

Abstract
Clinical studies have demonstrated that local expression of the cytokine IL-12 drives interferon-gamma expression and recruits T cells to the tumor microenvironment, ultimately yielding durable systemic T cell responses. Interrogation of longitudinal biomarker data from our late-stage melanoma trials identified a significant on-treatment increase of intratumoral CXCR3 transcripts that was restricted to responding patients, underscoring the clinical relevance of tumor-infiltrating CXCR3+ immune cells. In this study, we sought to understand if the addition of DNA-encodable CXCL9 could augment the anti-tumor immune responses driven by intratumoral IL-12. We show that localized IL-12 and CXCL9 treatment reshapes the tumor microenvironment to promote dendritic cell licensing and CD8+ T cell activation. Additionally, this combination treatment results in a significant abscopal anti-tumor response and provides a concomitant benefit to anti-PD-1 therapies. Collectively, these data demonstrate that a functional tumoral CXCR3/CXCL9 axis is critical for IL-12 anti-tumor efficacy. Furthermore, restoring or amplifying the CXCL9 gradient in the tumors via intratumoral electroporation of plasmid CXCL9 can not only result in efficient trafficking of cytotoxic CD8+ T cells into the tumor but can also reshape the microenvironment to promote systemic immune response.
Keywords: IL-12, tavokinogene telseplasmid, electroporation, CXCL9, CXCR3, chemotaxis, CT26, 4T1, anti-PD-1, DNA-encodable
Graphical abstract
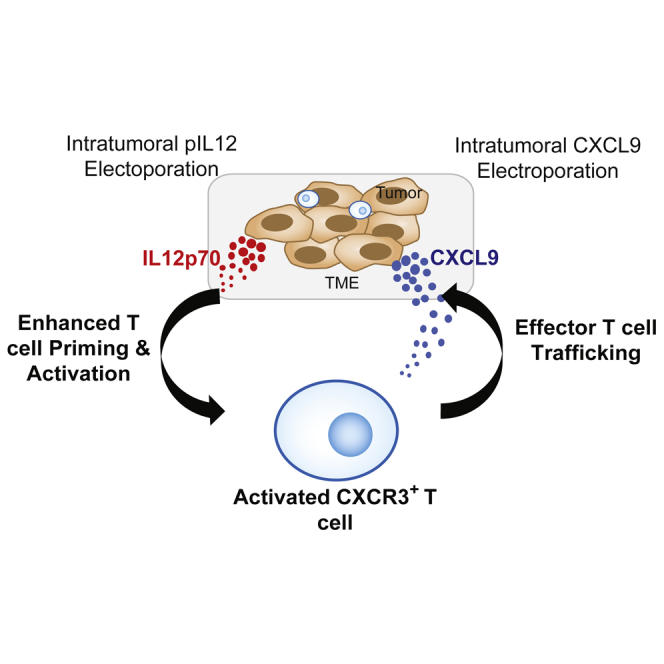
We demonstrate that the CXCR3/CXCL9 chemokine axis is critical for IL-12 therapy and that temporal and spatial control of this axis via intratumoral expression of CXCL9 leads to productive immune responses with potent anti-tumor effects. Expression of IL-12 and CXCL9 significantly improved anti-PD-1 response, providing a strong rationale for clinical testing.
Introduction
The overall tumor immune contexture characterized by the location, density, and functional organization of immune cells in the tumor microenvironment is widely recognized as an important determinant of clinical outcome.1 T cell inflamed tumors are often characterized by transcriptional profiles associated with T cell markers, pro-inflammatory cytokines, and chemokines. These profiles often correspond with clinical benefit to various immunotherapies, including checkpoint blockade and vaccines.2,3 Several immune cells (MDSCs, TAMs, TANs, Tregs, suppressive B cells), endothelial cells, and stromal cells can infiltrate the tumor during its progression. These cells act to limit T cell function and trafficking via direct contact or secretion of suppressive molecules, rendering a poorly immunogenic tumor microenvironment (TME).4, 5, 6, 7, 8, 9, 10 Therapies that can reprogram this TME from a suppressive T cell-excluded state to a productive T cell-inflamed state are actively being developed in research and clinical settings.
T cell trafficking is a tightly orchestrated multistep process involving the loss of L-selectin, (CD62L), and CCR7 on primed T cells, allowing them to egress from the lymph node while gaining the expression of homing molecules such as PSGL-1 (CD162). These activated lymphocytes will then tether and roll along endothelium expressing E-selectin, slowing down from the hemodynamic shear forces experienced in blood flow. This braking system allows for effective interactions between chemokine receptors (e.g., CXCR3, CCR5, CCR2) and their respective chemokines (e.g., CXCL9/10/11, CCL2/3/4/5) that are expressed in the vasculature, infected tissue, or tumor. Subsequently, integrins are activated, instigating lymphocyte firm adhesion to the endothelium, and leading to extravasation and chemotaxis to the microenvironment of need.11 Several studies have underscored the importance of chemokine axes for T cell infiltration into the tumors and for associated anti-tumor responses.12,13 Despite the existence of multiple chemokine/receptor pairs, the CXCR3-CXCL9/10/11 axis is often a critical checkpoint that can dictate a productive homing process.14
Both pre-clinical and clinical studies have featured the importance of attracting leukocytes to the TME to promote strong anti-tumor responses.14, 15, 16, 17, 18 Furthermore, chemokine gradients have been shown to be important in facilitating efficient in situ priming and differentiation of T cells, including newly described stem cell-like T cells (Tscm expressing TCF7+), by fostering interaction with antigen-presenting cells.19, 20, 21 The CXCR3-CXCL9/10/11 axis mainly regulates migration, differentiation, and activation of immune cells. CXCR3 is expressed on multiple immune cell types, including CD4+ T cells, CD8+ T cells, natural killer (NK), and NKT cells.22
Recent studies suggest that interferon-gamma (IFN-γ) is a critical driver for intratumoral T cell infiltration via upregulation of its target genes CXCL9/10/1123 and may improve the likelihood of clinical response to anti-PD-1 therapies, including pembrolizumab.24 We and others have previously shown, in both clinical and pre-clinical studies, that intratumoral expression of pro-inflammatory cytokines such as IL-12 can modulate expression of chemokines, like CXCL9/10/11, in the tumor through a positive feedforward loop with IFN-γ.25 Furthermore, IL-12 delivery to the TME results in robust immune activation, epitope spreading, and increased sensitivity to anti-PD-1 treatment.25,26 Given the substantial role of CXCR3 and associated chemokines in enabling anti-PD-1 therapeutic responses,19,20 it is possible that a CXCL9 gradient, established secondarily to IFN-γ via intratumoral IL-12 expression, is foundational for an effective anti-PD-1 response.
Tumor microenvironments often show variable expression of CXCR3 ligands, which influences their capacity to recruit T cells.27 This raises the possibility of therapeutic modalities that restore or augment intratumoral expression of CXCR3 ligands to not only traffic, retain, and engage primed cytotoxic CD8+ T cells in the tumor, but, if positioned correctly, to also promote in situ priming.28,29 Previously published longitudinal biomarker data from patients with late-stage melanoma treated with intratumoral (IT) plasmid IL-12 (pIL12) (tavokinogene telseplasmid, or Tavo) with electroporation (EP), IT-pIL12-EP, identified key immunological components associated with an effective therapy that included a significant on-treatment increase of intratumoral CXCR3 expression.30 Therefore, it was hypothesized that maximizing the intratumoral chemokine gradient to augment trafficking of CXCR3+ lymphocytes generated via IT-pIL12-EP would result in a more productive immune response. To investigate this, we first identified and utilized a tumor model in which the IL-12 response is CXCR3 dependent. By use of the colorectal CT26 tumor model, it was shown that IT-pIL12-EP leads to an increase in CXCR3+ lymphocytes in the local lymph node, indicative of increased priming. In addition, blocking CXCR3+ cells can completely abrogate tumor regression, confirming the significance of CXCR3 involvement. This local cytokine/chemokine therapy can drive broad immunological changes beyond the treated tumor, yielding systemic anti-tumor responses that can augment PD-1 checkpoint inhibition. This study suggests that the CXCR3-CXCL9 chemokine axis is critical for IL-12 therapy and that temporal/spatial control of the axis via intratumoral expression of CXCL9 may represent a key addition to intratumoral cytokine therapies in the clinic.
