

Abstract
Mutations in the TP53 tumour suppressor gene are found in ~50% of human cancers [1–6]. TP53 functions as a transcription factor that directly regulates the expression of ~500 genes, some of them involved in cell cycle arrest/cell senescence, apoptotic cell death or DNA damage repair, i.e. the cellular responses that together prevent tumorigenesis [1–6]. Defects in TP53 function not only cause tumour development but also impair the response of malignant cells to anti-cancer drugs, particularly those that induce DNA damage [1–6]. Most mutations in TP53 in human cancers cause a single amino acid substitution, usually within the DNA binding domain of the TP53 protein. These mutant TP53 proteins are often expressed at high levels in the malignant cells. Three cancer causing attributes have been postulated for mutant TP53 proteins: the inability to activate target genes controlled by wt TP53 (loss-of-function, LOF) that are critical for tumour suppression, dominant negative effects (DNE), i.e. blocking the function of wt TP53 in cells during early stages of transformation when mutant and wt TP53 proteins are co-expressed, and gain-of-function (GOF) effects whereby mutant TP53 impacts diverse cellular pathways by interacting with proteins that are not normally engaged by wt TP53 [1–6]. The GOF effects of mutant TP53 were reported to be essential for the sustained proliferation and survival of malignant cells and it was therefore proposed that agents that can remove mutant TP53 protein would have substantial therapeutic impact [7–9]. In this review article we discuss evidence for and against the value of targeting mutant TP53 protein for cancer therapy.
Subject terms: Tumour-suppressor proteins, Protein folding, Cancer
The attributes proposed for mutant TP53 to drive tumour development and possible approaches to target them. Model depicting the three attributes by which mutant TP53 is thought to promote tumour development: loss-of-function (LOF), dominant negative effects (DNE) over wt TP53 and neomorphic gain-of-function (GOF) effects.
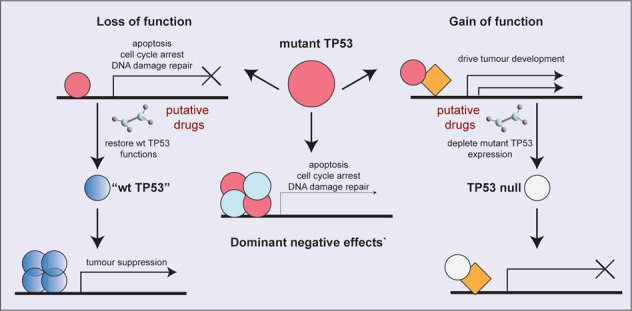
Facts
Mutations in the tumour suppressor TP53 are common in diverse human cancers (~50%) and are frequently associated with poor responses to anti-cancer therapy.
Mutations in TP53 block its tumour suppressive functions by preventing it from binding to target DNA sequences and upregulating genes that mediate several cellular processes, including apoptosis, cell cycle arrest and senescence.
Mutant TP53 proteins are also reported to have gain-of-function properties that can contribute to tumour growth.
Open questions
Are the reported gain-of-function effects of mutant TP53 proteins really critical for the sustained growth and therapy resistance of malignant cells?
Do the compounds that were reported to specifically target mutant TP53 proteins indeed kill malignant cells by acting on mutant TP53?
What are the best approaches for treating cancers expressing mutant TP53 protein?
Wild-type (wt) TP53 functions as a tumour suppressor
TP53 is a master transcription factor which directly regulates the expression of ~500 genes involved in diverse cellular responses [1, 2]. The TP53 protein contains several functional domains. Two acidic transactivation domains are located at the N-terminus, and they are critical for TP53 to interact with transcriptional co-activators and co-repressors. An unstructured basic regulatory domain is found at the C-terminus, which assists in the binding of TP53 to DNA and the stabilisation of TP53-DNA complexes. A sequence-specific DNA-binding domain is located in the centre of the TP53 protein. Finally, TP53 functions as a tetramer and the tetramerisation domain is located near the C-terminus [10].
In unstressed cells the TP53 protein is present at only low levels, mostly owing to a negative feedback loop: TP53 can transcriptionally induce MDM2 (called HDM2 in humans), an E3 ubiquitin ligase that ubiquitinates TP53, thereby priming it for proteasomal degradation [11]. When cells are subjected to stress, such as DNA damage, deprivation of metabolites or oncogene activation, the MDM2-TP53 interaction is inhibited as a consequence of several upstream signalling events that are not discussed here (for reviews, see [2–4]), and this results in the stabilisation of the TP53 protein. Stabilised TP53 accumulates in the nucleus where it binds as a homo-tetramer in a sequence-specific manner to target genes to regulate their expression, most likely in conjunction with additional transcription factors [12, 13]. In this way TP53 regulates the cellular responses that are critical for tumour suppression.
TP53 induces cell cycle arrest at the G1/S boundary and cell senescence mainly by direct transcriptional activation of the gene for the cyclin dependent kinase (CDK) inhibitor, p21 [14, 15] (Fig. 1). In addition, 14-3-3σ and GADD45 were reported to be critical for TP53-induced cell proliferation arrest by blocking the G2/M transition [16, 17].
Fig. 1. The functions of wt TP53.

Model depicting the target genes activated by wt TP53 and the cellular responses in which their protein products function in wt TP53 mediated tumour suppression.
TP53 induces cell death by activating the intrinsic apoptotic pathway that is regulated by the BCL-2 protein family which contains three types of proteins with different functions: the pro-survival family members (e.g. BCL-2, MCL-1) to prevent apoptosis, the pro-apoptotic BH3-only proteins (e.g. BIM, PUMA, NOXA) to initiate apoptosis, and the effectors of apoptosis (BAX, BAK) to kill cells [18, 19]. TP53 can directly transcriptionally activate the genes encoding the pro-apoptotic BH3-only proteins PUMA and NOXA [20–22] that are required for TP53-induced apoptosis, for example after exposure to DNA damage inducing agents [23–25] (Fig. 1). Notably, lymphoid cells from Puma/Noxa double knockout mice are as resistant to these agents as those from Trp53 knockout mice [26, 27], demonstrating that direct transcriptional activation of Puma and Noxa accounts for all the apoptosis inducing action of TP53 (at least in these cell types).
TP53 was also shown to activate genes that function in the coordination of DNA damage repair [28]. TP53 can orchestrate the nucleotide excision repair (NER) of UV-induced DNA damage through direct transcriptional activation of the DDB2 (encoding p48) and XPC genes [29, 30]. TP53 was also reported to transcriptionally activate several genes involved in DNA mismatch repair, including MLH1, MSH2 and PMS2 [31, 32]. This DNA repair process is thought to be important in tumour suppression since Mlh1, Msh2 as well as Pms2 gene deficient mice spontaneously develop tumours (Fig. 1) [33].
Several other cellular responses have also been reported to be activated by wt TP53, including the coordination of metabolism (for an expert review see [34]) and the silencing of large parts of the genome, thereby controlling retrotransposons [35].
Which cellular processes activated by TP53 and which TP53 target genes are critical for the prevention of tumour development?
