with increasing AFP levels needs further research.
Next, we conducted DTA-1 therapy experiments in orthotopic liver tumor model. Unfortunately, although we verified that the Treg infiltration decreased significantly in tumor microenvironment, neither activation of CD8+ T cell nor anti-tumor efficacy was observed. Tregs are widely believed to have strong immunosuppressive effects and can achieve significant antitumor benefits in anti-CD25 therapy or in conditional Treg knockout models29–31 but cannot be approved for clinical application due to autoimmune disease like immune dysregulation, enteropathy, and X-linked (IPEX) syndrome and even lethal side effect.32,33 In our study, Treg showed different expression characteristics of GITR in tumors, para-cancer, and normal tissues, and DTA-1 did not cause the imbalance of treg ratio in peripheral normal liver tissues and spleen in mice. Meanwhile, the safety of DTA-1 had been verified in phase I clinical trials.34,35 These results indicated that DTA-1 could be a promising drug if the antitumor effect made a breakthrough. Considering that immunosuppressive cells in tumor microenvironment not only include Treg, but also Macrophage, MDSC, etc.11 Liver macrophage owned the character of high plasticity and adapt their phenotype according to signals derived from hepatic microenvironment.21 It is noteworthy that we found macrophages play an important role in DTA-1 drug resistance, and increased M2 polarization becomes the main mechanism of DTA-1 resistance.
Our study found that the expression profile of GITR was mainly concentrated in T cells rather than macrophages, and in vitro macrophage GITR-ligand activation assay also verified that it had no direct effect on alternative macrophage polarization. Meanwhile, studies have reported that DTA-1 can increase Th2 reactivity and increase IL-4 secretion.19,23 Based on the above information, Rag1-KO T cell-deficienct mice and Foxp3DTR conditional Treg depletion mice were applied to verify the potential mechanism of M2 polarization. We found that M2 polarization mainly depends on T cells rather than Treg cells. In vitro experiments showed that GITR-ligand can directly increase Th2 differentiation and reduce Foxp3 expression. In summary, DTA-1 treatment mainly causes the increase of Th2 reactivity and thus enhances M2 polarization to establish immunosuppressive environment, undermining the benefits derived from Ti-Treg reduction (Figure 6).
Figure 6.
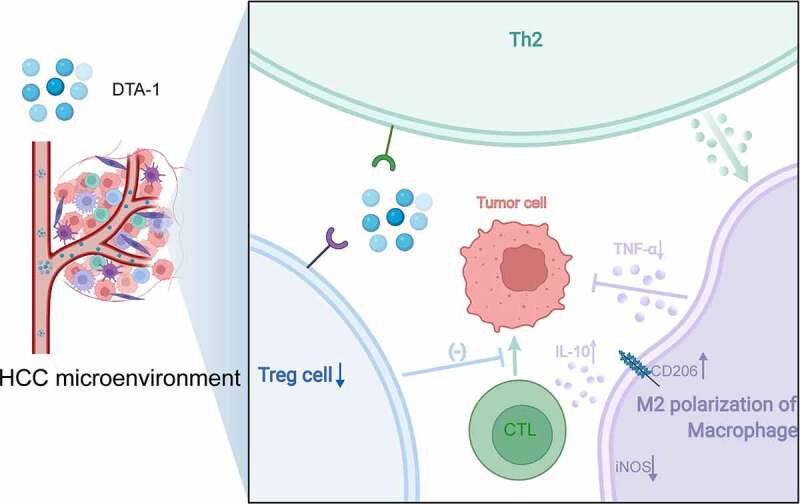
Schematic diagram showing that M2 polarization of macrophage attributed to the IL-4 elevation induced by Th2 immune activation in the treatment of DTA-1, resulting in DTA-1 resistance.
In conclusion, Th2-mediated M2 polarization and Ti-Treg reduction are the main biological reactions caused by DTA-1 therapy, and the former may be the main mechanism of DTA-1 resistance, while the latter may serve as a potential force for antitumor effects. In order to address the DTA-1 resistance in HCC, M2 polarization were revealed to become a key regulatory target. Distinguished from other studies that Th2-mediated Th9 and Th21 play a key role in the anti-tumor response,19,36 our finding showed that DTA-1 resistance mainly involves around Th2 response-mediated M2 polarization. We proposed the use of TLR4 agonist to reverse M2 polarization. To abrogate the double-suppressive microenvironment, combination with TLR4 agonist provide a promising therapeutic strategy to solve DTA-1 resistance alone.