Results
Clinical response to intratumoral electroporation of plasmid IL-12 plus pembrolizumab combination therapy positively correlates with intratumoral CXCR3 expression
Longitudinal biospecimens including tumor biopsies and isolated peripheral blood mononuclear cells (PBMCs) were collected during a phase 2 clinical trial evaluating IT-Tavo-EP in combination with pembrolizumab to treat patients with advanced melanoma predicted to not respond to anti-PD-1 checkpoint inhibitors based on a low density of tumor-infiltrating lymphocyte (TIL), specifically, a low frequency of intratumoral checkpoint-positive cytotoxic lymphocytes (<25% CD8+ PD1hiCTLA-4hi).31 Analysis of isolated PBMCs from these patients highlighted that this combination could drive an on-treatment increase in the frequency of proliferating CD8+ T cells in the periphery (Figure 1A, left) regardless of clinical response (Figure 1A, right). Interestingly, intratumoral expression of CXCR3 was significantly increased only in patients responding to the combination therapy (Figure 1B). Furthermore, CXCR3 gene expression positively correlated with both CD8 and CD4, but not with CD163, suggesting T cell focused CXCR3 expression (Figure S1A). These observations coupled with recently published studies focused on CXCR3-mediated anti-tumor immunity27, 28, 29 provided the rationale to ask whether CXCR3 and its associated chemokines could play a critical role in enabling intratumoral IL-12 therapeutic responses.
Figure 1.

Response to IL-12 plus pembroluzimab associates with intratumoral CXCR3 expression
(A) Flow cytometric analysis of proliferating (Ki-67+) CD8+ cells in the peripheral blood (Singlets > Live > CD56- CD16- CD14- > CD3+ > CD8+ CD4- > Ki67+) of patients’ pre-treatment and post-treatment (first day of the second treatment cycle, C2D1; left). Further breakdown of the percent increase in Ki-67 expression pre- and post-treatment from responding patients compared with non-responding patients (C2/Screen: Values at C2D1 divided by screen; right) (n = 7 per group); p value for unpaired Mann-Whitney t test is shown. (B) Comparison of post-treatment and pre-treatment (C2D1) levels of intratumoral CXCR3 mRNA transcript between responders (n = 5) and non-responders (n = 8). ∗p < 0.05 for paired Wilcoxin t tests are shown.
CXCR3-mediated signaling is required for intratumoral pIL12 electroporation therapy in a CT26 murine model
A CT26 murine tumor model was utilized to investigate the role of CXCR3 in intratumoral pIL12-EP-mediated anti-tumor responses. Membranous expression of CXCR3 is a dynamic process that shifts between endocytosis-mediated recycling and de novo synthesis,32 making its quantification by flow cytometric analysis a challenge. Nevertheless, interrogation of tumor-draining lymph nodes provided evidence that IT-pIL12-EP can drive not only intratumoral CXCR3 expression but also a higher frequency and mean fluorescent intensity of CXCR3+ CD8+ T cells in the local lymph nodes (Figure 2A). To better understand how IL-12-driven CXCR3+ T cells traffic in response to CXCL9, a surrogate chemotaxis assay was developed to quantify the frequency of cells migrating toward a CXCL9 gradient. CXCR3-blocking antibody (anti-CXCR3) was used to confirm the specificity of the CXCR3-CXCL9 interaction in mediating chemotaxis. CT26 tumors were injected with pIL12 or pUMVC3 empty vector (EV) and electroporated; after 4 days, the draining lymph nodes were harvested, and the lymphocytes were used in the chemotaxis assay (Figure 2B, left). Significantly higher numbers of lymphocytes migrated toward the CXCL9 gradient when isolated from the IT-pIL12-EP treated mice compared with when isolated from the empty vector control-treated mice (Figure 2B, right). This IL-12-driven migration was completely blocked by pre-incubating lymphocytes with an anti-CXCR3 antibody, confirming CXCR3-dependent chemotaxis. To further verify the specificity and efficacy of CXCR3-mediated chemotaxis, supernatants from HEK293 cells transfected with mouse pCXCL9 were quantified for mouse CXCL9 (mCXCL9) expression (Figure S2A) and assayed for their chemotactic potential on stimulated CXCR3+ OT1-GFP splenocytes (Figure S2B). Transfection-conditioned medium containing mCXCL9 enhanced chemotaxis relative to transfection control. This chemotaxis was significantly reduced with the addition of an anti-CXCL9 neutralizing antibody.
Figure 2.

Intratumoral IL-12 response is dependent on CXCR3-chemokine axis
(A) Representative flow cytometric analysis and the frequency of CXCR3 expression on CD8+ T cells from the draining lymph node collected 4 days after IT-EP with EV or pIL12 (50 μg per treatment; ∗∗p < 0.01, unpaired t test). (B) Left: chemotaxis schematic of draining lymph node cells collected 4 days post-IT-EP with EV or pIL12 (50 μg per treatment). Right: transfection-derived mouse CXCL9 (250 ng/mL)-induced chemotaxis. Chemotaxis effect was abrogated when cells were pre-incubated with anti-mCXCR3 monoclonal antibody. ∗∗∗∗p < 0.0001, one-way ANOVA. (C) CT26 contralateral subcutaneous tumor model was intratumorally electroporated with pIL12 (50 μg) with or without concomitant anti-CXCR3 antibody treatment. Growth of primary electroporated (left) and untreated contralateral (right) CT26 lesions after IT-EP with EV (gray), pIL12 + isotype control (black), and pIL12 + anti-mCXCR3 (black hatched) are shown (n = 10; data shown is a cumulative plot of two experiments with five animals in each group; statistical significance determined using two-way ANOVA with Bonferroni correction, ∗p < 0.02, ∗∗p < 0.006, ∗∗∗∗p < 0.0008, ns: not significant). (D) Kaplan-Meir curve showing the survival difference between IT-pIL12-EP + anti-mCXCR3 and IT-pIL12-EP groups. ∗p < 0.05, log rank (Mantel-Cox) test.
In agreement with the above-mentioned data, blockade of CXCR3-mediated homing in vivo resulted in loss of this anti-tumor effect of IT-pIL12-EP therapy. In a previously described CT26 bilateral tumor model,33 significant differences were observed in both control of contralateral tumor volume (Figure 2C, right) and survival (Figure 2D). Since CXCR3 is expressed on various immune subsets including NK cells and CD8+ T cells, we investigated which cell types were fundamental to this IL-12 response in the CT26 model. In the contralateral lesion, NK depletion resulted in a mild loss of tumor regression, while depletion of CD8+ cells had the most deleterious effect in controlling tumor growth, suggesting a primary role for CD8+ T cells in mediating a systemic anti-tumor response (Figure S3). Collectively, these pre-clinical data align with clinical observations associating intratumoral CXCR3 expression with IL-12 combination therapy (Figure 1) whereby the IT-pIL12-EP stimulates expansion of CXCR3+ lymphocytes in the tumor, the draining lymph node and the periphery, which are critical to mediating the anti-tumor response.
Combination of intratumoral pCXCL9 and pIL12 with electroporation increases tumor immunogenicity by modulating the TME and expanding the pool of antigen-specific T cells
Given that IT-pIL12-EP generates CXCR3+ lymphocytes in the lymph node and the periphery, we hypothesized that additional intratumoral expression of CXCL9 would lead to more efficient trafficking of lymphocytes into the tumor and ultimately remodeling of the TME. CT26 tumors were sequentially treated with a low but immunologically active dose of pIL12 (2 μg; Figure S4) on day 0 (1st treatment) followed by 100 μg of pCXCL9 on day 4 (2nd treatment) and day 7 (3rd treatment) (Figure 3A, schematic of treatment). Two days after the last treatment, treated tumors were excised, and total RNA was extracted from the tumor tissue. The immune-related transcriptome was assessed using the NanoString nCounter platform, which demonstrated enrichment of pathways related to G protein-coupled receptors (GPCRs) like CXCR3, as well as interleukin and interferon signaling (Figure 3B). In addition, pIL12 plus pCXCL9 treatment augmented antigen presentation and active T cell receptor (TCR) signaling, indicative of increased immunogenicity and T cell activation. Relatedly, productive changes beyond the TME were also observed in the periphery, with this combination driving an increase of AH1 antigen-specific CD8+ T cells (Figure 3C).
Figure 3.

