

Abstract
The tumour suppressor TP53 is a master regulator of several cellular processes that collectively suppress tumorigenesis. The TP53 gene is mutated in ~50% of human cancers and these defects usually confer poor responses to therapy. The TP53 protein functions as a homo-tetrameric transcription factor, directly regulating the expression of ~500 target genes, some of them involved in cell death, cell cycling, cell senescence, DNA repair and metabolism. Originally, it was thought that the induction of apoptotic cell death was the principal mechanism by which TP53 prevents the development of tumours. However, gene targeted mice lacking the critical effectors of TP53-induced apoptosis (PUMA and NOXA) do not spontaneously develop tumours. Indeed, even mice lacking the critical mediators for TP53-induced apoptosis, G1/S cell cycle arrest and cell senescence, namely PUMA, NOXA and p21, do not spontaneously develop tumours. This suggests that TP53 must activate additional cellular responses to mediate tumour suppression. In this review, we will discuss the processes by which TP53 regulates cell death, cell cycling/cell senescence, DNA damage repair and metabolic adaptation, and place this in context of current understanding of TP53-mediated tumour suppression.
Subject terms: Tumour-suppressor proteins, Cancer
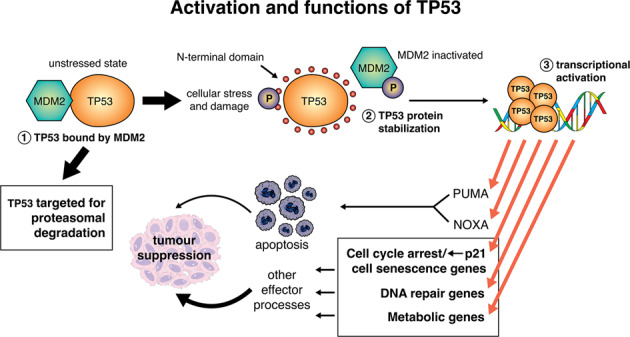
Activation of TP53 can stimulate diverse cellular responses. In unstressed cells, the TP53 protein is bound and ubiquitinated by its negative regulator, the E3 ubiquitin ligase MDM2 (called HDM2 in humans). This targets the TP53 protein for ubiquitin-dependent proteasomal degradation. When cells are exposed to stress, such as DNA damage, MDM2 is phosphorylated and is then no longer able to ubiquitinate TP53, allowing for TP53 stabilisation and functional activation. Of note, TP53 can be phosphorylated at the N-terminus, and this also interferes with MDM2 binding and its ability to ubiquitinate TP53, and thereby prime it for proteasomal degradation. Once activated, TP53 can transcriptionally induce ~500 direct target genes, with some of them known to be critical for the induction of diverse cellular responses, including apoptotic cell death via induction of PUMA and NOXA, as well as genes involved in cell cycle arrest and cell senescence, (e.g., p21), DNA repair (e.g., MLH1, MSH2) and metabolism (e.g., TIGAR), to implement tumour suppression.
Facts
TP53 is critical for tumour suppression.
TP53 functions as a transcription factor that can directly regulate the expression of approximately 500 target genes and indirectly many more.
Some of the direct transcriptional targets of TP53 encode proteins that are critical for TP53-induced apoptotic cell death, cell cycle arrest, cell senescence, DNA repair or adaptation of cellular metabolism.
The absence of TP53 causes spontaneous tumour development in 100% of mice, but the combined absence of Puma, Noxa and p21, the direct TP53 target genes that are critical for TP53-induced apoptosis and cell cycle arrest, does not cause spontaneous tumour development in mice.
Open Questions
Which of the cellular processes activated by TP53 are critical for tumour suppression in which cell types, and in the context of which oncogenic drivers?
Which combinations of TP53-activated processes must be defective for spontaneous tumour development to occur?
Are there currently unrecognised cellular responses activated by TP53 that are critical for tumour suppression?
The tumour suppressor TP53
The TP53 gene encodes a homo-tetrameric tumour suppressor protein, known as TP53 in humans and TRP53 in mice (we use TP53 throughout this text when referring in the same sentence to both the human and mouse protein), which acts as a transcription factor to regulate several cellular responses that cooperate to prevent tumorigenesis [1]. The TP53 protein contains an N-terminal transactivation and Mouse Double Minute 2 (MDM2; called HDM2 in humans) binding domain, a centrally located sequence-specific DNA binding domain, a tetramerisation domain and a C-terminal DNA binding regulatory domain [2, 3]. Diverse stress conditions lead to the activation of TP53 via several post-translational processes [3]. In unstressed cells, the E3 ubiquitin ligase MDM2 causes ubiquitination of TP53, targeting it for proteasomal degradation, thereby keeping TP53 protein levels and activity low [4, 5]. MDM4 (also called MDMX) can bind to the same region as MDM2 at the N-terminus of TP53 and thereby represses TP53 function. Unlike MDM2 however, MDM4 does not ubiquitinate TP53 and thereby prime it for proteasomal degradation [6]. The absence of MDM2 or MDM4 causes aberrant TP53-mediated apoptosis and embryonic lethality, which is prevented by the concomitant loss of TP53 [7]. This shows that TP53 activity is stringently controlled by MDM2 and MDM4 during embryonic development [8]. Experiments with mice in which Mdm2 was deleted in select cell types post-natally revealed that MDM2 must also have functions in addition to regulating TP53, because certain defects were not prevented by concomitant loss of TP53 [9].
DNA damage, nutrient deprivation and many other stressors stimulate signalling pathways that lead to post-transcriptional modifications (e.g., phosphorylation, acetylation) in TP53 and MDM2 [10, 11]. These processes converge to prevent MDM2 from binding and ubiquitinating TP53, thereby causing an increase in TP53 protein levels and activity. Following such activation, TP53 can bind to specific DNA sequences in the promoters of ~500 direct target genes, mostly promoting an increase in their expression (Graphical Abstract) [3, 12].
Approximately 50% of human cancers have mutations in the TP53 gene, most of which are point mutations in the DNA binding domain [1]. The mutant TP53 proteins have a reduced capacity to bind to specific DNA sequences in direct TP53 target genes (loss-of-function effect). This impairs normal TP53-regulated cellular responses, leading to immortalisation and neoplastic transformation of cells [13].
How does TP53 prevent tumour development?
The TP53 protein regulates several cellular processes that contribute to preventing tumorigenesis. The cyclin dependent kinase (CDK) inhibitor, p21Waf1/Cip1, encoded by a direct TP53 target gene, is critical for cell cycle arrest [14, 15]. Cell cycle arrest allows cells with DNA lesions to halt proliferation, allowing them to repair the damaged DNA, and thereby preventing the acquisition of oncogenic mutations that can drive tumorigenesis [15]. TP53 plays a direct role in DNA repair through transcriptional activation of several genes involved in various DNA repair pathways [16], including base excision repair [17], nucleotide excision repair [18], mismatch repair [19] and homologous recombination [17–20].
TP53 and programmed cell death
Apoptosis is a genetically programmed process for cell killing that removes cells that are no longer needed, damaged or infected. By removing cells with potentially oncogenic lesions that are at early stages of neoplastic transformation, apoptosis contributes to the suppression of tumour development [3]. TP53 controls the expression of some of the key initiators of apoptotic cell death. There are two distinct but ultimately converging pathways to apoptosis: the BCL-2 regulated (aka mitochondrial or intrinsic) pathway [21] and the death receptor induced (aka extrinsic) pathway [22]. The BCL-2-regulated pathway is controlled by the BCL-2 protein family, which contains three subgroups: the pro-survival proteins (BCL-2, MCL-1, BCL-XL, BCL-W, A1/BFL1), the pro-apoptotic BH3-only proteins (BIM, PUMA, BID, BMF, BIK, BAD, HRK, NOXA) and the effectors of apoptosis (BAX, BAK, BOK) [23–25]. Apoptosis can be induced by diverse stresses, such as nutrient deprivation or DNA damage, and is initiated by the transcriptional and/or post-transcriptional upregulation of the pro-apoptotic BH3-only proteins, which are critical for the initiation of apoptosis [26, 27]. The pro-survival BCL-2 proteins are bound and inhibited by the BH3-only proteins [23] and this unleashes the cell death effectors BAX and BAK that in healthy cells are restrained by the pro-survival BCL-2 proteins (Fig. 1). Upon activation, BAX and BAK cause mitochondrial outer membrane permeabilisation (MOMP) [28], which constitutes the point-of-no-return in apoptosis signalling (Fig. 1). The apoptosome (involving APAF-1) forms and the caspase cascade becomes activated, dismantling the cells (Fig. 1). Upregulation of the genes for BAX and APAF-1 by TP53 may enhance apoptosis, but the TP53-mediated upregulation of these genes is not essential for apoptosis, at least in hematopoietic cells [29, 30].
