

Abstract
Genome-wide association studies (GWASs) have identified thousands of risk loci for psychiatric and substance use phenotypes, however the biological consequences of these loci remain largely unknown. We performed a transcriptome-wide association study of 10 psychiatric disorders and 6 substance use phenotypes (GWAS sample size range, N = 9725–807,553) using expression quantitative trait loci data from 532 prefrontal cortex samples. We estimated the correlation of genetically regulated expression between phenotype pairs, and compared the results with the genetic correlations. We identified 393 genes with at least one significant phenotype association, comprising 458 significant associations across 16 phenotypes. Overall, the transcriptomic correlations for phenotype pairs were significantly higher than the respective genetic correlations. For example, attention deficit hyperactivity disorder and autism spectrum disorder, both childhood developmental disorders, had significantly higher transcriptomic correlation (r = 0.84) than genetic correlation (r = 0.35). Finally, we tested the enrichment of phenotype-associated genes in gene co-expression networks built from human prefrontal cortex samples. Phenotype-associated genes were enriched in multiple gene co-expression modules and the implicated modules contained genes involved in mRNA splicing and glutamatergic receptors, among others. Together, our results highlight the utility of gene expression data in the understanding of functional gene mechanisms underlying psychiatric disorders and substance use phenotypes.
Subject terms: Gene expression, Transcriptomics
Introduction
Psychiatric and substance use disorders are a leading cause of disease burden and account for 28.5% of global years lived with disability [1]. Genome-wide association studies (GWASs) have identified hundreds of genomic regions that are linked to psychiatric disorders and substance use behaviours (“mental health” phenotypes) [2]. These GWAS also provide novel insights into the genetic architecture and shared genetic risk factors across mental health phenotypes. For example, the Brainstorm Consortium used GWAS data to estimate genetic correlations (rg) across ten psychiatric disorders, revealing considerable sharing of common genetic risk [3]. However, relatively few studies have explored the common pathological or molecular mechanisms which contribute to these disorders. The majority (~93%) of disease-associated genetic variants are located in non-protein coding regions of the genome [4] suggesting they act through the regulation of gene expression rather than by directly altering the protein product. In the present study, we will integrate genetic and transcriptomic information from the brain to explore common transcriptomic mechanisms across 16 mental health phenotypes.
Previous studies have integrated genetic and gene expression data to gain biological insights into psychiatric disorders. For example, Gandal et al. compared levels of differential gene expression in postmortem brain samples from patients with autism (ASD), schizophrenia (SCZ), bipolar disorder (BD), depression (DEP), and matched healthy controls, and revealed significant overlap of disease-related signatures between the disorders [5]. Transcriptomic changes were most severe in ASD and least severe in DEP, with SCZ and BD showing intermediate levels of severity. While this study provided important new insights into the sharing of molecular mechanisms across mental health disorders, comparison of observed levels of gene expression is susceptible to reverse causation, where traits may affect gene expression levels [6]. Imputation of gene expression levels based on whole-genome and RNA sequence reference data from healthy participants provides a unique way to investigate how the genetically regulated component of gene expression is shared across phenotypes [6].
We and others have shown that functional networks are critical for understanding pathway convergence of manifold genetic risk variants in psychiatric diseases [7]. Gene co-expression networks model correlated levels of gene expression and provide a way to explore how the activity of multiple biologically related genes within the same co-expression network influence disease risk. We have previously generated co-expression networks in 13 brain tissues from healthy GTEx donors and report an association between four co-expression networks and Major Depressive Disorder, suggesting a role for synaptic signalling and neuronal development pathways [7]. In their study of post-mortem gene expression in patients vs. healthy controls, Gandal et al. explored module-level differential expression and showed that a module strongly enriched for microglial markers was upregulated specifically in ASD while several other modules were downregulated across ASD, SCZ, and BD [8].
In the present study, we conduct a comprehensive exploration of differences in genetically regulated levels of gene expression across 16 mental health phenotypes, including 10 psychiatric disorders (attention-deficit/hyperactivity disorder [ADHD], anorexia nervosa [AN], anxiety [ANX], autism spectrum disorder [ASD], bipolar disorder [BIP], depression [DEP], obsessive compulsive disorder [OCD], post-traumatic stress disorder [PTSD], schizophrenia [SCZ], and Tourette syndrome [TS]) and 6 substance use phenotypes (age of smoking initiation [AgeSmk], lifetime cannabis use, [CanInit], cigarettes per day [CigDay], drinks per week [DrnkWk], smoking cessation [SmkCes], and smoking initiation [SmkInit]). First, we integrate GWAS summary statistics with gene expression data from the prefrontal cortex of 532 healthy (i.e. control) PsychENCODE donors. Second, we perform a systematic exploration of differences in genetically regulated levels of gene expression for the 16 individual phenotypes and delineate genetic and transcriptomic overlap across phenotypes. Third, we generate co-expression networks and explore enrichment of GWAS association signals within network modules.