Intratumoral CXCL9 and IL-12 combination increases tumor immunogenicity by modulating the TME, leading to expansion of splenic antigen-specific CD8 cells
(A) Schematic showing sequential treatment regimen to assess tumor regression and monitor immune response. CT26 tumor-bearing mice were intratumorally electroporated with a low but immunologically active dose of pIL12 (2 μg, day 0) followed by IT-EP of 100 μg of either pCXCL9 or EV (days 4 and 7). Tumor and splenocytes were harvested 2 days after last EP (i.e., day 9) for NanoString and flow-based analysis. (B) Gene expression changes in electroporated CT26 lesions were assessed by NanoString nCounter technology (mouse PanCancer IO360 panel) with pathway scores. Pathway scores follow the assumptions of equal variance and normal distribution of t scores. Ordinary one-way ANOVA was used to calculate significance compared with the empty-vector-treated group (∗p < 0.05, ∗∗p < 0.009). GPCR, G protein-coupled receptor; MHC1, major histocompatibility complex 1. (C) Left: representative plots demonstrating enrichment of antigen-specific CD8 T cells (AH1+CD8+) in mice 9 days after treatment with pIL12 + pCXCL9 compared with pIL12 alone or EV on Singlets > Live > CD3+CD4- splenocytes. Right: increase in the percentage of AH1+ CD8+ T cells compared with empty-vector control (n = 3, 3–5 animals/group; ∗p < 0.05, one-way ANOVA).
Intratumoral electroporation of pCXCL9 augments IL-12 mediated abscopal anti-tumor responses
Considering that intratumoral treatment with pCXCL9 and pIL12 produced effective shifts in tumor immunogenicity with associated peripheral T cell responses, we investigated whether these dynamic immune markers could impact systemic tumor growth and survival. A CT26 bilateral tumor model was used to assess tumor regression and survival upon treatment (Figure 4A). Mice receiving a sequence of intratumoral pIL12 and pCXCL9 with electroporation demonstrated significantly reduced growth of both treated and untreated lesions on day 12 compared with mice receiving pIL12 only (Figures 4B and 4C). In addition, mice electroporated with pIL12 and subsequently pCXCL9 survived significantly longer than mice receiving only pIL12 treatment (Figure 4D). To eliminate the potential variability with electroporating a cocktail of plasmids to achieve this therapeutic combination, we explored whether expressing mIL-12 and mCXCL9 from a single multicistronic plasmid would result in a similar anti-tumor effect (Figure 4E). We first confirmed the expression and potency of each component expressed from the plasmid (Figure S5A). As expected, whereas there was substantial IL-12 expression from this pIL12-CXCL9 plasmid, it was approximately 5-fold lower than the expression from the pIL12 plasmid alone (Figure S5A). Despite exhibiting decreased expression, IL-12p70 and CXCL9 protein were detectable in CT26 and B16-F10 tumor extracts for up to 7 days post-EP (Figures S6A and S6B). Potencies of the resulting IL-12 and CXCL9 were confirmed by using an IL-12 reporter cell line and a chemotaxis assay, respectively (Figures S5B and S5C). For subsequent tumor regression studies, we normalized the plasmid doses to accommodate the difference in IL-12 expression levels. Consistent with the sequential treatment of pIL12 and pCXCL9, the electroporation of the pIL12-CXCL9 multigene plasmid in CT26 tumors resulted in a significantly better abscopal response (Figure 4G) and survival compared with mice treated with IL-12 alone (Figure 4H). Furthermore, electroporation of the multigene plasmid led to increases in the percentage of CXCR3+ CD8+ T cells and in the MFI of CXCR3 expression (Figure S5D). These results suggest that increasing the tumoral CXCL9 gradient augments IL-12 anti-tumor response.
Figure 4.

Intratumoral CXCL9 and IL-12 combination therapy augments abscopal anti-tumor response
(A) Schematic illustrating treatment regimen for sequential therapy to assess tumor regression. By using a contralateral tumor model, CT26-bearing mice were electroporated with low-dose pIL12 (1–2 μg, day 0) followed by either IT-EP of pCXCL9-mCherry or pUMVC3-mCherry (pCXCL9-C and EV-C, 100 μg, days 4 and 7). Tumor volumes were measured three times a week for regression and survival studies. (B and C) Growth of primary electroporated (B) and untreated contralateral CT26 lesions (C) after IT-EP of pIL12 + EV-C (red) and pIL12 + pCXCL9-C (blue) are shown. (n = 5; 5–10 animals/experiment), Statistical significance was determined using two-way ANOVA. ∗p < 0.05, ∗∗∗∗p < 0.0001. (D) Kaplan-Meir curve comparing the survival of the pIL12 + pCXCL9-C cohort with that of pIL12 + EV-C. ∗∗∗p < 0.0001 by log rank (Mantel-Cox) test. (E) Schematic of the pIL12-CXCL9 plasmid construct and treatment regimen to assess tumor regression of CT26 contralateral tumor model upon treatment with EV, pIL12, or pIL12-CXCL9. The dose of pIL12 plasmid was normalized to the amount of mIL-12 produced from the pIL12-CXCL9 plasmid by ELISA. (F and G) At 12 days post- treatment, contralateral tumors from the pIL12-CXCL9 cohort were significantly smaller. No significant difference was observed in primary tumor growth (n = 2; 7–10 animals/experiment; statistical significance determined using two-way ANOVA, ∗p < 0.05). (H) Kaplan-Meir curve showing the survival of the pIL12-CXCL9 cohort to be significantly better than that of pIL12. ∗p < 0.05, log rank (Mantel-Cox) test.
Intratumoral electroporation of pIL12-CXCL9 in a murine 4T1 model augments IL-12 anti-tumor effects
The murine mammary carcinoma 4T1 model was used to evaluate whether the abscopal anti-tumor effects of pIL12-CXCL9/EP observed in CT26 tumors extended to other indications and spontaneous metastases (Figure 5). Mice received three intratumoral electroporations on days 0, 4, and 7 with empty vector, pIL12, or pIL12-CXCL9 (Figure 5A). Compared with empty vector, mice electroporated with pIL12 alone showed significant tumor growth control in treated lesions at day 17 (Figure 5B, red versus blue bar). Electroporation of pIL12-CXCL9 significantly improved the anti-tumor effects seen with pIL12 alone (Figure 5B, green versus red bar). Metastatic lung nodules were enumerated when mice reached tumor burden (Figure 5C). Similar to treated lesions, electroporation of pIL12 alone reduced the number of lung nodules compared with EV (Figure 5B, red versus blue bar), while electroporation of pIL12-CXCL9 further reduced the number of lung nodules significantly beyond the effect seen with pIL12 (Figure 5B, green versus red bar). Taken collectively with the CT26 tumor data, these results suggest that the addition of CXCL9 to an intratumoral IL-12 therapy augments anti-tumor immunity, including control of distant untreated lesions and lung metastases in multiple models.
Figure 5.

Intratumoral electroporation of pIL12 and pIL12-CXCL9 in a 4T1 model demonstrates significant tumor growth inhibition and decrease in metastatic lung nodules
(A) Schematic showing treatment regimen to assess tumor regression. 4T1 tumor-bearing mice were intratumorally electroporated on days 0, 4, and 7 with EV (80 μg), pIL12 (20 μg), or bicistronic plasmid encoding pIL12-CXCL9 (80 μg). (B) Tumor volumes of treated lesion were measured three times a week for regression (n = 10–15 animals per group). (C) 4T1 metastatic lung nodules were enumerated across the three treatment groups, with lungs being collected when tumor volumes reached 1,000 mm3 (n = 25–30 animals per group pooled from two experiments; statistical significance determined using two-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
Intratumoral electroporation of pIL12 and pCXCL9 improves anti-PD-1 therapy
Recent studies have demonstrated a critical role for CXCL9 in facilitating anti-PD-1 checkpoint therapeutic responses.19,20,34 To investigate this potential relationship, a CT26 tumor model was used to determine whether this increased intratumoral chemokine gradient, particularly in the context of IL-12, could enhance anti-PD-1 therapy. Systemic anti-PD-1 checkpoint inhibitor or an isotype control was combined with a single intratumoral electroporation of pIL12 with or without pCXCL9 (Figure 6A). Mice receiving anti-PD-1 therapy with either empty vector or pIL12 saw no increase in survival in this CT26 tumor model (Figure 6B). However, the addition of CXCL9 to the combination of IL-12 and checkpoint inhibition yielded robust systemic immunity allowing for a significant improvement in survival (median survival: 31 days, right graph; versus 21 days with IL-12 ± anti-PD-1, middle graph; versus 17 days with EV ± anti-PD-1, left graph; Figure 6B). Furthermore, all mice in the group treated with pIL12, pCXCL9, plus anti-PD-1 (Figure 6C, right pie chart) were either tumor free (28%, green) or had a partial response (72%, red; partial response defined as mice surviving beyond day 17). In contrast, the pIL12 plus pCXCL9 treatment group (Figure 6C, left pie chart) had no tumor-free mice, with only partial responders (79%, red) or no response (21% of mice not responding to treatment, tan). These results highlight the clinical potential of combining checkpoint therapy with intratumoral electroporation of IL-12 and CXCL9.