Fig. 1. Induction of the BCL-2-regulated apoptotic pathway by TP53.
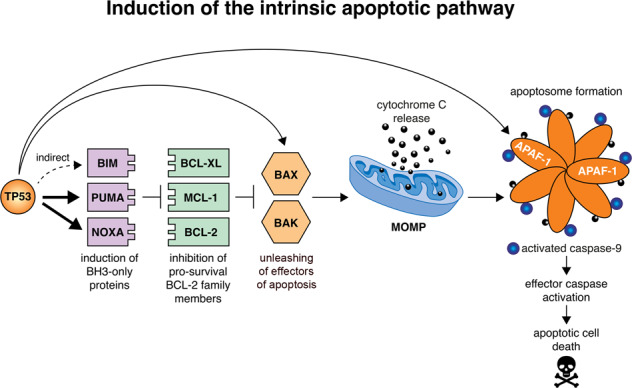
This model depicts the mechanism by which activated TP53 induces the BCL-2-regulated apoptotic pathway. When TP53 is activated, it directly transcriptionally activates the genes encoding the pro-apoptotic BH3-only proteins, PUMA and NOXA, and indirectly induces expression of the gene encoding the BH3-only protein BIM. The pro-survival BCL-2 family members (e.g., BCL-XL, MCL-1 and BCL-2) are repressed by binding of the pro-apoptotic BH3-only proteins. This permits the activation of the apoptosis effectors, BAX and BAK, which in healthy cells are kept in check by binding to the pro-survival BCL-2 proteins. Activated BAX and BAK can oligomerise at the mitochondrial outer membrane, forming pores, thereby causing mitochondrial outer membrane permeabilisation (MOMP) which releases cytochrome c and other apoptogenic factors. Following MOMP, the apoptosome (composed of the adaptor protein APAF-1, dATP, pro-caspase-9 and cytochrome c) forms, which leads to the activation of caspase-9 that triggers activation of downstream effector caspases (e.g., caspases -3, -6 and -7). This caspase cascade causes proteolysis of hundreds of cellular proteins to orchestrate the ordered dismantling of the dying cells. Thick arrows indicate the direct upregulation of target genes by TP53. The thin arrow represents the indirect activation of BIM by TP53. Of note, in the absence of TP53, BAX and APAF-1 are still expressed at levels that are sufficient for effective induction of apoptosis, for example after treatment of lymphoid cells with glucocorticoids, which does not require TP53 for apoptosis induction. Upregulation of BAX and APAF-1 by TP53 may serve to enhance the efficiency of apoptosis but the TP53-mediated upregulation of their genes is not essential for induction of apoptosis, at least in haematopoietic cells.
What is the evidence that TP53 prevents tumour development by inducing apoptosis?
Several early studies linked TP53 to the activation of apoptosis and it was soon proposed that this is critical for TP53-mediated tumour suppression [13, 31, 32]. Apoptosis triggered by enforced TP53 expression was prevented by over-expression of anti-apoptotic BCL-2 [29, 30]. This led to the search for TP53-activated initiators of apoptosis. Puma/Bbc3 and Noxa/Pmaip1, two genes that are directly transcriptionally upregulated by TP53, are critical for the initiation of apoptosis [33, 34]. Notably, lymphoid cells from PUMA/NOXAdouble knock-out mice are as resistant to apoptosis induced by the MDM2 inhibitor nutlin-3a, γ-irradiation or other agents that activate TP53, both in vitro and in vivo, as the corresponding cells from Trp53 knockout mice [35]. This demonstrates that transcriptional induction of these two BH3-only proteins accounts for all TP53-induced apoptosis, at least in these cell types [36, 37].
Interestingly, in malignant MYC-driven lymphoma cells, only the combined loss of the three BH3-only proteins PUMA, NOXA and BIM (the latter is probably indirectly upregulated by TP53) provided similar protection from DNA damage inducing chemotherapeutics as did loss of TP53 itself [38, 39]. This indicates that TP53-mediated apoptosis can be more complex in malignant cells than in non-transformed cells.
There is evidence that induction of apoptosis is critical for TP53-mediated tumour suppression in certain settings, but there is also evidence that in other settings, this is not essential. In Eµ-Myc transgenic mice, the over-expression of MYC causes a pre-leukaemic expansion of B cell precursors (pre-B cells), which progresses to malignant clonal pre-B/B lymphoma [40–42]. Approximately 20–30% of these lymphomas are selected for mutations and/or loss of TRP53 [43], demonstrating that TRP53 plays a critical role in restraining MYC-driven lymphomagenesis. In Eµ-Myc mice, loss of PUMA causes a further expansion of pre-leukaemic B lymphoid cells and substantially accelerates lymphoma development [43]. Another study showed that ~75% of lymphomas that arise in Eµ-Myc mice had selected against the expression of PUMA [44]. Interestingly, PUMA expression is not detected in ~40% of human Burkitt lymphomas [44], which are also driven by deregulated c-MYC expression. Moreover, in Burkitt lymphomas, the BIM gene is often epigenetically silenced by hyper-methylation of the promoter region and deacetylation of histone 3 by histone deacetylases (HDACs), and this is thought to contribute to chemotherapeutic resistance [45]. In mantle cell lymphoma, silencing of NOXA using RNA interference resulted in significant rescue from apoptosis that was induced by chemotherapeutic drugs [46]. Collectively, these findings reveal that PUMA and NOXA, as well as BIM, contribute to TP53-dependent killing of malignant cells induced by DNA damage inducing anti-cancer agents.
Trp53−/− mice spontaneously develop tumours, on a C57BL/6 background mostly thymic T cell lymphomas, at a 100% incidence within ~270 days [47]. If TP53-induced apoptosis was essential to prevent spontaneous tumour development, mice lacking PUMA plus NOXA, the essential mediators of TP53-induced apoptosis, should also develop tumours. Remarkably however, neither Puma−/−, Noxa−/− nor Puma−/−;Noxa−/− mice and not even Puma−/−;Noxa−/−;p21−/− mice (whose cells also are defective in TP53-induced G1/S boundary cell cycle arrest and senescence) developed tumours [48]. Similarly, mice with certain mutations in TRP53 that impair transcriptional activation of p21, Puma and Noxa, but retain TRP53-induction of certain other target genes, are also not prone to spontaneous tumour development [48–50]. These findings demonstrate that the combination of TRP53-mediated induction of apoptosis via PUMA and NOXA, plus TRP53-induced cell cycle arrest at the G1/S boundary and cell senescence via p21 are dispensable for TRP53-mediated suppression of spontaneous tumour development [48]. Thus, other TRP53 target gene driven cellular responses must also be required for TRP53 to suppress spontaneous tumour development.
In addition to regulating apoptosis for tumour suppression, TP53/TRP53 has also been reported to suppress tumorigenesis by inducing ferroptosis, a lytic form of programmed cell death (Table 2) [51, 52]. Acetylation of TRP53 at specific amino acid residues was reported to be critical for this [51, 53]. It must, however, also be noted that another group provided evidence that TP53/TRP53 actually inhibits ferroptosis [54], which indicates that ferroptosis may not contribute to TP53/TRP53-mediated tumour suppression.
Table 2.
TP53-regulated metabolic genes and their roles in glucose metabolism, glycolysis, oxidative phosphorylation, autophagy, ferroptosis and other metabolic processes.
| Pathway | GENE | TP53 regulation | Result of TP53 regulation | Reference |
|---|---|---|---|---|
| Glucose metabolism and glycolysis | TIGAR | Up | Repression of pentose phosphate pathway & glycolysis | [109] |
| RRAD | Up | Repression of glycolysis under hypoxic conditions | [115] | |
| Oxidative phosphorylation | sCO2 | Up | Upregulation of cytochrome c oxidase → Upregulation of oxidative phosphorylation | [154] |
| Autophagy | Tsc1 | Up | Repression of RHEB → Inhibition of mTOR → Induction of autophagy | [127] |
| Tsc2 | Up | Repression of RHEB → Inhibition of mTOR → Induction of autophagy | [127] | |
| DRAM | Up | Induction of autophagy | [131] | |
| ULK1 | Up | Induction of autophagy | [132] | |
| Atg7 | Up | Induction of autophagy | [135] | |
| Ferroptosis | GLS2 | Up | Induction of ferroptosis | [51] |
| Other | Zmat3 | Up | Alternative RNA splicing → Modulation of exon inclusion (e.g., in the genes encoding the TP53/TRP53 inhibitors MDM2 and MDM4) | [149] |
What is the evidence that TP53 prevents tumour development by inducing cell cycle arrest and cellular senescence?
