

Abstract
The metabolism of phthalic acid (PA) and di-(2-ethylhexyl)phthalate (DEHP) in sludge-amended agricultural soil was studied with radiotracer techniques. The initial rates of metabolism of PA and DEHP (4.1 nmol/g [dry weight]) were estimated to be 731.8 and 25.6 pmol/g (dry weight) per day, respectively. Indigenous microorganisms assimilated 28 and 17% of the carbon in [14C]PA and [14C]DEHP, respectively, into microbial biomass. The rates of DEHP metabolism were much greater in sludge assays without soil than in assays with sludge-amended soil. Mineralization of [14C]DEHP to 14CO2 increased fourfold after inoculation of sludge and soil samples with DEHP-degrading strain SDE 2. The elevated mineralization potential was maintained for more than 27 days. Experiments performed with strain SDE 2 suggested that the bioavailability and mineralization of DEHP decreased substantially in the presence of soil and sludge components. The microorganisms metabolizing PA and DEHP in sludge and sludge-amended soil were characterized by substrate-specific radiolabelling, followed by analysis of 14C-labelled phospholipid ester-linked fatty acids (14C-PLFAs). This assay provided a radioactive fingerprint of the organisms actively metabolizing [14C]PA and [14C]DEHP. The 14C-PLFA fingerprints showed that organisms with different PLFA compositions metabolized PA and DEHP in sludge-amended soil. In contrast, microorganisms with comparable 14C-PLFA fingerprints were found to dominate DEHP metabolism in sludge and sludge-amended soil. Our results suggested that indigenous sludge microorganisms dominated DEHP degradation in sludge-amended soil. Mineralization of DEHP and PA followed complex kinetics that could not be described by simple first-order equations. The initial mineralization activity was described by an exponential function; this was followed by a second phase that was described best by a fractional power function. In the initial phase, the half times for PA and DEHP in sludge-amended soil were 2 and 58 days, respectively. In the late phase of incubation, the apparent half times for PA and DEHP increased to 15 and 147 days, respectively. In the second phase (after more than 28 days), the half time for DEHP was 2.9 times longer in sludge-amended soil assays than in sludge assays without soil. Experiments with radiolabelled DEHP degraders suggested that a significant fraction of the 14CO2 produced in long-term degradation assays may have originated from turnover of labelled microbial biomass rather than mineralization of [14C]PA or [14C]DEHP. It was estimated that a significant amount of DEHP with poor biodegradability and extractability remains in sludge-amended soil for extended periods of time despite the presence of microorganisms capable of degrading the compound (e.g., more than 40% of the DEHP added is not mineralized after 1 year).
Phthalate esters are used in industrial production of lubricants, glues, insect repellents, dielectric fluids, and plastics (6). Among the phthalate esters, di(2-ethylhexyl)phthalate (DEHP) is one of the most frequently used additives in the manufacture of flexible polyvinyl chloride. DEHP is used as a plasticizer because of its stability, fluidity, and low volatility (24). The annual global production of DEHP has been estimated to be approximately 106 tons (13). DEHP has been classified as a priority pollutant with relatively low acute toxicity but suspected mutagenic and carcinogenic effects (6, 13, 20). DEHP and its metabolites may also affect humans in other ways, including potential effects on reproduction (e.g., xenoestrogenic effects) (5, 6, 13, 23).
Microbial degradation is believed to be the principal sink for DEHP in aquatic and terrestrial systems, such as sewage, soils, sediments, and surface waters (24). Abiotic hydrolysis of DEHP is thought to be negligible in these environments (24). Significant amounts of DEHP are released annually into aquatic and terrestrial environments from wastewater treatment plants. DEHP is found in both effluents from treatment plants and dewatered sludge. The concentrations may exceed 120 mg of DEHP/kg (dry weight) of sludge (10). Soil systems may contain elevated DEHP concentrations due to the use of sewage sludge as a soil fertilizer. DEHP is degraded slowly in soil by indigenous microorganisms, but the regulating mechanisms are not well understood. In addition, very little is known about control of DEHP degradation in soils that have been amended with sewage sludge from wastewater treatment plants. This is unfortunate as sewage sludge has been used extensively in many countries as a convenient soil fertilizer in forestry and agriculture.
In the present study, we examined the microbial degradation of DEHP and the degradation intermediate phthalic acid (PA) in agricultural soil amended with sewage sludge. The results were compared with findings obtained in degradation studies performed with pure cultures of DEHP degraders. Transformation of DEHP and PA was characterized by using 14C-labelled substrates in combination with different isotope techniques.
MATERIALS AND METHODS
Chemicals.
Analytical grade DEHP and PA were obtained from Merck (Darmstadt, Germany). [U-14C-ring]DEHP (188.7 MBq/mmol; purity, >99%) and [U-14C-ring]PA (469.9 MBq/mmol; purity > 99%) were obtained from Sigma Chemical Co. (St. Louis, Mo.). [U-14C]glucose (10.8 GBq/mmol; purity > 99%) was obtained from Amersham Life Science (Amersham, United Kingdom). Stock solutions of 14C-labelled DEHP, PA, and glucose were dissolved in hexane, methanol, and water, respectively.
All chemicals used for extraction and dilution of DEHP and PA were chromatographic grade. The chemicals were analyzed by capillary gas chromatography (GC) with flame ionization detection to verify the presence of low background concentrations. All glassware was heated at 550°C prior to use. All other equipment was washed with analytical grade hexane prior to use (Fisher Scientific, Pittsburgh, Pa.).
Soil and sludge samples.
Agricultural soil from the plough layer (depth, 0 to 20 cm) was collected at the agricultural research center at Foulum, Denmark. The soil was a sandy loam that had a pH of 5.9 and contained 2.5% (wt/wt) organic matter. The soil had never been amended with sewage sludge prior to the present study. Soil samples were sieved (2-mm mesh), dried to a water content of 7% (wt/wt), and then stored at 5°C until they were used.
Dewatered sewage sludge was collected at the municipal wastewater treatment plant at East Aalborg, Denmark. The sludge had a water content of 490% (wt/wt) and an organic matter content of 28.5% (wt/wt). The dewatered sludge from the treatment plant consisted of 50% dewatered active sludge and 50% dewatered sludge that had been stabilized anaerobically at approximately 40°C. The natural background level of extractable DEHP in the sewage sludge was 82.9 μg/g (dry weight).
Isolation of DEHP degraders.