Materials and methods
Description of the GWAS summary statistics
We included 10 psychiatric disorders and 6 substance use phenotypes (which we collectively refer to as “mental health” phenotypes) in our analyses. We selected only mental health phenotypes with significant SNP-based heritability (Z-score > 2). Details on the individual GWAS samples, including references, sample sizes and SNP-based heritability estimates, are provided in Table 1 and Supplementary Table 1.
Table 1.
Sample descriptions and SNP-based heritabilities of 16 mental health traits.
| Phenotype (abbreviation) | PMID | Sample size (N) | SNP-based heritability | TWAS FUSION (N) | ||||
|---|---|---|---|---|---|---|---|---|
| Case | Control | Total | h2snp (se) | Z | Genes | Novel | ||
| Anorexia Nervosa (AN) | 28494655 | 16,992 | 55,525 | 72,517 | 0.429 (0.0282) | 15.21 | 74 | 20 |
| Attention Deficit Hyperactivity Disorder (ADHD) | 30478444 | 19,099 | 34,194 | 53,293 | 0.217 (0.0141) | 15.41 | 53 | 12 |
| Autism Spectrum Disorder (ASD) | 30804558 | 18,382 | 27,969 | 46,351 | 0.112 (0.0097) | 11.54 | 54 | 13 |
| Anxiety Disorders (ANX) | 26754954 | 31,977 | 82,114 | 114,019 | 0.125 (0.0090) | 13.92 | 42 | 41 |
| Bipolar Disorder (BIP) | 31043756 | 20,352 | 31,358 | 51,710 | 0.200 (0.0101) | 19.83 | 171 | 58 |
| Depression (DEP) | 30718901 | 246,363 | 561,190 | 807,553 | 0.073 (0.0025) | 29.00 | 185 | 37 |
| Obsessive Compulsive Disorder (OCD) | 28761083 | 2688 | 7037 | 9725 | 0.280 (0.0432) | 6.48 | 15 | 15 |
| Post Traumatic Stress Disorder (PTSD) | 31594949 | 23,212 | 151,447 | 174,659 | 0.053 (0.0095) | 5.53 | 18 | 13 |
| Schizophrenia (SCZ) | 29483656 | 40,675 | 64,643 | 105,318 | 0.234 (0.0083) | 28.17 | 597 | 112 |
| Tourette’s Syndrome (TS) | 30818990 | 4819 | 9488 | 14,307 | 0.213 (0.0248) | 8.59 | 27 | 15 |
| Drinks Per Week (DrnkWk) | 30643251 | – | – | 537,349 | 0.049 (0.0021) | 23.19 | 260 | 62 |
| Smoking Initiation (SmkInit) | 30643251 | 311,629 | 321,173 | 632,802 | 0.104 (0.0033) | 31.64 | 312 | 72 |
| Cigarettes Per Day (CigDay) | 30643251 | – | NA | 263,954 | 0.073 (0.0069) | 10.52 | 163 | 36 |
| Smoking Cessation (SmkCes) | 30643251 | 92,573 | 220,248 | 312,821 | 0.060 (0.0039) | 15.36 | 61 | 19 |
| Age of Smoking Initiation (AgeSmk) | 30643251 | – | – | 262,990 | 0.047 (0.0028) | 16.93 | 70 | 23 |
| Cannabis Use Initiation (CanInit) | 30150663 | 43,380 | 118,702 | 162,082 | 0.118 (0.0075) | 15.69 | 74 | 24 |
PsychENCODE RNAseq data
We obtained a gene expression data measured from the prefrontal cortex in 532 healthy control subjects from the PsychECODE project (http://resource.psychencode.org/) [9]. PsychENCODE provides the largest available resource for the interrogation of genetically regulated gene expression in human brain, and includes uniformly processed samples from multiple sources, including GTEx (v8) and the CommonMind Consortium. An overview of RNAseq data preparation is provided in Supplementary Material 1.
TWAS FUSION gene annotation
We used TWAS FUSION [10] (web resources, Supplementary Material 2) to integrate eQTL information from the PsychENCODE project with GWAS summary statistics for 16 mental health phenotypes (Table 1) to identify genes whose genetically predicted expression levels are associated with each phenotype. We used expression weights generated by the PsychENCODE consortium [11], and Linkage Disequilibrium information from the 1000 Genomes Project Phase 3 [12]. These data were processed with the beta coefficients or odds ratios from each GWAS to estimate the expression-GWAS association statistic. For each phenotype, we adjusted for multiple testing using Bonferroni correction. We performed empirical Brown’s test [13] to combine TWAS FUSION P values to rank order genes based on their strength of association across the 16 phenotypes. The Brown’s test was performed on genes for which TWAS FUSION association statistics results were available for all 16 phenotypes.