Senescence is a process by which cells undergo irreversible cell cycle arrest. Telomere erosion induces a DNA damage response that causes TP53-induced cell cycle arrest, in part through the induction of p21, thereby acting as a barrier to cell immortalisation and tumorigenesis (Fig. 2) [55, 56]. Cell senescence can also be induced by cellular aging, DNA lesions and the expression of certain oncogenes, such as mutant RAS (Fig. 2) [57, 58]. The activation of TP53 in response to such stress can cause cell cycle arrest at the G1/S or G2/M boundaries of the cell cycle, from where cells can enter a senescent state [59, 60]. Induction of cellular senescence is in part due to the ability of TP53 to directly transcriptionally upregulate the gene for p21, although other effectors of cell senescence are likely to also play critical roles. During cellular senescence, the retinoblastoma (RB) tumour suppressor protein is activated and represses the transcription of genes encoding proteins that are critical for cell cycle progression (Fig. 2).
Fig. 2. Induction of cell senescence by TP53.
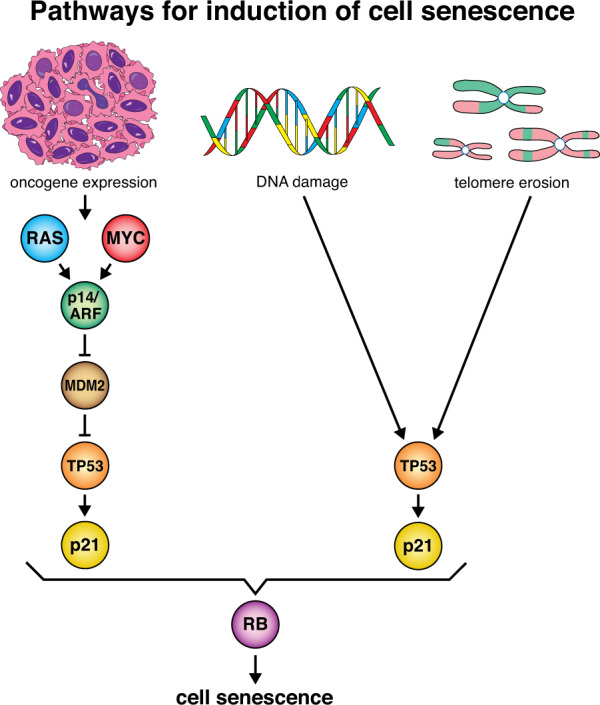
A variety of cellular stresses, such as oxidative damage, DNA damage, telomere shortening and downstream effects of oncogene activation, such as replication stress, can induce cellular senescence via the activation of TP53. This is in part due to the ability of TP53 to directly transcriptionally upregulate the gene for p21, although other effectors of cell senescence are likely to also play critical roles. During cellular senescence, the retinoblastoma (RB) tumour suppressor protein is activated and represses the transcription of genes encoding proteins that are critical for cell cycle progression.
Cellular senescence can occur in either a DNA damage-dependent or -independent manner (Fig. 2) [55]. Mice lacking DNA repair genes, such as DNA ligase IV and Brca1 exhibit premature aging, and mouse embryonic fibroblasts (MEFs) from these animals undergo senescence [61, 62]. The inactivation of TRP53 prevented these senescent phenotypes [61, 62]. These findings reveal a critical role for TP53 in DNA-damage induced cellular senescence.
TP53 also plays a role in oncogene-induced cell senescence (OIS) [58, 63]. In primary mouse and human cells with wt TP53, the expression of oncogenic mutant RAS rapidly caused permanent senescence-like G1 cell cycle arrest [64]. Loss of Trp53 prevented mutant RAS induced cell senescence in mouse models, thereby promoting tumorigenesis [64, 65]. Moreover, c-MYC over-expression can cause OIS through the transcriptional induction of p14ARF, which in turn leads to the activation of TP53 through the inhibition of MDM2 [66, 67] (Fig. 2).
So, what is the evidence that TP53 suppresses tumorigenesis through the induction of cell cycle arrest and cell senescence? MEFs lacking the direct TRP53 target gene p21 exhibit a profound defect in G1 cell cycle arrest in response to DNA damage, albeit to a lesser extent than MEFs lacking TRP53 [68]. This reveals that TP53-mediated induction of p21 is critical for this process, but that other direct or indirect TP53 target genes may also be involved. BTG2, another TP53-regulated gene, encodes an anti-proliferative protein that halts the cell cycle at the G1/S boundary in response to DNA damage [69]. Interestingly, knockdown of BTG2 in breast cancer cells resulted in increased tumour growth and metastases in vivo [70, 71]. Moreover, GADD45a, which is also encoded by a direct TP53 target gene, can cause the displacement of PCNA from the Cyclin D1 complex to inhibit DNA replication and can also reduce the activity of CDK1 by preventing its association with Cyclin B1 [72, 73]. Loss of Gadd45a resulted in failure of proliferating mouse lymphocytes to undergo cell cycle arrest at the G2/M-phase following treatment with DNA damage inducing agents [74].
A study by Liu et al., (2004) [75] showed that cells from mice expressing the TRP53 mutant, R172P, were unable to initiate apoptotic cell death, but were still able to undergo cell cycle arrest [75]. Homozygous R172P Trp53 mutant mice escaped early onset of thymic lymphomas, in contrast to Trp53−/− mice. The retention of chromosomal stability was therefore suggested to be crucial for the suppression of early onset tumorigenesis by allowing cells to undergo cell cycle arrest [75].
Elegant mouse models were developed that allow development of tumours driven by the absence of TRP53 plus tissue restricted expression of an oncogene, and then later on enabled wt TRP53 expression to be induced in the malignant cells. In the case of c-MYC-driven lymphomas, the expression of wt TRP53 caused tumour regression through induction of apoptosis [76], but in mutant RAS driven liver cancers, cessation of tumour expansion was associated with induction of cell senescence [77, 78]. These findings indicate that therapeutic activation of wt TP53 in malignant cells can cause tumour regression through induction of apoptosis or cell senescence, depending on cell type. This does not, however, mean that induction of cell cycle arrest and/or cell senescence is indispensable for TP53-mediated suppression of tumorigenesis. In fact, loss of p21, which is critical for TP53-induced G1/S boundary cell cycle arrest and cell senescence, does not cause spontaneous tumour development, and loss of p21 does not prominently accelerate oncogene-induced tumorigenesis [79].
What is the evidence that TP53 prevents tumour development by coordinating DNA repair?
DNA repair is a crucial cellular process, whereby various genetically programmed pathways are activated to maintain genomic integrity after exposure of cells to DNA-damaging agents [80], thereby preventing tumorigenesis (Fig. 3). Nucleotide excision repair (NER) removes helix-distorting lesions that are commonly caused by UV irradiation [81]. Base excision repair (BER) eliminates oxidised or alkylated bases that are modified by reactive oxygen species (ROS) (Fig. 3) [82]. DNA double stranded breaks (DSBs) that are induced by ionising radiation (IR) are repaired through homologous recombination (HR) or non-homologous end joining (NHEJ), and mismatch repair (MMR) scans DNA strands for nucleotides that have been erroneously inserted during the DNA replication process (Fig. 3) [83]. TP53 is thought to play an important role in coordinating DNA damage repair processes by halting the cell cycle, thereby allowing time for DNA repair [83]. Of note, TP53 can also directly transcriptionally activate certain DNA damage repair genes, thereby directly impacting the activity of DNA repair pathways [83, 84].
Fig. 3. Regulation of DNA damage repair by TP53.

Activated TP53 can transcriptionally upregulate an array of direct target genes involved in a range of DNA damage repair pathways, including base excision repair, non-homologous end joining, homologous recombination, nucleotide excision repair and mismatch repair. Thick arrows indicate the type of DNA damage repair pathway that is needed for the repair of a specific type of DNA lesion. The genes to the side of the arrows are genes that are upregulated by TP53 to orchestrate that particular damage repair pathway.
Several studies provided evidence that coordination of DNA repair is critical for TP53-mediated tumour suppression. The XPC, GADD45a, PCNA and DDB2 genes from the NER pathway are all direct transcriptional targets of TP53 [85–87]. Defects in some of these genes can promote tumorigenesis [74, 88]. Loss of one Trp53 allele in XPC-deficient mice led to the formation of more aggressive UV-radiation induced skin tumours, classified as higher-grade squamous cell carcinomas, compared to UV-irradiated XPC-knockout mice that were wt Trp53 [89] (Table 1). These findings suggest that XPC is critical for the suppression of tumours induced by DNA damage and that TP53-regulated processes independent of the induction of XPC must also contribute to the suppression of UV-induced skin cancer development [89]. GADD45a, encoded by a TP53 target gene, plays a role in the NER pathway [74]. Studies using mouse models revealed that loss of Gadd45a causes genomic instability, driving tumorigenesis (Table 1) [90].
