

Abstract
Background
Hepatocellular carcinoma (HCC) occurs in a well-defined high-risk patient population, but better screening tests are needed to improve sensitivity and efficacy. Therefore, we investigated the use of urine circulating tumour DNA (ctDNA) as a screening test.
Methods
Candidate markers in urine were selected from HCC and controls. We then enrolled 609 patients from five medical centres to test the selected urine panel. A two-stage model was developed to combine AFP and urine panel as a screening test.
Results
Mutated TP53, and methylated RASSF1a, and GSTP1 were selected as the urine panel markers. Serum AFP outperformed the urine panel among all cases of HCC, but the urine panel identified 49% of HCC cases with low AFP < 20 ng/ml. Using the two-stage model, the combined AFP and urine panel identified 148 of the 186 HCC cases (79.6% sensitivity at 90% specificity), which was 30% more than the cases detected with serum AFP alone. It also increased early-stage HCC detection from 62% to 92% (BCLC stage 0), and 40% to 77% (BCLC stage A).
Conclusion
Urine ctDNA has promising diagnostic utility in patients in HCC, especially in those with low AFP and can be used as a potential non-invasive HCC screening test.
Subject terms: Cancer screening, Hepatocellular carcinoma
Introduction
Despite the implementation of specific hepatocellular carcinoma (HCC) screening recommendations in a well-defined high-risk population, early detection of HCC remains challenging [1, 2]. HCC is the 2nd most rapidly rising cancer in the US and remains a leading cause of global cancer mortality [2–4]. As the prognosis of HCC depends on the tumour stage, early HCC diagnosis is especially critical. HCC patients often have underlying cirrhosis with worsening liver function over time, limiting them from treatments or clinical trials. Patients with early HCC can receive curative treatments such as resection or liver transplantation which have a 5-year survival rate of >70% compared to <10% for patients diagnosed with advanced disease.
Serum AFP has been the most widely recognised and universally used biomarker that is clinically validated for HCC screening. However, modest sensitivities (ranging from 40 to 60%), limit its efficacy as a screening tool [2, 5–7], particularly for early-stage disease [2]. AFP elevations are also associated with high alanine aminotransferase (ALT) level in viral hepatitis [8]). Ultrasound (US)-based screening is also operator-dependent with a variable 40–90% sensitivity range and technically limited for the detection of tumours <2 cm, especially in obese patients and in nodular livers (i.e., cirrhosis) [2]. Early detection requires a screening approach that can be implemented frequently in a non-invasive manner. Thus, better-performing biomarkers that improve early detection are urgently needed.
HCC is a heterogeneous disease caused by many etiologies with multiple genetic alterations; thus, it requires a panel of multiple markers to obtain high screening sensitivity [9]. Studies by our group and others have shown that urine contains low-molecular-weight (LMW) DNA (~1–2 nucleosome-sized) or cell-free DNA (cfDNA) derived from apoptotic cells throughout the body [10–17]. We have demonstrated that DNA from urine of patients with cancers, including HCC contains cancer-specific DNA signatures, including both genetic mutations and aberrant DNA methylation [13, 14, 16, 18, 19].
In this study, we selected a panel of DNA markers that arise in HCC [20–22], and are detectable in urine, and developed a non-invasive urine-based circulating tumour DNA (ctDNA) biomarker panel as a screening test with higher sensitivity than AFP alone.
Materials and methods
Patient enrolment and sample collection
We performed a multicenter case-control study patients enrolled at five medical centres (Thomas Jefferson University Hospital, The John Hopkins Hospital, University of Pennsylvania Hospital, Buddhist Tzu Chi Medical Center, and the National Cheng-Kung University Medical Center) between April 2013 and July 2019 under HIRB Project #171201-174. The study was performed in compliance and after approval from all sites’ respective institutional review boards. Each participant signed a consent form for participation in the study prior to data, blood, and urine collection. Since 2018, all urine samples were collected from subjects with no liquid uptake for at least 2 h to avoid liquid dilution effect on cfDNA yield in order to obtain cfDNA at least 1 ng/mL urine to be included in the study. As outlined in the flowchart (Fig. 1), after biomarker assay analysis, data was sent to the respective clinical sites for disease category unblinding. For the initial biomarker prescreening, shown in Fig. 2, archived, non-identifiable urine samples previously collected from patients with hepatitis, cirrhosis or HCC were used as previously described [23].