It remains unclear which of the many processes activated by TP53 are critical for tumour suppression and it is possible that the relative contributions of these processes to tumour suppression may vary depending on cell type and on which oncogenes are active in a given cell. While the absence of TP53 leads to lymphoma or certain other cancers with 100% penetrance in mice within ~250–300 days [36, 37], the loss of individual TP53 target genes that are critical for a given cellular response does not usually cause spontaneous tumour development in mice. For example, mice lacking p21, which is essential for TP53-induced G1/S boundary cell cycle arrest [14] and also plays a major role in TP53-induced cell senescence [38], are not tumour prone on a C57BL/6 background [15]. However, compared to wt controls an increased incidence of several types of cancers was observed on a mixed C57BL/6x129SV background [39] (a genetic background with higher tumour predisposition compared to C57BL/6). Moreover, mice lacking PUMA, NOXA or both of these BH3-only proteins that are essential for TP53-induced apoptosis [27, 40], are not tumour prone [40], although the absence of PUMA did accelerate c-MYC-driven lymphomagenesis [26] and increased the severity of carcinogen induced colon cancer development [41]. Remarkably, even Puma/Noxa/p21 triple knockout mice (on a C57BL/6 background) do not spontaneously develop cancer over at least 18 months even though their cells are defective in TP53-induced apoptosis, G1/S boundary cell cycle arrest and cell senescence [42]. In striking contrast, loss of certain genes that function in DNA damage repair and are reported to be directly regulated by TRP53 (e.g. Mlh1, Msh2) causes a marked predisposition to spontaneous tumour development in mice and also accelerated c-MYC-driven lymphomagenesis [32]. These findings and others [43] indicate that coordination of DNA repair may be the most important TP53 regulated process for tumour suppression.
The role of mutant TP53 in tumorigenesis
TP53 is the most frequently mutated gene (~50%) in human cancers [44]. Although some TP53 mutations lead to the complete loss of TP53 protein (mimicked experimentally by Trp53 knockout mice), most are missense mutations that cause single amino acid substitutions, usually within the TP53 DNA-binding domain (Fig. 2). The levels of such mutant TP53 proteins are generally very high in malignant cells although not in mutant TP53 expressing pre-malignant cells in mice in vivo [44, 45]. Mutant TP53 proteins have been proposed to drive malignant transformation and sustain tumour growth through several processes. Of note, the Li-Fraumeni cancer predisposition syndrome in humans, characterised by diverse cancers (e.g. osteosarcoma, acute leukaemia, breast cancer and brain cancer) often arising at a young age, is in ~70% of cases caused by inherited mutations in one allele of TP53 with loss of the wt TP53 allele (loss of heterozygosity, i.e. LOH) apparent in the malignancies that arise in these patients (for an expert review see [46]).
Fig. 2. Localisation of point mutations in TP53.

Model depicting the location and relative frequencies of point mutations found in human cancers. Data are derived from [44, 132].
Loss of function (LOF)
The TP53 mutations either change the conformation of TP53 proteins (structural mutants) or affect amino acids involved in DNA binding (contact mutants) [47]. Both types of TP53 mutants are unable to transcriptionally activate wt TP53 target genes and thereby cannot induce the essential mediators of apoptosis, cell cycle arrest, cell senescence and DNA damage repair, the processes thought to be critical for wt TP53 mediated tumour suppression.
Elegant experimental systems were developed in which tumour development could be initiated in mice by the absence of wt TP53, but expression of this tumour suppressor could be restored in the malignant cells. This revealed that restoration of wt TP53 in TP53-deficient cancers resulted in tumour regression owing to the activation of apoptosis or cell senescence in lymphomas or solid cancers, respectively [48–50]. Why restoration of wt TP53 causes apoptosis in lymphomas but cell senescence in solid cancers remains an intriguing question. This could be due to differences between these cell types and/or differences in the other oncogenic lesions that drive these malignancies. These factors could impact post-translational modification of TP53 and thereby influence its potency in activating different subsets of its direct target genes (i.e. cell cycle regulating genes vs cell death inducing genes), for example by attracting different co-activators or influencing its binding to target sequences in DNA (reviewed in [51]). Regardless, these findings reveal that sustained LOF of TP53 is required for continued tumour expansion.
Dominant negative effects (DNE) of mutant TP53
Whilst advanced tumours that have mutations in TP53 have often selected for loss of the WT TP53 allele (loss of heterozygosity (LOH)), at the early stages of malignant transformation, mutant TP53 often co-exists with wt TP53 in the nascent neoplastic cells. The mutant TP53 prevents wt TP53 from exerting its tumour suppressive functions due to the formation of mixed wt/mutant TP53 hetero-tetramers [44]. These mixed tetramers have significantly reduced ability to induce gene expression and cellular responses that are normally driven by tetramers containing only wt TP53 [52, 53]. Initial evidence for a dominant negative effect (DNE) of mutant TP53 came from co-transfection experiments in vitro, showing that processes known to be activated by wt TP53 could be inhibited by concomitant expression of mutant TP53 [54, 55]. A DNE of mutant TP53 was also observed in cells from Trp53R172H/+, Trp53R246S/+ and Trp53R270H/+ mutant knock-in mice. After treatment with stimuli that activate wt TP53 (e.g. γ-radiation), cells from these mice underwent considerably less apoptosis or cell cycle arrest compared to cells from wt (Trp53+/+) mice [56, 57]. The DNE of various TP53 mutants could also be seen when they were expressed in haematopoietic stem and progenitor cells (HSPCs) from Trp53+/− or Eμ-Myc;Trp53+/+ mice but, as expected, not when expressed in HSPCs from Trp53−/− mice since the latter do not express wt TP53 on which mutant TP53 could exert its DNE [43, 58]. The loss of the wt TP53 allele during advanced stages of tumorigenesis (LOH) [59] indicates that the complete loss of wt TP53 function provides further advantages for tumour expansion, even when mutant TP53 is expressed. Interestingly, the levels of mutant TP53 protein are much higher in tumour cells with LOH compared to cells co-expressing both wt TP53 and mutant TP53 [60]. This demonstrates that wt TP53 can somehow repress the levels of mutant TP53 protein.
Gain-of-function (GOF) effects of mutant TP53
Mutant TP53 has also been reported to be able to exert neomorphic gain-of-function (GOF) properties [44], i.e. functions that wt TP53 cannot exert. Removal of mutant TP53, and hence its GOF effects, by using siRNA was reported to inhibit the growth of certain tumour cells in culture and in vivo and to increase their sensitivity to cytotoxic drugs [9, 61]. The GOF effects of mutant TP53 were also reported to enhance tumour metastasis by impacting transcription factors that control the epithelial-mesenchymal transition (EMT) [62–64]. The GOF effects of mutant TP53 were shown to assist malignant cells in metabolic reprogramming to adapt to changes in the availability of growth factors and nutrients by activating glycolysis [65], promoting lipid synthesis [66] and nucleotide synthesis [67].
It has been postulated that mutant TP53 exerts its alleged GOF effects mainly through interactions with other transcription factors to upregulate or downregulate the expression of genes that are not controlled by wt TP53. Some TP53 mutants were reported to inhibit the functions of TP63 and TP73, the two family members of TP53 that can also regulate many of the known TP53 target genes [68–71]. Mutant TP53 was also reported to increase the transcriptional transactivation activity of NF-kB, E2F1, ETS1/ETS2 and YAP1, which are all implicated in promoting tumour growth [72–74] (Fig. 3). Many mechanisms have been proposed to be responsible for these reported GOF effects of mutant TP53 proteins, mostly including protein-protein interactions in which wt TP53 does not engage. It is also important to bear in mind that different mutant TP53 proteins may engage in different protein-protein interactions and thereby exert different GOF effects (for a review see [75]).
Fig. 3. The mechanisms proposed for mutant TP53 to exert its alleged GOF effects.

Model depicting the proposed interactions with other transcription factors that mutant TP53 proteins engage in to drive the development and expansion of tumours.