Table 1.
DNA repair genes that are directly regulated by TP53 and whose knock-out can promote spontaneous, UV-B- or γ-radiation induced tumour development in mice.
| Gene | DNA damage repair pathway | Gene knockout | Treatment | Outcome | Reference |
|---|---|---|---|---|---|
| Xpc | NER | −/− | UV-B Irradiation | Skin squamous cell carcinoma | [89] |
| Gadd45a | NER | −/− | Ionising Radiation | Lymphoma | [90] |
| Ddb2 | NER | −/− | UV-B Irradiation | Skin carcinoma | [96] |
| Mlh1 | MMR | −/− | – | Lymphoma and gastro-intestinal tumours | [100] |
| Msh2 | MMR | −/− | – | Colon carcinoma and lymphoid tumours | [97, 99] |
TP53 directly regulates additional genes involved in NER, such as PCNA [91]. PCNA, which was shown to interact with GADD45a, is involved in both DNA replication and DNA repair, with roles in both NER and MMR [92, 93]. Moreover, DDB2, which is transcriptionally regulated by TP53, binds to UV-induced DNA lesions, suggesting a role in NER [94, 95]. Accordingly, DDB2-deficient mice show reduced removal of cyclobutane pyrimidine dimers in skin cells and are more susceptible to UV-induced carcinogenesis compared to wt mice (Table 1) [96].
TP53 has also been reported to directly regulate the expression of the genes for MSH2 and MLH1, which play critical roles in MMR [20, 97, 98]. Notably, mutations in MSH2 are a cause of hereditary non-polyposis colorectal cancer (HNPCC) in humans. MSH2-deficient mice spontaneously develop lymphomas that exhibit microsatellite instability [97] and are prone to oncogene-induced development of colonic carcinomas (Table 1) [99]. Mlh1+/− as well as Mlh1−/− mice are also predisposed to developing lymphomas and tumours in the gastrointestinal tract [100]. Moreover, knockdown of Mlh1, Msh2 and certain other TP53-regulated DNA repair genes accelerated c-MYC-driven lymphomagenesis to a similar extent as loss of TRP53 itself [79]. Collectively, these findings indicate that TP53-regulated expression of genes involved in diverse DNA repair processes are critical for TP53-mediated tumour suppression.
What is the evidence that TP53 plays a role in the coordination of angiogenesis?
Angiogenesis is a process where factors released from cells signal for the formation of new blood vessels via the migration, growth and differentiation of endothelial cells [101]. Under normal conditions, the signalling for blood vessel formation is balanced through stimulation and inhibition to produce vessel growth only when necessary, such as during wound healing. Aberrant angiogenesis can provide solid tumours with nutrients and oxygen, thereby enabling their expansion and metastasis [101]. TP53 plays a role in the control of angiogenesis by inhibiting the production of pro-angiogenic molecules and by increasing the production of inhibitors of angiogenesis [102]. Interestingly, tumours carrying mutations in TP53 are often more vascularised compared to tumours expressing wt TP53.
What is the evidence that TP53 prevents tumour development by regulating metabolism or other cellular responses?
To maintain metabolic homoeostasis in cells exposed to stress, TP53 regulates diverse metabolic pathways, and this is thought to help cells adapt to stress conditions [103]. The production of energy and substrates from metabolic pathways enables synthesis of macro-molecules to support normal cellular life and its proper regulation is thought to contribute to tumour suppression [104]. The synthesis of proteins and nucleic acids that are needed to support cell proliferation relies on nutrient availability. Nutrient-poor conditions trigger responses to restrict cell growth and proliferation. These responses enable the breakdown of macro-molecules and even entire organelles to allow energy production for cell survival [104]. Notably, certain cancer cells are able to acquire nutrients from nutrient-poor environments, thereby enabling their expansion while simultaneously impairing the function of neighbouring non-transformed cells, including T lymphocytes that might otherwise attack the malignant cells [103, 105].
TP53 can promote mitochondrial oxidative phosphorylation, a metabolic process that produces adenosine triphosphate (ATP), using energy that is released by nutrients in the tricarboxylic acid cycle (TCA). TP53 reportedly directly regulates cytochrome c oxidase 2 (sCO2) gene expression, thereby enhancing oxidative phosphorylation (Table 2) [103, 106]. The gene for the TP53-induced glycolysis and apoptosis regulator (TIGAR) is also directly regulated by TP53. TIGAR decreases the amount of fructose-2,6-bisphosphate and blocks the glycolytic pathway, thereby pushing cells towards oxidative phosphorylation (Table 2) [107]. Reactive oxygen species (ROS) are toxic by-products of oxidative phosphorylation, which play major roles in tissue damage and tumorigenesis [108]. To combat oxidative stress within cells, the induction of TIGAR by TP53 enhances the pentose phosphate pathway, generating glutathione (GSH) that protects cells against ROS and the resulting damage to their genome [109].
Human cancers often experience hypoxia, indicating that the adaptation of malignant cells to hypoxic environments is crucial for sustaining tumour growth [110]. When oxygen levels are low, ATP is produced through glycolysis rather than through oxidative phosphorylation; therefore, enhanced glycolysis is a characteristic metabolic feature of many cancer cells [104, 111]. Because glycolysis produces 2 ATP molecules in contrast to the 36 ATP molecules produced by oxidative phosphorylation, cancer cells often exhibit high uptake and use of glucose compared to non-transformed cells, to facilitate the necessary rate of glycolysis [112]. Defects in TP53 cause a reduction in oxidative phosphorylation and enhanced glycolysis, and this was proposed to drive the development of metastatic cancer cells by engendering them with the ability to survive in hypoxic environments [113, 114]. Collectively, these findings suggest that TP53 helps to maintain cellular oxidative phosphorylation. When TP53 function is lost, cells are pushed toward the aerobic glycolytic pathway for energy production, which is thought to promote the survival and metastasis of malignant cells.
RAS Related Glycolysis Inhibitor and Calcium Channel Regulator (RRAD) is encoded by a direct TP53 target gene and plays a role in the control of glycolysis [115]. Hypoxia-stimulated glycolysis is repressed by TP53 in cancer cells through the upregulation of RRAD, which inhibits the translocation of the glucose transporter, GLUT1, to the plasma membrane (Table 2) [115]. These findings suggest that TP53 plays an important role in the control of cellular glucose metabolism and that when TP53 is absent or mutated, cells can adapt their metabolism by switching to aerobic glycolysis. However, to our knowledge, there is no evidence that the loss of TP53 target genes that function in glucose metabolism can cause tumour development. In mice, the loss of TIGAR actually slowed tumour development in both lymphoma and intestinal cancer models [116, 117]. Moreover, RRAD-deficient mice are also not tumour-prone [118].
Autophagy is a process for the degradation of macro-molecules and even entire organelles, that allows cells to gain energy and metabolites to survive when subject to nutrient deprivation. During autophagy, intracellular components are sequestered into double-membrane vesicles called autophagosomes [119]. When autophagosomes fuse with lysosomes, hydrolytic enzymes degrade the contents, which are then released into the cytoplasm to be recycled in biosynthetic pathways [120]. There is evidence that autophagy can be controlled by TP53-regulated genes. Interestingly, autophagy can also suppress TP53 activity in a negative feedback process [121]. When AMP-activated protein kinase (AMPK) is upregulated by TP53, it promotes autophagy through the transcriptional activation of SESTRIN 1 and 2. Of note, AMPK can also upregulate TP53 via phosphorylation of MDMX [122]. SESTRIN 1 and 2 activation causes activation of AMPK and subsequent phosphorylation of the tuberous sclerosis proteins TSC1 (hamartin) and TSC2 (tuberin), which form a heterodimeric tumour suppressor protein complex. This complex is an important component of the mammalian target of rapamycin (mTOR) signalling pathway, which receives signal input from nutrients, growth factors and cellular energy levels, and functions as a master regulator that sets the balance between anabolism and catabolism (Table 2) [123, 124].
The TSC1-TSC2 complex represses the activity of RHEB, resulting in the inhibition of mTOR activity, which promotes metabolic processes [125]. Notably, defects in TSC1 or TSC2 can promote tumorigenesis [126]. Eμ-Myc mice with loss of only one allele of Tsc2 developed lymphomas significantly faster than control (Tsc2+/+) Eμ-Myc mice [126]. These lymphomas had been selected for loss of the remaining wt Tsc2 allele, but none had acquired mutations in Trp53, whereas 20–30% of lymphomas in control Eμ-Myc mice selected for mutations in Trp53 [126]. Moreover, mice with loss of one allele of Tsc1 were found to develop renal tumours and hepatic hemangiomas [127]. These findings suggest that loss of TSC1 or TSC2 results in sustained upregulation of mTOR and its downstream signalling pathways. This is thought to promote tumour development through the upregulation of metabolic processes, and possibly also through changes in autophagic flux [126, 127]. This TSC1/TSC2-regulated process may play a critical role in TP53-mediated suppression of tumorigenesis.