Fig. 1.

Flow diagram showing outline of the study.
Fig. 2. Detection of HCC-associated DNA markers in urine from patients with HCC and controls (hepatitis and cirrhosis).

The distribution of each biomarker is shown in a scatter plots by disease group and evaluated using the non-parametric independent samples Wilcoxon rank-sum test comparing HCC versus non-HCC (hepatitis and cirrhosis). The number of patient samples analysed per marker per disease category is indicated in each panel. M and UM represent qualitative measurements of methylated and unmethylated DNA.
HCC was defined by histological examination or the appropriate imaging characteristics as defined by accepted guidelines at each clinical site. Clinical diagnosis of viral hepatitis and cirrhosis was determined by the expert opinion of the liver specialists at each centre. Tumour staging was determined by the Barcelona Clinic Liver Cancer staging system (BCLC) at each site. Detailed clinicopathological information at the time of sample collection is summarised in Table 1. For the control group, eligible patients (≥18 years) with cirrhosis from any aetiology and/or chronic hepatitis B (including co-infection with hepatitis C) who are recommended for routine HCC screening by expert society guidelines were included [2, 24, 25]. AFP levels were quantified by partnering centres in their clinical laboratories using Food and Drug Administration (FDA) approved AFP tests.
Table 1.
Clinical characteristics of patients in this study.
| Patient cohort (n = 609) | |||
|---|---|---|---|
| Diagnosis | Hepatitis B (n = 279) | Cirrhosis (n = 144) | HCC (n = 186) |
| Median age (IQR range), years | 55 (25–79) | 58.5 (29–89) | 64 (26–85) |
| Gender (M:F) | 153:126 | 98:46 | 141:45 |
| Aetiology | |||
| HBV | 271 | 87 | 74 |
| HCV | 0 | 10 | 41 |
| HBV/HCV | 8 | 7 | 16 |
| Non-viral | 0 | 34 | 46 |
| Unknown | 0 | 6 | 9 |
| BCLC stage (n) | |||
| 0 | 13 | ||
| A | 73 | ||
| B | 51 | ||
| C | 42 | ||
| D | 7 | ||
| AFP (ng/mL) | |||
| <20 | 276 | 136 | 98 |
| ≥20 | 3 | 8 | 88 |
BCLC Barcelona Clinic Liver Cancer staging, AFP alpha-fetoprotein, HBV hepatitis B, HCV hepatitis C.
Patient demographic and clinical data.
Urine DNA isolation and bisulfite treatment
Freshly collected urine was immediately mixed with 0.5 mol/L EDTA, pH 8.0, to a final concentration of 50 mmol/L EDTA to inhibit the possible nuclease activity in urine, and stored at −20 °C within 4 h of collection. To isolate urine DNA, a frozen urine sample was thawed at room temperature and then placed immediately in ice before DNA isolation. Thawed urine was processed for DNA isolation within an hour as described previously [12, 16] or by JBS urine cfDNA isolation kit (JBS Science Inc., Doylestown, PA) per manufacture’s specification. Bisulfite (BS) treatment was performed using the EZ DNA Methylation-Lightning™ Kit (Zymo Research, Irvine, CA) following the manufacturer’s guidelines.
Urine DNA biomarker quantification
PCR assays were tailored for short templates (≤87 bp amplicons) to detect circulation-derived genetic alterations in urine [15]. Eight candidate markers, mutated codon 249 TP53 (TP53 249) and CTNNB1 codons 32–37 (CTNNB1 32–37), and aberrantly methylated DNA of six genes (RASSF1A, GSTP1, CDKN2A, SFRP1, TFPI and MGMT), were selected because they are commonly present in HCC and can be detected using assays that amplify LMW DNA templates [26]. Three biomarker assays, the one for the TP53 codon 249 mutations, the aberrantly methylated RASSF1A (mRASSF1A) and the aberrantly methylated GSTP1 (mGSTP1) were selected for further development. Assays were performed in a blinded fashion. The measurement used for the mutated TP53 249 mutation was the percentage of total DNA, while for the methylation markers, mRASSF1A and mGSTP1 the measurement used was copies/mL urine. The kits for TP53, mRASSF1A and mGSTP1 assays were obtained from JBS Science, Inc. (Doylestown, PA) and performed as per the manufacturer’s guidelines in duplicates. The quantitative short amplicon methylation-specific PCR assays for CDKN2A, SFRP1, TFPI, and MGMT developed for this study are detailed in Supplemental Table 1.