Transcriptome-wide correlation analysis
We estimated the genome-wide genetic correlation between each pair of mental health phenotypes as a function of the predicted gene expression effect from TWAS FUSION using RhoGE [14]. Transcriptome correlations were performed using both the full distribution of FUSION results and after excluding the MHC region. RhoGE estimates the mediating effect of genetically regulated gene expression before calculating the correlation of effect sizes between pairs of phenotypes. The approximate sample sizes for each GWAS were extracted from the respective publicly available summary statistics, or calculated as the sum of cases and controls. The false discovery rate (FDR < 0.05) was used in multiple hypothesis testing to correct for multiple comparisons.
Genetic correlations and estimates of h2SNP
LD Score Regression was used to estimate SNP-based heritability (h2SNP) and genetic correlations between each pair of the 16 phenotypes, before and after the exclusion of the MHC region. SNP-based heritability for case-control phenotypes was estimated on the liability scale (see Supplementary Table 1 for the sample and population prevalence of each phenotype). The false discovery rate (FDR < 0.05) was used in multiple hypothesis testing to correct for multiple comparisons.
Hierarchical cluster analysis
We performed hierarchical clustering analysis for both transcriptomic and genetic correlations in order to examine the underlying genetic and transcriptomic structure between the 16 phenotypes. Complete-linkage clustering was implemented using the hclust function in R [15], where dissimilarity between phenotype pairs was defined as one minus the (genetic or transcriptomic) correlation.
Gene co-expression network analysis
We constructed a gene co-expression network from 532 prefrontal cortex control samples collected by the PsychENCODE project, using the weighted gene co-expression network analysis (WGCNA) package in R [16]. A signed pairwise correlation matrix using Pearson’s product moment correlation coefficient was calculated. A “soft-thresholding” value of 14 was selected by plotting the strength of correlation against a series (range 2–20) of soft threshold powers. The correlation matrix was transformed into an adjacency matrix, which was subsequently was normalised using a topological overlap function. Hierarchical clustering was performed using average linkage, with one minus the topological overlap matrix as the distance measure. The hierarchical cluster tree was cut into gene modules using the dynamic tree cut algorithm, with a minimum module size of 30 genes. We merged modules if the correlation between their eigengenes – defined as the first principal component of their genes’ expression values – was greater or equal to 0.8.
Gene-set analysis of co-expression networks
To identify gene co-expression networks enriched with candidate risk genes for each mental health phenotype, we performed gene-set analysis of TWAS FUSION results in tissue-specific gene co-expression networks using the gene-set analysis function in MAGMA v1.08 [17]. First, we generated an annotation (.annot) file, which contains SNPs mapped to (protein-coding) genes, for each phenotype using the default --annot function in MAGMA. Second, we generated raw (.raw) files from a gene-based analysis using the --snp-wise = mean function, which calculates an association statistic for each gene using the weighted sum of P values for a predefined genomic window (5 kilobases upstream and 1.5 kilobases downstream). The 1000 Genomes European reference panel (Phase 3) [12] was used to account for Linkage Disequilibrium between SNPs. Third, we modified the intermediary.raw files by replacing the MAGMA Z scores with TWAS FUSION Z scores for each gene. Finally, we ran a gene-level analysis to test the enrichment of TWAS FUSION association signals in the gene co-expression network modules from prefrontal cortex.
Characterisation of gene co-expression modules
Gene expression modules enriched with mental health phenotype GWAS association signals were assessed for biological pathways using g:Profiler (https://biit.cs.ut.ee/gprofiler/) [18]. Ensembl gene identifiers within enriched gene modules were used as input; we tested for the over-representation of module genes in Gene Ontology (GO) biological process terms, as well as KEGG [19] and Reactome [20] gene pathways. Multiple testing correction was done using g:SCS; this approach accounts for the correlated structure of GO terms and biological pathways, and corresponds to an experiment-wide threshold of α = 0.05.
Results
Transcriptome-wide association study
We calculated the association between imputed genetically regulated gene expression from prefrontal cortex and 16 psychiatric phenotypes using TWAS FUSION (Supplementary Table 2). We identified 393 genes significantly associated with at least one phenotype, comprising 458 significant associations across the 16 phenotypes. Of these, 264 were related to psychiatric disorders and 194 to substance use phenotypes. Within psychiatric phenotypes, the largest number of significant TWAS FUSION associations was observed for schizophrenia (N = 168) followed by depression (N = 41), while smoking initiation (N = 76) and drinks per week (N = 73) accounted for the largest number of substance use associations. After the removal of genes identified through proximity-based gene mapping, 303 genes (66%) associations were unique to TWAS FUSION (Supplementary Material 3; Supplementary Table 3). Results excluding the MHC region are presented in Supplementary Table 4.
We conducted empirical Brown’s test to rank-order genes of which the imputed gene expression levels are most strongly associated across the 16 phenotypes (Fig. 1). Interestingly, 8 of the top 20 most strongly associated genes across all phenotypes showed a significant association with concordant effects in depression and schizophrenia. It should be noted, however, that most of the associations were linked to the MHC region. Full results are presented in Supplementary Table 5 (including MHC) and Supplementary Table 6 (excluding MHC).