It was also reported that loss of TP53 increases autophagy, as evidenced by the accumulation of autophagosomes and autolysosomes and the aggregation of LC3 in several mammalian cell lines in vitro, as well as in diverse mouse tissues in vivo [128]. This study also showed that cells lacking TP53 had a higher probability of surviving under starvation conditions, and that this survival advantage was decreased when autophagy-related genes (ATG) were deleted [128]. The Damage-Regulated Autophagy Modulator (DRAM) is encoded by a direct TP53 target gene. DRAM is a lysosomal protein that promotes autophagy through the accumulation of autophagosomes (Table 2) [129, 130]. Knockdown of DRAM reduced autophagy and triggered lysosomal membrane permeabilisation and cell death [131]. The UNC-51 Like Autophagy Activating Kinase 1 (ULK1) is also encoded by a direct TP53 target gene [132]. ULK1 promotes autophagy in response to DNA damage, enhancing the fusion of autophagosomes and lysosomes [132, 133]. ULK1 has been implicated in organelle clearance during the maturation of immature red blood cells and in autophagy of mitochondria (mitophagy) [134]. Autophagy Related 7 (ATG7) is another direct TP53 target gene involved in autophagy (Table 2) [135].
The role of autophagy in cancer is complex, with evidence for both tumour-suppressive, as well as pro-tumorigenic roles. In ~50% of human pancreatic ductal adeno-carcinomas (PDACs), TP53 is mutated [136]. PDAC is usually preceded by pre-cancerous pancreatic intraepithelial neoplasia (PanIN). Mutant KRAS driven PanINs from mice lacking ATG7 did not progress to PDAC. However, mice expressing mutant KRAS and lacking both ATG7 and TP53 formed tumours despite defects in autophagy [137]. Tumours lacking TP53 had fewer autophagosomes in comparison to tumours expressing wt TP53, and cell lines derived from TP53-deficient tumours had a decrease in autophagic flux. Furthermore, KrasG12D;Trp53−/−;Atg7−/− mice developed PDACs more rapidly compared to KrasG12D;Trp53−/−;Atg7+/+ mice. This suggests that ATG7-dependent autophagy suppresses tumour development in this setting [137].
Ionising radiation (IR) is a common treatment for cancer and is known to induce autophagy in malignant cells [138, 139]. Although TP53-mediated autophagy was shown to promote tumour cell killing [140, 141], it can also contribute to protection against radiotherapy [142, 143]. TP53-mediated autophagic cell death was observed in MCF-7 breast cancer cells following γ-irradiation [144]. Conversely, TP53-mediated autophagy was reported to protect cells against γ-radiation by cleaning up damaged organelles and recycling amino acids [145]. Accordingly, HN30 head and neck cancer cells expressing wt TP53 were sensitised to γ-radiation by inhibition of autophagy, whereas HN30 cells with knockdown of TP53 were not sensitised to γ-radiation after treatment with inhibitors of autophagy [143]. Collectively, these findings show that autophagy can be induced by γ-radiation through a TP53-dependent process and that this can protect both malignant as well as non-transformed cells from death. Thus, inhibitors of autophagy may enhance the response to radiotherapy in wt TP53 expressing cancer cells [146].
It has also been suggested that malignant cells may exploit autophagy to promote their survival when subject to stress, and that this can enhance tumour growth. TP53 was shown to promote long-term survival of HCT116 colon cancer cells under nutrient deprivation [147]. During chronic starvation, the levels of LC3, which is critical for autophagy, were decreased in HCT116 cells with wt TP53, but not in their TP53-deficient derivatives [147]. Following replenishment of nutrients, rapid outgrowth of wt TP53 cells, but not of TP53-deficient HCT116 cells was observed [147]. This indicates that TP53-dependent regulation of autophagy allows cells to deal better with starvation than TP53-deficient cells. As a result of not halting cell cycling, the latter may sustain metabolic catastrophe and therefore die. These pro-survival effects of wt TP53 under starvation indicates that regulation of autophagy by TP53 can be pro-tumorigenic, and this may be a driver for retaining wt TP53 in certain cancers.
Can we learn more about the cellular processes important for TP53-mediated tumour suppression through the functional identification of critical TP53 target genes?
Both in vitro and in vivo genetic screens have identified several TP53/TRP53 target genes as critical for tumour suppression, including for example Zmat3 [79, 148–150]. ZMAT3 was shown to regulate gene splicing (inclusion or exclusion of certain exons) and to thereby impact the expression of isoforms of genes that impact tumorigenesis, such as the genes for the TP53/TRP53 inhibitors, MDM2 and MDM4 (Table 2) [149]. Moreover, silencing of ZMAT3 expression was shown to cause inclusion of certain exons of CD44, which encodes a protein involved in cell adhesion [151]. Notably, silencing of ZMAT3 only promoted tumorigenesis in combination with the absence of p21 and Puma [79], indicating that it regulates a cellular process that can provide back-up for tumour suppression when TP53/TRP53-induced cell cycle arrest and apoptotic cell death are disabled. With improved screening, we expect that more genes important for TP53 tumour suppression will be identified, and further research will be required to identify which cellular processes these genes influence.
Summary and perspectives
In conclusion, the tumour suppressor TP53 plays a key role in the suppression of tumorigenesis through the coordinated regulation of several different cellular processes, including apoptotic cell death, cell senescence, cell cycling, DNA repair and metabolism. Remarkably, combined loss of TP53-induced apoptosis and loss of TP53-induced G1/S boundary cell cycle arrest and cell senescence does not lead to spontaneous tumour development in mice [48, 49, 152]. These findings support the notion that several TP53 controlled cellular processes must be disabled simultaneously to recapitulate the spontaneous tumour development seen in TRP53-deficient mice. It is conceivable that certain TP53-regulated cellular processes might be more important for tumour suppression than induction of apoptosis, cell cycle arrest and cellular senescence. In vivo shRNA library screening and validation of hits using CRISPR/Cas9 gene editing technology demonstrated that the loss of several TP53 target genes involved in DNA repair processes can cause spontaneous tumour development and accelerate c-MYC oncogene driven lymphomagenesis [79]. Future work could focus on examining the overall impact of combinations of loss of TP53-regulated DNA repair genes, such as MLH1 or MSH2, plus TP53 target genes that encode regulators of cell cycle arrest, cell senescence, cell death or metabolism.
Secondly, we believe that it is time to go beyond focusing on knocking out entire TP53-regulated genes to investigate their role in the suppression of tumorigenesis, and move to selectively mutating the TP53 binding sites within these target genes of interest (Fig. 4). We consider it a “holy grail” in TP53 research to generate mice carrying defects in TRP53 binding sites in one or several direct TP53 target genes that will result in the development of spontaneous tumours. This would provide unambiguous proof that TP53-mediated control of the expression of these genes is essential for tumour suppression. Given the discussions above, it appears likely that mutation of the TRP53 binding sites in several target genes operating in diverse cellular processes will be required for the mice to spontaneously develop tumours (Fig. 4). It will also be interesting to cross mice with mutations in the TRP53 binding sites in target genes (e.g. Puma) with mice carrying tumour promoting oncogenic drivers. Complete loss of the Puma gene substantially accelerates c-MYC driven lymphoma development [43, 153] and it is tacitly assumed that this is due to the loss of TRP53-mediated induction of Puma expression in cells over-expressing c-MYC. However, it remains possible that TRP53-independent induction of Puma may also be critical for delaying c-MYC driven lymphoma development. If mutations in the TRP53 binding sites in the Puma gene will accelerate c-MYC driven lymphomagenesis to the same extent as complete loss of the Puma gene, this would prove that the tumour suppressive function of PUMA in this context relies solely on its transcriptional regulation by TRP53. Similar studies should be conducted to clarify with greater precision the mechanisms that are critical for the TP53-dependent effects of anti-cancer agents, particularly those that induce DNA damage. Such insight is predicted to spearhead translational work for the development of improved therapies for cancer patients.
Fig. 4. Proposed gene-targeting experiments to determine whether TP53-mediated upregulation of a specific direct TP53 target gene is responsible for tumour suppression.