Statistics
To assess the potential of candidate DNA biomarkers for HCC screening, scatter plots by disease group were constructed, and P value was provided by Wilcoxon rank-sum test for HCC and non-HCC (hepatitis + cirrhosis) comparison. A logistic regression model was used to distinguish HCC with the serum AFP, and/or urine ctDNA biomarkers. Because the biomarker data distributions were heavily skewed, biomarker data were log-transformed before analysis was conducted. Three models were built: (1) logistic model with AFP alone, (2) logistic model with ctDNA panel alone, and (3) two-stage model with HCC distinguished by AFP ≥ 20 ng/mL followed by AFP and ctDNA combined logistic model on the subpopulation of lower AFP (<20 ng/mL). The statistical comparison of each model is detailed in Supplemental Table 2. Comparing the model coefficients, it is observed that, α ~ a0 + a1, βk ~ bk, k = 0, 1, 2, 3, which indicates that urine ctDNA biomarkers do provide useful information to improve the HCC detection. The combined model can be interpreted as below.
Denote pHCC = probability of classified as HCC and logit(p) = log[(p/(1-p)) for 0 ≤ p ≤ 1. Consider the initial logistic model with serum APF alone
logit(pHCC) = a0 + b0 log(AFP) + e0, where the error term e0 can be interpreted by urine ctDNA biomarker as below
e0 = a1 + b1log(TP53) + b2log(mRASS) + b3log(mGSTP) + e1.
It follows that the combine the HCC model was built with
logit(pHCC) = α + β0 log(AFP) + β1log(TP53) + β2log(mRASS) + β3log(mGSTP) + e1, where α = a0 + a1, and βk = bk, k = 0, 1, 2, 3.
Receiver-operating characteristic (ROC) curves for all models were constructed [27] and compared statistically [28]. Under the condition of at least 90% specificity, the sensitivities of the models were also be compared. The analyses were conducted using SAS 9.4 (SAS, Cary, NC).
Validation
To evaluate the performance of our models with respect to accuracy and robustness, we employed tenfold cross-validation with bootstrap sampling (n = 1000). The process is described below. Step 1: The dataset of 609 patients were randomly split as ten equal subsets, S1, S2, … S10; Step 2: Subset S1 was used for prediction, and the other 9 subsets were used for modelling. Step 3: Repeat this process for all ten subsets from S1 to S10, and all records had been predicted independently because the data did not provide any information in the modelling process. Step 4: Repeat Step 1–Step 3 1000 times. Mean AUROC, standard error and 95% CI are reported. Mean sensitivity and specificity are reported together with the 5th and 95th percentiles of the 1000 bootstrap samples with a fixed cut-off of HCC prediction probability as pre-determined in the model building.
Results
Selection of potential ctDNA modifications for development as biomarkers for HCC screening
To first identify potential urine DNA markers for HCC, we analysed archived urine DNA isolated from patients with chronic liver disease, cirrhosis, or HCC for previously reported HCC-associated hotspot mutations in the TP53 and CTNNB1 genes and for aberrant methylation of six genes (GSTP1, RASSF1A, CDKN2A, SFRP1, TFP1 and MGMT). The distributions of the urine markers identified in each disease category are depicted with scatter plots (Fig. 2). Since the marker values were not normally but skewedly distributed, the Wilcoxon rank-sum test was used to select markers that were significantly higher in HCC group than that in non-HCC (hepatitis B, hepatitis B/C and cirrhosis). Patients with HCC had significantly higher levels of mutated TP53 249, mRASSF1A and mGSTP1 in urine than non-HCC (hepatitis + cirrhosis) (P < 0.001, by Wilcoxon rank-sum test). No significant differences were seen in the levels of mutated CTNNB1 codon 32–37 (P = 0.496), methylated SFRP1 (P = 0.798) and MGMT (P = 0.158) levels in urine DNA between HCC and non-HCC groups. Thus, we selected three (TP53 mutation, mRASSF1A and mGSTP1) of the eight tested to develop a new method for HCC screening.
