

Abstract
Inherited errors of mitochondrial fatty acid β-oxidation (FAO) are life threatening, even with optimum care. FAO is the major source of energy for heart and is critical for skeletal muscles especially during physiologic stress. Clinical trials revealed that triheptanoin (commercially known as Dojolvi; C7G), improved heart function and decreased hypoglycemia in long chain FAO disorders, but other symptoms including rhabdomyolysis persisted, suggesting suboptimal tissue distribution/utilization of heptanoic acid (C7) conjugates and/or rapid liver breakdown. In this study, medium branched chain fatty acids were tested as potential anaplerotic treatments in fibroblasts from patients deficient in very long chain acyl-CoA dehydrogenase (VLCAD), long chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), trifunctional protein (TFP), and carnitine palmitoyltransferase II (CPT II). Cells were cultured to near confluency and treated with C7, 2,6-dimethylheptanoic acid (dMC7), 6-amino-2,4-dimethylheptanoic acid (AdMC7), or 4,8-dimethylnonanoic acid (dMC9) for 72 h and targeted metabolomics performed. The profile of TCA cycle intermediates was improved in cells treated with these branched chain fatty acids compared with C7. Intracellular propionate was higher in AdMC7 treated cells compared with C7 in VLCAD, LCHAD, and TFP deficient cell lines. With AdMC7 treatment, succinate was higher in CPT II and VLCAD deficient cells, compared with C7. Malate and glutamate were consistently higher in AdMC7 treated VLCAD, LCHAD, TFP, and CPT II deficient cells compared with the C7 treatment. The results provide the impetus to further evaluate and consider branched chain fatty acids as viable anaplerotic therapy for fatty acid oxidation disorders and other diseases.
Keywords: CPT II deficiency, Dojolvi, LC-FAOD, LCHAD deficiency, long chain fatty acid oxidation disorders, MCT oil, medium branched chain fatty acids, TFP deficiency, triheptanoin, VLCAD deficiency
1 |. INTRODUCTION
Traditionally, long chain fatty acid oxidation disorders have been treated with medium chain triglyceride oil (MCT; most often trioctanoylglycerol, C8G) to bypass the block in the β-oxidation pathway, but patients continue to show episodes of metabolic decompensation that include hypoglycemia, rhabdomyolysis, and cardiomyopathy. Triheptanoin (commercially known as Dojolvi; C7G), has been approved as an anaplerotic drug treatment for long chain fatty acid β-oxidation (FAO) disorders and has also been investigated as a treatment for other diseases including glucose transporter type I deficiency G1D, epilepsy, and others.1–21 Clinical trials have shown that patients with FAO disorders (FAODs) treated with C7G have a lower frequency of these metabolic decompensation events; however, with both C7G and C8G treatments, rhabdomyolysis requiring hospitalization and other neurological symptoms persist. This provides the impetus to explore alternatives to heptanoic acid (C7) that compensate for the deficiency in energy while providing increased levels of TCA cycle intermediates to meet various physiological demands.
Biochemically, following the release of C7 from its glycerol backbone by intestinal lipases, it undergoes passive transport into mitochondria where it is esterified to the active metabolic form, heptanoyl-CoA, and undergoes two cycles of β-oxidation. The apparent physiologic advantage of C7 over octanoic acid (C8) is in providing a propionyl-CoA in addition to two acetyl-CoAs. Propionyl-CoA is an anaplerotic molecule that replenishes the TCA cycle by converting to succinyl-CoA, which has other crucial functions including a role in modulating activity of a large number of proteins and being the precursor of porphyrins (see Scheme 1). However, rapid incomplete liver utilization of C7 is easily demonstrable by the accumulation of valeryl (C5) and propionyl (C3) conjugates in blood of patients at the dosage tested. This rapid breakdown of C7 in the liver can be attributed to the abundance of medium chain acyl-CoA dehydrogenase (MCAD) and short chain acyl-CoA dehydrogenase (SCAD) in liver that catalyze the key first step of heptanoyl-CoA oxidation.
SCHEME 1.
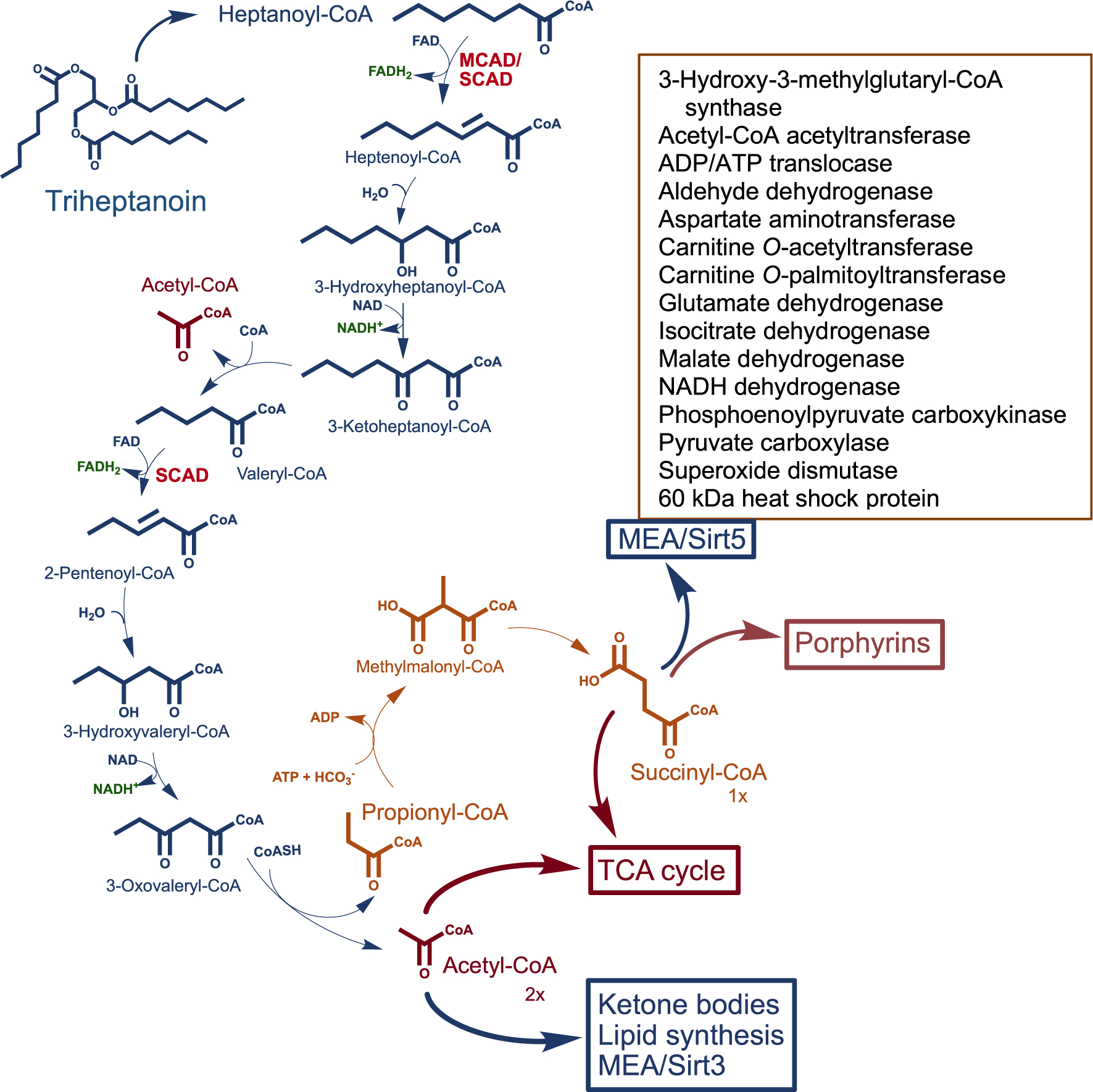
Proposed metabolic pathway of triheptanoin (triheptanoylglycerol). Ultimately two acetyl-CoAs and one succinyl-CoA are expected to be the anaplerotic contribution of the drug to the TCA cycle. Acetyl-CoA enters the TCA cycle to form citrate but has other functions including ketone body formation and used in protein acetylation for modulating enzyme activity (MEA), and lipid synthesis. Succinyl-CoA is a TCA cycle intermediate and other crucial functions including protein succinylation for MEA (box inset lists examples of enzymes modulated by succinylation/Sirtuin 5 desuccinylation), generating one GTP per cycle, and porphyrin synthesis
Reasons why C7G does not remedy all disease symptoms in patients with long chain FAODs are yet to be determined. One possibility is that with a rapid metabolism by the liver, suboptimum tissue distribution of C7 and its metabolites does not provide adequate anaplerosis (ie, replenishment of TCA cycle intermediates) to rescue the high demand of TCA cycle intermediates in other tissues, including muscle. In this context, it is reasonable to consider other alternatives to C7 as anaplerotic compounds that might not be metabolized as rapidly in the liver. Medium branched chain acyl-CoA esters are substrates for long chain acyl-CoA dehydrogenase (LCAD), which is less abundant in human liver than either MCAD or SCAD, and thus attractive alternatives as anaplerotic compounds. Among the acyl-CoA esters previously examined as substrates for LCAD, 2,6-dimethylheptanoyl-CoA was reported to be most active substrate for LCAD.22
In this study, three medium branched chain fatty acids, namely, 2,6-dimethylheptanoic acid (dMC7), 4,8-dimethylnonanoic acid (dMC9), and 6-amino-2,-4-dimethylheptanoic acid (AdMC7), were tested as potential anaplerotic molecules in fibroblasts from patients deficient in VLCAD, LCHAD, TFP, and CPT II. The levels of TCA cycle intermediates in cells treated with these fatty acids were compared with their levels in cells treated with C7 to normalize cellular response. We demonstrated that these three medium branched chain fatty acids can be utilized by the patient cells and can be viable components of a more effective treatment.
2 |. MATERIALS AND METHODS
The fatty acids for this study were obtained as follows: heptanoic acid (C7) and D,L-2,6-dimethylheptanoic acid (dMC7) were obtained from Matreya LLC, State College, PA. 6-Amino-2,4-dimethylheptanoic acid (AdMC7) was obtained from Sigma, St. Louis, MO. 4,8-Dimethylnonanoic acid (dMC9) was obtained from Rieke Metals, Lincoln, NE. This article does not contain any studies with human subjects performed by the any of the authors.
