

Abstract
The p53 protein is structurally and functionally divided into five domains. The proline-rich domain is localized at amino acids 55–100. 319 missense mutations were identified solely in the proline domain from human cancers. Six hotspot mutations were identified at amino acids 72, 73, 82, 84, 89, and 98. Codon 72 contains a polymorphism that changes from proline (and African descent) to arginine (with Caucasian descent) with increasing latitudes northward and is under natural selection for pigmentation and protection from UV light exposure. Cancers associated with mutations in the proline domain were considerably enriched for melanomas and skin cancers compared to mutations in other p53 domains. These hotspot mutations are enriched at UV mutational signatures disrupting amino acid signals for binding SH-3-containing proteins important for p53 function. Among the protein–protein interaction sites identified by hotspot mutations were MDM-2, a negative regulator of p53, XAF-1, promoting p53 mediated apoptosis, and PIN-1, a proline isomerase essential for structural folding of this domain.
Subject terms: Tumour-suppressor proteins, Cancer genetics
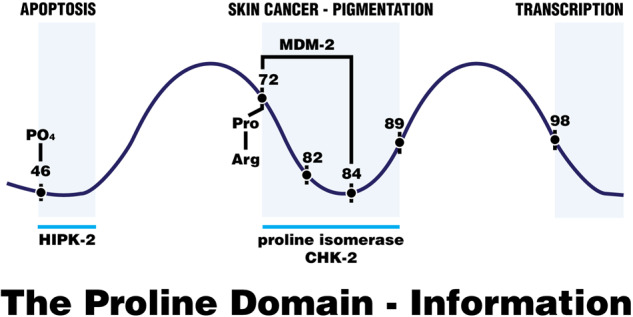
FACTS
It has been known that the p53 proline domain is located between amino acids 55–100 out of 393 amino acids; codon 72 is polymorphic, either arginine or proline, and these differences are under natural selection and vary by latitude, UV light exposure, and pigmentation.
This manuscript demonstrates that in cancerous tissues from humans the proline domain contains 6 hotspot mutations: at codons 72, 73, 82, 84, 89, and 98, that mutations in the proline domain are highly enriched for melanoma and skin cancer, and that the mutational signatures for these mutations in these cancers are derived from UV light exposure.
This analysis demonstrates that the codon 72 polymorphism undergoes mutations that produce a third amino acid. Cancer-associated mutations at a polymorphic site are unusual.
Mutations in all hotspot codons likely either inhibit p53 transcriptional activity or disrupt protein-protein interactions required for p53 functions.
The p53 protein interactions are with MDM-2, XAF-1, CHEK-2, and Pin-1.
QUERIES
Hotspot codon 73 is yet to be assigned a function, although it is possible that it has an impact upon the adjacent hot spot codon, 72.
Codon 49 adjacent to the proline domain is also a hotspot mutation and some mutations at this codon inhibit transcriptional activity of p53. What is its function?
The portion of the p53 gene that encodes the proline domain between codons 47 and 72, with introns 2 and 3, contains polymorphisms that differ between Caucasians and individuals of African descent and is in linkage disequilibrium. What are the haplotypes in individuals of African descent (proline) or of Caucasian descent (arginine) formed by this selection pressure that reduces recombination?
Was the proline domain acquired in the p53 gene during evolutionary changes over 800 million years to respond to tissue-specific cancer types?
Introduction
The human p53 protein is a transcription factor [1] and the 393 amino acids in this protein are structurally and functionally divided into five domains. Amino acids 1–55, starting from the N-terminus, contain two different transactivation sequences at residues 25, 26 and 53, 54 [2, 3] and this is followed by a proline rich domain involved in protein-protein interactions from amino acids 55–100 [4, 5]. The majority of the protein is composed of a DNA binding domain encompassing amino acids 102–292 [6] followed by a tetramerization domain from amino acid 323 to 356 [7] and then a carboxy-terminal domain from about amino acids 360–393 termed the regulatory domain because amino acid modifications (methylation and acetylation) of lysines in this domain regulate p53 activity [8]. The great majority of human cancers contain either mutations in the Tp53 gene or harbor wild type p53 proteins that are inactivated by a variety of physiological functions [1, 6]. About seventy-five percent of the Tp53 mutations are missense mutations localized in the DNA binding domain of the p53 protein [6]. Different missense mutations in the DNA binding domain occur at frequencies that range over four logs in different cancer tissue types and they disrupt the binding of the p53 protein to DNA and transcription of p53 regulated genes to varying extents [1, 6]. Eight of these mutations, occurring at frequencies of 1.5–8% of the mutational spectra, are termed hotspot mutations. Although these hotspots are only eight mutations out of a total of some 350 observed mutations in the DNA binding domain of the Tp53 gene, the eight hot spot mutations account for 33% of cancers with Tp53 mutations and the remaining Tp53 mutations are present at much lower frequencies in 67% of cancers with p53 mutations [6].
This review documents 319 single missense mutations found only in the proline domain (amino acids 55–100) of the p53 protein in a wide variety of spontaneous cancers and another 57 mutations localized in the adjacent nine residues (amino acids 46–54) to the proline domain. Codons with mutations that encode amino acids 49, 72, 73, 82, 84, 89, and 98 each contributed between 2.7–8% of the mutations observed in the proline and adjacent domain in patients with spontaneous cancers. Collectively mutations in these seven codons contribute approximately 33% of the mutations in these two regions of the protein (122 mutations/a total of 376 in the proline domain and the adjacent sequences). The DNA binding domain has 8 hotspot mutations that contribute to 33% of the cancers and the proline domain has 7 hotspot mutations that contribute to 33% of the cancers. These are remarkably similar distributions of amino acids composing a third of the cancers that appear to be driven by these alterations.