Aerobic DEHP degraders were isolated from soil and sludge by direct spread plating onto inorganic minimal medium agar containing 0.1% DEHP (1 g/liter). The inorganic medium (pH 7) contained 10 mM NH4Cl, 30 mM Na2HPO4, 20 mM KH2PO4, 0.8 mM Na2SO4, 0.2 mM MgSO4, 50 μM CaCl2, 25 μM FeSO4, 0.1 μM ZnCl2, 0.2 μM CuCl2, 0.1 μM NaBr, 0.05 μM Na2MoO2, 0.1 μM MnCl2, 0.1 μM KI, 0.2 μM H3BO3, 0.1 μM CoCl2, and 0.1 μM NiCl2. DEHP was added to the medium before autoclaving. Soil and sludge samples (10 cm3) were diluted in 90 ml of inorganic medium and homogenized in a blender. The homogenized samples were diluted serially, and subsamples (0.1 ml) were transferred to agar plates containing DEHP. The plates were incubated for 6 weeks in the dark. The following three aerobic, gram-positive isolates were selected for further analysis: strains SDE 2 and SDE 21 from dewatered sludge and strain JDE 9 from soil. Strains SDE 2, SDE 21, and JDE 9 were capable of utilizing both DEHP and PA as sole carbon and energy sources. These strains also grew on glucose and complex media, such as Trypticase soy broth.
PA and DEHP degradation by strain SDE 2.
Strain SDE 2 was grown to the late exponential phase on DEHP and then harvested by centrifugation (10,000 × g, 15 min). The cells were washed three times with inorganic medium and resuspended to a density of 109 cells/ml. Subsamples (10 ml) were transferred to 26-ml serum bottles to which radiolabelled PA or DEHP had been added. Some vials also contained dried soil or dried sludge (0.1 g [dry weight] of soil containing 5.95% organic matter; 0.1 g [dry weight] of sludge containing 28.5% organic matter) as a potential sorbent of DEHP. The soil and sludge were dried at 105°C and then sieved through a 125-μm-pore-size mesh. Samples of the throughfall were used in the assays performed with soil or sludge. Radiolabelled DEHP and PA were added to the assay vials in 0.1 ml of inorganic medium 24 h prior to the addition of strain SDE 2. This allowed time for sorption of [14C]DEHP in assays in which dried soil or sludge had been added. During this 24-h equilibration period, all of the vials were incubated at 4°C with an N2 atmosphere to prevent degradation of PA and DEHP. No mineralization of PA or DEHP was detected prior to the addition of strain SDE 2. The initial concentration of PA or DEHP was 5.2 μM. After addition of 10 ml of strain SDE 2 (109 cells/ml), 14CO2 evolution was determined by flushing the vial headspaces continuously with sterile air. Each culture vial was connected to a CO2 trap consisting of a glass scintillation vial in which 14CO2 was trapped in a 10-ml mixture of ethanolamine and ethyleneglycolmonomethylether (1:7, vol/vol). The vial containing SDE 2 and the CO2 trap were incubated on a shaker at 100 rpm to ensure that maximum gas transfer occurred in both systems.
PA and DEHP degradation in soil and sludge.
Degradation studies were performed with soil and sludge samples by using the incubation system described by Madsen et al. (11). This system consists of a 55-ml glass vial for incubation of an environmental sample and a 20-ml glass scintillation vial for trapping the 14CO2. The vials were connected by a stainless steel column (inside diameter, 5 mm) to allow gas diffusion from the vial with the sample to the vial with the CO2 trap (containing 2 ml of 1 M NaOH). The two vials were sealed with rubber stoppers through which the steel column was inserted. This simple leak-proof setup allowed rapid replacement of the CO2 trap (glass scintillation vial), followed by direct counting of the contents after addition of a scintillation cocktail.
The masses of the soil and sludge samples added were 21 g (fresh weight) (19.66 g [dry weight]) and 2.0 g (wet weight) (0.34 g [dry weight]), respectively. This resulted in a total sample weight of 20 g (dry weight) and a soil/sludge ratio of 58:1 (dry weight/dry weight). The samples were mixed manually with a spatula. All soil samples had a water content that was 75% of the field capacity after addition of the sludge to the soil samples. In assays with soil alone (21 g [fresh weight]), the water content was adjusted to 75% of the field capacity with autoclaved H2O. In assays with sludge alone (2 g [wet weight]), the sludge was mixed with sterile fine granular quartz particles (Merck), which served as an artificial surface. The quartz particles (19.66 g [dry weight] per sample) were heated at 600°C and washed with distilled water prior to use. In degradation studies in which bacteria were inoculated, DEHP degraders from cultures were washed with inorganic medium and added to a concentration of 109 cells per g (dry weight) of sample. Degradation studies performed with sludge slurries were carried out by using 1-liter Erlenmeyer flasks equipped with CO2 traps as described above (external glass scintillation vials containing 2 ml of 1 M NaOH). The sludge slurries each consisted of 2 g (wet weight) of sludge mixed with 200 ml of autoclaved distilled water. Each Erlenmeyer flask and CO2 trap were incubated at 120 rpm on a rotary shaker. Unless stated otherwise, all degradation studies were carried out at 20°C, and the controls consisted of samples that had been autoclaved three times.
In all incubations with sludge, [14C]DEHP and [14C]PA were mixed with the sludge before other material (soil, quartz, or water) was added. The label (approximately 100,000 dpm) was dissolved in 10 μl of solvent that was subsequently evaporated in a fume hood (hexane and methanol were used as carriers of [14C]DEHP and [14C]PA, respectively).
Oxic conditions were ensured in all experiments by regularly replacing the CO2 traps (containing 20 ml of air) and by directly injecting O2 in assays in which there was elevated oxygen consumption (assay mixtures containing sludge). In the latter experiments, 10 ml of pure O2 was injected weekly. The oxygen status of parallel control assay mixtures without radiolabel was evaluated by GC analysis.
The time courses for DEHP or PA depletion could be divided into two phases and were fitted by exponential and fractional power equations as described by Madsen et al. (11). The initial substrate depletion was described by an exponential function, whereas the mineralization activity later in the experiment was described better by a fractional power function.
The relative substrate depletion in the initial phase was calculated as follows (t ≤ tα):
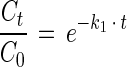 |
1 |
where t is time (days), tα is the time when the mineralization shifts from first-order kinetics to fractional power kinetics (days), Ct is the substrate concentration at time t (nanomoles per gram [dry weight]), C0 is the initial substrate concentration (nanomoles per gram [dry weight]), and k1 is the first-order rate coefficient (days−1).
In the initial phase (t ≤ tα), the time needed to deplete one-half of the substrate (t0.5) was calculated as follows:
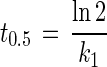 |
2 |
The relative substrate depletion in the late phase (t > tα) was calculated as follows:
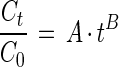 |
3 |
where A and B are model parameters.