Development of the urine ctDNA panel for HCC screening
As outlined in Fig. 1, prospectively collected urine DNA samples from 609 patients (186 HCC, 144 cirrhosis and 279 hepatitis B) contained at least 1 ng/mL of DNA, thus, were subjected to the 3-marker urine ctDNA panel quantification and analysed statistically for performance for HCC screening including serum AFP values, as described in “Materials and methods”. Three models were built: (1) logistic model with AFP alone, (2) logistic model with ctDNA panel alone, and (3) two-stage model with HCC distinguished by AFP ≥ 20 ng/mL, followed by a combined AFP and ctDNA combined logistic model on a subpopulation with lower AFP (<20 ng/mL). The statistical comparison of each model is detailed in Supplemental Table 2. All predict variables (serum AFP and three urine ctNDA markers) are statistically significant at 0.01 level. Next, we constructed a ROC (receiver-operating characteristic) curve and calculated the area under the curve (AUROC) for the urine ctDNA panel using logistic regression to distinguish HCC from the controls (Fig. 3a). AFP alone had AUROC (95%CI) of 0.8546 (0.8184–0.8908) compared to urine ctDNA with AUROC (95%CI) 0.7440 (0.7026–0.7854). Although AFP performed better than the urine panel overall, the diagnostic cut-off for serum AFP at 90% specificity was 5.8 ng/ml, which is significantly less than the widely accepted AFP value of ≥20 ng/mL [2, 24, 29], as patients with cirrhosis and chronic hepatitis are well-known to have variable baseline AFP levels with often higher than 5.8 ng/ml [7]. Based on this finding, we evaluate the utility of the urine panel further in a more practical clinical setting by investigating the performance of the urine panel in HCC cases with “low AFP” defined as <20 ng/ml.
Fig. 3. Performance of urine ctDNA markers for distinguishing HCC from non-HCC.

a Receiver-operating curves (ROC) of serum AFP alone and urine ctDNA markers alone. b Distribution of patients stratified by AFP cut-off of 20 ng/mL. The marker values are summarised in Supplemental Table 5. Each box represents a patient sample, and those with positive urine ctDNA biomarker detection are filled, based on the cut-off set at 90% specificity. AFP was positive (≥20 ng/mL) in 47.3% (88/186) of all HCC cases. Urine ctDNA panel was positive in 44.1% (82/186) of all HCC cases which included 48.9% (48/98) of the low AFP (<20 ng/mL) HCC group. c ROC of the two-stage model. d Comparison of three ROC curves as indicated.
Performance of urine panel in low-AFP-producing HCC and early-stage disease
First, we plotted the AFP distribution at 20 ng/mL threshold and the urine ctDNA panel with a cut-off value of 90% specificity for each patient (Fig. 3b). Of the 186 patients with HCC, 98 (53%) patients had low AFP, a rate consistent with previous studies [7, 30]. Among these 98 “low AFP” patients with HCC, the urine panel alone correctly identified 48 (49%) additional patients with HCC. This data suggests that the performance of urine ctDNA panel is independent of AFP value for predicting HCC from non-HCC which is consistent with the numerical results from the modelling processes. This indicates that the information provided by serum AFP and three urine ctDNA markers in HCC detection is independent (Supplemental Table 2). As serum AFP test is currently the most used biomarker with high specificity (97.4% in this cohort), we next proposed a Two-Stage model to first distinguish HCC by current AFP cut-off, 20 ng/mL, then combine AFP and urine ctDNA values as a combined panel to build a logistic model to distinguish HCC from the patients with low AFP (<20 ng/mL). As shown in Fig. 3c, the two-stage model had an AUROC of 0.9118 which was significantly higher than that of AFP alone (p < 0.0001 by using a contrast matrix [28]), as summarised in Supplemental Table 3. The two-stage model correctly identified 60 (60%) additional HCC patients with “low AFP” at 90% specificity. Overall, 148 of the 186 HCC cases (79.6% sensitivity at 90% specificity), were detected by the two-stage model, which is 30% more cases detected with serum AFP alone using the 20 ng/mL cut-off.