These hotspot mutations could guide us to residues in the proline domain that may well affect protein-protein interactions and Tp53 functions that help to prevent cancers from arising just as is observed with mutations in the DNA binding domain [6]. Previous experiments deleting the regions containing these hot spot mutations in the proline domain have been shown to reduce p53 apoptotic activity and therefore reduce p53 tumor suppression [4, 5]. One of these hot spot mutations is at codon 72, which is a naturally occurring polymorphism (proline to arginine change), that is associated with geographical and, therefore, racial differences [9] and has a demonstrated impact upon the age of onset of cancers in individuals with inherited Tp53 mutations [10–13] as well as responses to cancer treatments and overall survival in breast cancers with spontaneous Tp53 mutations [14]. Thus, examining the phenotypes of these hotspot mutations in the proline domain could well uncover important aspects of the functions of the p53 protein.
There are several reasons why we extended this search for mutations in the proline domain to the adjacent amino acids from codons 46–54. First phosphorylation of the serine at amino acid 46 is required or preferred for the p53 protein to initiate apoptosis in a cancer cell [15]. Second, amino acid 47 is a polymorphism (proline or serine) that is serine in most individuals of African descent and proline in individuals of Caucasian descent [16]. Third, the coding region of the p53 protein from amino acid 46 to 72, which includes introns 2 and 3 in the Tp53 gene, is strongly in linkage disequilibrium suggesting that this region forms a set of complex haplotypes that are different from each other in different populations depending upon the latitude, ultraviolet light exposure, altitude, and pigmentations [9, 17−19] of the individuals from different geographical locations. These mutations may well help define the functional properties of each haplotype. The hotspot mutations in the amino acids 49, and 72, and 73 may well play a dominant role in mediating these functions.
Databases of Tp53 mutations in the proline domain
Five different databases, IARC R20 (N = 21,732) [20], TCGA (N = 2764) [21], MSK-IMPACT (N = 16,244) [22], cBio-Genie (N = 17,817) [23], and COSMIC (N = 33,176) [24], with the Tp53 gene sequenced from cancers (N = 73,857 sequences) were screened for somatic Tp53 missense mutations, solely in either the proline domain (amino acids 55–100) or the adjacent section of nine amino acids (amino acids 46–54). Synonymous mutations, splice site mutations, deletions, chain termination codons, frameshift mutations, and inversions were eliminated. We also made sure not to record the same patient cancer sample from more than one of the five different databases. This resulted in the identification of 376 mutant Tp53 patient DNA sequences in the proline and the adjacent domain. 57 mutations were found in the adjacent segment (amino acids 46–54) to the proline domain, 22 of which were at amino acid 49, which therefore was designated a hotspot mutation. In total, 319 single missense mutations were found only in the proline domain, 70,045 missense mutations were identified solely in the DNA binding domain, and 1071 mutations were identified solely in the tetramerization domain (Table 1). In each case these missense point mutations were the only amino acids altered in the entire protein. By dividing the number of these mutations by the number of amino acids in a domain, the cross-sectional target for a mutation, the observed mutation frequency per amino acid residue was 5.7 in the adjacent ten amino acids to the proline domain, 6.93 in the proline domain, 366.73 for the DNA binding domain, and 31.5mutations per domain for the tetramerization domain (Table 1).
Table 1.
The distribution of missense mutations in the proline domain, the DNA binding domain and the tetramerization domain.
| P53 domain | Observed mutations within domain | Residues within domain | Observed mutations/residue |
|---|---|---|---|
| ADJACENT AMINO ACIDS 46–54 | 57 | 10 | 5.7 |
| PROLINE | 319 | 46 | 6.93 |
| DNA-BINDING | 70045 | 191 | 366.73 |
| TETRAMERIZATION | 1071 | 34 | 31.5 |
73,857 missense mutations in the Tp53 gene were analyzed from different cancers and the observed number of mutations in the proline domain, the DNA binding domain, and the tetramerization domain are presented, along with the number of amino acids in each domain and the observed mutation frequency per amino acid residue (the target size).
Figure 1 identifies the percent frequencies of the 376 missense mutations in the proline domain and the adjacent region of nine amino acids from the cancers screened in this study. The results demonstrate that there are hotspot mutations, defined as greater than ten independent mutations for each codon (the 90th percentile of the mutation frequency within this region), which corresponds to mutations with frequencies between 2.66 and 7.98%. This distribution is biased and not derived from a uniform background distribution so that a two-sided Kolmogorov-Smirnov test gave a p value of 3.81e–96, clearly indicating a non-random distribution of these hotspot mutations. The presence of hotspot (repeated) mutations that are distributed non-randomly suggests a causal relationship with cancer but does not eliminate the possibility that these codons spontaneously mutate at a higher frequency because of their sequence context in the DNA.
Fig. 1. The distribution of the 376 missense mutations in the proline domain and adjacent region (amino acids 46–54) of the p53 protein.
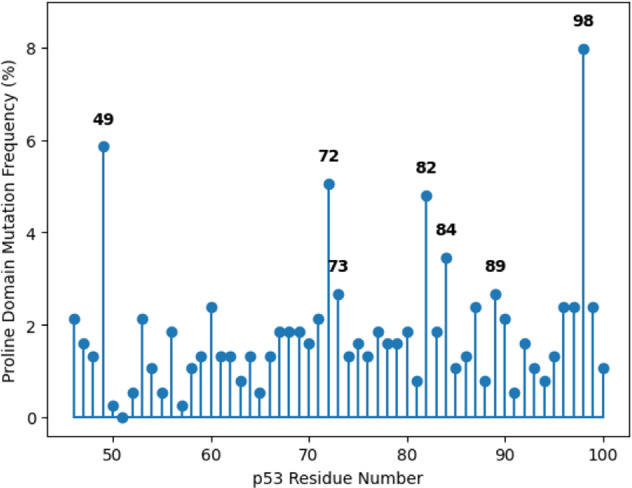
The 376 missense mutations are presented by the percentage of mutations at each residue between amino acids 46–100 in the p53 protein. Hotspot mutations are indicated by any codon with at least 2.66% mutation frequency. The mutational distribution is a non-uniform distribution (p = 3.81e–96). Amino acid 46 is required to be phosphorylated for maximal p53 apoptotic activity and this is accomplished by the XAF-1 protein [15], which binds to the proline domain [4, 5]. Amino acids 47 (proline to serine) and 72 (proline to arginine) are both polymorphisms differing by African and Caucasian descent.
