

Abstract
Extracellular cold-inducible RNA-binding protein (eCIRP) is a damage-associated molecular pattern (DAMP). Intercellular adhesion molecule-1 (ICAM-1) expressing neutrophils produce excessive amounts of neutrophil extracellular traps (NETs). We reveal that eCIRP generates ICAM-1+ neutrophils through triggering receptor expressed on myeloid cells-1 (TREM-1) and the ICAM-1+ neutrophils involve Rho GTPase to promote NETosis. Treatment of BMDN with rmCIRP increased the frequency of ICAM-1+ BMDN, while rmCIRP-treated TREM-1−/− BMDN or pretreatment of BMDN with TREM-1 inhibitor LP17 significantly decreased the frequency of ICAM-1+ neutrophils. The frequencies of ICAM-1+ neutrophils in blood and lungs were markedly decreased in rmCIRP-injected mice or septic mice treated with LP17. Coculture of ICAM-1−/− neutrophils or wild-type (WT) neutrophils with WT macrophages in the presence of a peptidylarginine deiminase 4 (PAD4) inhibitor reduced TNF-α and IL-6 compared to WT neutrophils treated with rmCIRP. Treatment of ICAM-1−/− neutrophils with rmCIRP resulted in reduced quantities of NETs compared to WT rmCIRP-treated neutrophils. Treatment of BMDN with rmCIRP-induced Rho activation, while blockade of ICAM-1 significantly decreased Rho activation. Inhibition of Rho significantly decreased rmCIRP-induced NET formation in BMDN. TREM-1 plays a critical role in the eCIRP-mediated increase of ICAM-1 expression in neutrophils, leading to the increased NET formation via Rho activation to exaggerate inflammation.
Keywords: eCIRP, ICAM-1, NETs, sepsis, TREM-1
1 |. INTRODUCTION
Sepsis is a life-threatening inflammatory disorder caused by a dysregulated host immune response to an infection.1 Its complex pathophysiology is still poorly understood. Neutrophils are the most abundant human blood leukocyte. Although these cells are fundamental in mounting effective immune responses against invading pathogens, they can also cause tissue damage that leads to lasting functional impairment.2,3 Previously, neutrophils were thought to be a single short-lived, homogeneous, and terminally differentiated cell population. Recent studies, however, have challenged the neutrophil homogeneity paradigm by identifying various subsets of neutrophils that differ both in surface protein expression and in function.4–6
One of the effector functions of neutrophils is the expulsion of neutrophil extracellular traps (NETs). NETs consist of chromatin containing not only DNA and histones, which act as damage-associated molecular patterns (DAMPs), but also enzymes such as myeloperoxidase (MPO) and neutrophil elastase (NE).2,7 NETs trap and kill bacteria, but excessive and uncontrolled release of NETs can exacerbate inflammation and cause tissue damage.2,7–9 We have recently described a unique neutrophil subpopulation that has increased NET formation and is characterized by the surface expression of intercellular adhesion molecule-1 (ICAM-1),10 an integrin known to be expressed on the surface of endothelial cells and epithelial cells.11 ICAM-1 is a ligand for lymphocyte function-associated antigen-1 (LFA-1), a receptor found on leukocytes.12 ICAM-1 initiates outside-in signaling that augments endothelial inflammatory responses, facilitates leukocyte transmigration, and increases vascular leak.12,13 The signaling downstream of ICAM-1 involves Rho GTPases, intracellular calcium (Ca2+), actin polymerization, and Src family kinases.13–17 Rho GTPases (Rho, Rac, and Cdc42) represent a family of small GTP-binding proteins involved in cell cytoskeleton organization, migration, transcription, and proliferation.18 During NETosis, neutrophils undergo dynamic alterations of their cellular morphology.19 We recently identified that ICAM-1+ neutrophils produce more NETs than ICAM-1− neutrophils.10 However, it remains unknown whether ICAM-1-expressing neutrophils can utilize Rho GTPases to promote NET formation.