Not all TP53 mutants are equivalent
Hundreds of different TP53 mutants have been identified in human cancers, and it appears likely that they do not all function in the same way in driving tumour development. Most TP53 mutations lead to LOF [44]. This may, however, not be universal, since some TP53 mutant proteins were reported to still retain part of the functions of wt TP53. For example, some mutations located in the acidic transactivation domains result in the production of a truncated form of TP53 that retains the ability to induce apoptosis [76]. Moreover, the DNA-binding domain mutant, TP53K120R, was reported to be only defective in the induction of apoptosis but was still able to induce cell cycle arrest and cell senescence [77].
Interestingly, different single-amino acid substitutions that affect the same residue were reported to have different impacts on TP53 function. R175C behaved like wt TP53 and could induce both cell cycle arrest and apoptosis. Conversely, the R175P mutant TP53 was defective in inducing apoptosis but retained the ability to induce cell cycle arrest, whereas R175D mutant TP53 showed loss of both functions [78].
Moreover, not all the TP53 mutants appear to be able to exert a DNE over endogenous wt TP53. Experiments using enforced expression of different TP53 mutants in colon cancer cells expressing endogenous wt TP53 first provided evidence for this notion. Only one hot-spot mutant that was tested, R273H, displayed a DNE, whereas the other TP53 mutants examined, V143A, R175H and R248W, were not able to repress all activities exerted by the endogenous wt TP53 [79]. In vivo experiments with mutant TP53 knockin mice confirmed and extended these findings. For example, Trp53R172H/+ and Trp53R270H/+ mutant knockin mice (codons R173H and R273H in human, respectively) displayed comparable tumour-free survival times when compared to Trp53+/− mice [56, 80]. However, these findings refer to spontaneous tumour development and it remains possible that in the context of the expression of certain oncogenes, the Trp53R172H/+ and Trp53R270H/+ knockin mice would exhibit significantly shorter tumour-free survival compared to the Trp53+/− mice.
Although LOF and DNE are widely accepted as critical outcomes of TP53 mutations, only some mutations, especially hot-spot mutations, are thought to give rise to GOF effects [81, 82]. Of note, even for different hot-spot mutations, the reported GOF effects are not equivalent. Trp53R172H/− and Trp53R270H/− (codons 175 and 273 in human, respectively) mutant knockin mice showed a different tumour spectrum compared to that seen in Trp53−/− mice but no shortening of tumour-free survival [56]. Humanised TP53R248Q/− mutant knockin mice not only developed different types of tumours but also showed shortened tumour-free survival times compared to Trp53−/− mice [83]. In contrast, enforced expression of five different TRP53 mutants (two of them hot-spot mutations) in HSPCs from Trp53−/− mice did not accelerate tumorigenesis or alter the tumour spectrum compared to control mice reconstituted with HSPCs from Trp53−/− mice that had been transduced with an empty vector [43]. Thus, in this experimental system no GOF effects of any of the TRP53 mutants tested could be detected.
Therapeutic targeting of mutant TP53 for cancer therapy
Depleting mutant TP53 in tumour cells
Since the GOF properties of mutant TP53 have been postulated to contribute to tumour development and metastasis and to impair the response of malignant cells to diverse anti-cancer therapeutics, approaches that could specifically deplete mutant TP53 protein levels are postulated to have promise for the treatment of diverse cancers (Fig. 4). The effects and specificities of the compounds described below are summarised in Table 1.
Fig. 4. Therapeutic approaches for targeting mutant TP53 protein for cancer therapy.

a Removal of mutant TP53 protein by using drugs or siRNA technology is expected to remove the GOF effects of mutant TP53 and to thereby prevent tumour expansion. b Restoring wt TP53 protein conformation in mutant TP53 proteins is expected to restore wt TP53 activated processes (e.g. cell cycle arrest, cell senescence, apoptotic cell death) and to thereby prevent tumour expansion.
Table 1.
List of the compounds discussed indicating their reported effects and specificities for different mutant TP53 proteins.
| Proposed mode of action | Drugs | Tumour type and TP53 state in which the drug was shown to be active |
|---|---|---|
| Depletion of mutant TP53 expression | SAHA | T-cell lymphoma [7] (mutant); breast cancer [86] (mutant);prostate cancer [87] (mutant) |
| FK228 | Neuroblastoma [88] (wt, mutant); non-small-cell lung cancer [89] (wt, mutant) | |
| Statins | Breast cancer, sarcoma, lung cancer, pancreatic cancer [96] (structural mutant); ovarian cancer [97] (wt, mutant) | |
| Gambogic acid | breast cancer [99] (mutant); prostate cancer [100] (null); lung cancer [101] (wt) | |
| siRNA/shRNA | breast cancer [102] (mutant); prostate cancer [103] (mutant); breast and colon cancer [8] (mutant) | |
| Restoration of wt TP53 functions | PRIMA-1/APR-246 | osteosarcoma, lung, ovarian and colon cancer [105] (mutant); multiple myeloma [106] (wt, mutant, null); espohageal cancer [107] (mutant); cholangiocancinoma [108] (mutant); sarcomas [113] (mutant, null); breast, colon cancer [116] (wt, mutant) |
| PEITC | breast and lung cancer [117] (R175H); prostate cancer [118] (mutant); breast cancer [119] (mutant) | |
| RITA | colon cancer [120] (wt); colon, lung, breast, skin cancer, Burkitt lymphoma [121] (mutant); neuroblastoma [122] (wt, mutant) | |
| CP-31398 | lung cancer, melanoma [123] (mutant); colon, breast cancer [124]: mutant; colon, lung, ovarian cancer [125] (wt, mutant); hepatocellular cancer [126] (mutant); colon cancer [127] (mutant); multiple myeloma [128] (wt, mutant, null) | |
| PK7088 | hepatocellular, gastric cancer [129] (Y220C) | |
| Arsenic Trioxide | acute lymphoblastic leukemia, lung, ovarian, pancreatic cancer, melanoma [130] (structural mutants) |
HSP90 and HDAC inhibitors
Heat shock protein 90 (HSP90) is a molecular chaperone that is involved in modulating the folding, stabilisation and degradation of many proteins including some oncogenic proteins, such as mutant TP53. HSP90 has been reported to impact tumour progression, metastasis and high levels of HSP90 are associated with poor prognosis in multiple cancers [84, 85]. The histone deacetylase (HDAC) inhibitor SAHA was reported to synergise with the HSP90 inhibitor 17AAG in degrading mutant TP53, thereby inducing apoptosis and decreasing tumour growth in xenografts [7]. FK228, another HDAC inhibitor, was also shown to inhibit growth and induce apoptosis in tumour cells. However, unlike SAHA, which was reported to kill tumour cells in a manner dependent on the removal of mutant TP53 [86, 87], FK228 induced cell death not only in mutant TP53 expressing tumour cells but also in those expressing wt TP53 [88, 89]. This raises the question of whether HDAC and HSP90 inhibitors, either on their own or in combination, really kill tumour cells by targeting mutant TP53 or through some other process.
Statins
Metabolic reprogramming is a hallmark of cancer [90]. The mevalonate pathway, which regulates the production of cholesterol and isoprenoids, was reported to be involved in the development and progression of cancer [91]. Mutant TP53 expressing tumours often present with over-activation of the mevalonate pathway [92]. Statins target the rate-limiting enzyme in cholesterol biosynthesis and are used as lipid-lowering agents [93]. Recently, the potential of statins in cancer treatment has been proposed based on the observation that these drugs can trigger apoptosis in certain tumour cells [94] and thereby increase their sensitivity to chemotherapeutic drugs [95]. Interestingly, statins were shown to cause the degradation of mutant TP53 by suppressing the interaction of mutant TP53 with the HSP40 family member, DNAJA1, and it was proposed that this process was responsible for their ability to reduce tumour growth [96]. It was reported that statins inhibit the growth of tumour cells expressing mutant TP53, while having only minimal impact on the growth of tumour cells expressing wt TP53. However, there are also studies showing that statins can kill certain tumour cells independent of their TP53 status [97]. Hence, the mechanisms by which statins kill tumour cells need further investigation.