Hotspot mutations and transcriptional activity of p53
The wild type p53 protein, as well as some mutant p53 proteins, function in normal and cancerous cells by acting as a transcription factor or by protein-protein interactions. Kato and his colleagues have employed a yeast assay with a wild type p53 c-DNA and a wide variety of mutant p53 c-DNAs producing proteins that transcribe a number of different p53 responsive elements from human genes [25]. The seven hotspot mutations observed in the proline domain and in the adjacent nine codon region with a hotspot mutation at codon 49 were examined for mutant transcriptional activity using this assay (Table 2). Rather clearly all of the 30 codon 98 mutations that were P98S, L, T and R mutations were defective in transcription (0–11.7% of wild type). Another mutation defective in transcription was the D49H mutant allele (9.7% of wild type) which was represented in 8 out of 22 mutations at codon 49. Amino acid 49 is called as an aspartic acid in the wild type protein but several publications suggest that this residue is polymorphic [26–28]. Based upon its transcriptional profile D49N, V and Y could be polymorphic and still have wild type like transcriptional activity. However, the D49H allele might well be a transcriptionally defective Li-Fraumeni inherited allele which would not be uncovered by sequencing of the DNA binding domain or a spontaneous mutation in the amino terminal domain contributing to cancers. The limitations of this assay are that it is carried out in yeast and not every possible p53 regulated gene is tested. As will be seen by subsequent analysis, codons 72, 82, 84, 89, and 98 participate in protein-protein interactions.
Table 2.
Transcriptional phenotypes of hotspot mutations in the proline domain and adjacent domain.
| Position | WT amino acid | MT amino acid | Mutation count | Lowest transcription factor activity (% of WT Function) |
|---|---|---|---|---|
| 49 | D | H | 8 | 9.7 |
| 49 | D | N | 8 | 86.9 |
| 49 | D | V | 3 | 41 |
| 49 | D | Y | 3 | 96.2 |
| 72 | P | A | 8 | 44 |
| 72 | P | S | 7 | 40.2 |
| 72 | P | H | 3 | 60.6 |
| 72 | P | T | 1 | 39.8 |
| 73 | V | M | 5 | 46.7 |
| 73 | V | L | 4 | 45.4 |
| 73 | V | E | 1 | 46.4 |
| 82 | P | L | 16 | 32.8 |
| 82 | P | T | 1 | 74.9 |
| 82 | P | S | 1 | 49.1 |
| 84 | A | V | 9 | 46 |
| 84 | A | G | 4 | 64.3 |
| 89 | P | S | 7 | 48 |
| 89 | P | L | 3 | 49.8 |
| 98 | P | S | 17 | 8 |
| 98 | P | L | 11 | 11.7 |
| 98 | P | T | 1 | 0 |
| 98 | P | R | 1 | 7.6 |
All observed missense mutations observed in the proline domain and adjacent domain are annotated with their mutation prevalence and their weakest transcriptional phenotype, defined as the minimum p53 response element transactivation activity as defined in reference [25].
Codon 72
It was a bit surprising that codon 72 was one of the hotspot mutations detected in the cancer databases. This is because the proline 72 arginine polymorphism (rs1042522) has a proline preferentially in individuals of African descent and an arginine residue preferentially in Caucasians [9]. These differences appear to occur under natural selection, changing in frequency from proline to arginine with increasing south to north latitudes from the equator [9]. There is abundant evidence that the codon 72 polymorphism in the Tp53 gene, its protein, and the pathway influences pigmentation in response to UV light exposure, altitude, as well as an association with transcription levels of the KIT ligand that is involved with neural crest migration and melanocyte distribution in the body [9, 17, 18]. Thus, both proline or arginine in codon 72 must be functioning well enough to help prevent cancers in the majority of individuals given their different environments, and both should be considered wild type based on the individual’s background. This brings up a problem because most datasets call proline the wild-type sequence [29] and so an arginine at codon 72 in some databases may be scored as a mutation when it is not one. It is a polymorphism adjusting to environmental changes. In this manuscript, a mutation at codon 72 is any amino acid that is neither proline nor arginine. Similarly, a mutation at codon 47 is any amino acid that is neither proline nor serine.
The mutational hotspots in Fig. 1 might identify amino acids that are functioning by employing one or more different protein-protein interactions in the same or different pathways to accomplish a goal. A variety of different publications have demonstrated that the negative regulator of p53 protein levels in a cell, MDM-2, binds to p53 in the transactivation domain around residues 25 and 26 [2] and in the proline domain between residues 62–92 [10–13]. It has been reported that the arginine codon 72 allele produces a p53 protein that binds with increased affinity to MDM-2 as compared to the proline codon 72 residue [10]. As a consequence, codon 72 Arg/Arg homozygotic individuals with Li-Fraumeni Syndrome and an inherited Tp53 mutation in the DNA binding domain have earlier onsets of first cancers when compared to individuals with a proline/proline polymorphism at codon 72 and an inherited Tp53 mutation in the DNA binding domain [10]. The tighter MDM-2 binding (higher affinity) to the wild type p53 protein in the cells of a Li-Fraumeni patient (Arg/Arg in the proline domain, with a Tp53 MT/Tp53 WT in the DNA binding domain) might well degrade the WT p53 protein more efficiently, lowering its tumor suppressive value, resulting in an earlier onset of cancer.