Fig. 1. Heatmap of TWAS associations for 20 top ranked genes across 16 mental health phenotypes.

Each square contains the TWAS Z score for the gene-phenotype association.
Genetic and transcriptomic correlations across 16 phenotypes
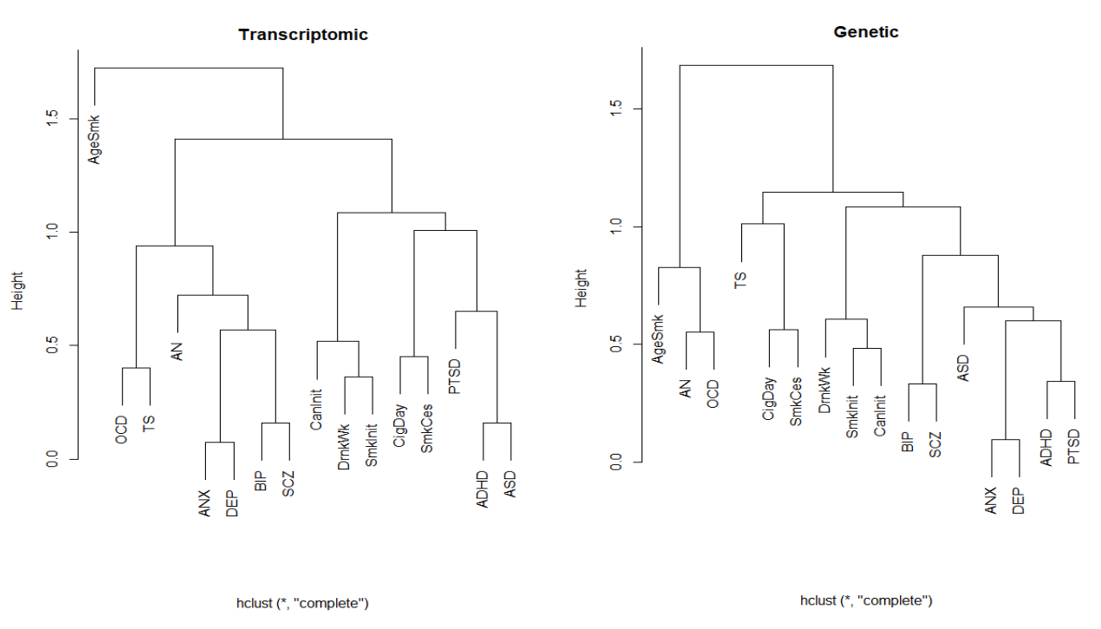
All phenotypes exhibited significant SNP-based heritability (Table 1 and Supplementary Table 7). We estimated correlations across the 16 pairs of phenotypes based on genetic variation (ρg) using LDSC (Fig. 2; below diagonal) and predicted expression (ρt) using RhoGE (Fig. 2, above diagonal), after excluding the MHC region. Tabulated data for genetic and transcriptomic correlations, including and excluding the MHC region, are shown in Supplementary Tables 8–11, and Supplementary Figure 1 shows the pairwise correlation differences between phenotypes. The genetic correlations (mean absolute ρg = 0.23; SD = 0.25) were significantly lower than the transcriptomic correlations (mean absolute ρt = 0.30; SD = 0.31). This was confirmed by a paired sample t-test; t-statistic = −3.48; P < 0.001 and by a test evaluating whether differences between Z-transformed ρ’s were significantly different from 0 (Z = −21.53, p < 2.2−e16; N = conservatively set at 1000 to calculate the population SD of the Z-scores based on SE = 1/sqrt (N = 3)). The genetic correlations explained a large proportion of the variance in transcriptomic correlations (Fig. 3; R2 = 0.7808; P < 2.2 × 10−16), with the most pronounced difference between ADHD and ASD, with ρg = 0.35 (SE = 0.05) and ρt = 0.84 (SE = 0.05). A hierarchical cluster analyses of genetic and transcriptomic correlations showed similar groupings between genetic and transcriptomic correlations (Supplementary Fig. 2). Both analyses, for example, were suggestive of strong sharing of genetic risk factors between anxiety and depression, and between bipolar disorder and schizophrenia. However, despite these similarities, some interesting differences were also revealed. For example, ADHD and ASD were grouped together in the transcriptomic cluster analysis but not the genetic cluster analysis, in line with the results from the genetic and transcriptomic correlation analysis.
Fig. 2. Heatmap of genetic and transcriptomic correlations between 16 mental health phenotypes.

Squares below the diagonal contain genetic correlations, while squares above the diagonal contain transcriptomic correlations. All correlations exclude the MHC region.
Fig. 3. Scatter plot of genetic and transcriptomic correlations across 16 mental health phenotypes.