Gambogic acid
Gambogic acid (GA) has been reported to have potential for the treatment of both solid as well as haematological cancers. GA was first reported to reduce the expression of MDM2, thereby activating wt TP53 and inhibiting tumour cell growth in a wt TP53-dependent manner [98]. Subsequent studies reported that GA also has the ability to reduce the levels of mutant TP53 by preventing the formation of HSP90/mutant TP53 complexes, thereby leading to ubiquitin/proteasome mediated degradation of mutant TP53 [99]. However, other studies found that different mechanisms, rather than effects on mutant TP53 protein, are critical for GA-induced killing of malignant cells. GA was reported to activate the intrinsic apoptotic pathway in TP53-deficient prostate cancer cells by inhibiting the MAPK pathway and the transcription factor c-FOS [100]. GA was also shown to activate ROS-induced endoplasmic reticulum (ER) stress, thereby inducing apoptosis in lung cancer cells [101]. Thus, GA appears to be able to inhibit tumour expansion by activating several cell growth inhibitory pathways, and the degradation of mutant TP53 is only one of the possible mechanisms that may contribute.
siRNA and shRNA
RNA interference (RNAi) can be a potential cancer therapeutic by silencing critical genes that are required for the proliferation and/or survival of tumour cells. Based on some of the considerations outlined above, it has been postulated that siRNA or shRNA mediated knockdown of mutant TP53 could be a promising strategy for inhibiting tumour expansion. Reducing the levels of mutant TP53 by using shRNA or siRNA was reported to elicit substantial apoptosis in mutant TP53 expressing breast cancer cells, but not in wt TP53 expressing cancer cells [102]. Knocking down mutant TP53 was shown to reduce the proliferation of prostate cancer cells by inducing cell cycle arrest at the G1/S or G2/M boundaries [103]. Furthermore, depletion of mutant TP53 by siRNA or shRNA was found to increase the sensitivity of certain tumour cells to anti-cancer drugs [8]. Collectively, these studies established the potential of the removal of mutant TP53 as a strategy for cancer therapy (Fig. 4). It must, however, be noted that a recent study pointed out significant off-target effects of RNAi technology: many putative cancer dependencies, and hence potential anti-cancer drug targets, identified from studies based on RNAi were found to be false leads as they could not be validated when using CRISPR technology to remove these proteins [104].
Restoring wt TP53 function in tumour cells expressing mutant TP53
Mutant TP53 proteins lack the ability to transactivate wt TP53 target genes, resulting in the loss of tumour suppressive functions. Restoring wt TP53 transcriptional activities to mutant TP53 in tumour cells is expected to lead to proliferation arrest, cellular senescence and apoptotic death with a consequent therapeutic benefit (Fig. 4), although this has not yet been proven using genetically engineered mice in which cells can be switched first from expressing wt TRP53 to mutant TRP53 and then, at will, back to wt TRP53.
Mutant TP53 reactivating agent (PRIMA-1) and APR-246
PRIMA-1 was identified in 2002 in a screen of a library of low-molecular-weight compounds [105]. PRIMA-1 was reported to restore wt TP53 conformation to several mutant TP53 proteins that were studied and to thereby reactive wt TP53 transcriptional activities in tumour cells expressing mutant TP53. This was shown to delay the growth and increase apoptosis of tumour cells with mutant TP53, but not of tumour cells containing wt TP53. APR-246, the methylated analogue of PRIMA-1, showed more efficient killing effects in tumour cells containing mutant TP53 and shared many characteristics with PRIMA-1. PRIMA-1 and APR-246 were able to reactivate the expression of wt TP53 target genes in tumour cells expressing mutant TP53 and these compounds were reported to kill tumour cells by inducing the expression of the pro-apoptotic BH3-only protein, NOXA [106, 107]. Moreover, PRIMA-1 and APR-246 were shown to activate the expression of p21 in mutant TP53 expressing tumour cells, thereby inducing cell cycle arrest and cell senescence [105, 108]. Importantly, these two compounds not only killed tumour cells in vitro, but they were also shown to delay tumour growth in vivo, thereby prolonging the survival of mice transplanted with cancer cells [105, 106]. Finally, both PRIMA-1 and APR-246 were shown to cooperate with certain chemotherapeutic drugs to kill tumour cells [109, 110]. Collectively, these findings established PRIMA-1 and APR-246 as promising anti-cancer drugs that target mutant TP53. APR-246 is currently being tested in several phase II clinical trials, including in TP53-mutant myeloid malignancies, high-grade serous ovarian cancer, oesophageal cancer and melanoma. However, PRIMA-1 was also shown to be able to kill tumour cells with wt TP53 [111], and even tumour cells that lack TP53 [112]. Similar observations were also reported for APR-246, which was shown to kill tumour cells irrespective of their TP53 status [113]. A critical underlying mechanism of APR-246 induced killing of malignant cells appears to involve reactive oxygen species (ROS). APR-246 was shown to cause an accumulation of intracellular ROS in a dose-dependent manner and this was reported to induce apoptosis in tumour cells irrespective of their TP53 status. This killing of tumour cells could be inhibited by scavenging intracellular ROS [114]. The ER stress/UPR-pathway was also reported to be involved in PRIMA-1 induced killing of malignant cells [115]. The unfolded and misfolded protein response pathways were significantly up-regulated in multiple myeloma (MM) cells in response to treatment with PRIMA-1, as demonstrated by the increased levels of HSP70, GADD34 and CHOP, all of which are markers of ER stress. Moreover, PRIMA-1 was found to cooperate with the UPR-inducing agent, bortezomib, in the killing of MM cells, and it could even re-sensitise bortezomib-resistant MM cells to this proteasome inhibitor [115]. Finally, PRIMA-1 was reported to induce autophagy in breast cancer cells as well as soft-tissue sarcoma cells, and this was independent of their TP53 status [113, 116]. Collectively, these observations indicate that the mechanisms of APR-246 induced killing of malignant cells still remain unclear. Recently it was reported that expression of SLC7A11 is a more reliable predictor of response to APR-246 that TP53 status in cancer cells [117]. The identification of the mechanisms that are responsible for APR-246 induced killing of tumour cells is predicted to inform the currently ongoing clinical trials of this drug.
Phenethyl Isothiocyanate (PEITC)
Phenethyl isothiocyanate (PEITC) is present at high levels in watercress and cruciferous vegetables, and this compound was reported to exert remarkable chemotherapeutic activity. Mutant TP53 was reported to be a target of PEITC. Unlike APR-246, which was reported to be able to restore wt TP53 function in all contact mutant TP53 proteins tested, PEITC was shown to delay proliferation and induce apoptosis only in tumour cells expressing one specific TP53 mutant protein, R175 [118]. PEITC was reported to restore wt TP53 conformation and transactivation functions to R175 mutant TP53. However, another study revealed that PEITC could exert significant anti-cancer activity not only in malignant cells expressing R175 mutant TP53, but also in cancer cells expressing any of the recognised structural TP53 mutants, including P223L, but it had no impact on tumour cells expressing contact TP53 mutants [119]. Mechanistically, it was reported that PEITC caused a reduction in the levels of mutant TP53 protein in tumour cells through a post-transcriptional process, whereas it had only minimal impact on the levels of wt TP53 in malignant cells [118, 120]. This suggests that PEITC may be able to target mutant TP53 through two different processes, reactivation of wt TP53 functions and a reduction of the levels of mutant TP53 protein. However, considerably more work is needed to validate PEITC as a promising compound for anti-cancer therapy and to identify which of its proposed mechanisms of action are critical for the killing of malignant cells.