In the late phase (t > tα), t0.5 was calculated as follows:
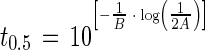 |
4 |
Substrate depletion curves were fitted by using KaleidaGraph 3.08 (Synergy Software) for Macintosh computers.
Extraction and analysis of DEHP.
DEHP was extracted from dried (50°C), homogenized soil and sludge samples in 38-ml glass centrifuge tubes. A sample (<1 g [dry weight]) was extracted four times with 15 ml of hexane for 30 min in an ultrasonic water bath. The combined hexane extract was concentrated to a volume of 5 ml by evaporating the hexane in a fume hood at 40°C with a flow of N2. Subsequently, a column chromatography cleanup procedure was carried out partially as described by Zurmühl (25). The concentrated hexane extract was loaded onto 20-ml glass syringe chromatography columns containing 10 g of neutral alumina (Merck). The alumina was deactivated with distilled H2O (15% [wt/wt] for 24 h in a closed vial) prior to packing of the columns. Each column was mounted on a vacuum manifold (Vac-Elut; Varian, Palo Alto, Calif.) and then eluted sequentially with 20 ml of hexane, 20 ml of 10% dichloromethane in hexane, and 20 ml of 50% dichloromethane in hexane. The last fraction contained the DEHP and was evaporated to dryness at 30°C under a flow of N2. The extracted DEHP was then redissolved in 0.5 ml of hexane for GC analysis. The recovery of DEHP from the deactivated alumina columns was above 95%.
DEHP and PA solutions were analyzed by capillary GC with flame ionization detection (model HP 5890 series II GC). PA and DEHP were dissolved in methanol and hexane, respectively. The GC was equipped with an HP Ultra 2 capillary column (50 m by 0.2 mm [inside diameter]) with a 5% phenylmethyl silicone film. Samples (2 μl) were analyzed in the splitless mode. Peaks were separated by using a 20-min temperature program, as follows: 1 min at 60°C, increase from 60 to 280°C at a rate of 30°C/min, 9 min at 280°C, increase from 280 to 300°C at a rate of 30°C/min, and 2 min at 300°C. The GC injector temperature was 270°C, the detector temperature was 300°C, and the initial column temperature was 60°C. H2 (initial flow rate, 1.6 ml/min) was used as the carrier gas; N2 (flow rate, 35 ml/min) was the make-up gas; and H2 and air (flow rates, 34 and 370 ml/min, respectively) were used for the flame ionization detector.
Phospholipid extraction from soil and sludge.
The amounts of radioactivity incorporated into microbial phospholipids were used to estimate the total biomass production during degradation of [14C]DEHP in soil and sludge samples (Fig. 1). The amount of radioactivity recovered in phospholipids relative to the amount assimilated into cell material was estimated by using conversion factors determined for pure cultures of a DEHP degrader (see below). Radiolabelled lipids were extracted from soil and sludge samples (5 g [dry weight]) with a mixture containing 10 ml of methanol and 5 ml of dichloromethane partially as described previously (19). The extractable lipids were separated into neutral lipids, glycolipids, and polar lipids by Si column chromatography (500 mg of SiO; Isolute, Hengoed, United Kingdom); the different lipid classes were eluted with 10 ml of chloroform, 10 ml of acetone, and 10 ml of methanol, respectively. DEHP was coextracted from the soil and sludge matrixes by using the methanol-dichloromethane solution. However, labelled DEHP was separated from labelled phospholipids by the Si column chromatography. The DEHP eluted in the chloroform fraction, whereas the phospholipids eluted in the methanol fraction. The amount of radioactivity in the phospholipid fraction was determined by liquid scintillation counting.
FIG. 1.

Methods used to characterize DEHP degradation in soil and sewage sludge. PLs, phospholipids.
Phospholipid content of DEHP degraders.
The phospholipid content of DEHP degraders was determined by studying the aerobic gram-positive strain SDE 2. The amount of radioactivity recovered in phospholipids relative to the amount assimilated into the cell material was used in calculations of total degradation rates for PA and DEHP in soil and sludge (Fig. 1). SDE 2 was grown to the late exponential phase in 100 ml of inorganic medium in 1-liter Erlenmeier flasks containing DEHP as the sole carbon and energy source. Each culture was harvested by centrifugation (10,000 × g, 15 min), washed twice, and resuspended in inorganic medium. Subsamples (10 ml) were transferred to 50-ml vials, and 5 × 105 dpm of [14C]glucose was added to each vial to label the bacterial biomass (glucose rather than DEHP was used to obtain more rapid labelling of strain SDE 2). Subsequently, the amount of [14C]glucose dissimilated was determined by monitoring 14CO2 production, whereas assimilation into bacterial biomass was determined by direct liquid scintillation counting of cells collected on 0.22-μm-pore-size filters. The amount of label assimilated into bacterial phospholipids was determined as described above for soil and sludge samples.
CrO3 oxidation of organic matter.
Oxidation of labelled soil organic matter to 14CO2 was carried out partially as described previously (4). This wet oxidation procedure provided an estimate of the total amount of 14C-labelled organic carbon that remained in a sample after incubation with a 14C-labelled substrate (Fig. 1). Dried soil or sludge (2 g [dry weight]) from samples incubated with [14C]DEHP were transferred to 160-ml serum vials. K2Cr2O7 (0.5 g) and Ag2SO4 (0.01 g) were mixed with each sample before a 2:1 mixture of concentrated H2SO4 and H3PO4 was added. The serum vials were sealed with Teflon-lined stoppers equipped with aluminum crimps and then incubated at 140°C for 60 min. The 14CO2 produced was flushed from the headspace and counted as described previously (18).
14C-PLFA fingerprinting.
Soil and sludge samples were incubated with [14C]DEHP or [14C]PA to specifically radiolabel the microorganisms capable of metabolizing these compounds. Subsequent to the incubation, whole-sample phospholipid ester-linked fatty acids (PLFAs) were extracted and analyzed. The abundance and distribution of labelled PLFAs were determined by radio GC analysis. The fingerprinting approach used has been described previously (19).