TP53 acts as a tumour suppressor through transcriptional induction of target genes. Binding of TP53 to TP53 binding sites in the promoter and intronic regions induces upregulation of a TP53 target gene. This allows the protein encoded by this TP53 target gene to be upregulated and exert its function that contributes to TP53-mediated tumour suppression. Mutation of TP53 binding sites in target genes can identify the critical genes important for TP53-mediated tumour suppression. Gene targeting can be used to mutate the TP53 binding site(s) in a gene of interest. In cells derived from such genetically modified mice, the TP53 target gene of interest cannot be upregulated by activated TP53, but it can still be regulated by other transcription factors that bind to other sequences in its promoter or intronic regions. If these genetically modified mice, for example after introducing a potent oncogene, such as c-MYC or mutant RAS, will show accelerated tumour development, this would demonstrate that TP53-mediated upregulation of this gene is critical for tumour suppression. If these genetically modified mice are not tumour prone, this would indicate that transcription factors in addition to TP53 can also induce the expression of this protein at levels sufficient for effective tumour suppression.
Acknowledgements
The authors thank the members of their laboratory and the Blood Cells and Blood Cancer Division for discussions, Catherine McLean for assistance with the preparation of this review and Peter Maltezos for drafting of figures. Work by the authors was supported by fellowships and grants from the Australian National Health and Medical Research Council (NHMRC) (Program Grant GNT1113133 to AS, Research Fellowships GNT1116937 to AS, Project Grants GNT1143105 to AS, Ideas Grants GNT 2002618 and GNT2001201 to GLK), the Leukemia & Lymphoma Society of America (Specialized Center of Research [SCOR] grant no. 7015-18 to AS and GLK), Victorian Cancer Agency (MCRF Fellowship 17028 to GLK), the estate of Anthony (Toni) Redstone OAM (AS and GLK), the Craig Perkins Cancer Research Foundation (GLK), and the Dyson Bequest (GLK). Work in the laboratories of the authors was made possible through Victorian State Government Operational Infrastructure Support (OIS) and Australian Government NHMRC Independent Research Institute Infrastructure Support (IRIIS) Scheme.
Author contributions
AFT, GLK and AS conceived ideas for this review article, planned for its content, drafted figures and wrote the text.
Data availability
This review article does not present any new primary data.
Competing interests
The authors declare no competing interests.
Ethics
This review article does not present new experimental results. Therefore, human and animal ethics statements are not required.
Footnotes
Edited by G. Melino
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Gemma L Kelly, Andreas Strasser.
References
- 1.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–13. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tafvizi A, Huang F, Fersht AR, Mirny LA, van Oijen AM. A single-molecule characterization of p53 search on DNA. Proc Natl Acad Sci. 2011;108:563–8. doi: 10.1073/pnas.1016020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boutelle AM, Attardi LD. p53 and tumor suppression: it takes a network. Trends Cell Biol. 2021;41:298–310. doi: 10.1016/j.tcb.2020.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 5.Shieh S-Y, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–34. doi: 10.1016/S0092-8674(00)80416-X. [DOI] [PubMed] [Google Scholar]
- 6.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–95. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 7.Liu G, Terzian T, Xiong S, Van Pelt C, Audiffred A, Box N, et al. The p53–Mdm2 network in progenitor cell expansion during mouse postnatal development. J Pathol. 2007;213:360–8. doi: 10.1002/path.2238. [DOI] [PubMed] [Google Scholar]
- 8.Xiong S. Mouse models of Mdm2 and Mdm4 and their clinical implications. Chin J Cancer. 2013;32:371–5. doi: 10.5732/cjc.012.10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bohlman S, Manfredi JJ. p53-independent effects of Mdm2. Sub Cell Biochem. 2014;85:235–46. doi: 10.1007/978-94-017-9211-0_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieschke E, Wang Z, Kelly GL, Strasser A. Discussion of some ‘knowns’ and some ‘unknowns’ about the tumour suppressor p53. J Mol Cell Biol. 2018;11:212–23. doi: 10.1093/jmcb/mjy077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Tavana O, Gu W. p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol. 2019;11:564–77. doi: 10.1093/jmcb/mjz060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 13.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030–40. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 14.El-Deiry WS, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–74. [PubMed] [Google Scholar]
- 15.Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26:1306–16. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- 16.Gatz SA, Wiesmüller L. p53 in recombination and repair. Cell Death Differ. 2006;13:1003–16. doi: 10.1038/sj.cdd.4401903. [DOI] [PubMed] [Google Scholar]
- 17.Oka S, Leon J, Tsuchimoto D, Sakumi K, Nakabeppu Y. MUTYH, an adenine DNA glycosylase, mediates p53 tumor suppression via PARP-dependent cell death. Oncogenesis. 2014;3:e121. doi: 10.1038/oncsis.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan T, Chu G. p53 Binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol Cell Biol. 2002;22:3247–54. doi: 10.1128/MCB.22.10.3247-3254.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warnick CT, Dabbas B, Ford CD, Strait KA. Identification of a p53 response element in the promoter region of the hMSH2 gene required for expression in A2780 ovarian cancer cells. J Biol Chem. 2001;276:27363–70. doi: 10.1074/jbc.M103088200. [DOI] [PubMed] [Google Scholar]
- 20.Arias-Lopez C, Lazaro-Trueba I, Kerr P, Lord CJ, Dexter T, Iravani M, et al. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006;7:219–24. doi: 10.1038/sj.embor.7400587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 22.Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–92. doi: 10.1016/j.immuni.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 24.Giam M, Huang DCS, Bouillet P. BH3-only proteins and their roles in programmed cell death. Oncogene. 2008;27:S128–S136. doi: 10.1038/onc.2009.50. [DOI] [PubMed] [Google Scholar]
- 25.Westphal D, Kluck RM, Dewson G. Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014;21:196–205. doi: 10.1038/cdd.2013.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erlacher M, Labi V, Manzl C, Böck G, Tzankov A, Häcker G, et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med. 2006;203:2939–51. doi: 10.1084/jem.20061552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 28.Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121:671–4. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Szekely L, Okan I, Klein G, Wiman KG. Wild-type p53-triggered apoptosis is inhibited by bcl-2 in a v-myc-induced T-cell lymphoma line. Oncogene. 1993;8:3427–31. [PubMed] [Google Scholar]
- 30.Strasser A, Harris AW, Jacks T, Cory S. DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell. 1994;79:329–39. doi: 10.1016/0092-8674(94)90201-1. [DOI] [PubMed] [Google Scholar]
- 31.Pfeffer CM, Singh ATK. Apoptosis: a target for anticancer therapy. Int J Mol Sci. 2018;19:448. doi: 10.3390/ijms19020448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez J, Tait SW. Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer. 2015;112:957–62. doi: 10.1038/bjc.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 34.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–82. doi: 10.1016/S1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 35.Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 2008;15:1019–29. doi: 10.1038/cdd.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–8. doi: 10.1016/S1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 37.Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 38.Valente LJ, Aubrey BJ, Herold MJ, Kelly GL, Happo L, Scott CL, et al. Therapeutic response to non-genotoxic activation of p53 by Nutlin3a is driven by PUMA-mediated apoptosis in lymphoma cells. Cell Rep. 2016;14:1858–66. doi: 10.1016/j.celrep.2016.01.059. [DOI] [PubMed] [Google Scholar]
- 39.Happo L, Cragg MS, Phipson B, Haga JM, Jansen ES, Herold MJ, et al. Maximal killing of lymphoma cells by DNA damage-inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood. 2010;116:5256–67. doi: 10.1182/blood-2010-04-280818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–8. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 41.Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in Eu-myc transgenic mice. Cell. 1986;47:11–18. doi: 10.1016/0092-8674(86)90361-2. [DOI] [PubMed] [Google Scholar]
- 42.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–69. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michalak EM, Jansen ES, Happo L, Cragg MS, Tai L, Smyth GK, et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009;16:684–96. doi: 10.1038/cdd.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garrison SP, Jeffers JR, Yang C, Nilsson JA, Hall MA, Rehg JE, et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol. 2008;28:5391–402. doi: 10.1128/MCB.00907-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richter JA, Rullan A, Beltran E, Agirre X, Calasanz MAJ, Roman-Gomez J, et al. Epigenetic silencing of BIM mediates chemotherapy resistance of patients with burkitt lymphoma that can be overcome by therapeutic reactivation of bim in mouse and human lymphoma models. Blood. 2008;112:607. doi: 10.1182/blood.V112.11.607.607. [DOI] [Google Scholar]