On the other hand, with spontaneous Tp53 mutations in the DNA binding domain in breast cancers, the inherited pro/pro genotype was associated with poorer disease-free survival with a multivariate Cox’s proportional hazards regression analysis of p = 0.047 and a risk ratio of recurrence of 1.67 [30]. In fact, many spontaneous cancers of different tissue types with pro/pro genotypes at codon 72 do more poorly with treatments for spontaneous mutations in their DNA binding domains of Tp53 [30]. An early onset of cancer with an inherited predisposition and the outcome of a treatment for a cancer with spontaneous Tp53 mutations are different phenotypes. This helps to explain why the ARG allele favors earlier onset of cancers in inherited Tp53 mutations whereas the PRO allele in spontaneous Tp53 mutant breast cancers, with only mutant alleles, gives rise to a poorer prognostic outcome after treatments with chemotherapy, two very different phenotypes with possibly distinct target genes and sets of protein-protein interactions. Of the mutations at codon 72 in the proline domain that are only associated with cancers, 19 mutations started with an inherited proline residue at this codon and were mutated to alanine (8 examples), serine (7 examples), histidine (3 examples), and threonine (1 example). These mutations would be expected to change the affinities of MDM-2 for the wild type and for the mutant p53 proteins.
Codons 72, 82, 84, 89, and 98: Protein- Protein Interactions
There are many PXXP (proline—any amino acid x 2—proline) sequence repeats in the proline domain which are binding sites for SH-3 receptors (protein-protein interactions) [31] and many PXXXP repeats and proline-alanine pairs throughout this domain. This could be the reason for the extensive network connectivity of the p53 protein with other signal transduction pathways [1]. A rather common SH-3 protein binding site is arginine followed by 3 or more amino acids followed by proline-X-X-proline [31]. This could help to explain the natural selection for the mutation from proline to arginine at codon 72 [9, 31], as it would change or enhance the SH-3 interactions. Figure 2 shows the amino acid sequences of the proline domain from codon 72 to 98. The polymorphic codon 72 is followed by five PXX*P signals (proline followed by at least two non-proline amino acids followed by proline), four of which contain hot spot mutations at codons 72, 73, 82, 84, 89, and 98. All of the proline domain hot spot mutations can be found in these SH-3 domain protein interaction sites. Four out of six are mutations of a proline. The arginine at codon 72 enhances the PXXP interaction site. These data are at least consistent with the idea that the proline domain carries out SH-3 mediated protein–protein interactions that influence the functions of the p53 protein in both normal and cancerous cells. Cancers that arise with Tp53 mutations localized solely in the proline domain harbor mutations that could disrupt these protein-protein interactions and, therefore, disrupt p53 homeostasis. An example of this could well be the MDM-2 protein, which has binding sites in this proline domain. The codon 72 polymorphisms and hotspot mutations are expected to alter the binding of MDM-2 to p53 proteins [10–13].
Fig. 2. The amino acid sequences in the proline domain and the SH-3 binding sites disrupted by the hot spot mutations in this domain.
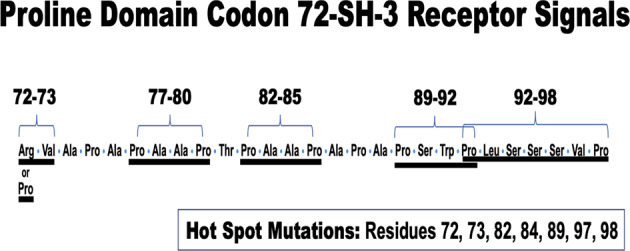
The underlined amino acids in this sequence of the proline domain point to PXX*P motifs which are protein-protein interaction sites for SH-3-containing proteins functioning with the p53 proline domain. The hotspot mutations disrupt many of these sites in cancers.
Another example that the hotspot mutations have phenotypes that result in cancers comes from previous research [32] that identified a region between amino acids 86–93 of p53 (Ala-Pro-Ala-Pro-Ser-Try-Pro-Leu) which is responsible for a loss of tetramer-stabilizing interactions producing dimers and monomers with a reduced level of DNA binding to p53 transcriptional activation sites. The observed mutations in this region, in particular at the hotspot at residue 89, reduces tetramer stability and therefore reduces p53 tumor suppressor function. Phosphorylation of serine-46 attracts the binding of Pin-1, a proline isomerase, which isomerizes prolines so as to enhance the proper structure of the proline domain for further protein-protein interactions. Mutations in the proline-82 hot spot reduce the proline isomerization and phosphorylation by CHK-2 that would ordinarily result in the activation of wild type p53 by Pin-1 and CHK-2. Therefore, the responses to DNA damage by p53 proline-82 mutations are much reduced [33]. The impaired CHK-2 binding and the mutated codon 82 PXXP motif reduces acetylation by p300, which is an essential co-activator for transcription by p53 [34, 35]. Thus, mutations in codons 72, 82, 84 and 89 clearly have phenotypes that impair protein-protein interactions that result in impaired structure and impaired protein modifications that reduce transcription and tumor suppression.