Sludge (0.3 g [wet weight]) and sludge-amended soil (0.3 g [wet weight] of sludge plus 3 g [fresh weight] of soil) were incubated with 0.37 MBq of [14C]DEHP or 0.33 MBq of [14C]PA. In incubations without soil, the sludge samples were mixed with sterile moist quartz particles (3 g [fresh weight]), which provided an inert surface. The sludge/soil ratio and the sludge/quartz ratio were each 1:55 (wt/wt). The samples were incubated in 55-ml vials equipped with external CO2 traps as described above. The experiments were terminated when more than 50% of the label added had been recovered as 14CO2. Extraction and analysis of 14C-PLFAs were carried out as described previously (19). The following modified 95-min GC temperature program was used to separate the 14C-PLFAs: 1 min at 60°C, increase from 60 to 170°C at a rate of 40°C/min, 0.5 min at 170°C, increase from 170 to 200°C at a rate of 0.4°C/min, increase from 200 to 300°C at a rate of 10°C/min, and 5.75 min at 300°C. Radiolabelled PLFAs were collected in 15 fractions on the basis of their retention times and equivalent chain lengths (Table 1).
TABLE 1.
Sampling scheme and equivalent chain lengths for radiolabelled PLFAs
| Fraction | Time (min) | Equivalent chain length |
|---|---|---|
| 1 | 0–15 | <13.4 |
| 2 | 15–20 | 13.4–14.4 |
| 3 | 20–26 | 14.4–15.5 |
| 4 | 26–30.3 | 15.4–16.0 |
| 5 | 30.3–34 | 16.0–16.4 |
| 6 | 34–38 | 16.4–16.9 |
| 7 | 38–44 | 16.9–17.4 |
| 8 | 44–51.5 | 17.4–18.0 |
| 9 | 51.5–57 | 18.0–18.4 |
| 10 | 57–63 | 18.4–18.8 |
| 11 | 63–72 | 18.8–19.3 |
| 12 | 72–79 | 19.3–19.7 |
| 13 | 79–85 | 19.7–20.7 |
| 14 | 85–90 | 20.7–21.7 |
| 15 | 90–95 | >21.7 |
Turnover of 14C-labelled biomass.
Radiolabelled DEHP degraders from pure cultures were added to sludge-amended soil in order to obtain an estimate of the turnover of 14C-labelled bacterial biomass (measured as 14CO2 production). Cultures of the aerobic DEHP degraders SDE 21 and JDE 9 were radiolabelled by adding [14C]PA to pure cultures. Washed cells (1 ml) were then added to sludge-amended soil (21 g [fresh weight] of soil plus 2 g [fresh weight] of sludge) and incubated in 55-ml vials equipped with external 14CO2 traps as described above. The initial levels of radioactivity in the bacterial cells added were 0.5 and 0.8 kBq for assays performed with SDE 21 and JDE 9, respectively.
Liquid scintillation counting.
The amounts of radioactivity associated with alkaline samples containing 14CO2 and extracts containing phospholipids were determined by using Packard Ultima Gold XR as the scintillation cocktail. The amounts of radioactivity in the 14CO2 traps used in the radio GC analysis of 14C-PLFAs and CrO3 oxidation of soil organic matter were determined by using Packard Instagel Plus and Packard Hionic Fluor as the scintillation cocktails, respectively. All samples were counted for 5 min with a Packard model 1600 TR liquid scintillation counter. The counts were corrected for quenching by using internal and external standards.
RESULTS
Mineralization of [14C]PA and [14C]DEHP in soil and sludge.
Mineralization of [14C]DEHP to 14CO2 was detected after a short lag period (24 h) both in sewage sludge slurries and in sewage sludge mixed with sterile quartz (Fig. 2A). The time courses of 14CO2 production were somewhat comparable, although slightly greater mineralization was observed with sludge-amended quartz (Fig. 2A). Approximately 45 to 50% of the [14C]DEHP was recovered as 14CO2 after 90 days. A similar time course was observed when sludge was mixed with agricultural soil (Fig. 2B). In all assays performed with [14C]DEHP, the apparent mineralization activity was low after recovery of about 40% of the added label (incubation time, >60 days). In contrast, much greater mineralization activity was observed with [14C]PA (Fig. 2B). The initial mineralization activity in sludge-amended soil was rapid, with 47% of the label recovered as 14CO2 after 3 days.
FIG. 2.

Mineralization of [14C]DEHP or [14C]PA to 14CO2 in sewage sludge and soil amended with sewage sludge. (A) Sludge slurries containing [14C]DEHP and sludge-amended sterile quartz containing [14C]DEHP. (B) Sludge-amended soil containing [14C]DEHP and sludge-amended soil containing [14C]PA. Data points represent the means of triplicate samples; error bars indicate the standard errors. d, day.
Degradation of [14C]PA and [14C]DEHP by strain SDE 2.
Aerobic gram-positive strain SDE 2 isolated from sewage sludge was capable of mineralizing both [14C]PA and [14C]DEHP to 14CO2 (Fig. 3). The mineralization rate during the initial 4 h of incubation was 2.5 times faster for [14C]PA than for [14C]DEHP. Apparent plateaus for PA and DEHP mineralization were reached when 47 and 24%, respectively, of the label had been converted to 14CO2.
FIG. 3.

Mineralization of [14C]PA and [14C]DEHP to 14CO2 by pure cultures of phthalate-degrading strain SDE 2. Sewage sludge or soil was added to examine the effects on the mineralization of DEHP. Dried (105°C) soil (0.01 g [dry weight]/ml; 6% organic C) and dried (105°C) sludge (0.01 g [dry weight]/ml; 29% organic C) were added to the culture media along with [14C]DEHP 24 h before strain SDE 2 was added (see text for details). Data points represent the means of triplicate samples.
The presence of material that may sorb DEHP (soil and sludge dried at 105°C) decreased the initial mineralization of [14C]DEHP by 61 and 94%, respectively (Fig. 3). Before strain SDE 2 was added, the dried sieved soil (0.01 g [dry weight]/ml; 6% organic C) and dried sieved sewage sludge (0.01 g [dry weight]/ml; 29% organic C) were equilibrated with [14C]DEHP for 24 h. After 20.8 h of incubation with strain SDE 2, only 14.8 and 3.4% of the [14C]DEHP were recovered as 14CO2 in cultures amended with dried soil and sludge, respectively.
Inoculation of soil and sludge with DEHP degraders.
Inoculation of soil and sludge samples with strain SDE 2 stimulated the mineralization of [14C]DEHP compared to samples containing only indigenous microorganisms (Fig. 2B and 4). Inoculation of 109 bacteria/g (dry weight) increased the initial mineralization rates in sludge and soil approximately fourfold. The elevated degradation potential compared to uninoculated samples was maintained for 27 days of incubation. However, the increased degradation potential due to inoculation with strain SDE 2 was maintained better in assay mixtures containing sludge than in assay mixtures containing soil (Fig. 4). The second addition of [14C]DEHP resulted in mineralization activities in samples containing sludge and soil that were 93 and 53%, respectively, of the initial values (the values obtained after the first [14C]DEHP addition).