Figure 6.

Intratumoral electroporation of pIL12 and pCXCL9 improves anti-PD-1 response
(A) Schematic showing treatment regimen for combination therapy to assess tumor regression. CT26 tumor-bearing mice were intratumorally electroporated on day 0 with low-dose EV or pIL12 (1–2 μg) followed by 100 μg of either pCXCL9 or EV on days 4 and 7. Once a week for three weeks, each group received i.p. injections of either isotype control (Rat IgG2a) or anti-mouse PD-1 starting (150 μg) at day 0. Tumor volumes were measured three times a week for regression and survival studies. (B) Kaplan-Meir curve comparing the survival of the pIL12 + pCXCL9 + anti-PD-1 cohort (right) with that of pIL12 + pCXCL9 + isotype. ∗∗p < 0.001, log rank (Mantel-Cox) test. (C) Pie charts illustrating the percentage of mice receiving complete, partial, or non-response diagnosis between treatment cohorts. Differentiation between non-response and partial response was determined by the median survival in the untreated group (17 days).
Discussion
Previously published biomarker data from a phase 2 trial of intratumoral electroporation of plasmid IL-12 plus pembrolizumab in patients with advanced melanoma selected to not respond to anti-PD-1 checkpoint inhibitors demonstrated a significant increase in transcripts associated with a robust innate and adaptive immune response.31 Further interrogation of this biomarker data revealed that while there were no significant differences in activated CD8+ T cells in the periphery, clinical responses were closely tied to intratumoral CXCR3 expression. Relatedly, others have reported that the density of CXCR3 expressed on activated TIL was higher in patients who responded to anti-PD-1 therapy compared with those who did not, emphasizing the clinical relevance of this chemokine axis within the TME.34 Furthermore, a significant increase of CXCL9 protein in the post-treatment serum was observed in non-responding patients (Figure S1B), underscoring that a tumor-focused chemokine gradient is critical to its immune function, which other pre-clinical and clinical studies have similarly demonstrated.29,34 Given the pivotal role of an intratumoral chemokine gradient in recruitment of cytotoxic CD8+ T cell into tumors,27 we hypothesized that combining intratumoral pIL12 with a DNA-encoded, locally secreted chemokine would further augment this axis and “license” a robust anti-PD-1 response beyond the treated lesion. In this study, we provide evidence for the functional relevance of the CXCR3 chemokine system for IL-12 response and demonstrate that establishing or fortifying this chemokine gradient in the tumor enhances not only IL-12 efficacy but also checkpoint inhibitor response.
Several studies have demonstrated that intratumoral IL-12 treatment leads to the generation and expansion of antigen-specific CD8+ T cells.25,35,36 To better understand potential immune subsets responsible for an anti-tumor response, NK cell and T cell depletion studies were performed. In untreated lesions, consistent with previous IT-pIL12-EP literature,36 NK depletion partially abrogated this response against CT26 tumors (similar to empty-vector-treated mice), whereas depletion of CD8+ cells completely eliminated IL-12-driven anti-tumor responses, emphasizing the role of adaptive cellular immunity. In the treated lesion, depletion of either NK or CD8+ cells resulted in partial abrogation of tumor growth. One possible explanation for this finding is that local IL-12 in the treated tumor activates intratumoral CD4+ T cells to control early tumor growth, as reported previously37 however, this localized IL-12-effect is unable to effectively prime the required adaptive (CD8+ T cell) response to control the contralateral tumor. Primed T cells and NK cells express CXCR3, which has been shown to be critical for trafficking from the lymph node to sites of inflammation and tumor.38 We found that in the CT26 model intratumoral IL-12 led to the generation of CXCR3-positive lymphocytes in the lymph node, typical of effective T cell priming. Furthermore, by use of a bilateral-tumor model, IT-pIL12-EP was able to elicit an abscopal response, partly due to dissemination of CXCR3+ CD8+ effector T cells from the lymph node to the distant contralateral tumor, which was eliminated upon antibody-mediated blockade of CXCR3. The CXCR3-blocking antibody was administered systemically just before and after therapy (days −3, 0, and 3), suggesting that this CXCR3 chemokine axis is active early in the efferent response as newly primed T cells from the lymph node circulate to the distant tumor. However, a study with an elongated time course of CXCR3 blockade may provide a more thorough understanding of the kinetic requirements of this axis post-treatment. Interestingly, there was a lack of disruption to the IL-12 response in electroporated tumors with blockade of CXCR3. While CT26 tumors are known to be relatively immunogenic and can respond to electroporation of empty vector, this lack of primary (treated) tumor growth with antibody blockade was likely attributed to a CXCR3-independent immune subset triggered by IT-pIL12-EP.
IFN-γ induces CXCL9/10/11, which have been shown to limit tumor growth by promoting trafficking of cytotoxic CD8+ T cells into the tumor.23,24 Whereas IL-12 increased induction of IFN-γ,25 we found that in the CT26 tumor model CXCL9/10/11 transcripts remained unchanged compared to empty vector control (data not shown). Various tumor intrinsic (e.g., epigenetic silencing)39 and extrinsic factors (e.g., chemokine-cleaving proteases)40 limit the expression of chemokines to promote immune evasion. Although any of these mechanisms might be entangled in these colon carcinomas, the CT26 model, when coupled with the robust 4T-1 mammary carcinoma model, collectively serves as an effective system to investigate the therapeutic advantages of augmenting the chemokine axis.
Although there are numerous studies reporting anti-tumor potential of individual chemokines when expressed as transgenes in the tumor,41, 42, 43 their principal mode of action (i.e., trafficking) suggests that they might work better in conjunction with cytokines.28,29,44 While originally evaluating other similar chemokines, this study focused on the immunological impact of CXCL9 in combination with IL-12. CXCL9 not only offers the advantage of attracting CD8+ cytotoxic cells45 but is also stringently sequestered by cell surface glycosaminoglycans (GAGs), creating a steeper gradient between tissue and blood stream.46 In contrast, CXCL11 traffics both cytotoxic and immunosuppressive regulatory T cells.45 Interestingly, CXCL10 is cleaved by CD26 peptidases, resulting in shortened half-life,47 dampening its potential therapeutic potential, which may explain the poor expression of early attempts of a CXCL10 therapeutic.
Similarly to our recently published data in triple-negative breast cancer,48 treatment of CT26 tumors with pIL12 and pCXCL9 may reshape the immune TME to promote DC licensing (enrichment of genes associated with antigen presentation machinery) and potentially more efficient cross presentation and activation of naive CD8+ cytotoxic T cells (upregulation in CD28 and CD8 activation score). In addition, we observed significant improvement in abscopal anti-tumor responses and survival upon combination treatment compared with IL-12 alone. These data underscore the importance of a restored CXCL9 gradient to elicit a strong systemic immune response in tumors like CT26, where the IFN-γ-CXCL9 feedforward loop can be dysfunctional. However, it is likely that exogenously expressed CXCL9 will only be able to modulate finite waves of trafficking of IL-12-driven CTLs into these tumors and might have to cooperate with other chemokines like CCL5 in orchestrating further rounds of T cell infiltration. The expression of CCL5 has been shown to correlate with anti-tumor immune responses and improved patient survival in the context of high CXCL9 expression.49 Interestingly, pIL12- and pCXCL9-treated mice demonstrated 3-fold higher expression of CCL5 compared with empty-vector control (data not shown). Alternatively, in tumor types where IL-12 drives suboptimal chemokine expression via an intact IFN-γ-CXCL9 feedforward loop, intratumoral CXCL9 will most likely result in a more durable anti-tumor response.
Other studies have advocated an additional role of chemokine gradient in the tumor. Chow et al., demonstrated that the CXCR3-chemokine axis was dispensable for early T cell trafficking.19 The study highlighted the role of the chemokine axis in repositioning T cells near CD103 dendritic cells for in situ priming. It is possible that such a mechanism might be at play in our study. Intratumoral pCXCL9 electroporation leads to secretion of the chemokine in the TME, which might favor in situ priming of pre-existing T cells in the tumor to initiate IFN-γ-driven Th1 effector mechanisms.