Early detection is critical for curative treatment options and overall survival. Therefore, we determined the overall performance of the AFP, the urine panel and the two-stage combined test stratified by tumour stage using the Barcelona Clinic Liver Cancer (BCLC) systems. AFP test alone (cut-off of ≥20 ng/mL) had sensitivities of 62%, 40%, 57%, 52% and 29% for stages 0, A, B, C and D, respectively. For the urine panel alone, at 90% specificity, the corresponding results were 23%, 49%, 39%, 43% and 71%. Encouragingly, the two-stage model demonstrated significantly improved sensitivities of 92%, 77%, 78%, 81% and 86% (Fig. 4). We noted the decrease in the sensitivity of AFP test in BCLC D group which is unexpected; this is likely due to the small sample size (n = 7) in this category. As early detection is key in screening tests, we then focused our attention to BCLC 0 and A. A closer inspection of the results shows that while the urine panel has a low 23% sensitivity in BCLC 0, all had AFP < 20 ng/mL, hence combining AFP and ctDNA values (two-stage) increased the sensitivity to 92%. Similarly, in BCLC A, the two-stage model raised the sensitivity to 77% with 90% specificity from 40%. Overall, the combination of AFP and urine panel in the two-stage model improved the sensitivity across all BCLC stages to above 75%, including early-stage cancer.
Fig. 4. Sensitivities of AFP ≥ 20 ng/ml, urine ctDNA and the two-stage model in different HCC stages per BCLC criteria.
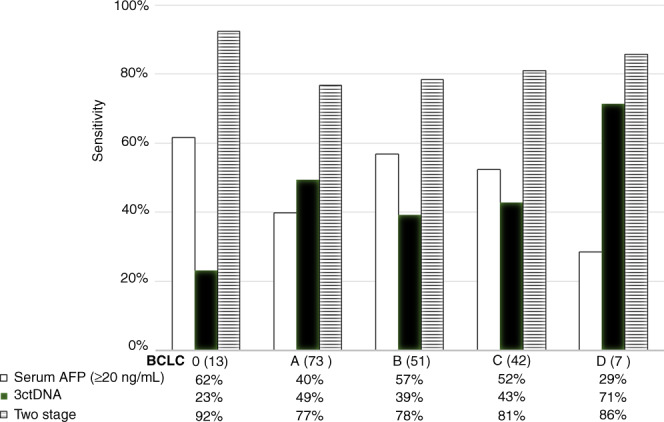
All tests were analysed at 90% specificity cut-off. The urine panel consistently selected additional HCC cases with low AFP, especially in early-stage BCLC stages 0 and A. BCLC Barcelona Clinic Liver Cancer staging. Patient numbers are shown in parenthesis in each stage.
In the control group, 11.8% and 7.5% of the patients with cirrhosis and HBV, respectively (total 38/423) were falsely positive by urine panel, compared to 4.9% and 1% using AFP ≥ 20 ng/ml alone. If ctDNA and AFP are combined with the two-stage model, false positivity was 18% (26/144) for cirrhosis and 5% (14/279) for the HBV group (Supplemental Fig. 1).
Cross-validation of urine panel and AFP
Tenfold cross-validation with bootstrap sampling (N = 1000) was used to examine the robustness of the model prediction, as detailed in “Materials and methods”. As summarised in Table 2, compared to the training set, the results from tenfold cross-validation with bootstrap sample (N = 1000) showed that the two-stage model is robust. The AUROC decreased from 0.912 in the training set to 0.902 (10.9%) in the validation, and the loss of sensitivity at 90% specificity (Supplemental Table 4) was only 1% from 79.6 to 78.6% which was not significant.
Table 2.
Biomarker performance evaluated by tenfold cross-validation.
| AUROC (95% CI) | Modelling | Validation |
|---|---|---|
| AFP | 0.854 (0.818–0.891) | 0.847 (0.810–0.885) |
| Urine ctDNA | 0.744 (0.703–0.785) | 0.715 (0.668–0.762) |
| Two-stage | 0.912 (0.884–0.939) | 0.902 (0.871–0.933) |
Discussion
In this study, we have developed urine-based screening test to screen for HCC among high-risk control patients with cirrhosis and hepatitis B. In the overall group, the urine panel did not perform better than AFP. Most strikingly, however, was that the urine panel performed independently from AFP and increased the screening sensitivity by identifying an additional 60 of 98 HCC cases with low AFP (<20 ng/mL) by the two-stage model. It is well-known that 40–60% of the patients with HCC have a normal or low AFP (<20 ng/mL), limiting AFP use alone as a HCC screening test. Our findings suggest that the urine panel may complement serum AFP as a more reliable non-imaging-based screening test. In early-stage disease, the combination of the urine panel and AFP using the Two-stage model increased the detection sensitivity from 62 to 92% in BCLC 0 and 40% to 77% in BCLC A stages.