FIG. 4.

Mineralization of [14C]DEHP to 14CO2 by soil and sludge samples inoculated with DEHP-degrading strain SDE 2. Bacteria were added at zero time at a density of 109 bacteria/g (dry weight). [14C]DEHP was added at zero time and again at day 27 (indicated by arrows). The levels of mineralization after the second [14C]DEHP addition were calculated relative to the second addition only. Data ponts represent the means of triplicate samples; error bars indicate the standard errors. d, day.
Depletion of DEHP and PA in soil and sludge.
Depletion curves for DEHP and PA in sludge and sludge-amended soil were estimated based on the complete mineralization (ultimate biodegradation) of the added [14C]DEHP or [14C]PA to 14CO2 (Fig. 5A). The mineralization curves each appeared to have an initial phase that could be described by an exponential decrease in the substrate concentration (linear decrease in ln or log plots) (Fig. 5B). The initial phase was succeeded by a second phase of low mineralization activity with kinetic characteristics different from those of a simple first-order decrease (Fig. 5B). This late phase was fitted best by a fractional power function (linear decrease in ln-ln or log-log plots). The entire depletion curves in Fig. 5A could not be fitted adequately by simple first-order equations (r2 < 0.9). As a result, the depletion curves were fitted by using the biphasic model described by equations 1 and 3 (see reference 11 for details). Depletion of [14C]DEHP in sludge alone could be described by the following equations:
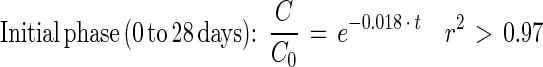 |
5 |
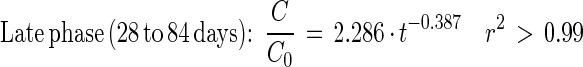 |
6 |
Depletion of [14C]DEHP in sludge-amended soil could be described by the following equations:
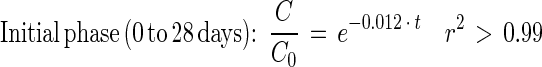 |
7 |
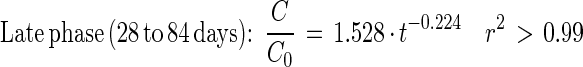 |
8 |
Depletion of [14C]PA in sludge-amended soil could be described by the following equations:
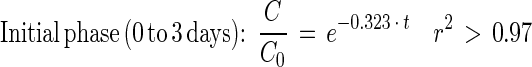 |
9 |
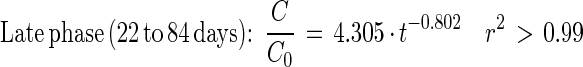 |
10 |
Fig. 5 and equations 5 through 8 show that the mineralization of DEHP in sludge-amended soil was slower than the mineralization of DEHP in sludge alone. Thus, mixing sewage sludge (2 g [wet weight]) with active agricultural soil (21 g [fresh weight]) did not stimulate mineralization activity compared to mixing sludge with inert quartz particles. The initial DEHP mineralization activity in sludge-amended soil was 27 times slower than the PA mineralization. During PA mineralization, the shift from true first-order kinetics to fractional power kinetics was coupled with an apparent transitional period (3 to 22 days) (Fig. 5B and C).
FIG. 5.

Depletion of DEHP and PA in sludge and sludge-amended soil, as estimated from mineralization of radiolabelled substrates. (A) Depletion curves. (B) Initial phase (exponential decrease). (C) Late phase (fractional power decrease). Data points represent the means of triplicate sampes. d, day.
Theoretical half times for the initial and late-phase mineralization activities in sludge and sludge-amended soil were calculated by using equations 2 and 4 (Table 2). The half time calculated for each combination of substrate and sample matrix was based on the coefficients estimated for the depletion curves (equations 5 through 10). The calculations revealed notable differences among the calculated half times in the initial and late phases of the incubations (Table 2). In sludge-amended soil, the half-time of DEHP increased 2.5-fold in the late phase of incubation (from 58 to 147 days). In sludge alone, the increase in half-time was only 1.3-fold (from 39 to 51 days). Using equation 8, we calculated that more than 40% of the DEHP in the sludge-amended soil should have escaped mineralization after 1 year of incubation. In contrast, mineralization of 90% of the PA in sludge-amended soil was expected in about 109 days (equation 10).
TABLE 2.
Degradation estimates for DEHP and PA in sludge and sludge-amended agricultural soila
| Sample | Substrate | Half time for initial phase (days) | Half time for late phase (days) | Initial rate of metabolism (pmol/g [dry wt]/day) |
|---|---|---|---|---|
| Sludge-amended quartz | DEHP | 39 | 51 | 45.4 |
| Sludge slurry | DEHP | 58 | 84 | 26.9 |
| Sludge-amended soil | DEHP | 58 | 147 | 25.6 |
| Sludge-amended soil + strain SDE 2 | DEHP | 9 | 35 | 219.4 |
| Sludge-amended soil | PA | 2 | 15 | 731.8 |
Three samples were examined in each experiment. The initial substrate concentration was 4.1 nmol of DEHP per g (dry weight) or 4.1 nmol of PA per g (dry weight). The concentration half times were estimated by using equations 2 and 4.
Degradation rates.
The initial rates of depletion of DEHP and PA in soil and sludge due to substrate mineralization (Fig. 5) were used to estimate the initial degradation rates. The initial degradation rates for DEHP and PA were calculated by determining the sum of the substrate dissimilation (14CO2 production) and the substrate assimilation (14C-labelled biomass production). The substrate assimilation was estimated from the amount of [14C]DEHP or [14C]PA incorporated into microbial phospholipids during the degradation process (Fig. 1). Radiolabelled phospholipids were extracted directly from soil and sludge samples incubated with [14C]DEHP or [14C]PA. Conversion from the 14C-labelled phospholipid content to total biomass production was based on the ratio of the amount of radiolabel assimilated into phospholipids to the amount of radiolabel assimilated into total microbial biomass estimated for aerobic phthalate degraders isolated from sludge. The mean phospholipid content of these organisms was 7.6% of the total biomass. Using the 14C-labelled phospholipid contents of samples incubated with [14C]DEHP or [14C]PA, we calculated the carbon conversion efficiencies for soil and sludge (biomass production relative to mineralization to CO2). The in situ carbon conversion efficiencies for DEHP and PA metabolism were estimated to be 17 and 28%, respectively. On the basis of these findings, the initial rate of substrate metabolism was calculated as follows: total DEHP or PA metabolism = mineralization estimated from CO2 production × Fa, where the conversion factor Fa corrects for carbon assimilation during substrate metabolism. Fa was estimated to be 1.20 and 1.39 for [14C]DEHP metabolism and [14C]PA metabolism, respectively. The corresponding initial degradation rates for DEHP and PA are shown in Table 2.