Multiple studies have highlighted the role of this CXCR3/chemokine axis in augmenting anti-PD-1 responses. Relatedly, we have demonstrated that intratumoral IL-12 therapy can effectively combine with anti-PD-1 checkpoint inhibitors to remodel the TME, resulting in IFN-γ-dependent licensing of DCs (in situ vaccine) and subsequent systemic immunity.25,26,31,48 Wang et al. demonstrated that the lack of response to PD-1 inhibitors in this non- or partially responding CT26 tumor model50 correlated with a limited density of infiltrating leukocytes and a gene signature representative of reduced T cell presence and activation. These observations suggest that anti-PD-1 treatment in a CT26 model would greatly benefit from remodeling of the TME with an influx of actively proliferating antigen-specific CD8+ T cells. Here, we show that intratumoral electroporation of pCXCL9 significantly improves anti-PD-1 response in pIL12-treated mice. Interestingly, no synergy was observed between anti-PD-1 therapy and pIL12 treatment in this model; nor was there an obvious therapeutic benefit of combining CXCL9 and IL-12 in this experiment. We believe that this variable therapy may relate to the above-described lack of IFN-γ-CXCL9 feedforward loop, but it is also worth noting that this model was designed to use a subtherapeutic dose of IL-12 to better observe the CXCL9-mediated effects, particularly in combination with anti-PD-1. However, independent of any lack of additive cytotoxicity, this IL-12/CXCL9 combination can drive the PD-1 axis and enhance CPI activity, as seen in other models.51 Other studies have additionally shown that the therapeutic effect of anti-PD-1 blockade was partially diminished upon loss of CXCL9 but not CXCL10, indicating a central role of CXCL9 in mediating anti-PD-1 immunotherapy.19 However, whereas this study mainly focused on immunological effects of combining IL-12 and CXCL9, one cannot rule out direct angiostatic effects of CXCL9 on tumor growth.52 Further studies are required to assess their contribution toward anti-tumor immunity.
In summary, our results support that the CXCR3/CXCL9 chemokine axis is critical for IL-12 therapy and that temporal and spatial control of this axis via intratumoral expression of CXCL9 leads to a more productive immune response with potent anti-tumor effects. Furthermore, intratumoral electroporation of plasmid IL-12 with plasmid CXCL9 significantly improved the anti-PD-1 response, which when coupled with current IT-pIL12-EP plus pembrolizumab combination clinical studies provides a strong rationale for advancing this enhanced platform to the clinic.
Materials and methods
Mice, tumor cell line, contralateral model
Female Balb/c mice, 6–8 weeks of age (Jackson Laboratories, Bar Harbor, ME, USA) were housed in accordance with AAALAC guidelines. CT26 and 4T1 (CRL-2638 and CRL-2539; ATCC, Manassas, VA, USA) were cultured in RPMI 1640 medium with 2 mM L-glutamine, (Thermo Fisher Scientific, Waltman, MA, USA) with 10% fetal bovine serum (Thermo Fisher Scientific) and 100 U/mL penicillin-streptomycin (Thermo Fisher Scientific). B16-F10 (CRL-6475, ATCC) were cultured in DMEM with 10% fetal bovine serum and 100 U/mL penicillin-streptomycin. Cells were harvested with 0.25% trypsin-EDTA (Thermo Fisher Scientific) and re-suspended in Hank’s balanced salt solution. Anesthetized mice were subcutaneously injected in the right flank with 1 million cells in a total volume of 0.1 mL per injection site. For contralateral experiments assessing abscopal anti-tumor responses in distant untreated lesions, the left flank was also injected with 0.25 million cells. Tumor growth was monitored by digital caliper measurements. Tumor volume (VT) was calculated using the formula VT = (a2× b)/2, where a is smallest diameter and b is perpendicular diameter. Mice with tumors ranging from 75 to 100 mm3 (right flank) and 20 to 50 mm3 (left flank) were randomized and divided into treatment groups. Tumor volumes were measured twice weekly. Mice were euthanized when either of the tumors reached 1,000 mm3 or the total tumor burden reached 2,000 mm3. All animal experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee at Explora BioLabs (San Diego, CA, USA).
Plasmids
The mouse IL-12 plasmid (pIL12) has been previously described.33 Briefly, pIL12 is a DNA plasmid that encodes for both mouse IL-12p35 and IL-12p40, separated by a P2A segment, and driven by a single CMV promoter. The mouse CXCL9 plasmid (pCXCL9) was generated from a DNA geneblock, obtained from Integrated DNA Technologies, Inc. (Coralville, IA, USA), encoding for mouse CXCL9 with flanking 5′ EcoR1 and 3′ Kpn1 restriction sites and a Kozak consensus initiation site. The geneblock was digested with EcoR1/Not1 and ligated into pUMVC3 plasmid (Aldveron, Fargo, ND, USA). To generate mouse CXCL9-P2A-mCherry plasmid (pCXCL9), the mCXCL9 sequence lacking a stop codon was PCR amplified from the above-mentioned geneblock with 5′ Xba1 and 3′ Not1 sites. A second geneblock encoding P2A-mCherry was designed with flanking 5′ Not1 and 3′ BglII and digested. Then, the two products were ligated together into pUMVC3, which was digested with Xba1 and BglII. The mouse IL-12-CXCL9 plasmid (pIL12-CXCL9) is a DNA plasmid driven by a single CMV promoter and encodes for mouse IL-12p35, IL-12-p40, and CXCL9, with a P2A segment in between each element. To construct pIL12-CXCL9, the stop codon was first removed from the 3′ end of IL-12p40 segment in pIL12 using QuikChange mutagenesis (Agilent, Santa Clara, CA, USA). Next, a geneblock encoding P2A-mCXCL9 was designed with flanking Not1 and Xba1 restriction sites. Finally, the pIL12-ΔSTOP and P2A-mCXCL9 were both digested with Not1 and Xba1, gel extracted, and ligated together. All constructs were validated using restriction enzyme digests and DNA sequencing. Large-scale endotoxin-free preps were purchased from Genewiz (South Plainfield, NJ, USA) or Aldevron and resuspended in sterile 0.9% saline.
Intratumoral electroporation and antibody treatment
Mice were anesthetized with 3% isoflurane for treatment. Two treatment paradigms were used for the studies. The first treatment strategy involved electroporating tumors with 2 μg of pIL12 or pUMVC3 empty vector (EV) on day 0 (1st treatment) followed by 100 μg of either EV or pCXCL9 on day 4 (2nd treatment) and day 7 (3rd treatment). The second strategy involved electroporating tumors with 50 μg of either EV, pIL12, or pIL12-CXCL9 on days 0, 4, and 7. With both strategies, circular plasmid DNA, diluted to the desired concentration in sterile 0.9% saline, was injected centrally into primary tumors, immediately followed by electroporation using a low-voltage generator optimized for gene electrotransfer33 that delivers eight unidirectional pulses (400 V/cm, 10 ms per pulse) with two-needle electrode array. Mice were treated with antibodies as indicated: anti-mouse CXCR3 (clone CXCR3-173; Bio X Cell, West Lebanon, NH, USA) 100 μg on days −3, 0, and 3; anti-Mouse PD-1 (clone RMP1-14; InvivoGen, San Diego, CA, USA) 150 μg (days 0, 7, and 14).
In vitro transfection and HEK293 cells
HEK293 cells (ATCC) were seeded at 1 million cells per 10-cm dish in DMEM with 4.5 mg/mL D-glucose, 4 mM L-glutamine, and 1 mM sodium pyruvate (Thermo Fisher Scientific). After 18 h, when cells were 70–80% confluent, cell medium was exchanged with Opti-MEM reduced-serum medium (Thermo Fisher Scientific), and cells were transfected using TransIT-LT1 transfection reagent (Mirus Bio, Madison, WI, USA). For a 10-cm dish, 16 μL of TransIT-LT1 reagent was incubated with 400 μL of Opti-MEM for 5 min at room temperature. This mixture was then allowed to complex with 4 μg of plasmid DNA for 15 min at room temperature. Resulting TransIT-LT1: DNA complexes were then added dropwise to the cell cultures and incubated for 96 h. Afterward, conditioned media were harvested, spun down to remove any cell debris, aliquoted, and stored at −80°C. Quantifications were done using DuoSet ELISA kits (R&D Systems, Minneapolis, MN, USA) specific to respective protein products, and analyzed with a Cytation 3 Multi-Mode Reader (BioTek) measuring absorbance at 450 nm with 570 nm correction.