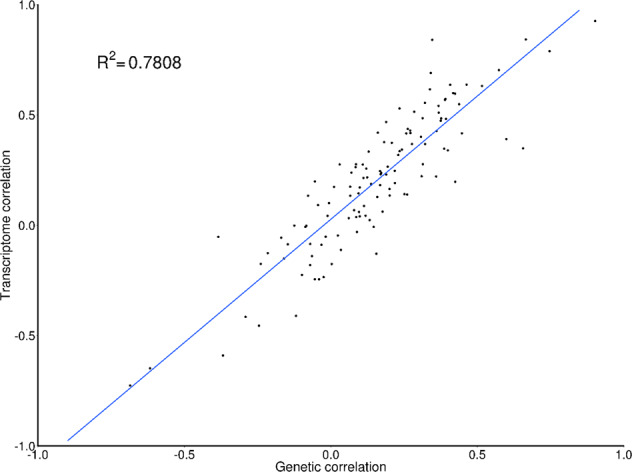
Correlations exclude the MHC region.
Co-expression network analysis
Gene co-expression network analysis organizes genes highly correlated genes into functionally related groups, and is therefore a useful molecular substrate for the biological interpretation of gene-based disease associations. Our gene co-expression expression network analysis identified 25 gene co-expression modules which ranged between 85 and 3042 genes in size. Biological pathway enrichment analysis showed each module contained genes involved in the same or similar biological pathways (for example, the immune response [module M9] or trans-synaptic signalling [M25]; Supplementary Table 12). Gene-set enrichment of TWAS FUSION associations found 6 modules were associated with at least one psychiatric disorder (FDR < 0.05) (Fig. 4). The strongest association was found between module M19, enriched with genes involved in mRNA splicing, and anxiety (FDR = 0.0063). Full results are provided in Supplementary Table 13 (including the MHC region) and Supplementary Table 14 (excluding MHC). We partitioned the heritability explained by the six modules (Supplementary Fig. 3) and showed 7 significant associations after Bonferroni correction for number of modules and phenotypes tested (P < 0.000054) (Supplementary Material 4; Supplementary Table 15). After including baseline functional annotations, a single module (module M10), enriched with nucleic acid and RNA metabolism pathways, remained significant in bipolar disorder and schizophrenia (Supplementary Table 16).
Fig. 4. Circos plot of TWAS Z scores, modular enrichments, and significant transcriptomic correlations across 16 mental health phenotypes.
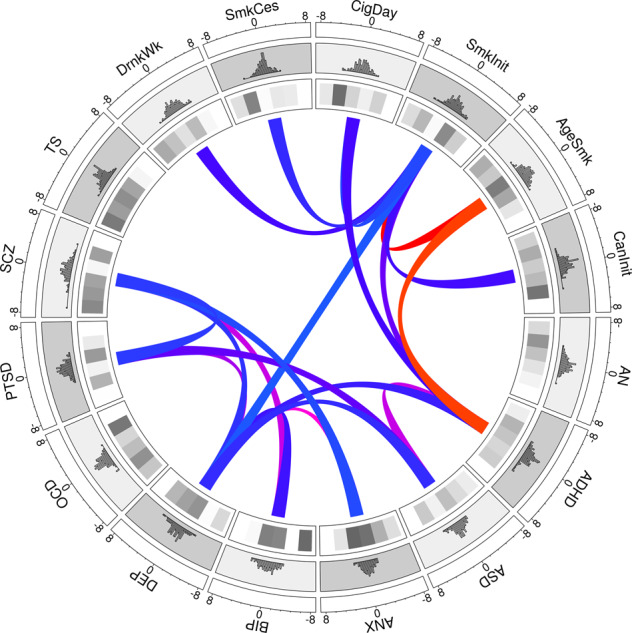
The outermost layer shows the distribution of TWAS Z scores for each phenotype, while the inner most layer shows the enrichment Z scores for each of the six significant co-expression modules in prefrontal cortex, with darker shading signifying greater enrichment. The inner ribbons represent significant transcriptomic correlations across phenotype pairs, with positive correlations colored blue and negative correlations colored red.
Discussion
We performed a systematic analysis of genetic and transcriptomic risk factors underlying psychiatric and substance use phenotypes. A transcriptome-wide association study identified 458 significant gene-phenotype associations, representing 393 unique genes. Many significant genes had concordant expression effect directions across phenotypes, suggesting extensive pleiotropy at the level of genetically regulated gene expression. The transcriptomic correlation for each phenotype pair was, on average, significantly larger than the corresponding genetic correlation. Transcriptomic associations for six psychiatric phenotypes were enriched in gene co-expression modules from prefrontal cortex and implicated multiple biological pathways in disease susceptibility. These data suggest the study of genetically regulated gene expression may reveal important information on biological pleiotropy underlying genetically correlated phenotypes.