Turnover of microbial 14C-labelled biomass.
DEHP degraders were radiolabelled and then added to sludge-amended soil samples in order to estimate the 14CO2 release from labelled microbial biomass in the absence of the radiolabelled substrates [14C]DEHP and [14C]PA (Fig. 6). DEHP-degrading strains SDE 21 and JDE 9 were isolated originally from sludge and soil, respectively. The levels of turnover of labelled microbial biomass (14CO2 production) were comparable for SDE 21 and JDE 9 added to sludge-amended soil (Fig. 6). After an initial more rapid turnover (from 0 to 2 days), metabolism of labelled microbial biomass followed an exponential decrease with a decay rate coefficient of 0.03 day−1 for both SDE 21 and JDE 9 (r2 ≥ 0.99).
FIG. 6.

Turnover of 14C-labelled microbial biomass in sludge-amended soil. DEHP-degrading strains SDE 21 and JDE 9 were radiolabelled and then mixed with sludge-soil samples. Data points represent the means of triplicate samples; error bars indicate the standard errors. d, day.
Direct fingerprinting of microorganisms that metabolize [14C]PA and [14C]DEHP.
The microorganisms that metabolized DEHP and PA in sludge and sludge-amended soil were characterized by analyzing radiolabelled PLFAs in samples after incubation with [14C]PA or [14C]DEHP. Assimilation of 14C into microbial biomass, including PLFAs, resulted in a radioactive fingerprint of the microorganisms actively involved in metabolism of the radiolabelled substrates (Fig. 7). Microorganisms metabolizing [14C]DEHP in sewage sludge produced 14C-PLFAs that were recovered in fractions 4, 5, 8, 10, and 14 (Fig. 7A). These fractions represented PLFAs with equivalent chain lengths of 15.4 to 16.4, 17.4 to 18.0, 18.4 to 18.8, and 20.7 to 21.7 (Table 1). Sludge samples incubated with [14C]PA produced a 14C-PLFA fingerprint that was different from the fingerprint produced by samples incubated with [14C]DEHP (Fig. 7A and B). The microorganisms dominating the metabolism of [14C]PA in sludge produced 14C-PLFAs that were recovered in fractions 3, 4, 5 through 8, and 11 (Fig. 7B). The microorganisms involved in the metabolism of [14C]DEHP in sludge samples (Fig. 7A) and in sludge-amended soil samples (Fig. 7C) produced 14C-PLFA fingerprints that were comparable; the fingerprints of the active DEHP-metabolizing populations were comparable despite the presence of significant amounts of agricultural soil in the latter samples (soil/sludge ratio, 55:1). A similar trend was observed with the microbial populations metabolizing [14C]PA in sludge and sludge-amended soil (Fig. 7B and D). In general, a slightly larger scatter of radioactivity among PLFA fractions was observed for sludge-amended soil assays than for sludge assays without soil (Fig. 7).
FIG. 7.

Direct 14C-PLFA fingerprints of the microorganisms involved in degradation of DEHP and PA in sludge and sludge-amended soil. (A) Sewage sludge incubated with [14C]DEHP. (B) Sewage sludge incubated with [14C]PA. (C) Sludge-amended soil incubated with [14C]DEHP. (D) Sludge-amended soil incubated with [14C]PA. Radiolabelled PLFAs were collected in 15 fractions, as shown in Table 1.
DISCUSSION
Biodegradation of organic pollutants in the environment requires the presence of microorganisms with enzymes capable of catalyzing the breakdown of the target molecules. Complete aerobic degradation of PA and phthalate esters (e.g., DEHP) involves several enzymes, including hydrolases, dehydrogenases, decarboxylases, and oxygenases (17). In the present study, the time courses of [14C]PA and [14C]DEHP mineralization in sludge-amended soil revealed that there were substantial differences in the biodegradabilities of the two compounds (Fig. 2). [14C]PA was mineralized much faster by the indigenous microorganisms than [14C]DEHP was. However, both compounds were labelled in the aromatic ring portions of their molecules ([U-14C]-ring]PA and [U-14C-ring]DEHP). These results suggest that the rate-limiting step in the enzymatic breakdown of DEHP in situ is not related to the aromatic portion of the molecule. This is consistent with studies which suggest that enzymatic cleavage of the ester bonds in metabolites of DEHP [e.g., mono-(2-ethylhexyl)phthalate] may constitute a biological bottleneck.
A factor that regulates the enzymatic degradation of hydrophobic organic pollutants in situ is the bioavailability of the substrate. The bioavailability of organic compounds in soils and sediments is affected strongly by sorption, partitioning, and diffusion (1, 3, 7, 8, 15, 21). The potential role of bioavailability in DEHP degradation is illustrated in Fig. 3. Mineralization of [14C]DEHP by strain SDE 2 was attenuated by the addition of dried soil (containing 6% organic matter) and dried sewage sludge (containing 29% organic matter). The presence of soil or sludge did not inhibit strain SDE 2 as the organisms survived well in other assays in the presence of soil or sludge organic compounds (Fig. 4). Although the low DEHP mineralization rate in the presence of soil or sludge may also be explained partially by facultative metabolism in the presence of alternative organic substrates, the results emphasize that the presence of organic material in situ regulates the bioavailability and degradation of DEHP substantially. Thus, a combination of restricted bioavailability and potential enzymatic limitations renders DEHP much less biodegradable in situ than PA.
Depletion curves for DEHP and PA in sludge and sludge-amended soil were determined based on mineralization of radiolabelled substrates to 14CO2 (Fig. 5). The mineralization activity was concentration dependent in soil and sludge and could not be described as a single exponential decrease (11). Exponential functions (pseudo first-order reactions) are often used as a convenient way to describe initial mineralization of xenobiotic compounds at low substrate concentrations (2, 14). In our studies, the best depletion fit (r2 > 0.97) was obtained by describing the initial activity with an exponential function, followed by a phase that was best described by a fractional power function (11). A shift in the apparent kinetics of DEHP mineralization was evident after approximately 28 days of incubation. In all assays, the relative mineralization activity was lowest in the late phase of the degradation experiment (fractional power kinetics). This translates into substantial increases in the estimated half times for the late phase compared to the initial phase (Table 2). For example, the theoretical half time for DEHP in sludge-amended soil increased from 58 to 147 days after 28 days of incubation (Fig. 5). A possible explanation for this phenomenon is that time-dependent immobilization of DEHP may occur in the soil-sludge matrix. This phenomenon has been described previously for a range of hydrophobic organic compounds in soil (1, 3, 7, 8, 15, 21). Several workers have concluded that physicochemical factors that determine the sequestration of the target molecules become increasingly important as regulators of mineralization activity with increasing pollution age (7, 8, 21).