Flow cytometric analysis
Murine lymphocytes, splenocytes, and tumor cells were isolated as described above and resuspended in FACS buffer (PBS without Ca2+ and Mg2+ with 2% FBS and 1 mM EDTA) to create a single-cell suspension. Cells were subsequently either stained or incubated with Golgi Block (BD Biosciences, Franklin Lakes, NJ, USA) at 37°C for 3 h for intracellular staining. For Dextramer staining, 2 million cells/sample were stained with Fixable Far Red Viability Stain (Thermo Fisher Scientific) at 1:1,000 dilution at room temperature followed by AH1 Dextramer (Immudex, Copenhagen, DK) at 10 μL/sample at room temperature, and all surface markers were stained on ice for 30 min. The following antibodies were used for the study: CD19-APC-Cy7, CD49b-PE, CD4-BUV805, CD45-BV510, CD3-BV421, CD45-FITC (BD Biosciences), and CD8-FITC (MBL, Woburn, MA, USA). Intracellular staining with IFN-γ-PerCP-Cy5.5 (BD Biosciences) was performed using an eBioscience FOXP3 transcription factor staining kit (Thermo Fisher Scientific). Patient blood (approximately 60 mL) was collected in sodium heparin or CPT vacutainer tubes (BD Biosciences), shipped overnight from clinical sites to Oncosec Medical, and processed into PBMCs as previously described.53 Cells were resuspended in CryoStor 10 (BioLife Solutions) and frozen in liquid nitrogen until use. PBMCs were analyzed for immune-cell subsets by flow cytometry using the following antibodies: CD56-APC-H7, PD1-BV480, Ki-67-PerCP-Cy5.5 (BioLegend, San Diego, CA, USA), CD3-BUV805, CD4-BUV395, CD8-BUV496, CD14-APC-H7, and CD19-APC-H7 (BD Biosciences). Flow cytometric samples were acquired on a LSRFortessa X-20 flow cytometer (BD Biosciences), and data analysis was done using FlowJo software (Becton-Dickinson, Ashland, OR, USA).
Chemotaxis assay
Splenocytes were isolated by pressing spleens from OT1-GFP mice through 70-μm filters, followed by red blood cell lysis with ACK lysing buffer (Thermo Fisher Scientific), and washed with PBS without Ca2+ and Mg2+ (Thermo Fisher Scientific) with 2% FBS. Isolated splenocytes were resuspended in R10 medium (RPMI 1640, 10% FBS, 1 mM sodium pyruvate, 10 mM HEPES, 70 μM β-mercaptoethanol, and 23 μg/mL gentamicin) at 1–2 million/mL. Splenocytes were pulsed with SIINFEKL peptide (Biomatik, Wilmington, DE, USA) at 1 μg/mL for 24 h, after which, cells were washed and allowed to recover in fresh R10 medium for 72 h, splitting the culture every 24 h. Similarly, draining lymph nodes were harvested from CT26-bearing mice 4 days after IT-EP treatment. Lymph nodes were pressed through 70-μm filters and washed with PBS without Ca2+ and Mg2+ with 2% FBS. To abrogate CXCR3 involvement, isolated cells were pre-incubated with CXCR3-blocking antibody (Bio X Cell) at 100 μg/mL for 1 h at 4°C. Prior to chemotaxis evaluation, cells were washed and resuspended in Hank’s balanced salt solution (Thermo Fisher Scientific) with 0.1% protease-free bovine serum albumin (Sigma) at 5 million cells/mL. Boyden chambers consisted of 24-well tissue culture plates and 6.5-mm-diameter inserts with polycarbonate membranes containing 5-μm pores (Corning, Corning, NY, USA). Below the inserts, 600 μL of 250 ng/mL mouse CXCL9 was added; on top of the inserts, 100 μL of the cell suspension was added (500,000 cells). Boyden chambers were incubated at 37°C, 5% CO2 for 2 h. Afterward, inserts were removed, and any migrated cells in the wells were washed with PBS without Ca2+ and Mg2+, fixed with 2% paraformaldehyde for 30 min, washed again with PBS, resuspended in a final volume of 250 μL of PBS, and counted on a Guava easyCyte 12HT flow cytometer (Millipore Sigma, Burlington, MA, USA), gating on GFP+ cells (for OT1-GFP) or on a lymphocyte forward scatter (FSC) versus side scatter (SSC) profile.
RNA extraction and gene expression analysis
Tumors were harvested from mice and flash-frozen in liquid nitrogen. Tissues were homogenized in TRIzol (Thermo Fisher Scientific) using the gentleMACS Octo Dissociator (Miltenyi, San Diego, CA, USA). Total RNA was isolated using TRIzol-chloroform extraction, and contaminating DNA was removed with RNase-free DNase I (Thermo Fisher Scientific). Gene expression profiling was performed using NanoString technology and the nCounter gene expression codeset Mouse PanCancer IO360 (digital profiling of 770 genes) for analysis (NanoString, Seattle, WA, USA). For each hybridization reaction, 100 ng of total RNA was mixed with capture and reporter probe and incubated at 65°C for 16 h. The probe set-target RNA complexes from each reaction were immobilized and processed on nCounter Cartridges using an nCounter MAX prep station, and transcripts were quantified on the Digital Analyzer (GEN 2). The nCounter Advanced Analysis software was used for detailed analysis. For clinical samples, patient tumor biopsies were obtained at screen and on the first day of treatment cycle 2 (C2D1), fixed in 10% neutral-buffered formalin, and embedded in paraffin. Tissue curls cut from these biopsies were deparaffinized, and RNA was extracted using RecoverAll Total Nucleic Acid Kit (Thermo Fisher Scientific) according to the manufacturer’s specifications. Isolated samples were measured on a NanoDrop 2000c and stored at −86°C. Transcriptomic analysis was performed using NanoString nCounter technology with Human Immunology v2 Gene Expression Panels. Normalized counts of RNA transcripts were determined, and data are reported relative to the average of pre-treatment (screen) counts.
IL-12 functional reporter cell assay
HEK-Blue IL-12 cells (InvivoGen), designed to detect bioactive human or mouse IL-12, were used to quantify the functionality of plasmid-derived IL-12 produced by the pIL12 and pIL12-CXCL9 plasmids. The bioassay was performed as described by the manufacturer, using a 2-fold dilution series of IL-12 ranging from 0 to 25.6 ng/mL. Briefly, HEK-Blue IL-12 cells at 50,000 cells/180 μL/well were incubated with 20 μL of IL-12 for 14–16 h at 37°C and 5% CO2. Afterward, 20 μL of the resulting conditioned media were added to 180 μL of freshly made QUANTI-Blue Solution (InvivoGen). Colorimetric assay was incubated for 30 min at 37°C and analyzed with a Cytation 3 Multi-Mode Reader (BioTek, Winooski, VT, USA) measuring absorbance at 630 nm.
Combination clinical study trial design
We previously described a TIL assay that predicts response to anti-PD-1 monotherapy based on the frequency of CD8+ CD45+ TIL that are PD1hi CTLA-4hi.31 With this assay, patients predicted to not respond to anti-PD-1 were enrolled in this trial if <25% of their CD8+ TIL were PD-1hi CTLA-4hi at screen. Tavo, or tavokinogene telseplasmid, as described by Canton et al.,54 encodes for both human IL-12p35 and IL-12p40, separated by an internal ribosome entry site (IRES), and driven by a single CMV promoter. Tavo was injected into accessible lesions at a dose of 0.25 lesion volume at a concentration of 0.5 mg/mL in PBS. Immediately following injection, the tissue was electroporated with six pulses at 300-ms intervals using a field strength of 1,500 V/cm and a pulse width of 100 μs. Pembrolizumab was administered as a flat dose of 200 mg delivered intravenously. Patients were administered pembrolizumab on the first day of every 3-week cycle. IT-pIL12-EP was given on days 1, 5, and 8 of every odd treatment cycle (every 6 weeks on cycles 1, 3, 5, 7, etc.). Data were broken out by clinical response, defined as ORR by RECIST.
Statistical analysis
All data are expressed as mean ± SEM unless otherwise indicated. Ordinary one-way ANOVA (Dunnett’s multiple comparisons test), two-way ANOVA (Sidak’s multiple comparisons test), and unpaired Mann-Whitney t test were used when applicable to calculate significance between treatment groups. Log rank (Mantel-Cox test) was performed for survival studies.
Acknowledgments
We want to extend our deepest appreciation for the work done by the OncoSec in vivo technical staff, including Chandra Inglis, Garrett Arauz, Taylor Congdon, Jose Diaz, Stephanie Paz, Nygel Oglesby, and Tarsicio Juarez, and the in vitro technical staff, including Kurt Sakurada and Vincent Wu. Funding to support the research was provided by OncoSec Medical Incorporated. Patient samples were from a multi-center, phase 2, open-label, single-arm trial in adults (≥18 years old) with metastatic or unresectable melanoma with accessible lesions (registered at cancer.gov as NCT02493361).