2.1 |. Fibroblast cells sources
Patient cells for the experiments were obtained in accordance with the approved guidelines and regulations of the experimental human protocols approved by the Institutional Review Board at the University of Pittsburgh, protocol #404017. Fibroblast cells had mutations in the following genes: ACADVL (Fb671, VLCADD; Fb833, VLCADD2), HADHA (Fb822, LCHADD), HADHB (Fb861, TFPD), CPT2 (Fb836, CPT IID). Table 1 details their genotype. Control fibroblast cell line, Fb826, (ATCC PCS201012, Primary Dermal Fibroblast; Normal, Human, Adult [HDFa]) was obtained from the American Type Culture Collection, Manassas, VA.
TABLE 1.
Fibroblast cell lines designation, genotype, and phenotype data used in this current study
| Fibroblast (this report designation) | Fibroblast, Coriell Institute designation | Defective protein | Genotype | Phenotype |
|---|---|---|---|---|
| FB671 VLCADD |
N/A | VLCAD | c.1619 T > C (p.L540P) c.1707–1716, 9 base insertion |
Recurrent rhabdomyolysis episodes |
| FB833 VLCADD2 |
N/A | VLCAD | Heterozygote c.520G > A (p.V174M) c.1825G > A (p.E609K) |
NBS: High C14:0, C14:1, and abnormal acylcarnitine |
| FB822 LCHAD |
N/A | LCHAD | Homozygote c.1528G > C p.E510Q |
Hypothermia;a hypoglycemia; LCHAD enzyme activity <6 times normal |
| FB861 TFPD |
N/A | LCKAT | c.693C-del >frame shift c.881C > G (p.P261R) |
LCKAT enzyme activity <8 times normal; LCHAD enzyme activity <3.5 times normal |
| FB836 CPT IID |
GM01763 | CPT II | Homozygote c.439C > T (p.S113L) |
Recurrent myoglobinuriaa |
As reported by Coriell Institute.
2.2 |. Cell culture and treatments
Fibroblast cells were grown in T175 flasks (six flasks for each treatment, n = 6) in regular culture medium consisting of Dulbecco’s Modified Eagle Medium (DMEM), Corning Life Sciences, Manassas, VA, containing 4.5 g/L glucose and supplemented with 10% fetal bovine serum (FBS), Gibco Life Technology, Grand Island, NY, and 100 IU penicillin and 100 μg/ml streptomycin, Corning Life Sciences, Manassas, VA. When the cultured cells were 85% to 90% confluent, the regular culture medium was changed in designated flasks to DMEM with either 4.5 g/L glucose and supplemented with 10% lipid stripped FBS, Sigma-Aldrich Co., St. Louis, MO, or medium devoid of glucose supplemented with 10% lipid stripped FBS, and 100 IU penicillin and 100 μg/ml streptomycin and incubated for 24 h at 37°C, 5% CO2 incubator. Following 24 h incubation, the cells were treated as follows: 2 mM heptanoic (C7), 2,6-dimethylheptanoic (dMC7), or 4,8-dimethylnonanoic (dMC9), or 144 μM of 6-amino-2,-4-dimethylheptanoic (AdMC7) acids in flasks devoid of glucose for 72 h. After 72 h, culture media samples were collected and frozen at −80°C for acylcarnitine profiling and cells were harvested by standard tissue culture method and cell pellets were frozen at −80°C. Cells were processed and extracts analyzed for TCA cycle intermediates at the Health Sciences Metabolomics and Lipidomics Core at the University of Pittsburgh.
2.3 |. Acylcarnitine profiling of cell culture medium
Acylcarnitine profiling was adapted from published methods23–25 with minor modifications. Briefly, aliquots (75 μl) of medium were mixed with methanol (20 μl) containing isotope labeled carnitine standards and the protein precipitated by addition of absolute ethanol (905 μl) and centrifugation (13,000 rpm, 10 min). A portion of the supernatant (50 μl) was dried under a stream of nitrogen gas and the acylcarnitine butyl esters generated by reaction (60°C for 15 min) in 100 μl of 3 N HCl in butanol. Dried residues were reconstituted in acetonitrile-water (80:20) for flow injection ESI-MS-MS analysis. Analysis was performed on a triple quadrupole API4000 mass spectrometer (AB Sciex, Framingham, MA) equipped with a ExionLC 100 HPLC system (Shimadzu Scientific Instruments, Columbia, MD). Analyst (V1.6.3, AB Sciex 2015) was utilized for data acquisition and ChemoView software (V2.0.3, AB Sciex 2014) for quantitation using isotope labeled carnitine standards. Acylcarnitine standards were purchased from Amsterdam UMC—VUmc (Amsterdam, NL) and Cambridge Isotope Laboratories, Inc. (Andover, MA). Acylcarnitines were measured using multiple reaction monitoring (MRM) for free carnitine (C0, m/z 218 > m/z 103) and acetylcarnitine (C2, m/z 260 > m/z 85) and Precursor Scan for precursor ions (Q1) of acylcarnitines (C3–C18, scan range m/z 270–502) that generated a product ion (Q3) at m/z 85.
2.4 |. Statistical analysis
Statistical significance was assessed by performing the unpaired and two-tailed Studenťs t-test or one-way analysis of variance (ANOVA) followed by Tukey multiple range test using GraphPad Prism version 7.04 for windows, GraphPad Software (La Jolla, CA, www.graphpad. com). Differences were considered significant when P < .05. The corresponding P values are provided in the figures’ legend.
Multivariate Principal Component Assay (PCA) and Sparse Partial Least Squares Discriminant Analysis (sPLS-DA), done using the “mixOmics” package in R was used to comprehensively analyze the data. Analysis of the metabolite expression for different disease models, that is, Control, VLCADD, VLCADD2, LCHADD, CPT-IID, TFPD by high dimensional spectral features using multivariate PCA and further discrimination of inter-class variance between the groups were made by the sPLS-DA. The variable importance in the projection (VIP) score for the metabolite profiles in sPLS-DA, summarizes contributions of the most prominent metabolites contributing to the observed variability, and allows the selection of the most predictive or discriminative features in the data to classify the samples accordingly.
3 |. RESULTS
3.1 |. TCA cycle intermediates content in control and FAOD cells
Levels of key TCA cycle intermediates, Figure 1A,B, and related metabolites, lactate, glutamate and aspartate, Figure 1C,D, among various long chain FAOD cell lines in the presence and absence of glucose are illustrated. Levels of these metabolites in the patient cells were compared statistically to their level in the control cell line. Absence of glucose in the media has affected the metabolites levels differently in each cell line. The following is a summary of such differences.
FIGURE 1.
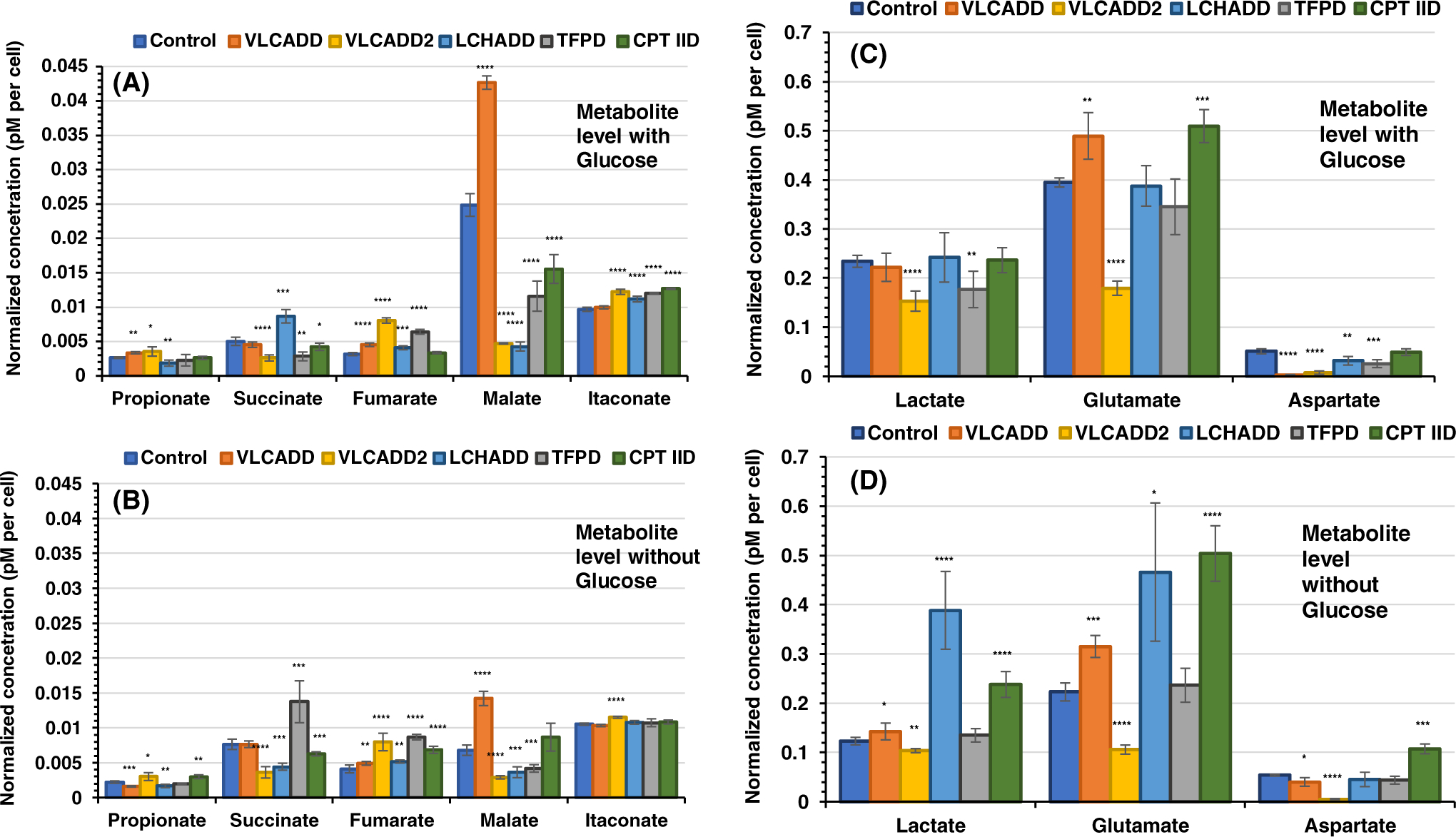
Measurements of selected TCA cycle intermediates in control and cells deficient in various β-oxidation proteins. Cells were counted and resuspended in processing extract buffer and MS assay performed with the final concentration expressed in μM, which was divided by the number of cells in the pellet and multiplied by 106. All media had no additional glutamine or pyruvate, and lipid stripped FBS was included instead of regular FBS in all. (A) All media included glucose and (B) all media was devoid of glucose. Data are presented as mean ± SD (SD) for replicates and analyzed using unpaired Student’s t test (using GraphPad Prism 7.04 software). *P < .05, **P < .01, ***P < .001, ****P < .0001, compared with control cells
3.1.1 |. Propionate
The pattern of intracellular propionate, primarily derived from the β-oxidation of the odd-numbered branched or straight chain fatty acids and valine and isoleucine catabolism level, among the various cell lines appear similar in the presence of glucose compared with its absence, with the exception of VLCADD. In VLCADD propionate is about 2-fold higher in the presence of glucose compared with in its absence, the level of propionate was not significantly different in the presence of glucose compared with no glucose, in the other five cell lines.