The XAF-1 protein (XIAP associated factor 1) is a 33.4 Kd protein with seven zinc fingers and was first shown to bind and inactivate the X-linked inhibitor of apoptosis protein in vivo [36] promoting apoptosis. Zinc finger 5 of this protein binds to the p53 protein in the proline domain at PXXP sites between amino acids 62 and 92 [15]. This displaces MDM-2 from the p53 protein, increasing the p53 concentration and the apoptotic frequency of p53 cell killing [15]. In addition, the XAF-1 protein binds to SIAH-2, a ubiquitin ligase that regulates the levels and activity of a protein kinase, HIPK-2 (homeobox interacting protein kinase-2) which, in turn, phosphorylates serine 46 of the p53 protein just adjacent to the proline domain. Serine 46 phosphorylation also promotes p53 mediated apoptosis [15]. Third, the XAF-1 protein binds to the E-3 ubiquitin ligase ZNF-313, which poly-ubiquitinates the p53-regulated p21 protein, that is then degraded and is yet a third pro-apoptotic event that cooperates with p53-induced apoptosis [15]. Deletions made in the proline domain that leave the rest of the p53 protein intact reduce p53’s ability to induce apoptosis in cells but leave several other growth restricting properties intact [4, 5]. A natural polymorphism in the XAF-1 gene, E134*, is a chain termination of that protein, which statistically occurs at a very high frequency (69%) along with an inherited mutation (R337H) in the tetramerization domain [19], and the loss of XAF-1 appears to enhance the penetrance and the appearance of multiple cancers in a Brazilian Li-Fraumeni cohort [19]. Thus, there appears to be at least three mechanisms that XAF-1 exerts that are proapoptotic in cooperation with p53 functions [15]. Additional studies would be welcome to confirm these phenotypes. Some of the hotspot mutations observed in Figs. 1 and 2 could weaken the binding of the XAF-1 protein to the proline domain and thus result in lower efficiencies of apoptosis, permitting some of the low penetrance/low frequency mutations in the DNA binding domain of p53 to produce a cancer, as first suggested by Pinto and Zambetti [19]. The XAF-1 protein is commonly inhibited at the level of transcription by epigenetic modifications of this gene in a number of cancers [15].
Cancer tissue type and sequence motif mutations in the proline domain
It is of some interest that 14.6% of the cancers with mutations solely in the proline domain and the adjacent nine amino acid sequences are either melanomas or skin cancers. This contrasts with only 0.03% of skin cancers in the DNA binding domain and 0.02% skin cancers from the tetramerization domain. This very large difference in the tissue type of cancers with mutations in different domains of the p53 protein is consistent with the functions of the p53 protein in pigmentation of the skin, UV light exposure, and melanocyte distribution resulting from transcriptional regulation of the KIT ligand by p53 [9, 18]. The mutational sequence motifs associated with different cancers [37] have identified several mutational signatures that are preferentially mutated after UV light exposures and are termed SBS7a, b, c, d, and SBS38. A total of 55 out of 376 mutations in the proline domain and adjacent region (amino acids 46–54) were identified in melanomas and skin cancers. Of these, 17 mutations (31%) were observed to alter proline residues in the PXXP sequences and 38 (69%) were not associated with those specific proline residues. Table 3 examines the expected number of PXXP and non-PXXP mutations in these cancers if the selection for mutations occurred uniformly across the domain, if there was an influence of dinucleotide pairing [38], and the predictions made by the five different UV signatures previously observed in many skin cancers and melanomas [37]. Table 3a contains the p value corresponding to the unconditional Boschloo test with the given background as a null distribution regarding mutations from just proline residues in the PXXP motifs. The UV SBS7b signature appears to be the one that is operative in selecting the mutations in the proline domain of p53 and contributes to skin cancers and melanomas to a greater extent than other signatures and chemical products produced by UV light (Table 3a). The Pearson (p = 0.031) and the Spearman (0.004) coefficients for this association are also statistically significant (Table 3b). Clearly then the selection of this mutational signature for producing the mutations in skin and melanocytes results in the increase in the proline domain and adjacent region’s mutations in these cancers. The great majority of Tp53 mutations in the DNA binding domain, as well as most of the hotspot mutations in that domain, occur at methylated CpG dinucleotides, which form codons for arginine [6]. Methylated CpG residues mutate spontaneously, producing a C to T transition, at approximately five-fold higher frequencies than do non-methylated CpG dinucleotides [38]. Thus, the mutational signature for hotspot mutations in the proline domain and the DNA binding domain differ from each other resulting in different cancer tissue types.
Table 3.
a, b (Boschloo test, Pearson and Spearman coefficients).
| 3a. | ||||
|---|---|---|---|---|
| PXXP | Not PXXP | p value (Boschloo) | ||
| UNIFORM | 1 | 54 | 1.94E–05 | |
| DINUCLEOTIDE | 4 | 51 | 0.0015 | |
| SBS7a | 6 | 49 | 0.011 | |
| SBS7b | 11 | 44 | 0.22 | |
| SBS7c | 2 | 53 | 0.00012 | |
| SBS7d | 1 | 54 | 1.94E–05 | |
| SBS38 | 0 | 55 | 2.03E–06 | |
| 3b | ||||
| Pearson r | Pearson p value | Spearman r | Spearman p value | |
| UNIFORM | −0.1 | 0.59 | −0.12 | 0.51 |
| DINUCLEOTIDE | 0.22 | 0.23 | 0.32 | 0.083 |
| SBS7a | 0.075 | 0.69 | 0.29 | 0.12 |
| SBS7b | 0.33 | 0.068 | 0.43 | 0.017 |
| SBS7c | 0.12 | 0.51 | 0.19 | 0.3 |
| SBS7d | −0.25 | 0.18 | −0.22 | 0.23 |
| SBS38 | −0.098 | 0.6 | 0.18 | 0.32 |
Observed = 17, Not Observed = 38.
The DNA sequence motifs giving rise to mutations generated by ultra-violet light. Of the 319 missense mutations identified in the proline domain, 55 mutations were found in melanomas and skin cancers. Of these, 17 mutations were proline-only mutations observed in the PXXP motifs and 38 were not found in those residues. The table shows what is expected from a uniform distribution of mutations, the distribution influenced from the dinucleotide frequencies in the domain [38], and five distinct classes of mutant sequence motifs that result from ultraviolet light exposures [37]. The p values indicate how well the predicted proline mutations in PXXP motifs compare to the observed frequencies of proline-only mutations in the PXXP motifs for a given null distribution. Only the SBS7b prediction of UV irradiated mutations is not statistically distinct from the observed mutation distributions.