Author contributions
J.Y.L. and B.N. designed and performed the experiments, analyzed the data, and wrote the manuscript. M.H., D.F., R.G., and J.S. performed experiments, analyzed data, and reviewed the manuscript. A.D. and A.A. provided patient material and reviewed the manuscript. D.A.C., E.B., J.Z., J.S., R.H., and L.S. contributed to patient sample processing, assay execution, and data analysis and reviewed the manuscript. A.M. and C.G.T. designed the experiments, interpreted, and evaluated the data, maintained oversight over the study, and wrote the manuscript.
Declaration of interests
A.D. receives research support from Merck, BMS, Genentech, Pfizer, Incyte, Checkmate, and OncoSec; is an advisory board member of Novartis, Merck, Genentech, and Pfizer; and owns shares in Neugogen and Trex. A.A. receives research support and is an advisory board member, consultant, shareholder, and honorarium recipient with OncoSec; is an advisory board member and stock shareholder for Valitor Biosciences, an advisory board member and honorarium recipient for Regeneron and Array, and receives research support from Acerta, Amgen, AstraZeneca, BMS, Dynavax, Genentech, Idera, Incyte, ISA, LOXO, Merck, Novartis, Sensei, and Tessa. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.04.005.
Supplemental information
References
- 1.Fridman W.H., Pagès F., Sautès-Fridman C., Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 2.Gajewski T.F., Corrales L., Williams J., Horton B., Sivan A., Spranger S. Cancer immunotherapy targets based on understanding the T cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv. Exp. Med. Biol. 2017;1036:19–31. doi: 10.1007/978-3-319-67577-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trujillo J.A., Sweis R.F., Bao R., Luke J.J. T cell–inflamed versus non-T cell–inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol. Res. 2018;6:990–1000. doi: 10.1158/2326-6066.cir-18-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim R., Emi M., Tanabe K., Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006;66:5527–5536. doi: 10.1158/0008-5472.can-05-4128. [DOI] [PubMed] [Google Scholar]
- 5.Lorenzo-Sanz L., Muñoz P. Tumor-infiltrating immunosuppressive cells in cancer-cell plasticity, tumor progression and therapy response. Cancer Microenviron. 2019;12:119–132. doi: 10.1007/s12307-019-00232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masucci M.T., Minopoli M., Carriero M.V. Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front. Oncol. 2019;9:1146. doi: 10.3389/fonc.2019.01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostrand-Rosenberg S., Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol. 2018;200:422–431. doi: 10.4049/jimmunol.1701019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paluskievicz C.M., Cao X., Abdi R., Zheng P., Liu Y., Bromberg J.S. T regulatory cells and priming the suppressive tumor microenvironment. Front. Immunol. 2019;10:2453. doi: 10.3389/fimmu.2019.02453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarvaria A., Madrigal J.A., Saudemont A. B cell regulation in cancer and anti-tumor immunity. Cell Mol. Immunol. 2017;14:662–674. doi: 10.1038/cmi.2017.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziani L., Chouaib S., Thiery J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front. Immunol. 2018;9:414. doi: 10.3389/fimmu.2018.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butcher E.C. Specificity of leukocyte-endothelial interactions and diapedesis: physiologic and therapeutic implications of an active decision process. Res. Immunol. 1993;144:695–698. doi: 10.1016/s0923-2494(93)80053-2. [DOI] [PubMed] [Google Scholar]
- 12.Viola A., Sarukhan A., Bronte V., Molon B. The pros and cons of chemokines in tumor immunology. Trends Immunol. 2012;33:496–504. doi: 10.1016/j.it.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Nagarsheth N., Wicha M.S., Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017;17:559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mikucki M.E., Fisher D.T., Matsuzaki J., Skitzki J.J., Gaulin N.B., Muhitch J.B., Ku A.W., Frelinger J.G., Odunsi K., Gajewski T.F., et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015;6:7458. doi: 10.1038/ncomms8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oghumu S., Varikuti S., Terrazas C., Kotov D., Nasser M.W., Powell C.A., Ganju R.K., Satoskar A.R. CXCR3 deficiency enhances tumor progression by promoting macrophage M2 polarization in a murine breast cancer model. Immunology. 2014;143:109–119. doi: 10.1111/imm.12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li K., Zhu Z., Luo J., Fang J., Zhou H., Hu M., Maskey N., Yang G. Impact of chemokine receptor CXCR3 on tumor-infiltrating lymphocyte recruitment associated with favorable prognosis in advanced gastric cancer. Int. J. Clin. Exp. Pathol. 2015;8:14725–14732. [PMC free article] [PubMed] [Google Scholar]
- 17.Mullins I.M., Slingluff C.L., Lee J.K., Garbee C.F., Shu J., Anderson S.G., Mayer M.E., Knaus W.A., Mullins D.W. CXC chemokine receptor 3 expression by activated CD8+ T cells is associated with survival in melanoma patients with stage III disease. Cancer Res. 2004;64:7697–7701. doi: 10.1158/0008-5472.can-04-2059. [DOI] [PubMed] [Google Scholar]
- 18.Reckamp K.L., Figlin R.A., Moldawer N., Pantuck A.J., Belldegrun A.S., Burdick M.D., Strieter R.M. Expression of CXCR3 on mononuclear cells and CXCR3 ligands in patients with metastatic renal cell carcinoma in response to systemic IL-2 therapy. J. Immunother. 2007;30:417–424. doi: 10.1097/cji.0b013e31802e089a. [DOI] [PubMed] [Google Scholar]
- 19.Chow M.T., Ozga A.J., Servis R.L., Frederick D.T., Lo J.A., Fisher D.E., Freeman G.J., Boland G.M., Luster A.D. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity. 2019;50:1498–1512.e5. doi: 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.House I.G., Savas P., Lai J., Chen A.X.Y., Oliver A.J., Teo Z.L., Todd K.L., Henderson M.A., Giuffrida L., Petley E.V., et al. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin. Cancer Res. 2020;26:487–504. doi: 10.1158/1078-0432.ccr-19-1868. [DOI] [PubMed] [Google Scholar]
- 21.Jansen C.S., Prokhnevska N., Master V.A., Sanda M.G., Carlisle J.W., Bilen M.A., Cardenas M., Wilkinson S., Lake R., Sowalsky A.G., et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. 2019;576:465–470. doi: 10.1038/s41586-019-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuo P.T., Zeng Z., Salim N., Mattarollo S., Wells J.W., Leggatt G.R. The role of CXCR3 and its chemokine ligands in skin disease and cancer. Front. Med. 2018;5:271. doi: 10.3389/fmed.2018.00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guirnalda P., Wood L., Goenka R., Crespo J., Paterson Y. Interferon γ-induced intratumoral expression of CXCL9 alters the local distribution of T cells following immunotherapy with Listeria monocytogenes. Oncoimmunology. 2013;2:e25752. doi: 10.4161/onci.25752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ayers M., Lunceford J., Nebozhyn M., Murphy E., Loboda A., Kaufman D.R., Albright A., Cheng J.D., Kang S.P., Shankaran V., et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/jci91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay A., Wright J., Shirley S., Canton D.A., Burkart C., Connolly R.J., Campbell J.S., Pierce R.H. Characterization of abscopal effects of intratumoral electroporation-mediated IL-12 gene therapy. Gene Ther. 2019;26:1–15. doi: 10.1038/s41434-018-0044-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garris C.S., Arlauckas S.P., Kohler R.H., Trefny M.P., Garren S., Piot C., Engblom C., Pfirschke C., Siwicki M., Gungabeesoon J., et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. 2018;49:1148–1161.e7. doi: 10.1016/j.immuni.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harlin H., Meng Y., Peterson A.C., Zha Y., Tretiakova M., Slingluff C., McKee M., Gajewski T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–3085. doi: 10.1158/0008-5472.can-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmer K., Hitt M., Emtage P., Gyorffy S., Gauldie J. Combined CXC chemokine and interleukin-12 gene transfer enhances antitumor immunity. Gene Ther. 2001;8:282–290. doi: 10.1038/sj.gt.3301386. [DOI] [PubMed] [Google Scholar]