3.1.2 |. Succinate
In general succinate levels were lower in cell lines with glucose (Figure 1A) compared with without glucose when comparing each cell line (Figure 1B). However, the pattern of succinate level among the six cell lines was not similar except control and VLCADD, where the succinate level was similar between these two cell lines whether in glucose or without. With glucose, succinate in LCHADD cells was about 2-fold higher than in its absence. This was in contrast to the level of succinate in TFPD cells, which was 4.8 higher in the absence of glucose than the presence of glucose.
3.1.3 |. Fumarate
The level of fumarate was similar in VLCADD, VLCADD2, and LCHADD cells with or without glucose with the latter two cell lines having the highest level compared with the other cell lines. In TFPD and CPT IID, fumarate level was about 1.4- and 2-fold higher in the absence of glucose compared with its level in the presence of glucose, respectively.
3.1.4 |. Malate
While the level of malate in cells incubated in the presence of glucose was about the same in VLCADD2 and LCHADD cells compared with its level in the absence of glucose, its level was significantly higher in control, VLCADD, TFPD, and CPT IID cells at 3.7-, 3.0-, 2.8-, and 1.8-fold of its level in the absence of glucose, respectively.
3.1.5 |. Itaconate
Itaconate level was slightly higher in VLCADD2, LCHADD, TFPD, and CPT IID cell lines compared with control in the presence of glucose but approximately the same without glucose.
3.1.6 |. Lactate
Lactate level in VLCADD2 and TFPD cells were lower than its level in control (Figure 1C). Without glucose, lactate level in control, VLCADD, VLCADD2, and TFPD was almost half of the level in media with glucose (Figure 1C,D). While lactate level without glucose was significantly higher in the LCHADD compared with that in the presence of glucose, its level in CPT IID was similar with or without glucose.
3.1.7 |. Glutamate
Glutamate level was significantly higher in VLCADD and CPT IID cell incubated in media with glucose compared with control (Figure 1C) but was significantly lower in VLCADD2. Glutamate level decreased significantly in control, VLCADD, VLCADD2, and TFPD when incubated without glucose, but was essentially similar LCAHDD and CPT IID with or without glucose.
3.1.8 |. Aspartate
Aspartate level was significantly low in VLCADD, and VLCADD2 compared with the control cells when grown in glucose, while it was slightly lower in VLCADD and significantly lower than control in VLCADD2 in the absence of glucose. In contrast, aspartate in CPT IID cells was similar to control when grown in the presence of glucose but higher when glucose was not present.
3.2 |. TCA cycle intermediates in FAOD cells treated with medium branched chain fatty acids
Medium branched chain fatty acids were selected based on their predicted metabolic pathways. To offset differences in cell viability and or growth condition among various cell lines, we used the C7 treatment as a reference fatty acid. For all comparisons, TCA cycle intermediates values resulting from the C7 treatment was set to 100%.
Since malate levels were the highest among the TCA cycle intermediates in the control cell line when grown in the presence or absence of glucose, we plotted the level of malate in the VLCADD, LCHADD, TFPD, and CPT IID cell lines relative to control as they responded to utilization of C7, dMC7, and AdMC7 as an alternative to glucose. In control cells, malate decreased significantly in the presence of all branched chain fatty acids compared with a glucose-free incubation (Figure 2). Malate decreased in VLCADD2 cell line with C7, dMC7, and AdMC7 treatments compared with a glucose-free incubation, and when treated with AdMC7, the malate level was close to the level in the absence of glucose. In LCHADD cell line, malate was comparable in the presence or absence of glucose or in the presence of the branched chain fatty acids. When glucose was replaced with dMC9, malate was at the lowest in all cell lines, except LCHADD cells. Malate was highest in VLCADD cells compared with the other deficient cell lines when treated with all branched chain fatty acids except in CPT IID cells with AdMC7 treatment or glucose-free incubations. The pattern of malate levels was similar in VLCADD and TFPD cell lines.
FIGURE 2.
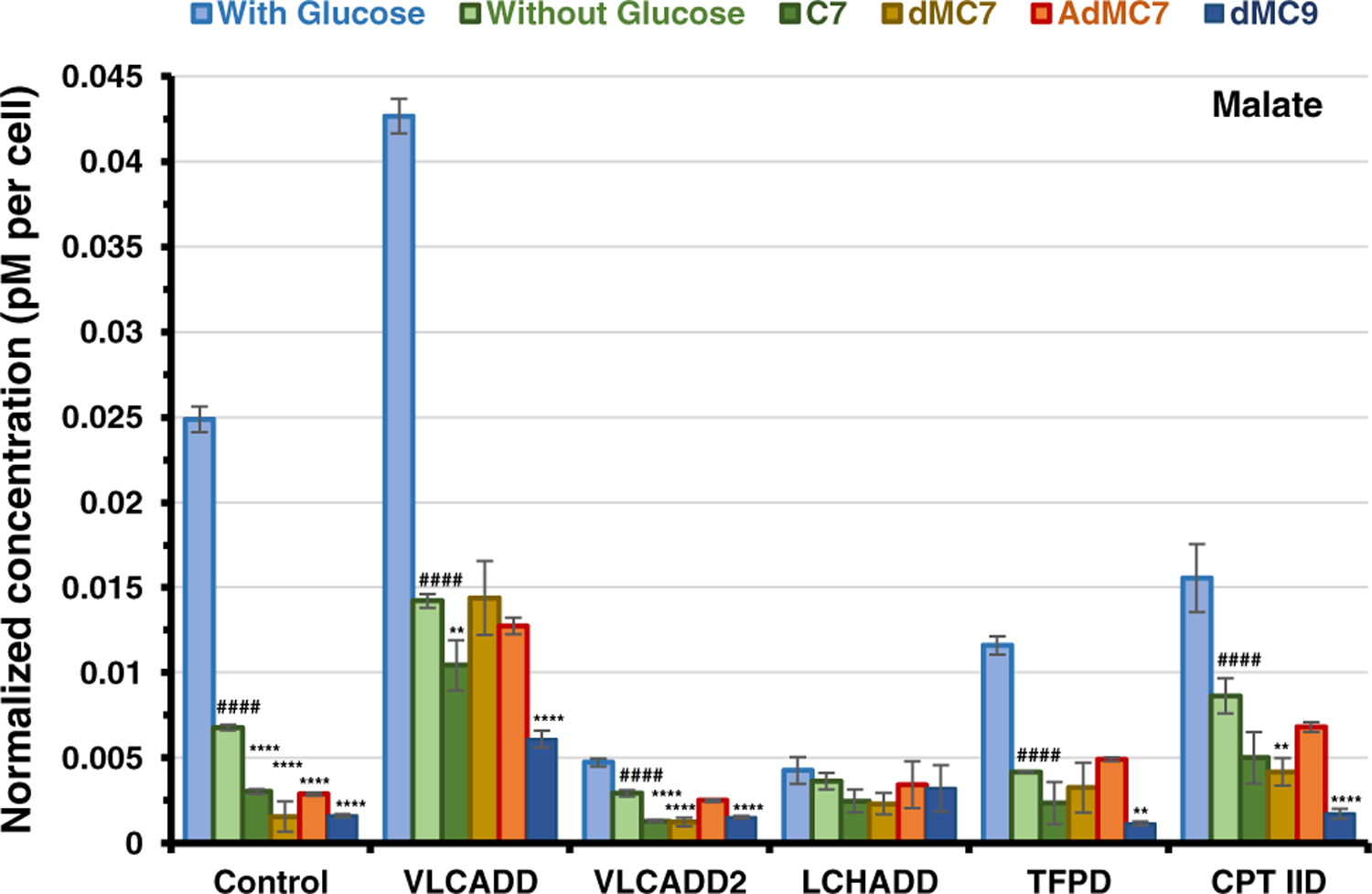
Measurements of malate in various cell lines in the absence of glucose and the presence of medium branched chain fatty acids. Sample preparation same as in Figure 1. All media had no additional glutamine or pyruvate, and lipid stripped FBS was included instead of regular FBS. Data are means ±SD; *P < .05, **P < .01,****P < .0001, treatment groups compared with respective untreated cells in the absence of Glucose; ####P < .0001 untreated cells in the absence of Glucose compared with respective cells in the presence of glucose, (Tukey multiple range test)
Figure 3A shows the relative level of intracellular propionate in the treated cells showed a similar pattern of response to dMC7 and AdMC7 in control, VLCADD, LCHADD, and TFPD cells when compared with C7 treatment, suggesting that the branched chain fatty acids were used with similar efficiency in each cell line. While dMC7 and AdMC7 treatment led to higher intracellular propionate in all cell lines except CPT IID, dMC9 led to lower amounts in TFPD and CPT IID compared with C7 suggesting that either of these two cell lines do not utilize this fatty acid as efficiently. Figure 3B–D illustrate the intracellular relative levels of three consecutive TCA cycle intermediates: succinate, fumarate, and malate in cells treated with the branched chain fatty acids compared with the C7 treatment. Both VLCAD deficient cell lines, VLCADD and VLCADD2, accumulated more succinate but similar or decreased fumarate when treated with dMC7, AdMC7, and dMC9 than with C7 (Figure 3B and C, respectively). Interestingly, LCHADD and TFPD cell lines with overlapping enzyme deficiencies had somewhat divergent results from each other, with dMC7 and AdMC7 leading to similar succinate but not fumarate or malate amounts comparable to C7 treatment. TFPD cell line did not appear to use dMC9 well, though LCHAD deficient cells accumulated more fumarate and malate when supplemented with this substrate than C7. Although CPT IID cells accumulated the most propionate when grown in C7, their levels of succinate and fumarate were similar with all the fatty acids. While malate levels were consistently higher in all cell lines treated with AdMC7 except control, malate levels were higher in VLCADD and TFPD cell lines treated with dMC7 compared with the C7 treatment. Malate levels on the other hand were higher in VLCADD2 and LCHADD cell lines treated with dMC9 but significantly lower in control, VLCADD, TFPD, and CPT IID cell lines (Figure 3D)
FIGURE 3.