We ranked genes whose expression is most strongly associated with multiple phenotypes. Increased expression of the most highly ranked gene, PSMA4, was significantly associated with schizophrenia, cigarettes per day, and smoking cessation. The gene PSMA4, located within the 15q25.1 gene cluster, is associated with nicotine dependence and lung cancer [21], and we recently linked its expression in multiple GTEx brain tissues to cigarettes per day and smoking cessation [22]. PSMA4 is also one of six “high confidence” genes in schizophrenia, based on probabilistic fine mapping approaches and observed expression profiles [23]. Using observed expression data, these authors found decreased PSMA4 expression in prefrontal cortex and hippocampus was associated with schizophrenia, while we reported the opposite effect direction with imputed (genetically regulated) gene expression in prefrontal cortex. Our association is consistent with previously reported PSMA4 TWAS associations for schizophrenia in brain [24]. It is possible the observed expression data (GSE21138) were confounded by a hidden variable, such as smoking status, which may explain the association with PSMA4 in schizophrenia cases compared to controls, rather than a causal disease process. This highlights an advantage of TWAS, which removes environmental noise by measuring the genetically regulated component of gene expression.
We found half of phenotype pairs had significant (FDR < 0.05) genome-wide genetic correlations at the level of predicted (genetically regulated) gene expression. While there was a strong relationship between the genetic and transcriptomic correlations (R2 = 0.78, P < 2.2 × 10−16), predicted expression was on average more strongly correlated than genetic variation. This difference may be explained by noise in the TWAS predictor (i.e. genetically regulated gene expression), which could, in turn, bias the transcriptomic correlations. However, TWAS maintains good control of the type-I error rate [25], suggesting any noise in the predictor would not inflate error rates in the transcriptomic correlations. Furthermore, noise in the predictor would bias the expression slope towards zero due to regression dilution, leading to an under-estimate (rather than inflation) of the transcriptomic correlations. Nonetheless, there is unresolved debate on the validity of the TWAS model and the extent to which the method can be used to reliably interpret GWAS summary statistics [26]. Future work using both simulated and observed data will be required to assess the accuracy of the test of the association between imputed gene expression and a phenotype.
The largest difference in genetic and transcriptomic correlations was observed for ASD and ADHD, two common childhood onset neurodevelopmental disorders. The strong transcriptomic correlation (ρt = 0.84) between ASD and ADHD is supported by their phenotypic similarity, where a large proportion of children (37–85%) with ASD have comorbid symptoms of ADHD [27]. While clinical guidelines dictate ASD cannot be diagnosed in the presence of ADHD, our data suggests the high co-occurrence of these disorders is due to a shared genetic regulation [28]. Furthermore, exome sequencing of children with ASD and ADHD indicated that they have a similar burden of rare protein-truncating variants [29]. This suggests the difference between the genetic and transcriptomic correlations may be due to the unaccounted effects of rare variation.
We can only speculate as to why the genetic correlations are generally lower than their respective transcriptomic correlations. First, phenotype pairs with highly correlated gene expression effects in cis (<0.5 Mb) regions but weakly correlated effects in trans-regions will have a higher transcriptomic correlation compared to their genetic correlation [14]. Second, it is possible the assumptions of LDSC, such as a highly polygenic genetic architecture underlying the investigated phenotypes, may be violated in our study. For example, LDSC may underestimate shared genetic regulation by incorrectly modelling the contribution of genomic regions more strongly enriched for heritability for some mental health phenotypes, while the transcriptomic correlation captures a truly high genetic overlap. Third, the difference between genetic and transcriptomic correlations may simply be a consequence of differences in GWAS sample size between phenotype pairs. Finally, highly discordant phenotype pairs such as ASD and ADHD (and to lesser extent SCZ) are known to have developmental origins and therefore adult gene expression may not be the most relevant molecular substrate for disease variant interpretation and correlation. Future studies may therefore leverage developmental eQTL data [30] to compare genetic and transcriptomic correlations over different stages of development.
A gene co-expression network analysis of prefrontal cortex tissue samples from PsychENCODE identified several gene modules enriched with gene-based associations for four psychiatric disorders (ANX, BIP, OCD, and ASD), and three substance use phenotypes (CPD, CanInit, and AgeSmk). The use of healthy brain samples to build the co-expression modules ensured the observed enrichments were unlikely to be affected by disease-associated expression signatures. The most strongly associated module was associated with ANX and strongly enriched in biological pathways associated with mRNA splicing. Splicing is genetically regulated [31] and can influence gene expression in particular tissues, giving rise to different functional effects such as altered neuronal connectivity and synaptic firing properties in the brain [32]. Alternative mRNA splicing events are associated with diverse psychiatric disorders, including SCZ [33], ASD [34], BIP [35], and DEP [36], highlighting the importance of alternative splicing in psychiatric disease susceptibility. Current genomic resources, such as GTEx [31], will help researchers better understand how genetic variants affect gene expression through alternative splicing events. Other phenotype-associated modules were enriched with biologically meaningful pathways. For example, the module M1 was associated with bipolar disorder and enriched with genes involved in the regulation of metabotropic glutamate receptors. Glutamatergic receptors are the primary effectors of glutamate, a critical excitatory neurotransmitter, and their dysregulation is implicated in many psychiatric disorders [37], including bipolar disorder [38]. Collectively, these data suggest gene co-expression networks may be used for the biological characterisation of genetic risk factors underlying psychiatric and substance use phenotypes.