Reactivation of TP53 and Induction of Tumour Cell Apoptosis (RITA)
RITA (reactivation of TP53 and induction of tumour cell apoptosis) is a small molecule that was identified in a screen of the National Cancer Institute library of compounds. It was initially reported to inhibit the TP53-MDM2 interaction and thereby activate wt TP53-driven anti-tumour effects [121]. However, subsequent studies reached the conclusion that RITA could also suppress proliferation and induce apoptosis in tumour cells expressing mutant TP53, since the compound was found to restore wt TP53 transcriptional activities in several hot-spot TP53 mutants. This was based on the observation that treatment with RITA led to the induction of wt TP53 target genes, including GADD45, BBC3, BAX and CKDN1A, in tumour cells expressing mutant TP53 [122]. However, similar to APR-246, other studies showed that RITA was also able to induce apoptosis in tumour cells expressing wt TP53 and even in TP53-deficient cancer cells [123]. Collectively, these studies indicate that the anti-cancer effects of RITA may not be specifically dependent on mutant TP53, and the mechanisms of RITA induced killing of tumour cells still need to be clarified.
CP-31398
CP-31398 is a small molecule that was identified in a screen of a synthetic compound library. It showed the ability to restore a functionally active conformation in mutant TP53 proteins, allowing it to exert wt TP53 transcriptional activity [124]. Subsequent studies revealed that the ability of CP-31398 to induce cell death was TP53-dependent, as this compound could only induce apoptosis in tumour cells expressing wt TP53 or mutant TP53, but not in those deficient for TP53 [125]. CP-31398 was found to increase the levels of wt TP53 by blocking its ubiquitination and proteasomal degradation and this allowed TP53 to activate its canonical cellular responses, including cell cycle arrest and apoptosis in tumour cells [126]. Conversely, CP-31398 was also reported to restore wt TP53 tumour suppressive function in TP53 mutant proteins independent of the nature of the TP53 mutation. CP-31398 was shown to delay the growth of hepatocellular cancer cells expressing R249S or Y220C mutant TP53 and colorectal cancer cells expressing R248Q or P309S mutant TP53, both in vitro and in vivo [127, 128]. The growth inhibitory effects were comparable between cancer cells expressing different TP53 mutants and cancer cells of different cellular origin. CP-31398 was also reported to cause an increase in ROS production and thereby trigger the intrinsic apoptotic pathway in MM cells, regardless of their TP53 status [129]. Collectively, these findings indicate that CP-31398 induced killing of malignant cells may not depend on the expression of mutant TP53 and the mechanisms responsible still require further investigation.
PK7088
The Y220C TP53 mutant protein is a paradigm for studying the restoration of wt TP53 function in a mutant TP53 protein, because it contains a unique surface crevice that is amenable to targeting by small molecules. The small molecule compound PK7088 was reported to bind to this surface crevice on Y220C mutant TP53 and thereby convert its structure from a mutant into the wt conformation with restoration of wt TP53 transcriptional activity [130]. PK7088 was shown to induce TP53-dependent cell cycle arrest and apoptosis by activating the expression of p21 and NOXA, respectively. These effects could be enhanced by addition of the MDM2 inhibitor nutlin-3a, which further indicated the successful restoration of wt TP53 structure and function in Y220C mutant TP53. This work raises the possibility that one specific small molecule might be needed to target each specific TP53 mutant protein, heralding a new paradigm for treating mutant TP53 expressing cancers.
Arsenic trioxide (ATO)
Arsenic trioxide (ATO) is a small molecule reported to be able to restore wt TP53 function in tumour cells expressing structural TP53 mutants [131]. ATO can bind to the DNA binding domain only in structural but not in contact TP53 mutants. It thereby induces the transcriptional activities that are characteristic of wt TP53, leading to the suppression of tumour growth both in vitro and in vivo. Thus, ATO may provide a promising therapy against mutant TP53 expressing cancers, but more studies are needed to validate its specificity and efficiency in treating mutant TP53.
Outlook
Many approaches have been tried in pre-clinical tests and even clinical trials to treat cancers by targeting mutant TP53, including reducing the levels of mutant TP53 protein or restoring wt TP53 functions in mutant TP53 proteins. However, for all of these approaches there is evidence in the published literature that the compounds tested can also kill malignant cells through processes that are independent of mutant TP53, or there are other significant limitations. Thus, targeting of mutant TP53 for anti-cancer therapy still remains a challenge that requires further investigation, and importantly, it should first be validated by generating mice in which cells can be sequentially switched from wt TRP53 to mutant TRP53 and then to a TRP53-deficient state that removal (and hence targeting) of mutant TRP53 (TP53) will actually have therapeutic impact.
Acknowledgements
The authors acknowledge the members of their laboratory and the Blood Cells and Blood Cancer Division at The Walter and Eliza Hall Institute (WEHI) for discussions.
Author contributions
ZW, AS and GLK studied the literature and wrote the review article. ZW prepared the figures and the table, which were edited by AS and GLK.
Funding
The work by the authors was supported by fellowships and grants from the Australian National Health and Medical Research Council (NHMRC) (Programme Grant 1113133 to AS, Investigator Grant 2007887 to AS, Synergy Grant 2010275 to AS, Research Fellowship 1116937 to AS, Project Grant 1143105 to AS, Ideas Grants 2002618 and 2001201 to GLK and 2001406), the Leukemia & Lymphoma Society of America (Specialised Centre of Research [SCOR] grant no. 7015-18 to AS and GLK), Victorian Cancer Agency (MCRF Fellowship 17028 to GLK), the estate of Anthony (Toni) Redstone OAM (AS and GLK), the Craig Perkins Cancer Research Foundation (GLK), The Jack Brockhoff Foundation (MAA) and the Dyson Bequest (GLK). Work in the laboratories of the authors was made possible through Victorian State Government Operational Infrastructure Support (OIS) and Australian Government NHMRC Independent Research Institute Infrastructure Support (IRIIS) Scheme.
Competing interests
The authors declare no competing interests.
Footnotes
Edited by G. Melino
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Andreas Strasser, Gemma L. Kelly.
Contributor Information
Andreas Strasser, Email: strasser@wehi.edu.au.