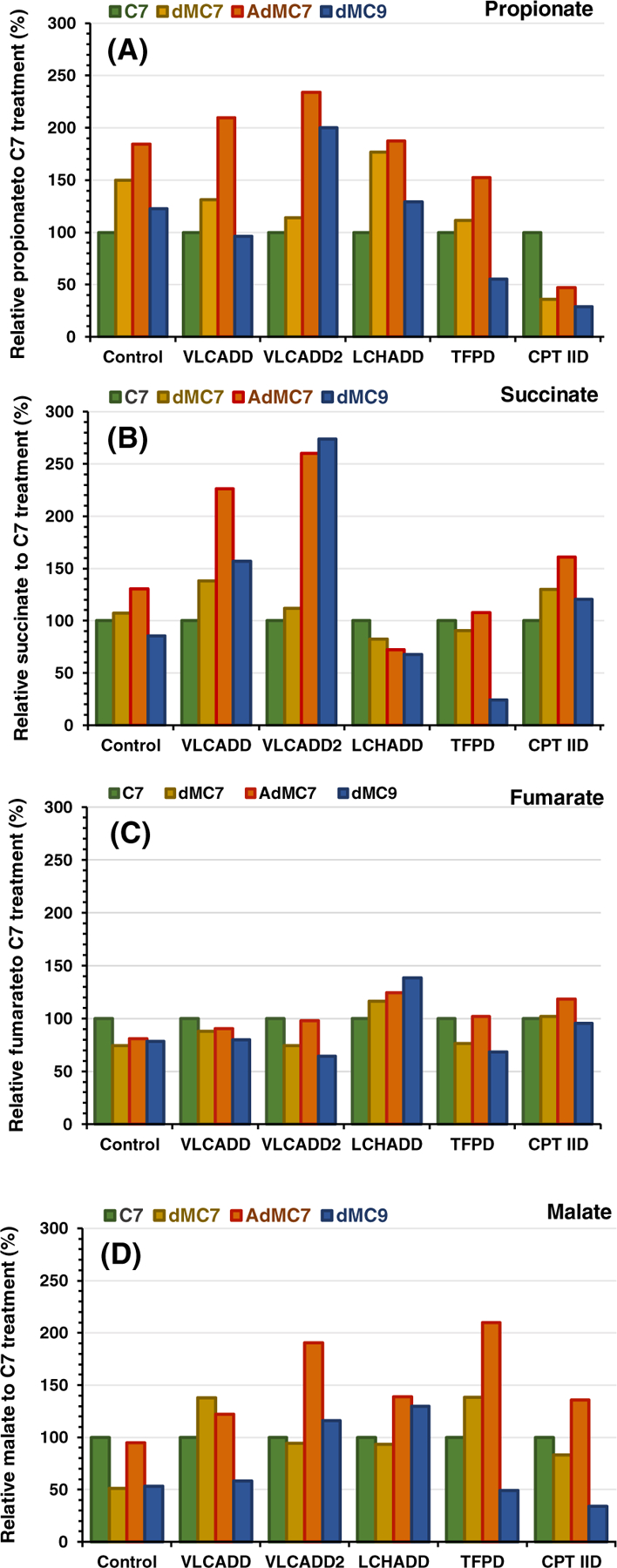
Relative intracellular presence of (A) propionate, (B) succinate, (C) fumarate, and (D) malate in control, VLCADD, VLCADD2, LCHADD, TFPD, and CPT IID cell lines (see Table 1 for genotype details) in the presence of 2 mM of C7, dMC7, or dMC9, or 144 μM AdMC7 acids with values of metabolites normalized to C7. Percentages plotted were obtained from measurements of metabolites in pellets from six flasks (n = 6) for each treatment, averaged and normalized to values obtained from the C7 treatments as reference to offset cell viability bias. Media contained lipid stripped FBS and no glucose or pyruvate
3.3 |. Itaconate is not elevated in cells grown in branched chain substrates compared with C7
Cells with VLCAD deficiency have previously been shown to have an increase in reactive oxygen species, a finding usually associated with inflammation.26–28 To assess the effect of various treatments on TCA cycle intermediates related to inflammation, itaconate levels were monitored in cellular extracts based on previous studies. Itaconate, a decarboxylation product of cis-aconitate, plays a role as anti-inflammatory metabolite acting via Nrf2 to limit inflammation and modulate type I interferons.29 While itaconate levels were similar in FAOD cells compared with control when grown without glucose (Figure 1B), its level varied slightly when cells were grown in the various medium chain fatty acid treatments (Figure 4A). When treated with dMC9, itaconate was lower relative to C7 in all cell lines except control and LCHADD cell lines. The subtle differences, that is, within a range of 10% relative to C7, are hard to discern with respect to their significance without a broader survey of other inflammation markers.
FIGURE 4.
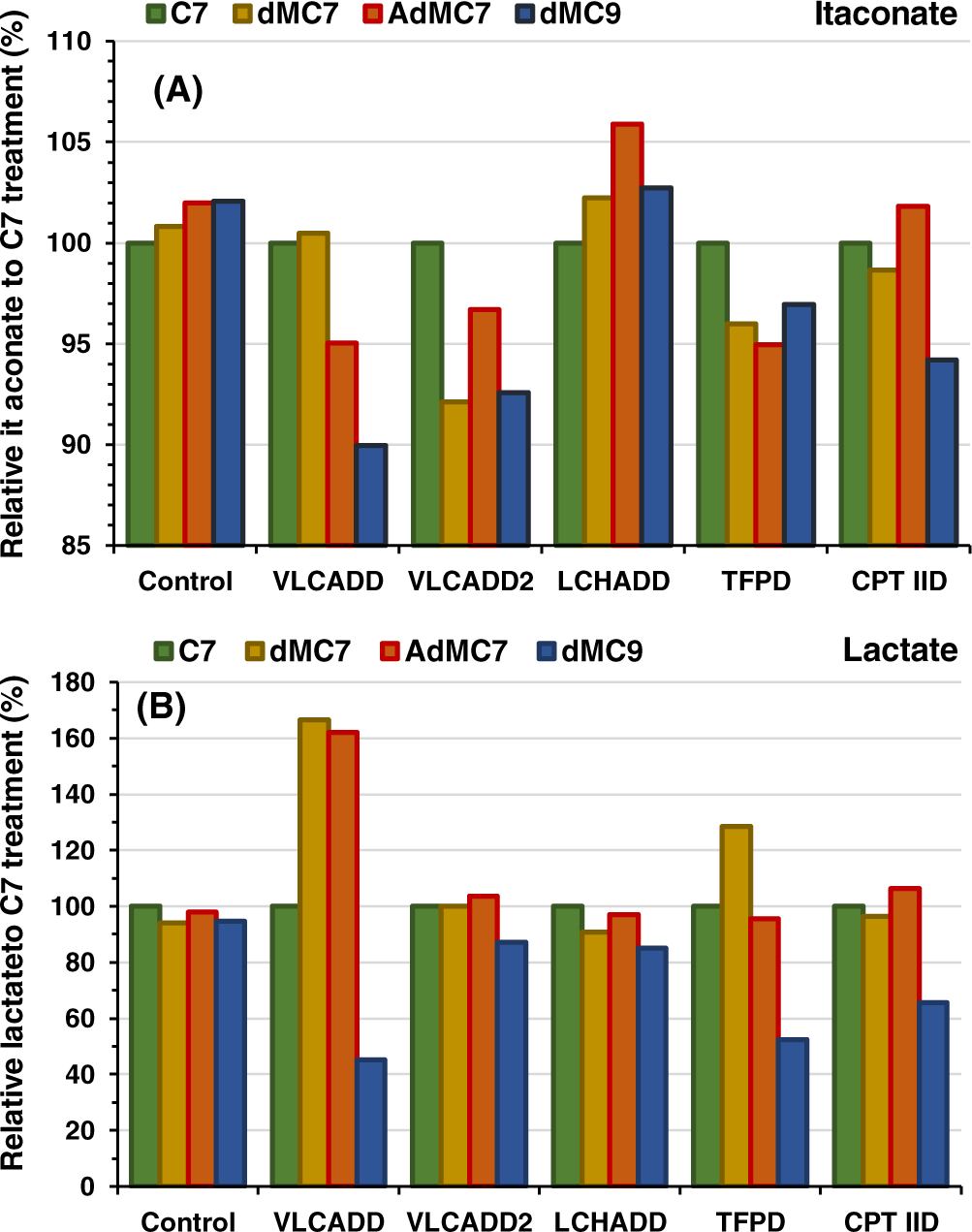
Relative intracellular presence of (A) itaconate and (B) lactate in control, VLCADD, VLCADD2, LCHADD, TFPD, and CPT IID cell lines in the presence of 2 mM of C7, dMC7, or dMC9, or 144 μM AdMC7 acids with values of metabolites normalized to C7 analyzed as in Figure 1. Media contained lipid stripped FBS and no glucose or pyruvate
3.4 |. Intracellular lactate varies across cells lines and medium chain fatty acids
Cells with fatty acid oxidation disorders often have a secondary impairment in oxidative phosphorylation, which is thought to be the reason for intermittent lactic acidosis in patients during times of metabolic stress.30 As a measure of this dysfunction, lactate levels were measured in cell extracts with and without glucose (Figure 1C,D). The lactate level rose in one VLCADD cell line when grown in dMC7 and AdMC7 compared with C7 (Figure 4B) but was the same or slightly lower with all others. Growth of the VLCADD, TFPD, and CPT IID cell lines in dMC9, led to lower levels of lactate than growth in C7.
3.5 |. Changes in intracellular concentrations of glutamate and aspartate
Glutamate and aspartate can be metabolized into the TCA intermediates oxaloacetate and 2-oxoglutarate, respectively. Thus, changes in their concentrations with therapy would augment a direct anaplerotic effect. Growth of all patient cells in AdMC7 raised intracellular glutamate by 18% to 50% more relative to C7 in all cell lines (Figure 5A), while growth in dMC9 lowered relative intracellular glutamate in all cell lines except for LCHADD. Aspartate was significantly higher in VLCADD grown in all the medium branched chain fatty acids compared with C7, but was unchanged in the VLCADD2, LCHADD, and TFPD cell lines. Aspartate was 50% higher in the CPT IID cells with media containing AdMC7 compared with media containing C7.
FIGURE 5.