This is the largest study to report the use of urine-based ctDNA biomarker to screen for HCC. This highlights the potential use of urine ctDNA as a new HCC screening tool for early detection. Urine collection requires little technical expertise without the need for phlebotomy. It would be better tolerated for patients with cirrhosis who have chronic anaemia. Diagnostic imaging using ultrasound will remain an important tool in HCC surveillance, but it also has its limitations. One of them is accessibility to an imaging centre which has been associated with the underuse of HCC screening [31]. Urine collection from home may provide a significant advantage with its easy accessibility to at-risk population, as we also see comparable marker performance between frozen urine samples and room-temperature stored sample (unpublished data). Increased feasibility and accessibility to screening test can improve early HCC detection in a population-level strategy for early detection of HCC.
The use of ctDNA in blood as a liquid biopsy for cancer detection has been studied extensively for decades but is limited by low sensitivity, particularly in early cancer stages [17, 26, 32–34]. Recently, studies have reported the detection of plasma ctDNA alterations and protein markers in serum to identify early-stage HCC [35–38], and although they have shown promise, these studies have utilised healthy controls. The choice of at-risk patient controls, as used in this study, is especially important with methylation biomarkers since they are also potentially detectable in patients with precancerous conditions such as liver cirrhosis [13, 32] and hepatitis [32, 33]. Consistent with previous studies [13, 29, 30], we also found the overlap in the quantity of methylated ctDNA detected in non-HCC group in this study.
Despite these encouraging results, some limitations merit consideration. First, even though the study included patients with early-stage HCC (BCLC stages 0, A and B), the sensitivity of our test may be less in the real-world screening setting, but it may not be significantly decreased because only 1% decrease of sensitivity from 79.6 to 78.6% if the two-stage model prediction is used as determined by the cross-validation. Further studies with an independent blinded validation set are needed to evaluate the test before clinically used in HCC screening. Furthermore, in this cross-sectional study, serial sample collections or additional imaging data were not obtained. These covariates may offer a valuable understanding of any trends in ctDNA detection and might include patients with precancerous lesions or HCC that are too small to be detected by standard screening imaging.
In summary, the urine ctDNA panel as a non-invasive test for HCC screening shows promising use in those with low AFP and early-stage liver disease. Its specificity remained high compared to patients with cirrhosis and could be used as an additional first-line screening tool in high-risk populations. An independent multicenter prospective study is underway to determine if this combinatorial approach using a non-invasive biomarker improves early detection of HCC.
Supplementary information
Acknowledgements
We thank Dmitry Goryunov for the support in proofreading and editing of this manuscript.
Author contributions
AKK and JPH contributed equally to this study. AKK contributed to formal analysis, original draft, patient recruitment and sample collection. JPH contributed to manuscript revision and editing, patient recruitment and sample collection. SYL contributed to conceptualisation, formal analysis, original draft and manuscript revision and editing. T-TC, H-WH, C-TH, Y-JL and TG contributed to patient recruitment and sample selection. YL contributed to formal analysis. GP and HL contributed to patient recruitment and sample selection and project administration. T-JL contributed to data curation. JW and DC contributed to data curation, formal analysis, and software support. MGG contributed to manuscript revision and editing. SJ contributed to conceptualisation, data curation, formal analysis and manuscript revision and editing. WS contributed to funding acquisition and project supervision. YHS contributed to lead conceptualisation, funding acquisition, original draft and manuscript revision and editing.
Funding
This work is supported by R43CA165312, R44CA165312, R01CA202769 and K08CA237624.
Data availability
The data generated and analysed during the study are available from the corresponding author on reasonable request.
Competing interests
AK is a consultant to AstraZeneca, WS, SJ and SL are shareholders of JBS Science Inc. YHS has received funding from JBS Science, Inc. The remaining authors declare no competing interests.
Ethics approval and consent to participate
The study received ethics approval from Heartland institutional review board (171201-173).
Consent to publish
Not applicable.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Amy K. Kim, James P. Hamilton.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-022-01706-9.
References
- 1.Davila JA, Morgan RO, Richardson PA, Du XL, McGlynn KA, El-Serag HB. Use of surveillance for hepatocellular carcinoma among patients with cirrhosis in the United States. Hepatology. 2010;52:132–41. doi: 10.1002/hep.23615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology. 2018;67:358–80.. doi: 10.1002/hep.29086. [DOI] [PubMed] [Google Scholar]
- 3.Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Altekruse SF, et al. SEER cancer statistics review, 1975–2013, based on November 2015 SEER data submission, posted to the SEER website. Bethesda, MD, USA: National Cancer Institute; 2016.