Supplementary Material
Funding Statement
This work was supported by the Health Commission of Zhejiang Province under Grant [JBZX-202004]; Research Unit Project of Chinese Academy of Medical Science under Grant [2019-I2M-5-030]; and Research Project of Jinan Microecological Biomedicine Shandong Laboratory under Grant [JNL-2022002A, JNL-2022008B]
Ethics approval and consent to participant
The present study was approved by the Ethical Committee of the First Affiliated Hospital of Zhejiang University School of Medicine [ID: IIT20210439A], and written informed consent was obtained from all patients.
Disclosure statement
There are no competing interests to declare.
Data and materials availability
All data needed to evaluate the conclusions in the paper are presented in the paper and/or the Supplementary Materials. Additional data related to this paper may requested from authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125(9):3335–11. doi: 10.1172/JCI83871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(17):1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng AL, Hsu C, Chan SL, Choo S-P, Kudo M.. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J Hepatol. 2020;72(2):307–319. doi: 10.1016/j.jhep.2019.09.025. [DOI] [PubMed] [Google Scholar]
- 5.Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, Su R, Hong L, Lu H, Zhang F, et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology. 2019;70(1):198–214. doi: 10.1002/hep.30593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waidmann O, Köberle V, Bettinger D, Trojan J, Zeuzem S, Schultheiß M, Kronenberger B, Piiper A. Diagnostic and prognostic significance of cell death and macrophage activation markers in patients with hepatocellular carcinoma. J Hepatol. 2013;59(4):769–779. doi: 10.1016/j.jhep.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Yao W, Yuan Y, Chen P, Li B, Li J, Chu R, Song H, Xie D, Jiang X, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66(1):157–167. doi: 10.1136/gutjnl-2015-310514. [DOI] [PubMed] [Google Scholar]
- 8.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 10.Mehla K, Singh PK. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer. 2019;5(12):822–834. doi: 10.1016/j.trecan.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019;79(18):4557–4566. doi: 10.1158/0008-5472.CAN-18-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, Movahedi K, Houbracken I, Schouppe E, Elkrim Y, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74(1):24–30. doi: 10.1158/0008-5472.CAN-13-1196. [DOI] [PubMed] [Google Scholar]
- 13.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9(4):271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dostert C, Grusdat M, Letellier E, Brenner D. The TNF Family of Ligands and Receptors: communication Modules in the Immune System and Beyond. Physiol Rev. 2019;99(1):115–160. doi: 10.1152/physrev.00045.2017. [DOI] [PubMed] [Google Scholar]
- 16.Patel M, Xu D, Kewin P, Choo-Kang B, McSharry C, Thomson N, Liew F. Glucocorticoid-induced TNFR family-related protein (GITR) activation exacerbates murine asthma and collagen-induced arthritis. Eur J Immunol. 2005;35(12):3581–3590. doi: 10.1002/eji.200535421. [DOI] [PubMed] [Google Scholar]
- 17.Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X, Wu K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. 2018;17(1):129. doi: 10.1186/s12943-018-0864-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi Y, Shi Y, Haymaker CL, Naing A, Ciliberto G, Hajjar J. T-cell agonists in cancer immunotherapy. J Immunother Cancer. 2020;8(2):e000966. doi: 10.1136/jitc-2020-000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim IK, Kim BS, Koh CH, Seok JW, Park JS, Shin KS, Bae EA, Lee GE, Jeon H, Cho J, et al. Glucocorticoid-induced tumor necrosis factor receptor-related protein co-stimulation facilitates tumor regression by inducing IL-9-producing helper T cells. Nat Med. 2015;21(9):1010–1017. doi: 10.1038/nm.3922. [DOI] [PubMed] [Google Scholar]