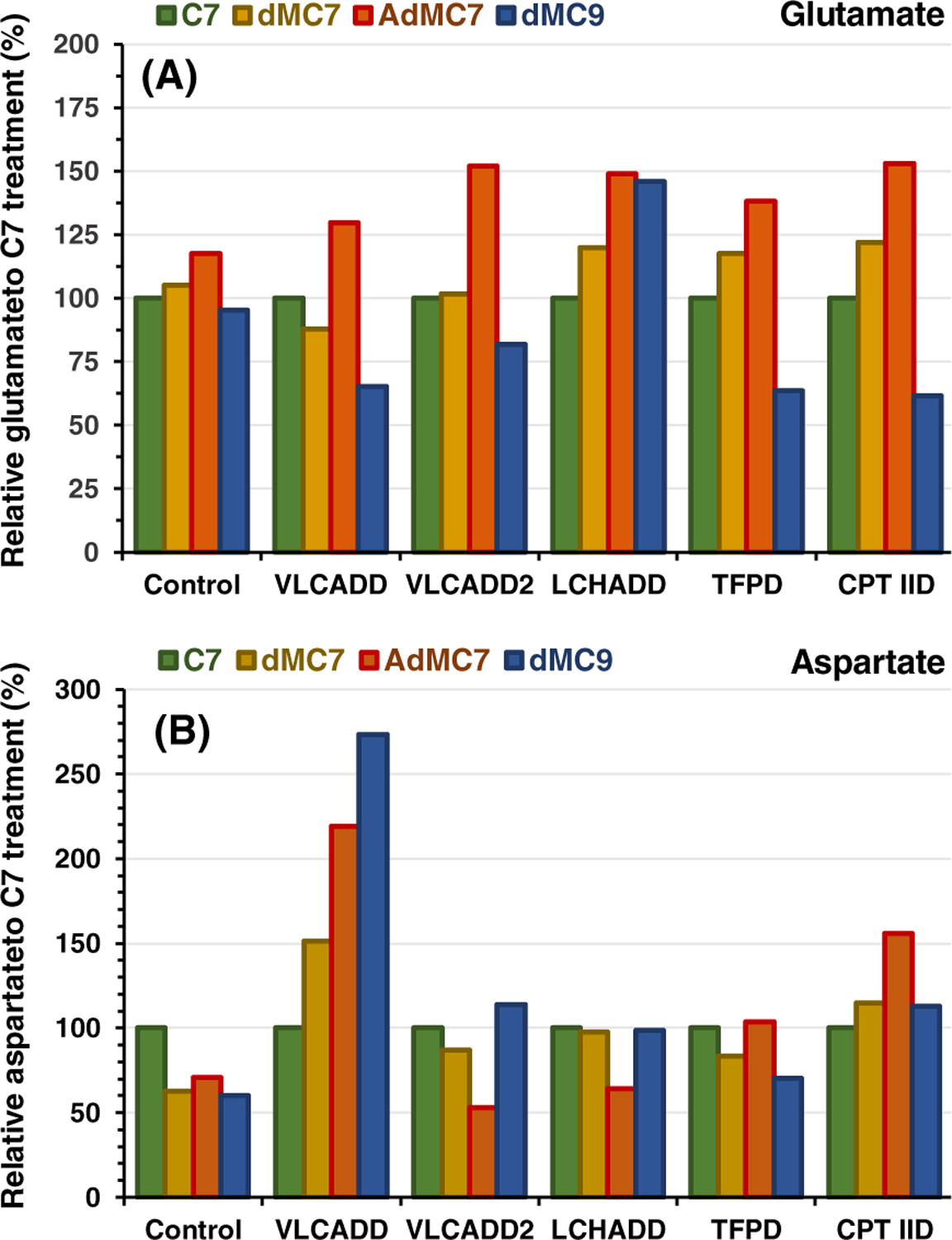
Relative intracellular presence of (A) glutamate and (B) aspartate in control, VLCADD, VLCADD2, LCHADD, TFPD, and CPT IID cell lines in the presence of 2 mM of C7, dMC7, or dMC9, or 144 μM AdMC7, with values of metabolites normalized to C7 analyzed as in Figure 1. Media contained lipid stripped FBS and no glucose or pyruvate
3.6 |. Sparse partial least squares-discriminant analysis
The intracellular metabolites data were also analyzed globally to investigate trends of the treatments in the overall cellular function. Scores plots of sPLS-DA were used to discriminate the samples in a supervised way and show the similarities and dissimilarities between the samples, Figure S1. sPLS-DA was further used to identify metabolites that mostly contribute to the discrimination of the treatments in each disease model and represented as a heatmap. The colors of each metabolite represent the degree of variation in the different treatments: red indicates increased levels, while blue indicates decreased levels.
3.6.1 |. Control
The score plot of control samples shows PC1 and PC2 values of 67% and 23% of data variation, respectively, to show the metabolic changes between the different treatment groups. The glucose and C7 treated samples show a distinct alteration, as observed in the scores plot. The metabolites glutamate, succinate, malate, and lactate contribute to the variation with glucose treatment whereas fumarate and aspartate contribute to the variation in the C7 treatment.
3.6.2 |. VLCADD
The metabolic variations in the VLCADD model show PC1 and PC2 values of 70%, and 12% of data variation, respectively, highlighting the metabolic variations between the treatments. Treatments with glucose show the most variation from the scores plot with the aspartate and itaconate contributing to it. The dMC9 also shows differences when compared with the other treatments with succinate, glutamate, lactate, propionate, and malate as the significant contributors.
3.6.3 |. VLCADD2
sPLS-DA indicates that the first two components attribute to 74% of the variation in the VLCADD2 model. Treatment with glucose shows significant variation in the scores plot with aspartate and itaconate as metabolites that have a negative association to the treatment. The scores plot also indicates that the treatment with AdMC7 and without glucose have no differences.
3.6.4 |. LCHADD
sPLS-DA score plot across the first two components do not appear to reveal dominant modes of variation between the treatments, with the first single component explaining only 26% of the variation in the data.
3.6.5 |. TFPD
The TFPD model indicates that the treatment with glucose and dMC9 show the most variation in the data, and contributes to 54%, and 18% of the variation in the first, and second, components, respectively. The metabolites accounting for the variation in dMC9 treatment include succinate, glutamate, and malate.
3.6.6 |. CPT-IID
The score plots indicate that treatment with glucose shows the most variation with aspartate, fumarate, propionate, and succinate as significant contributors.
3.7 |. Extracellular acylcarnitines accumulation in culture media
In patients, excess acyl-CoAs not metabolized in the mitochondria are exported to the cytoplasm where they can be further metabolized in other cellular pathways or conjugated to carnitine for excretion by the kidney. Media acetylcarnitine reflects intracellular acetyl-CoA levels, the end product of fatty acid oxidation. The TFPD cell line showed a significant increase in the relative extracellular acetylcarnitine when fed dMC7, AdMC7, and dMC9, while the media levels were modestly higher in VLCADD2 cell line compared with C7 (Figure 6A). Acetylcarnitine was modestly higher in media of LCHADD and CPT IID cells treated with dMC9 compared with C7. Its levels were similarly higher in media of CPT IID cells with AdMC7 and dMC9 compared with C7.
FIGURE 6.
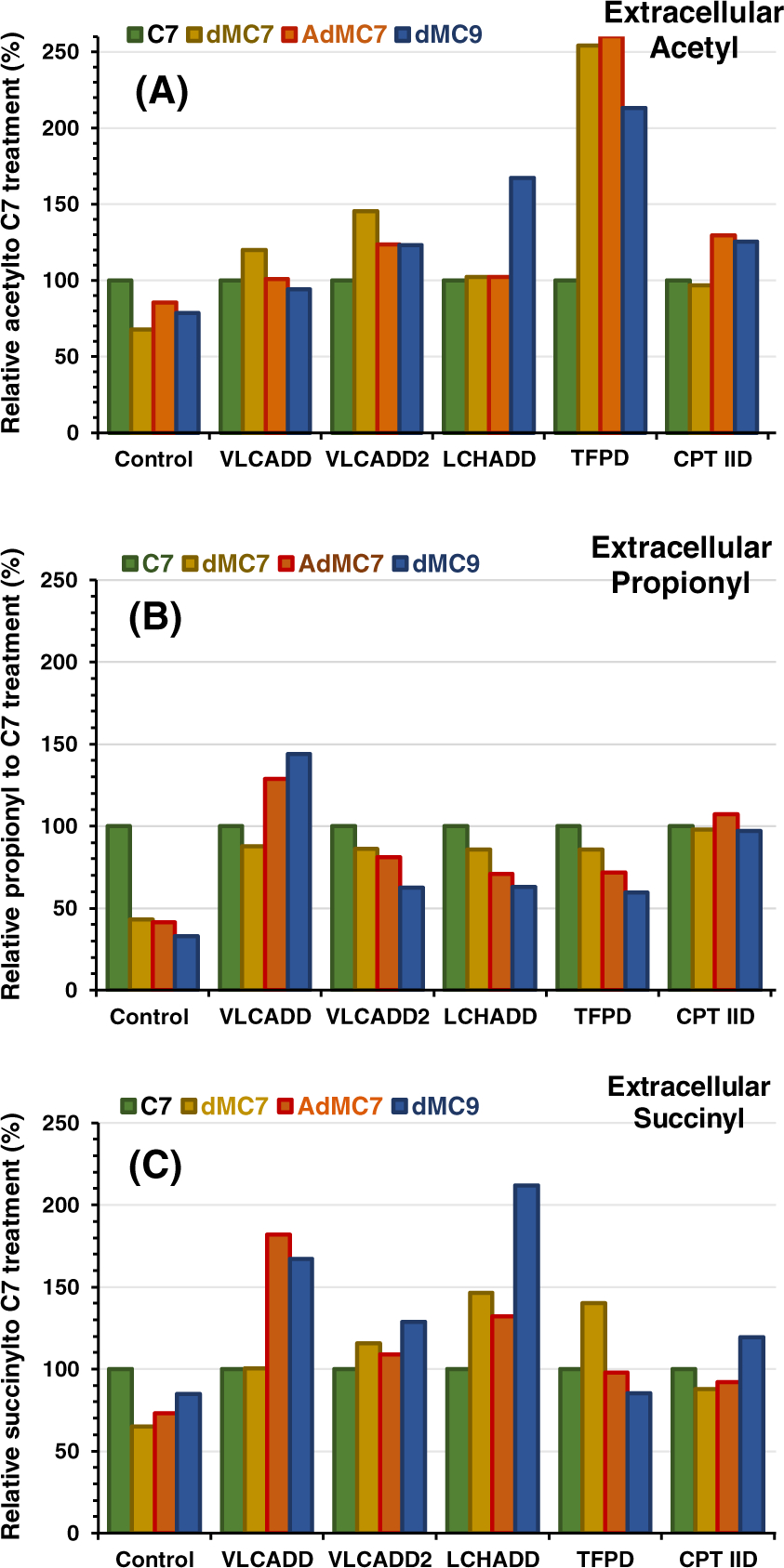
Relative extracellular presence of C2-, C3-, and C4DC-carnitine corresponding to acetylcarnitine, propionylcarnitine, and succinylcarnitine in media of control, VLCADD, VLCADD2, LCHADD, TFPD, and CPT IID cell lines in the presence of 2 mM C7, dMC7, or dMC9, or 144 μM AdMC7, with values of metabolites normalized to C7. Media contained lipid stripped FBS and no glucose or pyruvate
Treatment of patients with C7 leads to an increase in propionylcarnitine in blood and media.31 Control cells grown in the presence of any of the medium branched chain fatty acids led to a decrease in propionylcarnitine in media compared with C7 treatment (Figure 6B). Propionylcarnitine in the VLCADD media was similar with the dMC7 treatment while it was higher with treatments of AdMC7 and dMC9 compared with the C7 treatment. Excreted propionylcarnitine pattern was similar regardless of the supplement compared with C7 treatment, except in CPT IID cells where propionylcarnitine levels across all treatments were similar.