Gemma L. Kelly, Email: gkelly@wehi.edu.au
References
- 1.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170:1062–78. doi: 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 3.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 4.Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer. 2020;20:471–80. doi: 10.1038/s41568-020-0262-1. [DOI] [PubMed] [Google Scholar]
- 5.Oren M. p53: not just a tumor suppressor. J Mol Cell Biol. 2019;11:539–43. doi: 10.1093/jmcb/mjz070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–13. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. 2015;523:352–6. doi: 10.1038/nature14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bossi G, Lapi E, Strano S, Rinaldo C, Blandino G, Sacchi A. Mutant p53 gain of function: reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene. 2006;25:304–9. doi: 10.1038/sj.onc.1209026. [DOI] [PubMed] [Google Scholar]
- 9.Vikhanskaya F, Lee MK, Mazzoletti M, Broggini M, Sabapathy K. Cancer-derived p53 mutants suppress p53-target gene expression-potential mechanism for gain of function of mutant p53. Nucleic Acids Res. 2007;35:2093–104. doi: 10.1093/nar/gkm099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raj N, Attardi LD. The Transactivation Domains of the p53 Protein. Cold Spring Harb Perspect Med. 2017;7:a026047–64. [DOI] [PMC free article] [PubMed]
- 11.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 12.Kamada R, Toguchi Y, Nomura T, Imagawa T, Sakaguchi K. Tetramer formation of tumor suppressor protein p53: Structure, function, and applications. Biopolymers. 2016;106:598–612. doi: 10.1002/bip.22772. [DOI] [PubMed] [Google Scholar]
- 13.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 14.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–16. doi: 10.1016/0092-8674(93)90499-G. [DOI] [PubMed] [Google Scholar]
- 15.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–84. doi: 10.1016/0092-8674(95)90039-X. [DOI] [PubMed] [Google Scholar]
- 16.Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, et al. 14-3-3sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1:3–11. doi: 10.1016/S1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 17.Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, et al. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci USA. 1999;96:3706–11. doi: 10.1073/pnas.96.7.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Bio. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 19.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 20.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–8. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 21.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–94. doi: 10.1016/S1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 22.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–82. doi: 10.1016/S1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 23.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 24.Jeffers JR, Parganas E, Lee Y, Yang CY, Wang JL, Brennan J, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–8. doi: 10.1016/S1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 25.Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, et al. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17:2233–8. doi: 10.1101/gad.1103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michalak EM, Jansen ES, Happo L, Cragg MS, Tai L, Smyth GK, et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009;16:684–96. doi: 10.1038/cdd.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L, et al. BH3-only proteins Puma and Bim are rate-limiting for gamma-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106:4131–8. doi: 10.1182/blood-2005-04-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams AB, Schumacher B. p53 in the DNA-damage-repair process. Cold Spring Harb Perspect Med. 2016;6:a026070–84. [DOI] [PMC free article] [PubMed]
- 29.Smith ML, Chen IT, Zhan Q, O’Connor PM, Fornace AJ., Jr Involvement of the p53 tumor suppressor in repair of u.v.-type DNA damage. Oncogene. 1995;10:1053–9. [PubMed] [Google Scholar]
- 30.Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci USA. 1999;96:424–8. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, Sadowski I. Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements. Proc Natl Acad Sci USA. 2005;102:4813–8. doi: 10.1073/pnas.0407069102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janic A, Valente LJ, Wakefield MJ, Di Stefano L, Milla L, Wilcox S, et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med. 2018;24:947–53. doi: 10.1038/s41591-018-0043-5. [DOI] [PubMed] [Google Scholar]
- 33.Lee K, Tosti E, Edelmann W. Mouse models of DNA mismatch repair in cancer research. DNA Repair (Amst) 2016;38:140–6. doi: 10.1016/j.dnarep.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 35.Leonova KI, Brodsky L, Lipchick B, Pal M, Novototskaya L, Chenchik AA, et al. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc Natl Acad Sci USA. 2013;110:E89–98. doi: 10.1073/pnas.1216922110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 37.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/S0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 38.Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277:831–4. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- 39.Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001;61:6234–8. [PubMed] [Google Scholar]
- 40.Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 2008;15:1019–29. doi: 10.1038/cdd.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu W, Carson-Walter EB, Kuan SF, Zhang L, Yu J. PUMA suppresses intestinal tumorigenesis in mice. Cancer Res. 2009;69:4999–5006. doi: 10.1158/0008-5472.CAN-09-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, et al. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013;3:1339–45. doi: 10.1016/j.celrep.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 43.Aubrey BJ, Janic A, Chen Y, Chang C, Lieschke EC, Diepstraten ST, et al. Mutant TRP53 exerts a target gene-selective dominant-negative effect to drive tumor development. Genes Dev. 2018;32:1420–9. doi: 10.1101/gad.314286.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Gene Dev. 2012;26:1268–86. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Lozano G. p53: multiple facets of a Rubik’s Cube. Annu Rev Cancer Biol. 2017;1:185–201. doi: 10.1146/annurev-cancerbio-050216-121926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malkin D. Li-fraumeni syndrome. Genes Cancer. 2011;2:475–84. doi: 10.1177/1947601911413466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walerych D, Lisek K, Del Sal G. Mutant p53: one, no one, and one hundred thousand. Front in Oncol. 2015;5:00289–95. [DOI] [PMC free article] [PubMed]
- 48.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 49.Shchors K, Persson AI, Rostker F, Tihan T, Lyubynska N, Li N, et al. Using a preclinical mouse model of high-grade astrocytoma to optimize p53 restoration therapy. P Natl Acad Sci USA. 2013;110:E1480–E1489. doi: 10.1073/pnas.1219142110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carlsen L, El-Deiry WS. Differential p53-mediated cellular responses to DNA-damaging therapeutic agents. Int J Mol Sci. 2021;22:11828–43. [DOI] [PMC free article] [PubMed]
- 52.Willis A, Jung EJ, Wakefield T, Chen X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene. 2004;23:2330–8. doi: 10.1038/sj.onc.1207396. [DOI] [PubMed] [Google Scholar]
- 53.Chan WM, Siu WY, Lau A, Poon RYC. How many mutant p53 molecules are needed to inactivate a tetramer? Mol Cell Biol. 2004;24:3536–51. doi: 10.1128/MCB.24.8.3536-3551.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler KW, Vogelstein B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science. 1992;256:827–30. doi: 10.1126/science.256.5058.827. [DOI] [PubMed] [Google Scholar]
- 55.Sun Y, Dong Z, Nakamura K, Colburn NH. Dosage-dependent dominance over wild-type p53 of a mutant p53 isolated from nasopharyngeal carcinoma. FASEB J. 1993;7:944–50. doi: 10.1096/fasebj.7.10.8344492. [DOI] [PubMed] [Google Scholar]