- 4.Ferlay JSI, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 5.Daniele B, Bencivenga A, Megna AS, Tinessa V. α-fetoprotein and ultrasonography screening for hepatocellular carcinoma. Gastroenterology. 2004;127:S108–S12.. doi: 10.1053/j.gastro.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 6.Zhou L, Liu J, Luo F. Serum tumor markers for detection of hepatocellular carcinoma. World J Gastroenterol. 2006;12:1175–81. doi: 10.3748/wjg.v12.i8.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta S, Bent S, Kohlwes J. Test characteristics of α-fetoprotein for detecting hepatocellular carcinoma in patients with hepatitis C: a systematic review and critical analysis. Ann Intern Med. 2003;139:46–50. doi: 10.7326/0003-4819-139-1-200307010-00012. [DOI] [PubMed] [Google Scholar]
- 8.Yang JD, Dai J, Singal AG, Gopal P, Addissie BD, Nguyen MH, et al. Improved performance of serum alpha-fetoprotein for hepatocellular carcinoma diagnosis in HCV cirrhosis with normal alanine transaminase. Cancer Epidemiol Prevention Biomark. 2017;26:1085–92.. doi: 10.1158/1055-9965.EPI-16-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pillai A, Ahn J, Kulik L. Integrating genomics into clinical practice in hepatocellular carcinoma: the challenges ahead. Am J Gastroenterol. 2020;115:1960–9. doi: 10.14309/ajg.0000000000000843. [DOI] [PubMed] [Google Scholar]
- 10.Husain H, Melnikova VO, Kosco K, Woodward B, More S, Pingle SC, et al. Monitoring daily dynamics of early tumor response to targeted therapy by detecting circulating tumor DNA in urine. Clin Cancer Res. 2017;23:4716–23.. doi: 10.1158/1078-0432.CCR-17-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su Y-H, Wang M, Aiamkitsumrit B, Brenner DE, Block TM. Detection of K-ras mutation in urine of patients with colorectal cancer. Cancer Biomark. 2005;1:177–82. doi: 10.3233/CBM-2005-12-305. [DOI] [PubMed] [Google Scholar]
- 12.Su Y-H, Song J, Wang Z, Wang X, Wang M, Brenner DE, et al. Removal of high molecular weight DNA by carboxylated magnetic beads enhances the detection of mutated K-ras DNA in urine. Ann N Y Acad Sci. 2008;1137:82–91. doi: 10.1196/annals.1448.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain S, Xie L, Boldbaatar B, Lin SY, Hamilton JP, Meltzer SJ, et al. Differential methylation of the promoter and first exon of the RASSF1A gene in hepatocarcinogenesis. Hepatol Res. 2014;45:1110–23. doi: 10.1111/hepr.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hann H-W, Jain S, Park G, Steffen JD, Song W, Su Y-H. Detection of urine DNA markers for monitoring recurrent hepatocellular carcinoma. Hepatoma Res. 2017;3:105–11.. doi: 10.20517/2394-5079.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su Y-H, Wang M, Block TM, Landt O, Botezatu I, Serdyuk O, et al. Transrenal DNA as a diagnostic tool: important technical notes. Ann N Y Acad Sci. 2004;1022:81–9. doi: 10.1196/annals.1318.014. [DOI] [PubMed] [Google Scholar]
- 16.Su Y-H, Wang M, Brenner DE, Ng A, Melkonyan H, Umansky S, et al. Human urine contains small, 150 to 250 nucleotide-sized, soluble DNA derived from the circulation and may be useful in the detection of colorectal cancer. J Mol Diagnostics. 2004;6:101–7. doi: 10.1016/S1525-1578(10)60497-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su Y-H, Wang M, Brenner DE, Norton PA, Block TM. Detection of mutated K-ras DNA in urine, plasma, and serum of patients with colorectal carcinoma or adenomatous polyps. Ann N Y Acad Sci. 2008;1137:197–206. doi: 10.1196/annals.1448.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song BP, Jain S, Lin SY, Chen Q, Block TM, Song W, et al. Detection of hypermethylated vimentin in urine of patients with colorectal cancer. J Mol Diagnostics. 2012;14:112–9. doi: 10.1016/j.jmoldx.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen S, Zhao J, Cui L, Liu Y. Urinary circulating DNA detection for dynamic tracking of EGFR mutations for NSCLC patients treated with EGFR-TKIs. Clin Transl Oncol. 2017;19:332–40.. doi: 10.1007/s12094-016-1534-9. [DOI] [PubMed] [Google Scholar]