Succinylcarnitine was significantly increased in the media of VLCADD, VLCADD2, and LCHADD cells grown in dMC9 and AdMC7 compared with C7. Succinylcarnitine was also higher in the media of TFPD cell line grown in the presence of dMC7 and CPT IID cell line in the presence of dMC9 compared with C7 (Figure 6C).
4 |. DISCUSSION
The major aim of the study was to demonstrate that the branched chain fatty acids can be metabolized and utilized as anaplerotic molecules in cells deficient in long chain fatty acid oxidation and are comparable in performance to the clinical trial-tested C7. A secondary aim was to demonstrate that these MBCFAs can produce significant levels of propionyl-CoA, the precursor of succinyl-CoA. A deficiency of the latter is a key contributor to the pathophysiology of these disorders (see below) and its replenishment impact other TCA intermediates. While fibroblasts have a low level of basal metabolism, they have a long history of use in studies of FAODs and are routinely used as a surrogate for other more metabolically active and relevant tissues.
4.1 |. Variabilities in levels of TCA cycle intermediates among FAODs cell lines
Taken in isolation for each of the metabolites, the variability in the profile of intracellular propionate, succinate, and fumarate among the FAODs fibroblast cell lines when compared with control were not dramatically altered in the presence or the absence of glucose, except succinate without glucose in LCHADD and CPT IID cells and fumarate in VLCADD2, TFPD, and CPT IID cells. Succinate levels were decreased in the presence of glucose in VLCADD2, LCHADD and TFP cells. The variability may reflect the variability in phenotypes and the fact that different tissues are expected to have different physiological demands for each of the TCA cycle intermediates. Having a similar pattern for succinate in the absence of glucose, that is, cells under metabolic stress, compared with cells in the presence of glucose is notable, with the exception of LCHADD and CPT IID cells where succinate levels were higher compared with control in the absence of glucose. Variations in the level of malate was the most intriguing among the different FAODs cell lines with and without glucose. Malate levels were nearly 2-fold higher in the VLCADD cell line compared with control in the presence and absence of glucose while they were significantly reduced in VLCADD2 when compared with control, a finding worthy of additional studies.
The consistent dramatic decrease of aspartate in extracts of VLCADD2 deficient cells in the presence or absence of glucose may reflect diminished amounts of oxaloacetate in cells as well but it is unexplained given the milder presentation of the phenotype when compared with the VLCADD cell line, which is considered severe. The significant increase in lactate and aspartate in CPT IID may suggest a switch to glycolysis and protein use for energy. Similarly, LCHADD cells showed a significant increase in lactate and glutamate but not a significant increase in aspartate. Glutamate level was reduced significantly in control, VLCADD, VLCADD2, and TFPD cells in the absence of glucose, but not in LCHADD or CPT IID cells.
4.2 |. Comparing performance of MBCFAs
The levels of the TCA cycle intermediates are variable among the FAODs cell lines tested in the presence of MBCFAs compared with C7. Notable is that in the presence of dMC7 and AdMC7 the relative propionate level was higher in control, VLCADD, VLCADD2 and LCHADD cell lines. The relative succinate level paralleled the relative propionate level in control, VLCADD and VLCADD2 cell lines but not LCHADD cell line. In general, AdMC7 seem to have performed with consistency among the relative levels of the consecutive intermediates, propionate, succinate, fumarate, and malate, compared with C7, in control, VLCAD, VLCADD2, LCHADD, and CPT IID, albeit with lower levels of succinate in LCHADD and propionate in CPT IID. The level of glutamate was higher in all cell lines in the presence of AdMC7 as well. While the mode of transportation of AdMC7 may have provided an advantage in fibroblasts, it is likely that it will be as readily metabolized to its energy providing components, two propionyl-CoA and one pyruvate, in other tissues. dMC9 appears to have been least consistent across the various intermediates in vitro, and so perhaps least efficacious, testing in vivo will provide better assessment as various target tissues may utilize it differently.
4.3 |. Global statistical analysis
The sPLS-DA and heatmap models offer a global perspective on how different cell lines response to the imbalance created by the absence of activity of one specific enzyme in addition to possible physically associated proteins. The sPLDs plots show that glucose is distinct from all other medium chain fatty acids in all cell lines, except LCHADD where all treatments do not show variation. Additionally, C7 in control and dMC9 in VLCADD showed variation. This analysis implies that the metabolic fate and amount of intermediates outcome are generally similar among the cell lines with C7 and different MBCFA treatments.
4.4 |. Replenishing succinyl-CoA, key to efficacy
MBCFAs provide a rich source of succinyl-CoA, a key intermediate in cell physiology (Scheme 1). The evidence for this hypothesis is supported by clinical findings with treatment of long chain FAODs with C8G vs C7G. The use of medium chain C8 as the active drug ingredient in C8G was based on bypassing dysfunctional long chain FAO enzymes and the hypothesis that a deficit in the supply of acetyl-CoA is the major cause of pathogenesis as this impairs ATP production, generation of ketone bodies, gluconeogenesis, modulation of protein function through lysine acetylation, and synthesis of physiologically vital molecules such as steroids and complex lipids. However, clinical studies indicate C8G does not adequately address the hypoglycemia with recurrence of other symptoms. Despite reducing the number of acetyl-CoAs generated from C8G by half when using C7G, the latter essentially eliminates hypoglycemia in patients, highlighting the role of replenishment of succinyl-CoA and/or possible differences between C8 and C7 conjugates in their cellular utilization. Replenishing succinyl-CoA is likely key to eliminating the hypoglycemia by improving oxaloacetate availability for gluconeogenesis and sparing consumption of pyruvate to form oxaloacetate by pyruvate carboxylase, which is activated by excess acetyl-CoA as perhaps the case in C8G treatment. Moreover, replenishment of succinyl-CoA likely plays a key role in the substantial improvement of cardiomyopathy in patients with long chain FAODs. This conclusion is consistent with previous studies linking lysine succinylation/desuccinylation as regulatory mechanism of cardiac function,32–36 and cardiomyopathy presenting in propionic acidemia and methylmalonic aciduria patients where the deficit of succinyl-CoA is severe.32–36 Besides its use in lysine succinylation, succinyl-CoA is the precursor for the synthesis of δ-aminolevulinate, which in turn is the precursor of porphyrin synthesis. Multiple metabolic imbalances must therefore be considered in FAODs when designing therapeutic agents rather than focusing only on depletion in energy. MBCFAs surveyed in this study provide double the amount of succinyl-CoA/ molecule compared with C7 and can thus be more potent.
One concern related to long term excess intake of non-physiologic medium straight chain fatty acids in treating long chain FAODs is lipogenesis as has been demonstrated in mouse models.37–39 Rapid breakdown in liver mediated by abundant MCAD and SCAD likely leads to excess acetyl-CoA in mitochondria that, in the presence of relatively low oxaloacetate and pyruvate levels, can be carboxylated to generate malonyl-CoA, an inhibitor of succinate dehydrogenase. In the cytoplasm, excess conversion of acetyl-CoA to malonyl-CoA inhibits CPT I activity and promotes lipogenesis.40
4.5 |. Optimizing the composition of a medium chain triglyceride to treat long chain FAODs
The conclusion from the above discussion is that one critical design feature that an alternative fatty acid must include is the key component(s) that C7 introduces to provide improvements in patients’ phenotype. Furthermore, with the critical importance of replenishing succinyl-CoA, acetyl-CoA, and other TCA cycle intermediates, along with rapid metabolism of C7-CoA in the liver, it becomes important to consider the demands for TCA cycle intermediates and the anaplerotic effect of the drugs on each tissue individually, including skeletal muscle, heart, and neural peripheral tissue. In search for medium chain fatty acid derivatives with more favorable pharmacokinetic and pharmacodynamic properties, optimum peripheral tissue distribution, and metabolic balance, the aim now is to eliminate the biochemical causes of rhabdomyolysis and other neural peripheral symptoms in patients receiving C7G treatment. One potential approach to optimize tissue distribution and utilization of any potential alternative anaplerotic fatty acids is to take advantage of substrate specificities and tissue expression of the various mitochondrial acyl-CoA dehydrogenases (ACADs) that catalyze the rate limiting step of their breakdown. LCAD, a mitochondrial matrix enzyme typically not considered a major contributor to overall energy metabolism, has different tissue expression and substrate utilization profiles compared with other ACADs. It is expressed at a low level in skeletal muscle and heart and is significantly lower in liver than VLCAD. Additionally, LCAD utilizes medium, branched chain acyl-CoA substrates with which VLCAD shows no activity. Since whole body balance studies involving multiple tissues are complicated to model, we utilized fibroblasts derived from patients with various deficiencies in long chain FAO enzymes for an in vitro screening. Accordingly, our initial alternative candidate compounds are hypothesized to bypass rapid straight chain fatty acid metabolism in the liver. dMC9- and dMC7-CoA esters are consecutive downstream products of the catabolism of phytanic (tetramethylhexadecanoic) acid, a branched long chain fatty acid found in dairy products and ruminant animal fat. Phytanic acid is initially subjected to one α- and three β-oxidation cycles in the peroxisomes to form dMC9-CoA,22,41,42 which is then transported into the mitochondria for further β-oxidation as illustrated in Scheme 2. Unlike the rapid metabolism of C7-CoA, dMC9-CoA and dMC7-CoA metabolism is expected to be slower since LCAD, the first enzyme postulated to oxidize both compounds, is not as abundant in liver as MCAD and SCAD.