The evolution of the proline domain
These results reinforce the importance of the domain structure of the p53 protein not only in their structural and functional differences but also in the cell biological distinctions of the tissue types in the body that are protected by different domains in the p53 protein. It is possible that the evolution of the domain structure of p53 was assembled by adding domains to a tumor suppressor function as the exposure to diverse mutagens occurred in different animals. We can rather clearly observe this by exploring the impact of evolutionary forces upon the proline domain sequences (amino acids 72–98) of mammals that are diurnal versus those that are nocturnal and comparing these sequences with the human proline domain. From the activity cycles found in the COMBINE dataset [39], we find that diurnal and nocturnal mammals’ p53 proteins have quite different levels of DNA sequence similarity to the human proline domain (Fig. 3). The levels of similarity to the human proline domain in these animals also positively correlates with body mass, adult length, and longevity [39] (Fig. 3). While the proline domain is not necessarily responsible for all of these phenotypic differences, it coordinates with other evolutionary advantages optimizing the p53 protein to the needs and lifestyles of these different mammals. It would be of some interest to exchange a human p53 proline domain into a mouse p53 proline domain so as to observe the impact of this upon some of the phenotypes discussed here and to determine if this hybrid p53 proline domain has all the properties of this tumor suppressor protein in mice as compared to humans.
Fig. 3. Differences in diverse mammalian proline domains.

a–d Mammalian proline domains relate to activity cycle and body parameters. a The human proline domain corresponding to amino acids 72–98 was aligned to mammal p53 proteins downloaded from the UniProt [29] with the BLASTp algorithm. The maximum score was retrieved and the distributions of these scores were plotted for diurnal, nocturnal, and “other” mammals’ activity cycles derived from the COMBINE dataset [39]. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. b–d Alignment scores are plotted against the log10 of the adult mass in grams (b), the log10 of the adult body length in millimeters (c), and the log10 of the typical longevity in days derived from the COMBINE dataset [39].
Questions remain?
Codon 72 is an example of a polymorphism (Pro to Arg) that occurred under natural selection as humans arose in Africa and migrated into northern or southern climates with less direct exposure to sunlight then at the equator, acting as a mutagen. It impacts skin pigment and melanocyte migration during development protecting against mutations in the skin [9–13]. Both the proline and the arginine amino acids function as wild type in the environments in which they arose and mediate tumor suppression. It was a surprise to see 19 independent mutations arise at that codon with four different amino acids (A, S, H, and T) possibly contributing to the cancers. The phenotypes of codon 72 amino acid changes (MDM-2 binding, XAF-1 interactions, an altered SH-3 protein binding site) are perfectly consistent with the central functions of the p53 protein. In fact, these observations explain why the proline to arginine polymorphism arose, because the addition of an arginine residue enhances the MDM-2 and SH-3 binding sites at the P-X-X-P residues. What remains unclear is why codon 73 is also a hotspot mutation in the proline domain. The most likely reason for this is that codon 73, by being adjacent to codon 72, might disrupt the binding of proteins to this region of the proline domain in the p53 protein. Protein-protein interaction sites are commonly composed of several amino acids to gain enough kilocalories for the appropriate binding constants. So, codon 73 mutations could also play a role in protein-protein binding.
Mutations in codon 98 clearly disrupt the transcriptional ability of the p53 protein. This could be because codon 98 is in close proximity to the start of the DNA binding domain, whose structure it could then disrupt. Or it could be because that proline at codon 98 plays an important role in protein-protein interactions mediated by the P-X-X-P signals. Four of the six hot spots mutations in the proline domain occur at prolines in these sites.
We were surprised to see that codon 46 (serine) is not a major hotspot for mutation because its phosphorylation is much preferred so that the p53 protein can initiate apoptosis which is thought to be important in preventing cancers. However, the p53 protein can initiate cell death by five distinct mechanisms: apoptosis, ferroptosis, necroptosis mediated by Fas or TNF, and senescence [1], so perhaps other mechanisms of cell death predominate in these situations. Perhaps apoptosis is not the major form of cell death acting to enforce tumor suppression in particular cases.
In southern Brazil there is a large population of individuals (about 1/360 people) who inherit a Tp53 mutation, R377H, located in the tetramerization domain in the heterozygous state of the Tp53 gene. The penetrance of this mutation in this population is about 60% for cancer development. In this group there is a chain termination mutation in 1/125 individuals in the XAF-1 gene [19], which helps to promotes apoptosis in a p53-mediated fashion [15]. A study of the genotypes of these genes in this cohort by Pinto and Zambetti [19] demonstrated a significant enrichment of the compound genotypes, mutant p53 R377H and XAF-1 chain termination mutation, in individuals who developed sarcomas (p = 0.003). The XAF-1 gene is located on chromosome 17 just 2 megabases away from the Tp53 gene. The fact that there are selection pressures for the development of polymorphisms to arise in the Tp53 gene based upon geographical locations and therefore racial differences (observed from the XAF-1 gene to Tp53 codons 47 and 72 along with two introns of the Tp53 gene) makes it worthwhile to explore the haplotypes in the two megabases adjacent to the Tp53 gene contrasting diverse racial types and geographical locations. A finding of significant linkage disequilibrium in the region between the XAF-1 gene and the Tp53 gene would suggest those alleles have favorable and selectable phenotypes, so as to interact together, and therefore would persist as haplotypes. This would support the evidence presented here that protein-protein interactions between proteins encoded in this region of two megabases do interact in selectable ways.