- 56.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 57.Lee MK, Sabapathy K. The R246S hot-spot p53 mutant exerts dominant-negative effects in embryonic stem cells in vitro and in vivo. J Cell Sci. 2008;121:1899–906. doi: 10.1242/jcs.022822. [DOI] [PubMed] [Google Scholar]
- 58.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–34. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 59.Cavenee WK. Loss of heterozygosity in stages of malignancy. Clin Chem. 1989;35:B48–52. doi: 10.1093/clinchem/35.1.48. [DOI] [PubMed] [Google Scholar]
- 60.Xue Y, Raharja A, Sim W, Wong ES, Rahmat SA, Lane DP. The hot-spot p53R172H mutant promotes formation of giant spermatogonia triggered by DNA damage. Oncogene. 2017;36:2002–13. doi: 10.1038/onc.2016.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wong RPC, Tsang WP, Chau PY, Co NN, Tsang TY, Kwok TT. p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol Cancer Therapeutics. 2007;6:1054–61. doi: 10.1158/1535-7163.MCT-06-0336. [DOI] [PubMed] [Google Scholar]
- 62.Ali A, Wang Z, Fu J, Ji L, Liu J, Li L, et al. Differential regulation of the REGgamma-proteasome pathway by p53/TGF-beta signalling and mutant p53 in cancer cells. Nat Commun. 2013;4:2667. doi: 10.1038/ncomms3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dong P, Karaayvaz M, Jia N, Kaneuchi M, Hamada J, Watari H, et al. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2013;32:3286–95. doi: 10.1038/onc.2012.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roger L, Jullien L, Gire V, Roux P. Gain of oncogenic function of p53 mutants regulates E-cadherin expression uncoupled from cell invasion in colon cancer cells. J Cell Sci. 2010;123:1295–305. doi: 10.1242/jcs.061002. [DOI] [PubMed] [Google Scholar]
- 65.Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–58. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun. 2015;6:7389. doi: 10.1038/ncomms8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Inoue K, Fry EA. Alterations of p63 and p73 in human cancers. Subcell Biochem. 2014;85:17–40. doi: 10.1007/978-94-017-9211-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marin MC, Jost CA, Brooks LA, Irwin MS, O’Nions J, Tidy JA, et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat Genet. 2000;25:47–54. doi: 10.1038/75586. [DOI] [PubMed] [Google Scholar]
- 70.Marine JC, Berx G. Transforming growth factor-beta and mutant p53 conspire to induce metastasis by antagonizing p63: a (ternary) complex affair. Breast Cancer Res. 2009;11:304. doi: 10.1186/bcr2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 72.Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, Maor-Aloni R, et al. Mutant p53 enhances nuclear factor kappa B activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 2007;67:2396–401. doi: 10.1158/0008-5472.CAN-06-2425. [DOI] [PubMed] [Google Scholar]
- 73.Fontemaggi G, Dell’Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol. 2009;16:1086–93. doi: 10.1038/nsmb.1669. [DOI] [PubMed] [Google Scholar]
- 74.Kim MP, Lozano G. Mutant p53 partners in crime. Cell Death Differ. 2018;25:161–8. doi: 10.1038/cdd.2017.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alvarado-Ortiz E, de la Cruz-Lopez KG, Becerril-Rico J, Sarabia-Sanchez MA, Ortiz-Sanchez E, Garcia-Carranca A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front Cell Dev Biol. 2020;8:607670. doi: 10.3389/fcell.2020.607670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Phang BH, Othman R, Bougeard G, Chia RH, Frebourg T, Tang CL, et al. Amino-terminal p53 mutations lead to expression of apoptosis proficient p47 and prognosticate better survival, but predispose to tumorigenesis. Proc Natl Acad Sci USA. 2015;112:E6349–6358. doi: 10.1073/pnas.1510043112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–83. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ryan KM, Vousden KH. Characterization of structural p53 mutants which show selective defects in apoptosis but not cell cycle arrest. Mol Cell Biol. 1998;18:3692–8. doi: 10.1128/MCB.18.7.3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Williams AC, Miller JC, Collard TJ, Bracey TS, Cosulich S, Paraskeva C. Mutant P53 Is Not Fully Dominant over Endogenous Wild-Type P53 in a Colorectal Adenoma Cell-Line as Demonstrated by Induction of Mdm2 Protein and Retention of a P53 Dependent G1 Arrest after Gamma-Irradiation. Oncogene. 1995;11:141–9. [PubMed] [Google Scholar]
- 80.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–72. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 81.Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–17. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee MK, Teoh WW, Phang BH, Tong WM, Wang ZQ, Sabapathy K. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell. 2012;22:751–64. doi: 10.1016/j.ccr.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 83.Hanel W, Marchenko N, Xu S, Yu SXF, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013;20:898–909. doi: 10.1038/cdd.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017;18:345–60. doi: 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 85.Calderwood SK, Gong J. Heat shock proteins promote cancer: it’s a protection racket. Trends Biochem Sci. 2016;41:311–23. doi: 10.1016/j.tibs.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shi XY, Ding W, Li TQ, Zhang YX, Zhao SC. Histone deacetylase (HDAC) inhibitor, suberoylanilide hydroxamic acid (SAHA), induces apoptosis in prostate cancer cell lines via the Akt/FOXO3a signaling pathway. Med Sci Monit. 2017;23:5793–802. doi: 10.12659/MSM.904597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011;18:1904–13. doi: 10.1038/cdd.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Panicker J, Li Z, McMahon C, Sizer C, Steadman K, Piekarz R, et al. Romidepsin (FK228/depsipeptide) controls growth and induces apoptosis in neuroblastoma tumor cells. Cell Cycle. 2010;9:1830–8. doi: 10.4161/cc.9.9.11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen DM, Chen GA, et al. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst. 2002;94:504–13. doi: 10.1093/jnci/94.7.504. [DOI] [PubMed] [Google Scholar]
- 90.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–73. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 91.Thurnher M, Gruenbacher G, Nussbaumer O. Regulation of mevalonate metabolism in cancer and immune cells. Biochim Biophys Acta. 2013;1831:1009–15. doi: 10.1016/j.bbalip.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 92.Parrales A, Thoenen E, Iwakuma T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018;25:460–70. doi: 10.1038/s41418-017-0026-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nayor M, Vasan RS. Recent update to the US cholesterol treatment guidelines: a comparison with international guidelines. Circulation. 2016;133:1795–806. doi: 10.1161/CIRCULATIONAHA.116.021407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wong WWL, Dimitroulakos J, Minden MD, Penn LZ. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. 2002;16:508–19. doi: 10.1038/sj.leu.2402476. [DOI] [PubMed] [Google Scholar]
- 95.Kornblau SM, Banker DE, Stirewalt D, Shen D, Lemker E, Verstovsek S, et al. Blockade of adaptive defensive changes in cholesterol uptake and synthesis in AML by the addition of pravastatin to idarubicin + high-dose Ara-C: a phase 1 study. Blood. 2007;109:2999–3006. doi: 10.1182/blood-2006-08-044446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parrales A, Ranjan A, Iyer SV, Padhye S, Weir SJ, Roy A, et al. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat Cell Biol. 2016;18:1233–43. doi: 10.1038/ncb3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martirosyan A, Clendening JW, Goard CA, Penn LZ. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. 2010;10:103. doi: 10.1186/1471-2407-10-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gu H, Wang X, Rao S, Wang J, Zhao J, Ren FL, et al. Gambogic acid mediates apoptosis as a p53 inducer through down-regulation of mdm2 in wild-type p53-expressing cancer cells. Mol Cancer Ther. 2008;7:3298–305. doi: 10.1158/1535-7163.MCT-08-0212. [DOI] [PubMed] [Google Scholar]
- 99.Wang J, Zhao Q, Qi Q, Gu HY, Rong JJ, Mu R, et al. Gambogic acid-induced degradation of mutant p53 is mediated by proteasome and related to CHIP. J Cell Biochem. 2011;112:509–19. doi: 10.1002/jcb.22941. [DOI] [PubMed] [Google Scholar]