- 46.Dengler MA, Weilbacher A, Gutekunst M, Staiger AM, Vöhringer MC, Horn H, et al. Discrepant NOXA (PMAIP1) transcript and NOXA protein levels: a potential Achilles’ heel in mantle cell lymphoma. Cell Death Dis. 2014;5:e1013. doi: 10.1038/cddis.2013.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 48.Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, et al. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013;3:1339–45. doi: 10.1016/j.celrep.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 49.Brady Colleen A, Jiang D, Mello Stephano S, Johnson Thomas M, Jarvis Lesley A, Kozak, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–83. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–83. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang S-J, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 2016;17:366–73. doi: 10.1016/j.celrep.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy ME. Ironing out how p53 regulates ferroptosis. Proc Natl Acad Sci. 2016;113:12350–2. doi: 10.1073/pnas.1615159113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018;22:569–75. doi: 10.1016/j.celrep.2017.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.FdAd Fagagna, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 56.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14:501–13. doi: 10.1016/S1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 57.Mijit M, Caracciolo V, Melillo A, Amicarelli F, Giordano A. Role of p53 in the regulation of cellular senescence. Biomolecules. 2020;10:420. doi: 10.3390/biom10030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qian Y, Chen X. Senescence regulation by the p53 Protein Family. Methods Mol Biol. 2013;965:37–61. doi: 10.1007/978-1-62703-239-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/S0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 60.Sugrue MM, Shin DY, Lee SW, Aaronson SA. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc Natl Acad Sci. 1997;94:9648–53. doi: 10.1073/pnas.94.18.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ongusaha PP, Ouchi T, Kim KT, Nytko E, Kwak JC, Duda RB, et al. BRCA1 shifts p53-mediated cellular outcomes towards irreversible growth arrest. Oncogene. 2003;22:3749–58. doi: 10.1038/sj.onc.1206439. [DOI] [PubMed] [Google Scholar]
- 62.Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. doi: 10.1016/S1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- 63.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Senescence in premalignant tumours. Nature. 2005;436:642–642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 64.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 65.Ferbeyre G, Stanchina ED, Lin AW, Querido E, McCurrach ME, Hannon GJ, et al. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol. 2002;22:3497–508. doi: 10.1128/MCB.22.10.3497-3508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu C-H, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci. 2007;104:13028–33. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ko A, Han SY, Choi CH, Cho H, Lee M-S, Kim S-Y, et al. Oncogene-induced senescence mediated by c-Myc requires USP10 dependent deubiquitination and stabilization of p14ARF. Cell Death Differ. 2018;25:1050–62. doi: 10.1038/s41418-018-0072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deng C, Zhang P, Wade Harper J, Elledge SJ, Leder P. Mice Lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–84. doi: 10.1016/0092-8674(95)90039-X. [DOI] [PubMed] [Google Scholar]
- 69.Guardavaccaro D, Corrente G, Covone F, Micheli L, D’Agnano I, Starace G, et al. Arrest of G(1)-S progression by the p53-inducible gene PC3 is Rb dependent and relies on the inhibition of cyclin D1 transcription. Mol Cell Biol. 2000;20:1797–815. doi: 10.1128/MCB.20.5.1797-1815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takahashi F, Chiba N, Tajima K, Hayashida T, Shimada T, Takahashi M, et al. Breast tumor progression induced by loss of BTG2 expression is inhibited by targeted therapy with the ErbB/HER inhibitor lapatinib. Oncogene. 2011;30:3084–95. doi: 10.1038/onc.2011.24. [DOI] [PubMed] [Google Scholar]
- 71.Powell E, Shao J, Yuan Y, Chen H-C, Cai S, Echeverria GV, et al. p53 deficiency linked to B cell translocation gene 2 (BTG2) loss enhances metastatic potential by promoting tumor growth in primary and metastatic sites in patient-derived xenograft (PDX) models of triple-negative breast cancer. Breast Cancer Res. 2016;18:13–13. doi: 10.1186/s13058-016-0673-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hollander MC, Kovalsky O, Salvador JM, Kim KE, Patterson AD, Haines DC, et al. Dimethylbenzanthracene carcinogenesis in Gadd45a-null mice is associated with decreased DNA repair and increased mutation frequency. Cancer Res. 2001;61:2487–91. [PubMed] [Google Scholar]
- 73.Smith ML, Chen I-T, Zhan Q, Bae I, Chen C-Y, Gilmer TM, et al. Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science. 1994;266:1376–80. doi: 10.1126/science.7973727. [DOI] [PubMed] [Google Scholar]
- 74.Zhan Q. Gadd45a, a p53- and BRCA1-regulated stress protein, in cellular response to DNA damage. Mutat Res/Fundamental Mol Mechanisms Mutagenesis. 2005;569:133–43. doi: 10.1016/j.mrfmmm.2004.06.055. [DOI] [PubMed] [Google Scholar]
- 75.Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet. 2004;36:63–68. doi: 10.1038/ng1282. [DOI] [PubMed] [Google Scholar]
- 76.Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in β cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002;109:321–34. doi: 10.1016/S0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 77.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 78.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Janic A, Valente LJ, Wakefield MJ, Di Stefano L, Milla L, Wilcox S, et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med. 2018;24:947–53. doi: 10.1038/s41591-018-0043-5. [DOI] [PubMed] [Google Scholar]
- 80.Li L-Y, Guan Y-D, Chen X-S, Yang J-M, Cheng Y. DNA repair pathways in cancer therapy and resistance. Front Pharmacol. 2021;11:629266. doi: 10.3389/fphar.2020.629266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kusakabe M, Onishi Y, Tada H, Kurihara F, Kusao K, Furukawa M, et al. Mechanism and regulation of DNA damage recognition in nucleotide excision repair. Genes Environ. 2019;41:2. doi: 10.1186/s41021-019-0119-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Williams AB, Schumacher B. p53 in the DNA-damage-repair process. Cold Spring Harb Perspect Med. 2016;6:a026070. doi: 10.1101/cshperspect.a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meek DW. The p53 response to DNA damage. DNA Repair. 2004;3:1049–56. doi: 10.1016/j.dnarep.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 85.Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci. 2002;99:12985–90. doi: 10.1073/pnas.202485699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jin S, Mazzacurati L, Zhu X, Tong T, Song Y, Shujuan S, et al. Gadd45a contributes to p53 stabilization in response to DNA damage. Oncogene. 2003;22:8536–40. doi: 10.1038/sj.onc.1206907. [DOI] [PubMed] [Google Scholar]
- 87.Xu J, Morris GF. p53-mediated regulation of proliferating cell nuclear antigen expression in cells exposed to ionizing radiation. Mol Cell Biol. 1999;19:12–20. doi: 10.1128/MCB.19.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Q-e Wang, Zhu Q, Wani MA, Wani G, Chen, et al. AA. Tumor suppressor p53 dependent recruitment of nucleotide excision repair factors XPC and TFIIH to DNA damage. DNA Repair. 2003;2:483–99. doi: 10.1016/S1568-7864(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 89.Cheo DL, Meira LB, Hammer RE, Burns DK, Doughty AT, Friedberg EC. Synergistic interactions between XPC and p53 mutations in double-mutant mice: neural tube abnormalities and accelerated UV radiation-induced skin cancer. Curr Biol. 1996;6:1691–4. doi: 10.1016/S0960-9822(02)70794-X. [DOI] [PubMed] [Google Scholar]
- 90.Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, et al. Genomic instability in Gadd45a-deficient mice. Nat Genet. 1999;23:176–84. doi: 10.1038/13802. [DOI] [PubMed] [Google Scholar]
- 91.Morris GF, Bischoff JR, Mathews MB. Transcriptional activation of the human proliferating-cell nuclear antigen promoter by p53. Proc Natl Acad Sci. 1996;93:895–9. doi: 10.1073/pnas.93.2.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnson RE, Kovvali GK, Guzder SN, Amin NS, Holm C, Habraken Y, et al. Evidence for involvement of yeast proliferating cell nuclear antigen in DNA mismatch repair *. J Biol Chem. 1996;271:27987–90. doi: 10.1074/jbc.271.45.27987. [DOI] [PubMed] [Google Scholar]
- 93.Chen IT, Smith ML, O’Connor PM, Fornace AJ., Jr Direct interaction of Gadd45 with PCNA and evidence for competitive interaction of Gadd45 and p21Waf1/Cip1 with PCNA. Oncogene. 1995;11:1931–7. [PubMed] [Google Scholar]
- 94.Itoh T, Cado D, Kamide R, Linn S. DDB2 gene disruption leads to skin tumors and resistance to apoptosis after exposure to ultraviolet light but not a chemical carcinogen. Proc Natl Acad Sci. 2004;101:2052–7. doi: 10.1073/pnas.0306551101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci. 1999;96:424–8. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yoon T, Chakrabortty A, Franks R, Valli T, Kiyokawa H, Raychaudhuri P. Tumor-prone phenotype of the DDB2-deficient mice. Oncogene. 2005;24:469–78. doi: 10.1038/sj.onc.1208211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Reitmair AH, Schmits R, Ewel A, Bapat B, Redston M, Mitri A, et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet. 1995;11:64–70. doi: 10.1038/ng0995-64. [DOI] [PubMed] [Google Scholar]