It is noteworthy that DEHP (and PA) in soil and sludge can be transformed to various intermediates (primary biodegradation) at a rate that may exceed the mineralization rates (ultimate biodegradation). However, calculating DEHP biodegradation rates from direct measurements of substrate disappearance is complicated because immobilization of hydrophobic compounds may render these compounds resistant to conventional chemical extraction (1, 7, 26). Zurmühl et al. (26) concluded that a significant fraction of the DEHP added to soil columns became irreversibly sorbed with time. Thus, time-dependent immobilization of nonextractable DEHP could subsequently complicate direct concentration measurements and lead to overestimates of microbial transformation activity.
A process that contributes to 14CO2 production in mineralization assays performed with radiolabelled substrates is turnover of radiolabelled microbial biomass (i.e., 14CO2 production from 14C-labelled cell constituents). This biomass turnover may be falsely interpreted as a low level of mineralization of the radiolabelled compound that was added initially. In some assays, 14CO2 may be released from bacterial biomass long after the added 14C-labelled precursor has been degraded completely or has become unavailable for biodegradation. In the present study, radiolabelled DEHP degraders inoculated into sludge-amended soil released 14CO2 exponentially with a rate coefficient of 0.03 day−1 (Fig. 6). This translates into an estimated half time for labelled microbial biomass of 23 days. This turnover time is roughly comparable to the estimated half time for [14C]PA and [14C]DEHP mineralization after inoculation with SDE 2 (Table 2, late phase). In these assays, a large amount of substrate was metabolized initially, which could have supported substantial biomass labelling and the subsequent slow release of significant amount of 14CO2 during endogenous and exogenous carbon turnover. This phenomenon may complicate proper interpretation of the degradation kinetics in assays with poor bioavailability and result in half time estimates that are too optimistic. As a result, the DEHP half times estimated in the present study (Table 2) probably are minimal.
The measured initial mineralization rates for DEHP and PA were corrected for assimilation of substrate-derived 14C into microbial biomass to provide an estimate of the initial metabolism of the substrates (Table 2). The assimilation factor used in estimating the metabolism was based on carbon conversion efficiencies (metabolic efficiencies) estimated for the degradation of DEHP and PA in sludge and sludge-amended soil. Radiolabelled bacterial phospholipids were used as indicators of bacterial biomass production during degradation of [14C]-DEHP and [14C]PA. The phospholipid content relative to the total microbial biomass was estimated on the basis of phthalate ester degraders isolated from soil and sludge. The mean phospholipid content, 7.6%, was comparable to the mean phospholipid contents reported for other bacteria (12, 16). On the basis of the direct extractions, the in situ carbon conversion efficiencies for DEHP and PA metabolism were estimated to be 17 and 28%, respectively. These values are somewhat lower than the metabolic efficiencies reported for DEHP degradation in a soil slurry reactor (40 to 50%) (9). However, because metabolic efficiencies depend strongly on environmental factors (22), variations among experiments are expected. As a result, direct estimates of the metabolic efficiency for DEHP appear to be necessary if these values are to be included in degradation estimates. Simply assuming high metabolic efficiencies (e.g., 50%) could be associated with nontrivial errors.
Substrate-specific radiolabelling followed by analysis of 14C-PLFAs has been used for direct fingerprinting of active methane and phenanthrene degraders in environmental samples (19). In the present study, active DEHP and PA degraders in sludge and sludge-amended soil were compared by using this approach, and the direct radiolabelling technique provided simple radioactive PLFA fingerprints (14C-PLFA fingerprints) of the organisms that metabolized [14C]DEHP or [14C]PA (Fig. 7). The results suggest that microbial populations with different PLFA compositions dominated [14C]PA and [14C]DEHP metabolism in the samples (Fig. 7). In contrast, phenotypically comparable populations dominated [14C]DEHP metabolism in sludge and sludge-amended soil (Fig. 7A and C) and [14C]PA metabolism in sludge and sludge-amended soil (Fig. 7B and D). Thus, addition of agricultural soil to sludge samples at a ratio of 55:1 (wt/wt) did not result in a labelling pattern that was different from the patterns obtained in sludge assays without soil. This is consistent with results from the mineralization experiments which indicated that addition of soil to sludge samples did not increase the mineralization of [14C]DEHP (Fig. 2). In fact, depletion of DEHP due to microbial mineralization was appreciably slower in assays performed with sludge-amended soil than in sludge assays performed without soil (Fig. 5 and Table 2). In the late phase of the experiment, the half time for DEHP degradation in sludge-amended soil was approximately three times greater than the half time in sludge (Table 2). The limited contributions of the soil microorganisms to the degradation of DEHP in sludge-amended soil were probably due to immobilization of DEHP and its metabolites in the sludge matrix. Collectively, the mineralization and fingerprinting data suggest that indigenous microorganisms in the sewage sludge were responsible for the majority of the DEHP degradation in the sludge-amended soil.
The findings described above suggest that a significant amount of DEHP (and its metabolites) with poor biodegradability and extractability (e.g., bound residues) may remain in sludge-amended soil for extended periods of time despite the presence of microorganisms that are capable of degrading the compounds. However, it is uncertain whether this old DEHP poses a potential health risk in situ (e.g., when the pollution age is more than 100 days). Alexander (1) has suggested that the risk due to the presence of hydrophobic pollutants may decrease substantially over time because the compounds become sequestered in inaccessible soil microsites. Future studies will show whether this is true for DEHP that enters terrestrial environments due to fertilization with sewage sludge.
In summary, DEHP degradation in sludge-amended soil is regulated strongly by factors that affect the bioavailability of the compound in the sludge-soil matrix. The degradation kinetics are complex, and DEHP appears to become increasingly less bioavailable during incubation for more than 4 weeks. Our results also suggest that DEHP degradation as estimated from addition and extraction of nonlabelled DEHP may significantly overestimate the mineralization rate. On the basis of radiotracer techniques, we concluded that indigenous microorganisms in sludge rather than indigenous microorganisms in soil are responsible for the majority of DEHP mineralization in sludge-amended soils. Model predictions suggest that more than 40% of the DEHP in sludge-amended soil is not mineralized after 1 year.