This analysis of the genetics and the phenotypes of the proline domain of the p53 protein (45 amino acids) demonstrates the complexity and impact that the amino acid sequences from approximately 10% of the p53 protein can impart upon the functions of this tumor suppressor protein. The Tp53 gene has been detected in some of the earliest multicellular animals whose origin, eight hundred million years ago, was to protect the germ line from mutations [1]. As the sea anemone comes to the surface of a body of water to feed on green plants and is exposed to sunlight, the p53 protein functions to kill germ line cells that incur DNA damage and preserve the DNA sequences in the species. Even the DNA sequences that the p53 protein binds to, so as to promote the transcription of genes involved in cellular apoptosis and death, in flies and worms, are conserved from invertebrates through humans [40]. The Tp53 gene functions are an excellent example of the tension between the selection to retain useful functions by eliminating mutations, and the entropic forces generating diversity to permit natural selection and change as the environment dictates. With the advent of stem cells regenerating tissues over a lifetime in the vertebrates it offered an innovative opportunity to repurpose the Tp53 gene and p53 protein from protecting against changes in the germ line in invertebrates to protecting against mutations in the somatic tissues from developing cancers.
Acknowledgements
We thank Suzanne Christen for help in the preparation of this manuscript.
Author contributions
DH performed all statistical analyses from datasets, contributed to the writing of the manuscript and assigning the phenotypes; BG contributed to the statistical information in the manuscript, and AJL conceived of ideas and wrote the manuscript.
Funding
This work was supported by a grant from the NCI, P01CA087497-20.
Data availability
All data is included in the Figures and Tables in the manuscript.
Five databases were screened for somatic Tp53 missense mutations, solely in either the proline domain (amino acids 55–100) or the adjacent section of nine amino acids (amino acids 46–54). The web addresses for the five databases are:
1. GENIE: https://genie.cbioportal.org/
2. COSMIC: https://cancer.sanger.ac.uk/cosmic
3. IARC R20: https://tp53.isb-cgc.org/
4. MSK-IMPACT: https://www.cbioportal.org/
5. TCGA: https://portal.gdc.cancer.gov/
The protein sequences were downloaded from UniProt: https://www.uniprot.org/. The COMBINE dataset is available in the reference in the paper.
Competing interests
DH has no competing interests. AL is a founder, shareholder, receives fees from, and is a member of the board of directors of PMV pharmaceutical company, which produces small molecules that reactivate mutant p53 proteins. He also chairs the SAB of Janssen Pharmaceutical company and is a member of the board of Meira GTX, a gene therapy company for restoring eye sight. He is also a member of the board of directors of GeneCentric, a RNA seq diagnostic company for cancer treatments. He also receives funding from a grant from the NCI, P01CA087497-20. BG is a founder, and advisor of ROME, a company that explores the role of repetitive DNA sequences in cancers. AL is on the SAB of ROME.
Footnotes
Edited by G Melino
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Levine AJ p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer. (2020). 10.1038/s41568-020-0262-1. [DOI] [PubMed]
- 2.Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 1994;8:1235–1246. doi: 10.1101/gad.8.10.1235. [DOI] [PubMed] [Google Scholar]
- 3.Mello SS, Attardi LD. Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol. 2018;51:65–72. doi: 10.1016/j.ceb.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker KK, Levine AJ. Identification of a novel p53 functional domain which is necessary for efficient growth suppression. Proc Natl Acad Sci, USA. 1996;93:15335–15340. doi: 10.1073/pnas.93.26.15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baptiste N, Friedlander P, Chen X, Prives C. The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene. 2002;21:9–21. doi: 10.1038/sj.onc.1205015. [DOI] [PubMed] [Google Scholar]
- 6.Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018;25:154–160. doi: 10.1038/cdd.2017.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeffrey PD, Gorina S, Pavletich NP. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science. 1995;267:14987–1502. doi: 10.1126/science.7878469. [DOI] [PubMed] [Google Scholar]
- 8.Zhu J, Dou Z, Sammons MA, Levine AJ, Berger SL. Lysine methylation represses p53 activity in teratocarcinoma cancer cells. Proc Natl Acad Sci USA. 2016;113:9822–9827. doi: 10.1073/pnas.1610387113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beckman G, Birgander R, Själander A, Saha N, Holmberg PA, Kivelä A, et al. Is p53 polymorphism maintained by natural selection? Hum Hered. 1994;44:266–70. doi: 10.1159/000154228. [DOI] [PubMed] [Google Scholar]
- 10.Guha T, Malkin D. Inherited TP53 mutations and the li-fraumeni syndrome. Cold Spring Harb Perspect Med. 2017;7:a026187. doi: 10.1101/cshperspect.a026187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toledo F, Krummel KA, Lee CJ, Liu CW, Rodewald LW, Tang M, et al. A mouse p53 mutant lacking the proline-rich domain rescues Mdm4 deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory network. Cancer Cell. 2006;9:273–285. doi: 10.1016/j.ccr.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Zilfou JT, Hoffman WH, Sank M, George DL, Murphy M. The corepressor mSin3a interacts with the proline-rich domain of p53 and protects p53 from proteasome-mediated degradation. Mol Cell Biol. 2001;21:3974–85. doi: 10.1128/MCB.21.12.3974-3985.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger M, Sionov RV, Levine AJ, Haupt Y. A role for the polyproline domain of p53 in its regulation by Mdm2. J Biol Chem. 2001;276.6:3785–3790. doi: 10.1074/jbc.M008879200. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Yao L, Ouyang T, Li J, Wang T, Fan Z, et al. Codon 72 polymorphism predicts the pathologic response to neoadjuvant chemotherapy in patients with breast cancer. Clin Canc Res. 2005;11:7328–7333. doi: 10.1158/1078-0432.CCR-05-0507. [DOI] [PubMed] [Google Scholar]