The findings of this study should be interpreted in view of the following limitations. First, the TWAS expression weights were estimated using bulk tissue gene expression data from prefrontal cortex. While brain is the pathogenic tissue of interest for the phenotypes included in our study, we will likely miss tissue or cell-type-specific effects on gene expression which, in turn, may influence the transcriptomic correlations. Second, the TWAS FUSION approach does not test whether gene expression and a phenotype are affected by the same causal SNP in a cis-eQTL region. As such, the approach does not provide direct evidence of causal relationship between expression and disease risk. Mendelian randomisation-based approaches, such as SMR [39] and MR-JTI [25], may refine our list of gene candidates by selecting genes most likely associated through pleiotropy, where gene expression and a phenotype are affected by the same causal variant. Finally, our gene co-expression analyses rely on the stability (i.e. robustness) of gene co-expression networks in prefrontal cortex. We built signed networks using similar parameters to those described by Gandal et al. [11]. Using a permutation procedure, these authors concluded their modules were robust to the influence of outlier samples on network architecture, providing confidence in the stability of our co-expression network.
Our study highlights the benefits of integrating GWAS and gene expression data to characterise biological pleiotropy and functional mechanisms underlying complex phenotypes. By integrating transcriptomic data from prefrontal cortex with GWAS data, we identified hundreds of genes associated with mental health phenotypes. Furthermore, many genes were associated with multiple phenotypes, highlighting extensive biological pleiotropy. We found a significant difference between transcriptomic and genetic correlations across all phenotype pairs, and the magnitude of the difference was particularly large for ADHD and ASD. These data suggest transcriptomic correlations may provide additional insight into the functional relationship between mental health phenotypes. Finally, we observed some enrichment of candidate risk genes for several phenotypes within co-expression networks from prefrontal cortex, suggesting our approach will prove useful in characterising the functional impact of phenotype-associated genetic variation. Future analyses could extend our approach by incorporating additional sources of genomic (for example, epigenetic marks) and statistical (e.g. SNP priors) information within co-expression networks.
Supplementary information
{kind=link}
{kind=link}
Acknowledgements
ERG is supported by NIH/NHGRI R35HG010718, NIH/NHGRI R01HG011138, NIH/NIA AG068026, and NIH/NIGMS R01GM140287. ERG, EMD, and ZFG are supported by NIH/NIA AG068026
Data availability
The datasets analysed for the current study are publicly available. A full list of GWAS used in the current study are listed in Table 1.
Competing interests
The authors declare no competing interests.
Ethical approval
No ethical approval was required. The study used de-identified summary-level (GWAS) and gene expression data.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41431-022-01037-6.
References
- 1.Whiteford HA, Ferrari AJ, Degenhardt L, Feigin V, Vos T. The global burden of mental, neurological and substance use disorders: an analysis from the global burden of disease study 2010. PLoS One. 2015;10:e0116820. doi: 10.1371/journal.pone.0116820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Børglum AD, Breen G, et al. Psychiatric genomics: an update and an agenda. Am J Psychiatry. 2018;175:15–27. doi: 10.1176/appi.ajp.2017.17030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L et al. Analysis of shared heritability in common disorders of the brain. Science. 2018; 360. 10.1126/science.aap8757. [DOI] [PMC free article] [PubMed]
- 4.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science. 2018;359:693 LP–697. doi: 10.1126/science.aad6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47:1091–8. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerring ZF, Gamazon ER, Derks EM. A gene co-expression network-based analysis of multiple brain tissues reveals novel genes and molecular pathways underlying major depression. PLOS Genet. 2019;15:e1008245. doi: 10.1371/journal.pgen.1008245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gandal MJ, Leppa V, Won H, Parikshak NN, Geschwind DH. The road to precision psychiatry: translating genetics into disease mechanisms. Nat Neurosci. 2016;19:1397–407. doi: 10.1038/nn.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362:eaat8464. doi: 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48:245–52. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science (80-) 2018;362:eaat8127. doi: 10.1126/science.aat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delaneau O, Marchini J, Consortium T. 1000 GP. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun. 2014;5:3934. doi: 10.1038/ncomms4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poole W, Gibbs DL, Shmulevich I, Bernard B, Knijnenburg TA. Combining dependent P-values with an empirical adaptation of Brown’s method. Bioinformatics. 2016;32:i430–i436. doi: 10.1093/bioinformatics/btw438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mancuso N, Shi H, Goddard P, Kichaev G, Gusev A, Pasaniuc B. Integrating gene expression with summary association statistics to identify genes associated with 30 complex traits. Am J Hum Genet. 2017;100:473–87. doi: 10.1016/j.ajhg.2017.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Müllner D. Fastcluster: fast hierarchical, agglomerative clustering routines for R and Python. J Stat Softw. 2013;53:1–18. doi: 10.18637/jss.v053.i09. [DOI] [Google Scholar]