- 29.Pan J., Burdick M.D., Belperio J.A., Xue Y.Y., Gerard C., Sharma S., Dubinett S.M., Strieter R.M. CXCR3/CXCR3 ligand biological Axis impairs RENCA tumor growth by a mechanism of immunoangiostasis. J. Immunol. 2006;176:1456–1464. doi: 10.4049/jimmunol.176.3.1456. [DOI] [PubMed] [Google Scholar]
- 30.Algazi A., Bhatia S., Agarwala S., Molina M., Lewis K., Faries M., Fong L., Levine L.P., Franco M., Oglesby A., et al. Intratumoral delivery of tavokinogene telseplasmid yields systemic immune responses in metastatic melanoma patients. Ann. Oncol. 2020;31:532–540. doi: 10.1016/j.annonc.2019.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Algazi A.P., Twitty C.G., Tsai K.K., Le M., Pierce R., Browning E., Hermiz R., Canton D.A., Bannavong D., Oglesby A., et al. Phase II trial of IL-12 plasmid transfection and PD-1 blockade in immunologically quiescent melanoma. Clin. Cancer Res. 2020;26:2827–2837. doi: 10.1158/1078-0432.ccr-19-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meiser A., Mueller A., Wise E.L., McDonagh E.M., Petit S.J., Saran N., Clark P.C., Williams T.J., Pease J.E. The chemokine receptor CXCR3 is degraded following internalization and is replenished at the cell surface by de novo synthesis of receptor. J. Immunol. 2008;180:6713–6724. doi: 10.4049/jimmunol.180.10.6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burkart C., Mukhopadhyay A., Shirley S.A., Connolly R.J., Wright J.H., Bahrami A., Campbell J.S., Pierce R.H., Canton D.A. Improving therapeutic efficacy of IL-12 intratumoral gene electrotransfer through novel plasmid design and modified parameters. Gene Ther. 2018;25:93–103. doi: 10.1038/s41434-018-0006-y. [DOI] [PubMed] [Google Scholar]
- 34.Han X., Wang Y., Sun J., Tan T., Cai X., Lin P., Tan Y., Zheng B., Wang B., Wang J., et al. Role of CXCR3 signaling in response to anti-PD-1 therapy. Ebiomedicine. 2019;48:169–177. doi: 10.1016/j.ebiom.2019.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greaney S.K., Algazi A.P., Tsai K.K., Takamura K.T., Chen L., Twitty C.G., Zhang L., Paciorek A., Pierce R.H., Le M.H., et al. Intratumoral plasmid IL12 electroporation therapy in patients with advanced melanoma induces systemic and intratumoral T-cell responses. Cancer Immunol. Res. 2020;8:246–254. doi: 10.1158/2326-6066.cir-19-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sin J.-I., Park J.-B., Lee I.H., Park D., Choi Y.S., Choe J., Celis E. Intratumoral electroporation of IL-12 cDNA eradicates established melanomas by Trp2180–188-specific CD8+ CTLs in a perforin/granzyme-mediated and IFN-γ-dependent manner: application of Trp2180–188 peptides. Cancer Immunol. Immunother. 2012;61:1671–1682. doi: 10.1007/s00262-012-1214-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tugues S., Burkhard S.H., Ohs I., Vrohlings M., Nussbaum K., vom Berg J., Kulig P., Becher B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015;22:237–246. doi: 10.1038/cdd.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Groom J.R., Luster A.D. CXCR3 in T cell function. Exp. Cell Res. 2011;317:620–631. doi: 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng H., Zhao W., Yan C., Watson C.C., Massengill M., Xie M., Massengill C., Noyes D.R., Martinez G.V., Afzal R., et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin. Cancer Res. 2016;22:4119–4132. doi: 10.1158/1078-0432.ccr-15-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bronger H., Magdolen V., Goettig P., Dreyer T. Proteolytic chemokine cleavage as a regulator of lymphocytic infiltration in solid tumors. Cancer Metastasis Rev. 2019;38:417–430. doi: 10.1007/s10555-019-09807-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feldman A.L., Friedl J., Lans T.E., Libutti S.K., Lorang D., Miller M.S., Turner E.M., Hewitt S.M., Alexander H.R. Retroviral gene transfer of interferon-inducible protein 10 inhibits growth of human melanoma xenografts. Int. J. Cancer. 2002;99:149–153. doi: 10.1002/ijc.10292. [DOI] [PubMed] [Google Scholar]
- 42.Francis L., Guo Z.S., Liu Z., Ravindranathan R., Urban J.A., Sathaiah M., Magge D., Kalinski P., Bartlett D.L. Modulation of chemokines in the tumor microenvironment enhances oncolytic virotherapy for colorectal cancer. Oncotarget. 2016;7:22174–22185. doi: 10.18632/oncotarget.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hensbergen P.J., Wijnands P.G.J.T.B., Schreurs M.W.J., Scheper R.J., Willemze R., Tensen C.P. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in vivo involving attraction of CD8+ T lymphocytes but not inhibition of angiogenesis. J. Immunother. 2005;28:343–351. doi: 10.1097/01.cji.0000165355.26795.27. [DOI] [PubMed] [Google Scholar]
- 44.Dilloo D., Bacon K., Holden W., Zhong W., Burdach S., Zlotnik A., Brenner M. Combined chemokine and cytokine gene transfer enhances antitumor immunity. Nat. Med. 1996;2:1090–1095. doi: 10.1038/nm1096-1090. [DOI] [PubMed] [Google Scholar]
- 45.Karin N., Wildbaum G. The role of chemokines in adjusting the balance between CD4+ effector T cell subsets and FOXp3-negative regulatory T cells. Int. Immunopharmacol. 2015;28:829–835. doi: 10.1016/j.intimp.2015.03.037. [DOI] [PubMed] [Google Scholar]
- 46.Liao F., Rabin R.L., Yannelli J.R., Koniaris L.G., Vanguri P., Farber J.M. Human Mig chemokine: biochemical and functional characterization. J. Exp. Med. 1995;182:1301–1314. doi: 10.1084/jem.182.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barreira da Silva R., Laird M.E., Yatim N., Fiette L., Ingersoll M.A., Albert M.L. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat. Immunol. 2015;16:850–858. doi: 10.1038/ni.3201. [DOI] [PubMed] [Google Scholar]
- 48.Telli M.L., Nagata H., Wapnir I., Acharya C.R., Zablotsky K., Fox B.A., Bifulco C.B., Jensen S.M., Ballesteros-Merino C., Le M.H., et al. Intratumoral plasmid IL12 expands CD8+ T cells and induces a CXCR3 gene signature in triple-negative breast tumors that sensitizes patients to anti–PD-1 therapy. Clin. Cancer Res. 2021;27:2481–2493. doi: 10.1158/1078-0432.ccr-20-3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dangaj D., Bruand M., Grimm A.J., Ronet C., Barras D., Duttagupta P.A., Lanitis E., Duraiswamy J., Tanyi J.L., Benencia F., et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell. 2019;35:885–900.e10. doi: 10.1016/j.ccell.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang S., Campos J., Gallotta M., Gong M., Crain C., Naik E., Coffman R.L., Guiducci C. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc. Natl. Acad. Sci. 2016;113:E7240–E7249. doi: 10.1073/pnas.1608555113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu C., Zhang Y., Rolfe P.A., Hernández V.M., Guzman W., Kradjian G., Marelli B., Qin G., Qi J., Wang H., et al. Combination therapy with NHS-muIL12 and avelumab (anti-PD-L1) enhances antitumor efficacy in preclinical cancer models. Clin. Cancer Res. 2017;23:5869–5880. doi: 10.1158/1078-0432.ccr-17-0483. [DOI] [PubMed] [Google Scholar]
- 52.Strieter R.M., Polverini P.J., Kunkel S.L., Arenberg D.A., Burdick M.D., Kasper J., Dzuiba J., Van Damme J., Walz A., Marriott D., et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis (∗) J. Biol. Chem. 1995;270:27348–27357. doi: 10.1074/jbc.270.45.27348. [DOI] [PubMed] [Google Scholar]
- 53.Bhatia S., Longino N.V., Miller N.J., Kulikauskas R., Iyer J.G., Ibrani D., Blom A., Byrd D.R., Parvathaneni U., Twitty C.G., et al. Intratumoral delivery of plasmid IL12 via electroporation leads to regression of injected and noninjected tumors in merkel cell carcinoma. Clin. Cancer Res. 2020;26:598–607. doi: 10.1158/1078-0432.ccr-19-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Canton D.A., Shirley S., Wright J., Connolly R., Burkart C., Mukhopadhyay A., Twitty C., Qattan K.E., Campbell J.S., Le M.H., et al. Melanoma treatment with intratumoral electroporation of tavokinogene telseplasmid (pIL-12, tavokinogene telseplasmid) Immunotherapy. 2017;9:1309–1321. doi: 10.2217/imt-2017-0096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.