SCHEME 2.
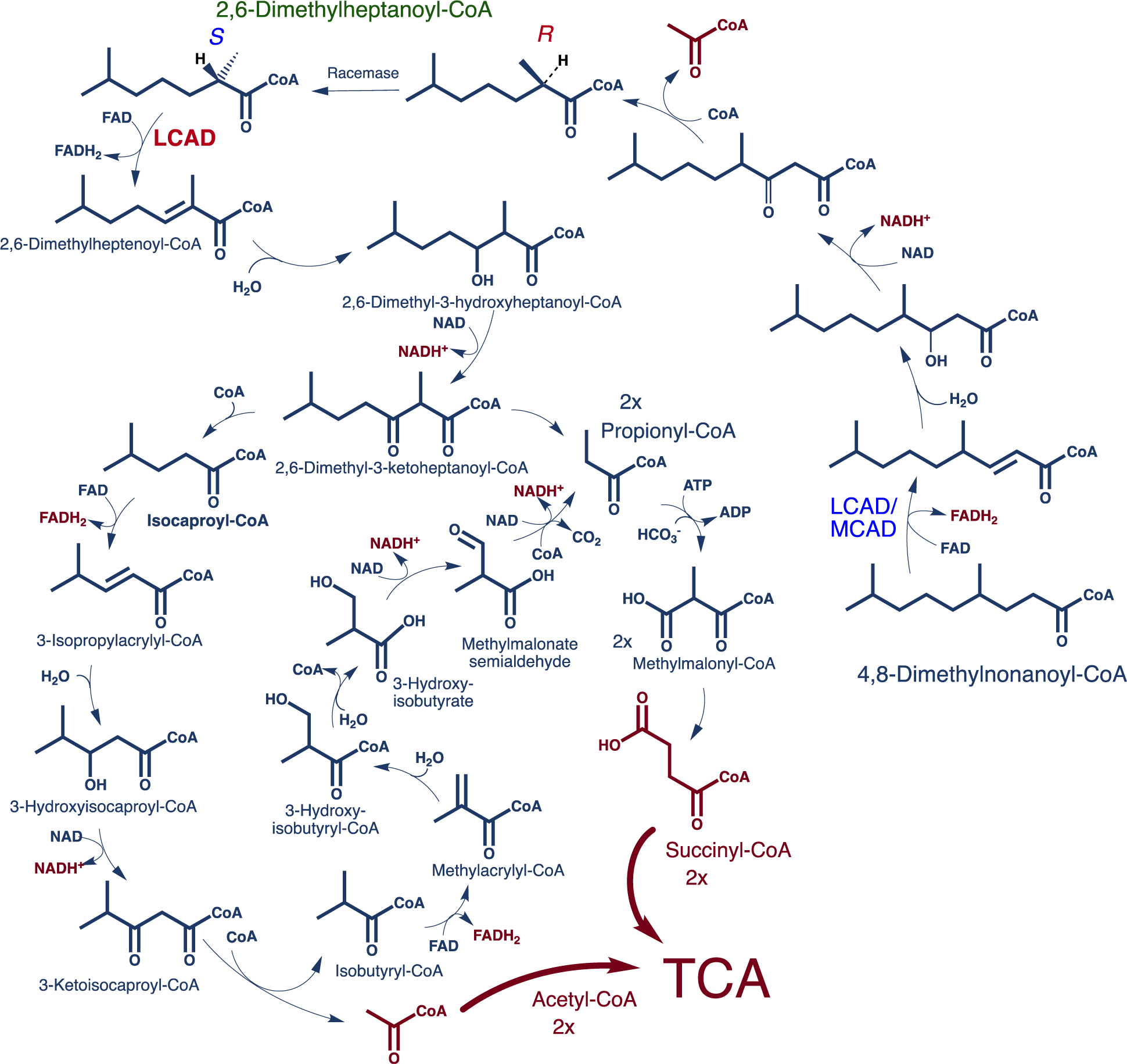
Proposed metabolic pathway of 4,8-dimethylnonanoyl-CoA, the CoA ester of dMC9. LCAD, and/or MCAD, catalyzes the first α,β-dehydrogenation of 4,8-dimethylnonanoyl-CoA in this pathway and medium chain TFP is hypothesized to catalyze the next three reactions. 2,6-Dimethylheptanoyl-CoA and acetyl-CoA are produced in this first β-oxidation cycle with the former using LCAD to start another β-oxidation cycle that continues to produce propionyl-CoA and isocaproyl-CoA as end products for additional metabolism by other pathways. Isocaproyl-CoA is hypothesized to be further oxidized by branched chain amino acid metabolism enzymes to release one acetyl-CoA and one propionyl-CoA. Reducing equivalents expected from the above pathways are five NADH+ and three FADH2. This is in addition of two acetyl-CoAs and two succinyl-CoAs that are the anaplerotic contribution to the TCA cycle
After considering the possible channeling of the above branched chain fatty acids to other metabolic pathways including lipogenesis, AdMC7 utilization as an anaplerotic amino fatty acid was examined. The structure of AdMC7, a water-soluble zwitterion, suggests a possible requirement for active transport across membranes, potentially leading to less first pass uptake by liver and increased delivery to other tissues. It also may have other metabolic advantages. The hypothetical metabolic pathway, Scheme 3, demonstrates that it is likely reliant on mostly mitochondrial matrix enzymes for its utilization. Being water-soluble, AdMC7 also has flexibility over oil-based hydrophobic fatty acids for alternative pharmaceutical formulations. Similar to the C7G design, it could also be formulated as a triglyceride or conjugated to an amino acid through a peptide bond.
SCHEME 3.
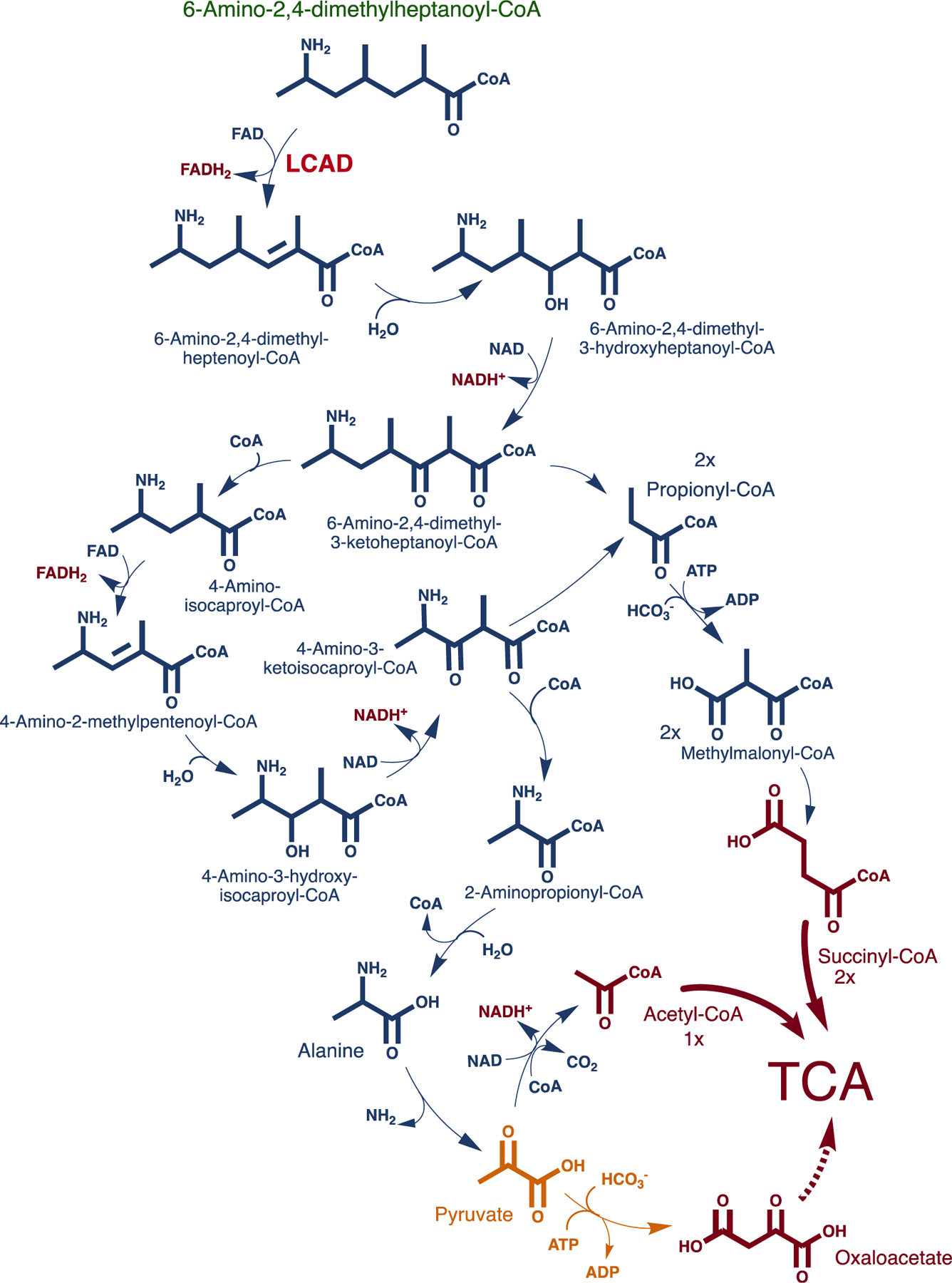
Proposed metabolic pathway of 6-amino-2,4-dimethylheptanoyl-CoA, the CoA ester of 6-amino-2,4-dimethylheptanoic acid. LCAD is expected to carry out the first α,β-dehydrogenation step of this CoA ester. Two succinyl-CoA and one pyruvate are expected as the active anaplerotic contribution of the amino fatty acid with possible channeling of the latter to other functions including gluconeogenesis via conversion to oxaloacetate by pyruvate carboxylase
In conclusion, while we recognize the variability in this study being high, we found that normalizing performances of the various MBCFAs, using C7 as the reference fatty acid, was a suitable tool to offset such variabilities that relate to the experimental limitations. Based on this comparative approach, this study identifies three medium branched chain fatty acids, with a postulated key methyl group at the C2 and/or C4 position, that can be considered as treatment alternatives to C7 or C8, in their triglyceride form, for long chain FAODs. Additional questions that should be addressed include whether these MBCFAs would: (1) have favorable tissue distribution profiles, and/or (2) be amenable to more tolerable formulations than C7G or MCT oil. Ultimately, a final optimized therapeutic drug may contain different anaplerotic moieties on the hydroxyl arms of the glycerol backbone, or even be composed of a mixture of conjugates of carriers other than glycerol.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to acknowledge Lorna Cropcho at the Biochemical Genetics Lab, UPMC Children’s Hospital of Pittsburgh, for material support and technical advice and methods transfer of the mass spectroscopy acylcarnitine assay to our lab. The authors would also like to thank Drs. Stacy G. Wendell and Steven Mullett at the Health Sciences Metabolomics & Lipidomics Core at the University of Pittsburgh for providing their expertise.