ACKNOWLEDGMENTS
We thank Kirsten Maagaard for excellent technical assistance. We also thank Per Møldrup for valuable suggestions and Mågens Åge for encouragement.
This work was supported by the Danish Environmental Research Programme (Center for Sustainable Land Use and Management of Contaminants, Carbon, and Nitrogen) and the Danish Technical Research Council.
REFERENCES
- 1.Alexander M. How toxic are toxic chemicals in soil? Environ Sci Technol. 1995;29:2713–2717. doi: 10.1021/es00011a003. [DOI] [PubMed] [Google Scholar]
- 2.Alexander M, Scow K M. Kinetics of biodegradation in soil. In: Sawney B L, Brown K, editors. Reactions and movement of organic chemicals in soils. Special Publication 22. Madison, Wis: Soil Science Society of America and American Society of Agronomy; 1989. pp. 243–269. [Google Scholar]
- 3.Bollag J-M. Decontaminating soil with enzymes. Environ Sci Technol. 1992;26:1876–1881. [Google Scholar]
- 4.Coughtrey P J, Nancarrow D J, Jackson D. Extraction of carbon-14 from biological samples by wet oxidation. Commun Soil Sci Plant Anal. 1986;17:393–399. [Google Scholar]
- 5.Davis B J, Maronpot R R, Heindel J J. Di(2-ethylhexyl)phthalate suppresses estradiol and ovulation cycling in rats. Toxicol Appl Pharmacol. 1994;128:216–223. doi: 10.1006/taap.1994.1200. [DOI] [PubMed] [Google Scholar]
- 6.Giam C S, Atlas E, Powers M A, Leonard J E. Phthalic acid esters. In: Hutzinger O, editor. The handbook of environmental chemistry. Berlin, Germany: Springer; 1984. pp. 67–142. [Google Scholar]
- 7.Hatzinger P B, Alexander M. Effect of aging of chemicals on the their biodegradability and extractability. Environ Sci Technol. 1995;29:537–545. doi: 10.1021/es00002a033. [DOI] [PubMed] [Google Scholar]
- 8.Hatzinger P B, Alexander M A. Biodegradation of organic compounds sequestered in organic solids or in nanopores within silica particles. Environ Toxicol Chem. 1997;16:2215–2221. [Google Scholar]
- 9.Irvine R L, Earley J P, Kehrberger G J, Delaney B T. Bioremediation of soils contaminated with bis-(2-ethylhexyl)phthalate (BEHP) in a soil slurry-sequencing batch reactor. Environ Prog. 1993;12:39–44. [Google Scholar]
- 10.Kjølholt J, Andersen H V, Poll C. Presence and effects of organic pollutants in sewage sludge. Copenhagen, Denmark: Danish Environmental Protection Agency; 1995. . (In Danish.) [Google Scholar]
- 11.Madsen, P. L., J. B. Thyme, K. Henriksen, P. Møldrup, and P. Roslev. Kinetics of di(ethylhexyl)phthalate (DEHP) mineralization in sludge-amended soil. Submitted for publication.
- 12.Neidhardt F C, Ingraham J L, Schaechter M. Physiology of the bacterial cell. Sunderland, Mass: Sinauer Associates; 1990. [Google Scholar]
- 13.Nielsen E, Larsen P B. Toxicological evaluation and limit values for DEHP and phthalates other than DEHP. Environmental Review Report 6/1996. Copenhagen, Denmark: Danish Environmental Protection Agency; 1996. [Google Scholar]
- 14.Nyholm N, Ingerslev F, Berg U T, Pedersen J P, Frimer-Larsen H. Estimation of kinetic rate constants for biodegradation of chemicals in activated sludge wastewater treatment plants using short term batch experiments and μg/l range spiked concentrations. Chemosphere. 1996;33:851–864. doi: 10.1016/0045-6535(96)00180-4. [DOI] [PubMed] [Google Scholar]
- 15.Pignatello J J, Xing B. Mechanisms of slow sorption of organic chemicals to natural particles. Environ Sci Technol. 1996;30:1–11. [Google Scholar]
- 16.Ratledge C, Wilkinson S G. Microbial lipids. Vol. 1. London, United Kingdom: Academic Press; 1988. [Google Scholar]
- 17.Ribbons D W, Keyser P, Kunz D A, Taylor B F, Eaton R W, Anderson B N. Microbial degradation of phthalates. In: Gibson D T, editor. Microbial degradation of organic compounds. New York, N.Y: Marcel Dekker; 1984. pp. 371–397. [Google Scholar]
- 18.Roslev P, Iversen N, Henriksen K. Oxidation and assimilation of atmospheric methane by soil methane oxidizers. Appl Environ Microbiol. 1997;63:874–880. doi: 10.1128/aem.63.3.874-880.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roslev P, Iversen N, Henriksen K. Direct fingerprinting of metabolically active bacteria in environmental samples by substrate specific radiolabelling and lipid analysis. J Microbiol Methods. 1998;31:99–111. [Google Scholar]
- 20.Schmitzer J L, Scheunert I, Korte F. Fate of bis(2-ethylhexyl) [14C]phthalate in laboratory and outdoor soil-plant systems. J Agric Food Chem. 1988;36:210. [Google Scholar]
- 21.Scow K M, Alexander M. Effect of diffusion on the kinetics of biodegradation: experimental results with synthetic aggregates. Soil Sci Soc Am J. 1992;56:128–134. [Google Scholar]
- 22.Shen J, Bartha R. Metabolic efficiency and turnover of soil microbial communities in biodegradation tests. Appl Environ Microbiol. 1996;62:2411–2415. doi: 10.1128/aem.62.7.2411-2415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siddiqui A, Srivastava S P. Effect of di(2-ethylhexyl)phthalate administration on rat sperm count and on sperm metabolic enzymes. Bull Environ Contam Toxicol. 1992;48:115–119. doi: 10.1007/BF00197492. [DOI] [PubMed] [Google Scholar]
- 24.Staples C A, Peterson D R, Parkerton T F, Adams W J. The environmental fate of phthalate esters: a literature review. Chemosphere. 1997;35:667–749. [Google Scholar]
- 25.Zurmühl T. Development of a method for the determination of phthalate esters in sewage sludge including chromatographic separation from polychlorinated biphenyls, pesticides and polyaromatic hydrocarbons. Analyst. 1990;115:1171–1175. [Google Scholar]
- 26.Zurmühl T, Durner W, Herrmann R. Transport of phthalate-esters in undisturbed and unsaturated soil columns. J Contam Hydrol. 1991;8:111–133. [Google Scholar]