- 98.Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, et al. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet. 1998;18:276–9. doi: 10.1038/ng0398-276. [DOI] [PubMed] [Google Scholar]
- 99.Reitmair AH, Redston M, Cai JC, Chuang TC, Bjerknes M, Cheng H, et al. Spontaneous intestinal carcinomas and skin neoplasms in Msh2-deficient mice. Cancer Res. 1996;56:3842–9. [PubMed] [Google Scholar]
- 100.Edelmann W, Yang K, Kuraguchi M, Heyer J, Lia M, Kneitz B, et al. Tumorigenesis in Mlh1 and Mlh1/Apc1638N Mutant Mice. Cancer Res. 1999;59:1301–7. [PubMed] [Google Scholar]
- 101.Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2:213–9. doi: 10.2147/vhrm.2006.2.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Teodoro JG, Evans SK, Green MR. Inhibition of tumor angiogenesis by p53: a new role for the guardian of the genome. J Mol Med. 2007;85:1175–86. doi: 10.1007/s00109-007-0221-2. [DOI] [PubMed] [Google Scholar]
- 103.Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and metabolism. J Mol Cell Biol. 2019;11:284–92. doi: 10.1093/jmcb/mjy070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 105.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Won KY, Lim S-J, Kim GY, Kim YW, Han S-A, Song JY, et al. Regulatory role of p53 in cancer metabolism via SCO2 and TIGAR in human breast cancer. Hum Pathol. 2012;43:221–8. doi: 10.1016/j.humpath.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 107.Green DR, Chipuk JE. p53 and metabolism: inside the TIGAR. Cell. 2006;126:30–32. doi: 10.1016/j.cell.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 108.Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011;2:948–57. doi: 10.18632/oncotarget.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008;44:1529–35. doi: 10.1016/j.freeradbiomed.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ma W, Sung HJ, Park J, Matoba S, Hwang P. A pivotal role for p53: Balancing aerobic respiration and glycolysis. J Bioenerg Biomembranes. 2007;39:243–6. doi: 10.1007/s10863-007-9083-0. [DOI] [PubMed] [Google Scholar]
- 111.X-d Zhang, Z-h Qin, Wang J. The role of p53 in cell metabolism. Acta Pharmacol Sin. 2010;31:1208–12. doi: 10.1038/aps.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liang Y, Liu J, Feng Z. The regulation of cellular metabolism by tumor suppressor p53. Cell Biosci. 2013;3:9. doi: 10.1186/2045-3701-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci. 2011;108:16259–64. doi: 10.1073/pnas.1113884108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR. Hypoxia-modified cancer cell metabolism. Front Cell Dev Biol. 2019;7:4. doi: 10.3389/fcell.2019.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, et al. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget. 2014;5:5535–46. doi: 10.18632/oncotarget.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, et al. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev Cell. 2013;25:463–77. doi: 10.1016/j.devcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature. 2017;544:372–6. doi: 10.1038/nature22056. [DOI] [PubMed] [Google Scholar]
- 118.Li Y, Chang Y, Li X, Li X, Gao J, Zhou Y, et al. RAD-deficient human cardiomyocytes develop hypertrophic cardiomyopathy phenotypes due to calcium dysregulation. Front Cell Dev Biol. 2020;8:585879. doi: 10.3389/fcell.2020.585879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 120.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–8. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.White E. Autophagy and p53. Cold Spring Harb Perspect Med. 2016;6:a026120. doi: 10.1101/cshperspect.a026120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.He G, Zhang Y-W, Lee J-H, Zeng SX, Wang YV, Luo Z, et al. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol Cell Biol. 2014;34:148–57. doi: 10.1128/MCB.00670-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–5. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 124.Mallela K, Kumar A. Role of TSC1 in physiology and diseases. Mol Cell Biochem. 2021;476:2269–82. doi: 10.1007/s11010-021-04088-3. [DOI] [PubMed] [Google Scholar]
- 125.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–60. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci. 2008;105:10853–8. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kobayashi T, Minowa O, Sugitani Y, Takai S, Mitani H, Kobayashi E, et al. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc Natl Acad Sci. 2001;98:8762–7. doi: 10.1073/pnas.151033798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D’Amelio M, Djavaheri-Mergny M, et al. A dual role of p53 in the control of autophagy. Autophagy. 2008;4:810–4. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
- 129.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 130.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 131.Laforge M, Limou S, Harper F, Casartelli N, Rodrigues V, Silvestre R, et al. DRAM triggers lysosomal membrane permeabilization and cell death in CD4+ T cells infected with HIV. PLOS Pathog. 2013;9:e1003328. doi: 10.1371/journal.ppat.1003328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011;18:1598–607. doi: 10.1038/cdd.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang C, Wang H, Zhang D, Luo W, Liu R, Xu D, et al. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat Commun. 2018;9:3492. doi: 10.1038/s41467-018-05449-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kundu M, Lindsten T, Yang C-Y, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/S1535-6108(03)00309-X. [DOI] [PubMed] [Google Scholar]
- 137.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 138.Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol. 2011;99:287–92. doi: 10.1016/j.radonc.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 139.Lomonaco SL, Finniss S, Xiang C, DeCarvalho A, Umansky F, Kalkanis SN, et al. The induction of autophagy by γ-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer. 2009;125:717–22. doi: 10.1002/ijc.24402. [DOI] [PubMed] [Google Scholar]
- 140.Wilson EN, Bristol ML, Di X, Maltese WA, Koterba K, Beckman MJ, et al. A switch between cytoprotective and cytotoxic autophagy in the radiosensitization of breast tumor cells by chloroquine and vitamin D. Hormones Cancer. 2011;2:272–85. doi: 10.1007/s12672-011-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jo GH, Bögler O, Chwae Y-J, Yoo H, Lee SH, Park JB, et al. Radiation-induced autophagy contributes to cell death and induces apoptosis partly in malignant glioma cells. Cancer Res Treat. 2015;47:221–41. doi: 10.4143/crt.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gewirtz DA. Cytoprotective and nonprotective autophagy in cancer therapy. Autophagy. 2013;9:1263–5. doi: 10.4161/auto.25233. [DOI] [PubMed] [Google Scholar]
- 143.Chakradeo S, Sharma K, Alhaddad A, Bakhshwin D, Le N, Harada H, et al. Yet another function of p53-the switch that determines whether radiation-induced autophagy will be cytoprotective or nonprotective: implications for autophagy inhibition as a therapeutic strategy. Mol Pharmacol. 2015;87:803–14. doi: 10.1124/mol.114.095273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cui L, Song Z, Liang B, Jia L, Ma S, Liu X. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncol Rep. 2016;35:3639–47. doi: 10.3892/or.2016.4752. [DOI] [PubMed] [Google Scholar]
- 145.Patel NH, Sohal SS, Manjili MH, Harrell JC, Gewirtz DA. The roles of autophagy and senescence in the tumor cell response to radiation. Radiat Res. 2020;194:103–15. doi: 10.1667/RADE-20-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lim SM, Mohamad Hanif EA, Chin S-F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021;11:56. doi: 10.1186/s13578-021-00570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci. 2010;107:18511–6. doi: 10.1073/pnas.1006124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Younger ST, Kenzelmann-Broz D, Jung H, Attardi LD, Rinn JL. Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res. 2015;43:4447–62. doi: 10.1093/nar/gkv284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bieging-Rolett KT, Kaiser AM, Morgens DW, Boutelle AM, Seoane JA, Van Nostrand EL, et al. Zmat3 is a key splicing regulator in the p53 tumor suppression program. Mol Cell. 2020;80:452–69.e459. doi: 10.1016/j.molcel.2020.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Best SA, Vandenberg CJ, Abad E, Whitehead L, Guiu L, Ding S, et al. Consequences of Zmat3 loss in c-MYC- and mutant KRAS-driven tumorigenesis. Cell Death Dis. 2020;11:877. doi: 10.1038/s41419-020-03066-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Muys B, Anastasakis D, Claypool D, Pongor L, Li X, Grammatikakis I, et al. The p53-induced RNA-binding protein ZMAT3 is a splicing regulator that inhibits the splicing of oncogenic CD44 variants in colorectal carcinoma. Genes Dev. 2021;35:102–16. doi: 10.1101/gad.342634.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–83. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hemann MT, Zilfou JT, Zhao Z, Burgess DJ, Hannon GJ, Lowe SW. Suppression of tumorigenesis by the p53 target PUMA. Proc Natl Acad Sci. 2004;101:9333–8. doi: 10.1073/pnas.0403286101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sung HJ, Ma W, Wang P-y, Hynes J, O'Riordan TC, Combs CA, et al. Mitochondrial respiration protects against oxygen-associated DNA damage. Nat Commun. 2010;1:5. doi: 10.1038/ncomms1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This review article does not present any new primary data.