Funding information
Children’s Hospital of Pittsburgh Foundation, PA (US); National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Number: R01 DK78755; University of Pittsburgh
FUNDING
This study was partly funded by the Children’s Hospital of Pittsburgh Foundation, PA, and Jerry Vockley NIH R01 DK78755.
Footnotes
CONFLICT OF INTEREST
Al-Walid Mohsen, the corresponding author, has a patent application submitted to the US patent office pertaining compounds mentioned in this manuscript. Anuradha Karunanidhi, Clinton Van’t Land, Dhivyaa Rajasundaram, Mateus Grings, and Jerry Vockley, all have no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
Data archiving is not mandated but data will be made available on reasonable request.
REFERENCES
- 1.Borges K, Kaul N, Germaine J, Kwan P, O’brien TJ. Randomized trial of add-on triheptanoin vs medium chain triglycerides in adults with refractory epilepsy. Epilepsia Open. 2019;4: 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calvert S, Barwick K, Par M, Ni Tan K, Borges K. A pilot study of add-on oral triheptanoin treatment for children with medically refractory epilepsy. Eur J Paediatr Neurol. 2018;22:1074–1080. [DOI] [PubMed] [Google Scholar]
- 3.Gillingham MB, Heitner SB, Martin J, et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2017;40:831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hainque E, Caillet S, Leroy S, et al. A randomized, controlled, double-blind, crossover trial of triheptanoin in alternating hemiplegia of childhood. Orphanet J Rare Dis. 2017;12:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hainque E, Gras D, Meneret A, et al. Long-term follow-up in an open-label trial of triheptanoin in GLUT1 deficiency syndrome: a sustained dramatic effect. J Neurol Neurosurg Psychiatry. 2019;90:1291–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahapatra S, Ananth A, Baugh N, Damian M, Enns GM. Triheptanoin: a rescue therapy for cardiogenic shock in carnitineacylcarnitine translocase deficiency. JIMD Rep. 2018;39:19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDonald T, Hodson MP, Bederman I, Puchowicz M, Borges K. Triheptanoin alters [U-(13)C6]-glucose incorporation into glycolytic intermediates and increases TCA cycling by normalizing the activities of pyruvate dehydrogenase and oxoglutarate dehydrogenase in a chronic epilepsy mouse model. J Cereb Blood Flow Metab. 2019;40:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald T, Puchowicz M, Borges K. Impairments in oxidative glucose metabolism in epilepsy and metabolic treatments thereof. Front Cell Neurosci. 2018;12:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mochel F, Hainque E, Gras D, et al. Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J Neurol Neurosurg Psychiatry. 2016;87:550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ørngreen MC, Vissing J. Treatment opportunities in patients with metabolic myopathies. Curr Treat Options Neurol. 2017; 19:37. [DOI] [PubMed] [Google Scholar]
- 11.Roe CR, Brunengraber H. Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin: review of 15 years experience. Mol Genet Metab. 2015;116:260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiffmann R, Wallace ME, Rinaldi D, et al. A double-blind, placebo-controlled trial of triheptanoin in adult polyglucosan body disease and open-label, long-term outcome. J Inherit Metab Dis. 2018;41:877–883. [DOI] [PubMed] [Google Scholar]
- 13.Schwarzkopf TM, Koch K, Klein J. Reduced severity of ischemic stroke and improvement of mitochondrial function after dietary treatment with the anaplerotic substance triheptanoin. Neuroscience. 2015;300:201–209. [DOI] [PubMed] [Google Scholar]
- 14.Shoffner JM. Concerning "Triheptanoin vs trioctanoin for long-chain fatty acid oxidation disorders: A double blinded, randomized controlled trial" by Gillingham et al. J Inherit Metab Dis. 2019;42:394–395. [DOI] [PubMed] [Google Scholar]
- 15.Sklirou E, Alodaib AN, Dobrowolski SF, Mohsen A-WA, Vockley J. Physiological perspectives on the use of triheptanoin as anaplerotic therapy for long chain fatty acid oxidation disorders. Front Genet. 2021;11:1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan KN, Hood R, Warren K, et al. Heptanoate is neuroprotective in vitro but triheptanoin post-treatment did not protect against middle cerebral artery occlusion in rats. Neurosci Lett. 2018a;683:207–214. [DOI] [PubMed] [Google Scholar]
- 17.Tan KN, Simmons D, Carrasco-Pozo C, Borges K. Triheptanoin protects against status epilepticus-induced hippocampal mitochondrial dysfunctions, oxidative stress and neuronal degeneration. J Neurochem. 2018b;144:431–442. [DOI] [PubMed] [Google Scholar]
- 18.Vockley J, Burton B, Berry GT, et al. Results from a 78-week, single-arm, open-label phase 2 study to evaluate UX007 in pediatric and adult patients with severe long-chain fatty acid oxidation disorders (LC-FAOD). J Inherit Metab Dis. 2019;42: 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vockley J, Burton B, Berry GT, et al. UX007 for the treatment of long chain-fatty acid oxidation disorders: safety and efficacy in children and adults following 24 weeks of treatment. Mol Genet Metab. 2017;120:370–377. [DOI] [PubMed] [Google Scholar]
- 20.Vockley J, Charrow J, Ganesh J, et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol Genet Metab. 2016; 119:223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamada K, Taketani T. Management and diagnosis of mitochondrial fatty acid oxidation disorders: focus on very-long-chain acyl-CoA dehydrogenase deficiency. J Hum Genet. 2019; 64:73–85. [DOI] [PubMed] [Google Scholar]
- 22.Wanders RJ, Denis S, Ruiter JP, Ijlst IJ, Dacremont G. 2,6-Dimethylheptanoyl-CoA is a specific substrate for long-chain acyl-CoA dehydrogenase (LCAD): evidence for a major role of LCAD in branched-chain fatty acid oxidation. Biochim Biophys Acta. 1998;1393:35–40. [DOI] [PubMed] [Google Scholar]
- 23.Rinaldo P, Cowan TM, Matern D. Acylcarnitine profile analysis. Genet Med. 2008;10:151–156. [DOI] [PubMed] [Google Scholar]
- 24.Shen JJ, Matern D, Millington DS, et al. Acylcarnitines in fibroblasts of patients with long chain 3-hydroxyacyl-CoA dehydrogenase deficiency and other fatty acid oxidation disorders. J Inherit Metab Dis. 2000;23:27–44. [DOI] [PubMed] [Google Scholar]
- 25.Smith EH, Matern D. Acylcarnitine analysis by tandem mass spectrometry. Cut Protoc Human Genet. 2010;17:1–20. [DOI] [PubMed] [Google Scholar]
- 26.Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders: a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1066–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leipnitz G, Mohsen A-W, Karunanidhi A, et al. Evaluation of mitochondrial bioenergetics, dynamics, endoplasmic reticulum-mitochondria crosstalk, and reactive oxygen species in fibroblasts from patients with complex I deficiency. Sci Rep. 2018;8:1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCoin CS, Gillingham MB, Knotts TA, et al. Blood cytokine patterns suggest a modest inflammation phenotype in subjects with long-chain fatty acid oxidation disorders. Physiol Rep. 2019;7:e14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills EL, Ryan DG, Prag HA, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seminotti B, Leipnitz G, Karunanidhi A, et al. Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum Mol Genet. 2019;28:928–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roe CR, Sweetman L, Roe DS, David F, Brunengraber H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest. 2002;110:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Critelli K, Mckiernan P, Vockley J, et al. Liver transplantation for propionic acidemia and methylmalonic acidemia: perioperative management and clinical outcomes. Liver Transpl. 2018; 24:1260–1270. [DOI] [PubMed] [Google Scholar]
- 33.De Bie I, Nizard SD, Mitchell GA. Fetal dilated cardiomyopathy: an unsuspected presentation of methylmalonic aciduria and hyperhomocystinuria, cblC type. Prenat Diagn. 2009;29:266–270. [DOI] [PubMed] [Google Scholar]
- 34.Prada CE, Al Jasmi F, Kirk EP, et al. Cardiac disease in methylmalonic acidemia. J Pediatr. 2011;159:862–864. [DOI] [PubMed] [Google Scholar]
- 35.Riemersma M, Hazebroek MR, Helderman-Van Den Enden A, et al. Propionic acidemia as a cause of adult-onset dilated cardiomyopathy. Eur J Hum Genet. 2017;25:1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sadhukhan S, Liu X, Ryu D, et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci U S A. 2016;113: 4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Primassin S, Tucci S, Spiekerkoetter U. Hepatic and muscular effects of different dietary fat content in VLCAD deficient mice. Mol Genet Metab. 2011;104:546–551. [DOI] [PubMed] [Google Scholar]
- 38.Tucci S Very long-chain acyl-CoA dehydrogenase (VLCAD-) deficiency-studies on treatment effects and long-term outcomes in mouse models. J Inherit Metab Dis. 2017;40:317–323. [DOI] [PubMed] [Google Scholar]
- 39.Tucci S, Pearson S, Herebian D, Spiekerkoetter U. Long-term dietary effects on substrate selection and muscle fiber type in very-long-chain acyl-CoA dehydrogenase deficient (VLCAD (–/–)) mice. Biochim Biophys Acta. 2013;1832:509–516. [DOI] [PubMed] [Google Scholar]
- 40.Bowman CE, Wolfgang MJ. Role of the malonyl-CoA synthetase ACSF3 in mitochondrial metabolism. Adv Biol Regul. 2019; 71:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verhoeven NM, Roe DS, Kok RM, Wanders RJ, Jakobs C, Roe CR. Phytanic acid and pristanic acid are oxidized by sequential peroxisomal and mitochondrial reactions in cultured fibroblasts. J Lipid Res. 1998;39:66–74. [PubMed] [Google Scholar]
- 42.Wanders RJ, Komen J, Ferdinandusse S. Phytanic acid metabolism in health and disease. Biochim Biophys Acta. 2011;1811:498–507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data archiving is not mandated but data will be made available on reasonable request.