- 100.Pan H, Lu LY, Wang XQ, Li BX, Kelly K, Lin HS. Gambogic acid induces cell apoptosis and inhibits MAPK pathway in PTEN(−/−)/p53(−/−) prostate cancer cells in vitro and ex vivo. Chin J Integr Med. 2018;24:109–16. doi: 10.1007/s11655-017-2410-3. [DOI] [PubMed] [Google Scholar]
- 101.Zhu M, Jiang Y, Wu H, Shi W, Lu G, Cong D, et al. Gambogic acid shows anti-proliferative effects on non-small cell lung cancer (NSCLC) cells by activating reactive oxygen species (ROS)-induced endoplasmic reticulum (ER) stress-mediated apoptosis. Med Sci Monit. 2019;25:3983–8. doi: 10.12659/MSM.916835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lim LY, Vidnovic N, Ellisen LW, Leong CO. Mutant p53 mediates survival of breast cancer cells. Br J Cancer. 2009;101:1606–12. doi: 10.1038/sj.bjc.6605335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhu H, Mao Q, Lin Y, Yang K, Xie L. RNA interference targeting mutant p53 inhibits growth and induces apoptosis in DU145 human prostate cancer cells. Med Oncol. 2011;28:S381–387. doi: 10.1007/s12032-010-9679-9. [DOI] [PubMed] [Google Scholar]
- 104.Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Yuan ML, et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med. 2019;11:aaw8412. [DOI] [PMC free article] [PubMed]
- 105.Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–8. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 106.Saha MN, Jiang H, Yang Y, Reece D, Chang H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol Cancer Ther. 2013;12:2331–41. doi: 10.1158/1535-7163.MCT-12-1166. [DOI] [PubMed] [Google Scholar]
- 107.Furukawa H, Makino T, Yamasaki M, Tanaka K, Miyazaki Y, Takahashi T, et al. PRIMA-1 induces p53-mediated apoptosis by upregulating Noxa in esophageal squamous cell carcinoma with TP53 missense mutation. Cancer Sci. 2018;109:412–21. doi: 10.1111/cas.13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Piyawajanusorn C, Kittirat Y, Sa-Ngiamwibool P, Titapun A, Loilome W, Namwat N. PRIMA-1(MET) Induces Cellular Senescence and Apoptotic Cell Death in Cholangiocarcinoma Cells. Cancer Genomics Proteom. 2019;16:543–52. doi: 10.21873/cgp.20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Magrini R, Russo D, Ottaggio L, Fronza G, Inga A, Menichini P. PRIMA-1 synergizes with adriamycin to induce cell death in non-small cell lung cancer cells. J Cell Biochem. 2008;104:2363–73. doi: 10.1002/jcb.21794. [DOI] [PubMed] [Google Scholar]
- 110.Maslah N, Salomao N, Drevon L, Verger E, Partouche N, Ly P, et al. Synergistic effects of PRIMA-1Met (APR-246) and Azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica. 2020;105:1539–51. [DOI] [PMC free article] [PubMed]
- 111.Li XL, Zhou J, Chan ZL, Chooi JY, Chen ZR, Chng WJ. PRIMA-1met (APR-246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget. 2015;6:36689–99. doi: 10.18632/oncotarget.5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nahi H, Merup M, Lehmann S, Bengtzen S, Mollgard L, Selivanova G, et al. PRIMA-1 induces apoptosis in acute myeloid leukaemia cells with p53 gene deletion. Brit J Haematol. 2006;132:230–6. doi: 10.1111/j.1365-2141.2005.05851.x. [DOI] [PubMed] [Google Scholar]
- 113.Grellety T, Laroche-Clary A, Chaire V, Lagarde P, Chibon F, Neuville A, et al. PRIMA-1(MET) induces death in soft-tissue sarcomas cell independent of p53. BMC Cancer. 2015;15:684. doi: 10.1186/s12885-015-1667-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yoshikawa N, Shimizu Y, Yoshihara M, Nakamura K, Suzuki S, Kajiyama H, et al. Prima-1met induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of P53 status and chemo-sensitivity in epithelial ovarian cancer cells. Int J Gynecol Cancer. 2017;27:324–324. doi: 10.3892/or.2016.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Teoh PJ, Bi CL, Sintosebastian C, Tay LS, Fonseca R, Chng WJ. PRIMA-1 targets the vulnerability of multiple myeloma of deregulated protein homeostasis through the perturbation of ER stress via p73 demethylation. Oncotarget. 2016;7:61806–19. doi: 10.18632/oncotarget.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Russo D, Ottaggio L, Foggetti G, Masini M, Masiello P, Fronza G, et al. PRIMA-1 induces autophagy in cancer cells carrying mutant or wild type p53. Biochim Biophys Acta. 2013;1833:1904–13. doi: 10.1016/j.bbamcr.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 117.Fujihara KM, Corrales Benitez M, Cabalag CS, Zhang BZ, Ko HS, Liu DS, et al. SLC7A11 Is a Superior Determinant of APR-246 (Eprenetapopt) Response than TP53 Mutation Status. Mol Cancer Ther. 2021;20:1858–67. [DOI] [PubMed]
- 118.Aggarwal M, Saxena R, Sinclair E, Fu Y, Jacobs A, Dyba M, et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016;23:1615–27. doi: 10.1038/cdd.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Aggarwal M, Saxena R, Asif N, Sinclair E, Tan J, Cruz I, et al. p53 mutant-type in human prostate cancer cells determines the sensitivity to phenethyl isothiocyanate induced growth inhibition. J Exp Clin Cancer Res. 2019;38:307. doi: 10.1186/s13046-019-1267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang X, Di Pasqua AJ, Govind S, McCracken E, Hong C, Mi L, et al. Selective depletion of mutant p53 by cancer chemopreventive isothiocyanates and their structure-activity relationships. J Med Chem. 2011;54:809–16. doi: 10.1021/jm101199t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG. Masucci M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321–8. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 122.Zhao CY, Grinkevich VV, Nikulenkov F, Bao W, Selivanova G. Rescue of the apoptotic-inducing function of mutant p53 by small molecule RITA. Cell Cycle. 2010;9:1847–55. doi: 10.4161/cc.9.9.11545. [DOI] [PubMed] [Google Scholar]
- 123.Burmakin M, Shi Y, Hedstrom E, Kogner P, Selivanova G. Dual targeting of wild-type and mutant p53 by small molecule RITA results in the inhibition of N-Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin Cancer Res. 2013;19:5092–103. doi: 10.1158/1078-0432.CCR-12-2211. [DOI] [PubMed] [Google Scholar]
- 124.Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science. 1999;286:2507–10. doi: 10.1126/science.286.5449.2507. [DOI] [PubMed] [Google Scholar]
- 125.Takimoto R, Wang W, Dicker DT, Rastinejad F, Lyssikatos J, el-Deiry WS. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol Ther. 2002;1:47–55. doi: 10.4161/cbt.1.1.41. [DOI] [PubMed] [Google Scholar]
- 126.Wang W, Takimoto R, Rastinejad F, El-Deiry WS. Stabilization of p53 by CP-31398 inhibits ubiquitination without altering phosphorylation at serine 15 or 20 or MDM2 binding. Mol Cell Biol. 2003;23:2171–81. doi: 10.1128/MCB.23.6.2171-2181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.He XX, Zhang YN, Yan JW, Yan JJ, Wu Q, Song YH. CP-31398 inhibits the growth of p53-mutated liver cancer cells in vitro and in vivo. Tumour Biol. 2016;37:807–15. doi: 10.1007/s13277-015-3857-5. [DOI] [PubMed] [Google Scholar]
- 128.He X, Kong X, Yan J, Yan J, Zhang Y, Wu Q, et al. CP-31398 prevents the growth of p53-mutated colorectal cancer cells in vitro and in vivo. Tumour Biol. 2015;36:1437–44. doi: 10.1007/s13277-014-2389-8. [DOI] [PubMed] [Google Scholar]
- 129.Arihara Y, Takada K, Kamihara Y, Hayasaka N, Nakamura H, Murase K, et al. Small molecule CP-31398 induces reactive oxygen species-dependent apoptosis in human multiple myeloma. Oncotarget. 2017;8:65889–99. doi: 10.18632/oncotarget.19508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu X, Wilcken R, Joerger AC, Chuckowree IS, Amin J, Spencer J, et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013;41:6034–44. doi: 10.1093/nar/gkt305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen S, Wu JL, Liang Y, Tang YG, Song HX, Wu LL, et al. Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell. 2021;39:225–39 e228. doi: 10.1016/j.ccell.2020.11.013. [DOI] [PubMed] [Google Scholar]
- 132.Miller M, Shirole N, Tian R, Pal D, Sordella R. The evolution of TP53 mutations: from loss-of-function to separation-of-function mutants. J Cancer Biol Res. 2016;4:1091–103. [PMC free article] [PubMed]