- 16.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 2008;9:1–13. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLOS Comput Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, et al. g:Profiler—a web server for functional interpretation of gene lists (2016 update) Nucleic Acids Res. 2016;44:W83–W89. doi: 10.1093/nar/gkw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joshi-Tope G, Gillespie M, Vastrik I, D’Eustachio P, Schmidt E, de Bono B, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–22. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marees AT, Gamazon ER, Gerring Z, Vorspan F, Fingal J, van den Brink W, et al. Post-GWAS analysis of six substance use traits improves the identification and functional interpretation of genetic risk loci. Drug Alcohol Depend. 2019;206:107703. doi: 10.1016/j.drugalcdep.2019.107703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma C, Gu C, Huo Y, Li X, Luo X-J. The integrated landscape of causal genes and pathways in schizophrenia. Transl Psychiatry. 2018;8:67. doi: 10.1038/s41398-018-0114-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pividori M, Rajagopal PS, Barbeira A, Liang Y, Melia O, Bastarache L, et al. PhenomeXcan: mapping the genome to the phenome through the transcriptome. Sci Adv. 2020;6:eaba2083. doi: 10.1126/sciadv.aba2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou D, Jiang Y, Zhong X, Cox NJ, Liu C, Gamazon ER. A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat Genet. 2020;52:1239–46. doi: 10.1038/s41588-020-0706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Leeuw C, Werme J, Savage J, Peyrot W, Posthuma D. Reconsidering the validity of transcriptome-wide association studies. bioRxiv. 2021; https://www.biorxiv.org/content/10.1101/2021.08.15.456414v1.
- 27.Leitner Y. The co-occurrence of autism and attention deficit hyperactivity disorder in children—what do we know? Front Hum Neurosci. 2014;8:268. doi: 10.3389/fnhum.2014.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rommelse NNJ, Geurts HM, Franke B, Buitelaar JK, Hartman CA. A review on cognitive and brain endophenotypes that may be common in autism spectrum disorder and attention-deficit/hyperactivity disorder and facilitate the search for pleiotropic genes. Neurosci Biobehav Rev. 2011;35:1363–96. doi: 10.1016/j.neubiorev.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 29.Satterstrom FK, Walters RK, Singh T, Wigdor EM, Lescai F, Demontis D, et al. Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat Neurosci. 2019;22:1961–5. doi: 10.1038/s41593-019-0527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strober BJ, Elorbany R, Rhodes K, Krishnan N, Tayeb K, Battle A, et al. Dynamic genetic regulation of gene expression during cellular differentiation. Science. 2019;364:1287 LP–1290. doi: 10.1126/science.aaw0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318 LP–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vuong CK, Black DL, Zheng S. The neurogenetics of alternative splicing. Nat Rev Neurosci. 2016;17:265–81. doi: 10.1038/nrn.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flaherty E, Zhu S, Barretto N, Cheng E, Deans PJM, Fernando MB, et al. Neuronal impact of patient-specific aberrant NRXN1α splicing. Nat Genet. 2019;51:1679–90. doi: 10.1038/s41588-019-0539-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farini D, Cesari E, Weatheritt RJ, La Sala G, Naro C, Pagliarini V, et al. A dynamic splicing program ensures proper synaptic connections in the developing cerebellum. Cell Rep. 2020;31:107703. doi: 10.1016/j.celrep.2020.107703. [DOI] [PubMed] [Google Scholar]
- 35.Harrison PJ, Geddes JR, Tunbridge EM. The emerging neurobiology of bipolar disorder. Trends Neurosci. 2018;41:18–30. doi: 10.1016/j.tins.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le François B, Zhang L, Mahajan GJ, Stockmeier CA, Friedman E, Albert PR. A novel alternative splicing mechanism that enhances human 5-HT1A receptor RNA stability is altered in major depression. J Neurosci. 2018;38:8200 LP–8210. doi: 10.1523/JNEUROSCI.0902-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crupi R, Impellizzeri D, Cuzzocrea S. Role of metabotropic glutamate receptors in neurological disorders. Front Mol Neurosci. 2019;12:20. doi: 10.3389/fnmol.2019.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martí SB, Cichon S, Propping P, Nöthen M. Metabotropic glutamate receptor 3 (GRM3) gene variation is not associated with schizophrenia or bipolar affective disorder in the German population. Am J Med Genet. 2002;114:46–50. doi: 10.1002/ajmg.1624. [DOI] [PubMed] [Google Scholar]
- 39.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–7. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analysed for the current study are publicly available. A full list of GWAS used in the current study are listed in Table 1.