- 15.Lee MG, Han J, Jeong SI, Her NG, Lee JH, Ha TK, et al. XAF1 directs apoptotic switch of p53 signaling through activation of HIPK2 and ZNF313. Proc Natl Acad Sci USA. 2014;111:15532–7. doi: 10.1073/pnas.1411746111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barnoud T, Parris JLD, Murphy M. Common genetic variants in the Tp53 pathway and their impact on cancer. Jour Mol Cell Biol. 2019;11:578–585. doi: 10.1093/jmcb/mjz052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi H, Tan SJ, Zhong H, Hu W, Levine A, Xiao CJ, et al. Winter temperature and UV are tightly linked to genetic changes in the p53 tumor suppressor pathway in Eastern Asia. Am J Hum Genet. 2009;84:534–41. doi: 10.1016/j.ajhg.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeron-Medina J, Wang X, Repapi E, Campbell MR, Su D, Castro-Giner F, et al. A polymorphic p53 response element in KIT ligand influences cancer risk and has undergone natural selection. Cell. 2013;155:410–22. doi: 10.1016/j.cell.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinto EM, Figueiredo BC, Chen W, Galvao HCR, Formiga MN, Fragoso MCBV, et al. XAF1 as a modifier of p53 function and cancer susceptibility. Sci Adv. 2020;6:eaba3231. doi: 10.1126/sciadv.aba3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37:865–76. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- 21.Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, et al. Toward a shared vision for cancer genomic data. N Engl J Med. 2016;375:1109–12. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7:818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–7. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, et al. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci. 2003;100:8424–9. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murakami Y, Hayashi K, Hirohashi S, Sekiya T. Aberrations of the tumor suppressor p53 and retinoblastoma genes in human hepatocellular carcinomas. Canc Res. 1991;51:5520–5. [PubMed] [Google Scholar]
- 27.Kawamura M, Kikuchi A, Kobayashi S, Hanada R, Yamamoto K, Horibe K, et al. Mutations of the p53 and ras genes in childhood t (1; 19)-acute lymphoblastic leukemia. Blood. 1995;85:2546–52. doi: 10.1182/blood.V85.9.2546.bloodjournal8592546. [DOI] [PubMed] [Google Scholar]
- 28.Toguchida J, Yamaguchi T, Dayton SH, Beaughamp RL, Herrera GE, Ishizaki K, et al. Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med. 1992;326:1301–8. doi: 10.1056/NEJM199205143262001. [DOI] [PubMed] [Google Scholar]
- 29.UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–D489. doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toyama T, Zhang Z, Nishio M, Hamaguchi M, Kondo N, Iwase H, et al. Association of TP53 codon 72 polymorphism and the outcome of adjuvant therapy in breast cancer patients. Breast Cancer Res. 2007;9:R34. doi: 10.1186/bcr1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weng Z, Rickles RJ, Feng S, Richard S, Shaw AS, Schreiber SL et al. Structure-function analysis of SH3 domains: SH3 binding specificity altered by single amino acid substitutions. Mol Cell Biol. :5627–34 (1995). 10.1128/MCB.15.10.5627. [DOI] [PMC free article] [PubMed]
- 32.Natan E, Baloglu C, Pagel K, Freund SM, Morgner N, Robinson CV, et al. Interaction of the p53 DNA-binding domain with its n-terminal extension modulates the stability of the p53 tetramer. J Mol Bio. 2011;409:358–68. doi: 10.1016/j.jmb.2011.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger M, Stahl N, Del Sal G, Haupt Y. Mutations in proline 82 of p53 impair its activation by Pin1 and Chk2 in response to DNA damage. Mol Cell Biol. 2005;25:5380–8. doi: 10.1128/MCB.25.13.5380-5388.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D’Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol. 2002;4:11–9. doi: 10.1038/ncb714. [DOI] [PubMed] [Google Scholar]
- 35.Dornan D, Shimizu H, Burch L, Smith AJ, Hupp TR. The proline repeat domain of p53 binds directly to the transcriptional coactivator p300 and allosterically controls DNA-dependent acetylation of p53. Mol Cell Biol. 2003;23:8846–8861. doi: 10.1128/MCB.23.23.8846-8861.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liston P, Fong WG, Kelly NL, Toji S, Miyazaki T, Conte D, et al. Identification of XAF-1 as an antagonist of XIAP anti-Caspase activity. Nat Cell Biol. 2001;3:128–133. doi: 10.1038/3590550237. [DOI] [PubMed] [Google Scholar]
- 37.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lunter G, Hein J. A nucleotide substitution model with nearest-neighbour interactions. Bioinformatics. 2004;20:i216–i223. doi: 10.1093/bioinformatics/bth901. [DOI] [PubMed] [Google Scholar]
- 39.Soria CD, Pacifici M, DiMarco M, Stephen SM & Rondindi C COMBINE: a coalesced mammal database of intrinsic and extrinsic traits, Ecology. 10-1002/ecy3344. [DOI] [PubMed]
- 40.Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, et al. The origins and evolution of the P53 family of genes. Cold Spring Harb Perspect Biol. 2010;2:a001198. doi: 10.1101/cshperspect.a001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data is included in the Figures and Tables in the manuscript.
Five databases were screened for somatic Tp53 missense mutations, solely in either the proline domain (amino acids 55–100) or the adjacent section of nine amino acids (amino acids 46–54). The web addresses for the five databases are:
1. GENIE: https://genie.cbioportal.org/
2. COSMIC: https://cancer.sanger.ac.uk/cosmic
3. IARC R20: https://tp53.isb-cgc.org/
4. MSK-IMPACT: https://www.cbioportal.org/
5. TCGA: https://portal.gdc.cancer.gov/
The protein sequences were downloaded from UniProt: https://www.uniprot.org/. The COMBINE dataset is available in the reference in the paper.