- 20.Sabharwal SS, Rosen DB, Grein J, Tedesco D, Joyce-Shaikh B, Ueda R, Semana M, Bauer M, Bang K, Stevenson C, et al. GITR Agonism Enhances Cellular Metabolism to Support CD8+T-cell Proliferation and Effector Cytokine Production in a Mouse Tumor Model. Cancer Immunol Res. 2018;6(10):1199–1211. doi: 10.1158/2326-6066.CIR-17-0632. [DOI] [PubMed] [Google Scholar]
- 21.Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66(6):1300–1312. doi: 10.1016/j.jhep.2017.02.026. [DOI] [PubMed] [Google Scholar]
- 22.Ponte JF, Ponath P, Gulati R, Slavonic M, Paglia M, O’Shea A, Tone M, Waldmann H, Vaickus L, Rosenzweig M, et al. Enhancement of humoral and cellular immunity with an anti-glucocorticoid-induced tumour necrosis factor receptor monoclonal antibody. Immunology. 2010;130(2):231–242. doi: 10.1111/j.1365-2567.2009.03228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motta AC, Vissers JL, Gras R, Van Esch BC, Van Oosterhout AJ, Nawijn MC. GITR signaling potentiates airway hyperresponsiveness by enhancing Th2 cell activity in a mouse model of asthma. Respir Res. 2009;10(1):93. doi: 10.1186/1465-9921-10-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wanderley CW, Colón DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, Pereira JA, Silva CM, Silva CR, Silva RL, et al. Paclitaxel Reduces Tumor Growth by Reprogramming Tumor-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer Res. 2018;78(20):5891–5900. doi: 10.1158/0008-5472.CAN-17-3480. [DOI] [PubMed] [Google Scholar]
- 25.Kansy BA, Concha-Benavente F, Srivastava RM, Jie H-B, Shayan G, Lei Y, Moskovitz J, Moy J, Li J, Brandau S, et al. PD-1 Status in CD8+T Cells Associates with Survival and Anti-PD-1 Therapeutic Outcomes in Head and Neck Cancer. Cancer Res. 2017;77(22):6353–6364. doi: 10.1158/0008-5472.CAN-16-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, Vitaly K, Itahashi K, Maeda Y, Matsui S, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21(11):1346–1358. doi: 10.1038/s41590-020-0769-3. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S . Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3(2):135–142. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 29.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59:3128–3133. [PubMed] [Google Scholar]
- 30.Wei WZ, Jacob JB, Zielinski JF, Flynn JC, Shim KD, Alsharabi G, Giraldo AA, Kong YCM. Concurrent Induction of Antitumor Immunity and Autoimmune Thyroiditis in CD4+ CD25+ Regulatory T Cell–Depleted Mice. Cancer Res. 2005;65(18):8471–8478. doi: 10.1158/0008-5472.CAN-05-0934. [DOI] [PubMed] [Google Scholar]
- 31.Kurebayashi Y, Olkowski CP, Lane KC, Vasalatiy OV, Xu BC, Okada R, Furusawa A, Choyke PL, Kobayashi H, Sato N. Rapid Depletion of Intratumoral Regulatory T Cells Induces Synchronized CD8 T- and NK-cell Activation and IFNgamma-Dependent Tumor Vessel Regression. Cancer Res. 2021;81(11):3092–3104. doi: 10.1158/0008-5472.CAN-20-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 33.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22(1):531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 34.Piha-Paul SA, Geva R, Tan TJ, Lim DW, Hierro C, Doi T, Rahma O, Lesokhin A, Luke JJ, Otero J, et al. First-in-human phase I/Ib open-label dose-escalation study of GWN323 (anti-GITR) as a single agent and in combination with spartalizumab (anti-PD-1) in patients with advanced solid tumors and lymphomas. J Immunother Cancer. 2021;9(8):e002863. doi: 10.1136/jitc-2021-002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balmanoukian AS, Infante JR, Aljumaily R, Naing A, Chintakuntlawar AV, Rizvi NA, Ross HJ, Gordon M, Mallinder PR, Elgeioushi N, et al. Safety and Clinical Activity of MEDI1873, a Novel GITR Agonist, in Advanced Solid Tumors. Clin Cancer Res. 2020;26(23):6196–6203. doi: 10.1158/1078-0432.CCR-20-0452. [DOI] [PubMed] [Google Scholar]
- 36.Koh CH, Kim I-K, Shin K-S, Jeon I, Song B, Lee J-M, Bae E-A, Seo H, Kang T-S, Kim B-S, et al. GITR Agonism Triggers Antitumor Immune Responses through IL21-Expressing Follicular Helper T Cells. Cancer Immunol Res. 2020;8(5):698–709. doi: 10.1158/2326-6066.CIR-19-0748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.