- 20.Nault J-C, Zucman-Rossi J. Genetics of hepatocellular carcinoma: the next generation. J Hepatol. 2014;60:224–6. doi: 10.1016/j.jhep.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 21.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–4. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 22.Su Y-H, Kim AK, Jain S. Liquid biopsies for hepatocellular carcinoma. Transl Res. 2018;201:84–97. doi: 10.1016/j.trsl.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen D, Jain S, Su Y-H, Song W. Building classification models with combined biomarker tests: application to early detection of liver cancer. J Stat Sci Appl. 2017;5:91–103. doi: 10.17265/2328-224X/2017.0506.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Omata M, Cheng A-L, Kokudo N, Kudo M, Lee JM, Jia J, et al. Asia–Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol Int. 2017;11:317–70.. doi: 10.1007/s12072-017-9799-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanwal F, Singal AG. Surveillance for hepatocellular carcinoma: current best practice and future direction. Gastroenterology. 2019;157:54–64. doi: 10.1053/j.gastro.2019.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su Y-H, Lin SY, Song W, Jain S. DNA markers in molecular diagnostics for hepatocellular carcinoma. Expert Rev Mol Diagnostics. 2014;14:803–17. doi: 10.1586/14737159.2014.946908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- 28.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–45. doi: 10.2307/2531595. [DOI] [PubMed] [Google Scholar]
- 29.Liver EAFTSOT. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69:182–236. doi: 10.1016/j.jhep.2018.03.019. [DOI] [PubMed] [Google Scholar]
- 30.Lok AS, Sterling RK, Everhart JE, Wright EC, Hoefs JC, Di Bisceglie AM, et al. Des-γ-carboxy prothrombin and α-fetoprotein as biomarkers for the early detection of hepatocellular carcinoma. Gastroenterology. 2010;138:493–502. doi: 10.1053/j.gastro.2009.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim AK, Singal AG. Health disparities in diagnosis and treatment of hepatocellular carcinoma. Clin liver Dis. 2014;4:143. doi: 10.1002/cld.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong X, Hou Q, Chen Y, Wang X. Diagnostic value of the methylation of multiple gene promoters in serum in hepatitis B virus-related hepatocellular carcinoma. Dis Markers. 2017;2017:1–6. doi: 10.1155/2017/2929381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohamed NA, Swify EM, Amin NF, Soliman MM, Tag-Eldin LM, Elsherbiny NM. Is serum level of methylated RASSF1A valuable in diagnosing hepatocellular carcinoma in patients with chronic viral hepatitis C? Arab J Gastroenterol. 2012;13:111–5. doi: 10.1016/j.ajg.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Chen VL, Xu D, Wicha MS, Lok AS, Parikh ND. Utility of liquid biopsy analysis in detection of hepatocellular carcinoma, determination of prognosis, and disease monitoring: a systematic review. Clin Gastroenterol Hepatol. 2020;18:2879–902.. doi: 10.1016/j.cgh.2020.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu R-H, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater. 2017;16:1155–61.. doi: 10.1038/nmat4997. [DOI] [PubMed] [Google Scholar]
- 36.Howell J, Atkinson SR, Pinato DJ, Knapp S, Ward C, Minisini R, et al. Identification of mutations in circulating cell-free tumour DNA as a biomarker in hepatocellular carcinoma. Eur J Cancer. 2019;116:56–66. doi: 10.1016/j.ejca.2019.04.014. [DOI] [PubMed] [Google Scholar]
- 37.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–30.. doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chalasani NP, Ramasubramanian T, Bhattacharya A, Olson MC, Roberts LR, Kisiel JB, et al. A novel blood-based panel of methylated DNA and protein markers for detection of early-stage hepatocellular carcinoma. Clin Gastroenterol Hepatol. 2020;3565:31224–6. doi: 10.1016/j.cgh.2020.08.065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated and analysed during the study are available from the corresponding author on reasonable request.