Cold-inducible RNA-binding protein (CIRP), a 19-kDa RNA chaperone, has been shown to be released outside the cells (known as extracellular CIRP or eCIRP) during sepsis, hemorrhagic shock, and ischemia-reperfusion (I/R) injury,20–22 where it acts as a DAMP to fuel inflammation. CIRP is expressed in macrophages and other cells and can be released outside these cells through passive release during necrotic cell death, or by active secretion by lysosomal exocytosis pathway during inflammation.20,21 We have recently discovered that eCIRP can induce NET-forming ICAM-1+ neutrophils.10 However, the precise molecular mechanism of NET-forming ICAM-1+ neutrophil formation and their activation by eCIRP are still poorly understood. eCIRP has been shown to activate macrophages via binding to Toll-like receptor 4 (TLR4)20 and, more recently, by recognizing triggering receptor expressed on myeloid cells-1 (TREM-1).23 TREM-1 is an innate immune receptor expressed primarily on neutrophils and macrophages24 whose activation leads to phosphorylation of DNAX-activating protein of 12 kDa (DAP12) and spleen tyrosine kinase (Syk), resulting in the production of cytokines and chemokines.25 Three other molecules including high mobility group box 1 (HMGB1), extracellular actin, and peptidoglycan recognition protein 1 (PGLYRP1) have been proposed as TREM-1 agonists.26–28 Furthermore, TREM-1 and TLR4 act synergistically to promote inflammation.24,29
We therefore hypothesized that eCIRP induces NET-forming ICAM-1+ neutrophils via the activation of TREM-1 on the surface of neutrophils. Indeed, in this article, we demonstrate that eCIRP activates TREM-1 on neutrophils, inducing the surface expression of ICAM-1 and promoting NET formation. We establish that TREM-1 induces NET-forming ICAM-1+ neutrophils both after eCIRP injection and during a preclinical model of sepsis. Furthermore, we identify a novel pathway of NET formation through ICAM-1-mediated Rho activation. Finally, using a pharmacologic inhibitor of TREM-1, LP17,30 we show the decreased number of this subset of neutrophils in the blood and lungs during sepsis, thus directing a novel therapeutic avenue in sepsis.
2 |. MATERIALS AND METHODS
2.1 |. Mice
Male 8–12 week-old wild-type (WT) C57BL/6 mice were purchased from Charles River (Wilmington, MA). TREM-1−/− mice (Trem1tm1(KOMP)Vlcg) were generated by the National Institutes of Health Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository University of California, Davis, CA. ICAM-1−/− mice (B6.129S4-Icam1tm1Jcgr/J) were purchased from the Jackson Laboratory (Bar Harbor, ME). All experiments were performed in accordance with the guidelines for the use of experimental animals by National Institutes of Health and were approved by our Institutional Animal Care and Use Committees (IACUC).
2.2 |. Reagents, peptides, and antibodies
EasySep mouse neutrophil enrichment kit (Cat. No.: 19762; STEMCELL, Vancouver, BC, Canada), EasySep mouse monocyte enrichment kit (Cat. No.: 19761; STEMCELL), Ca++ Mg++ free Hank’s balanced salt solution (HBSS) (Corning; Corning, NY), collagenase I (Worthington Biochemical, Lakewood, NJ), red blood cell (RBC) lysing buffer (BD Biosciences, San Jose, CA), LP17 (LQVTDSGLYRCVIYHPP), and LP17 scramble (TDSRCVIGLYHPPLQVY) peptides from GenScript (Piscataway, NJ), protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN), protein A/G plus agarose beads (Pierce Classic IP Kit, Thermo Fischer Scientific, Waltham, MA), peptidylarginine deiminase 4 (PAD4) inhibitor CI-amidine (Cat. No.: 506282, EMD Millipore, Burlington, MA), Rho kinase inhibitor Y-27632 (Cat. No.: ab120129, Cambridge, MA), active Rho detection kit (Cat. No.: 8820, Cell Signaling Technology, Danvers, MA), TNF-α (Cat. No.: 558538), and IL-6 (Cat. No.: 555240) ELISA kits (BD Bioscience), NETs assay kit (Item No.: 601010; Cayman Chemical, Ann Arbor, MI). APC-rat anti-mouse Ly-6G Ab (clone 1A8; Biolegend, San Diego, CA), anti-TREM-1 Ab (clone: 174021, R&D systems, Minneapolis, MN), anti-phosphotyrosine Ab (Cat. No.: 05–321; clone: 4G10; EMD Millipore), anti-DAP12 Ab (Cell Signaling Technology), anti-pSyk Ab (Abcam), anti-Syk Ab (Cell Signaling Technology), anti-β-actin Ab (Sigma-Aldrich, St Louis, MO), anti-ICAM-1 Ab (clone: 3E2, BD Biosciences, San Jose, CA), anti-MPO Ab (clone: mAb 8F4; Hycult Biotech, Uden, The Netherlands), anti-NE Ab (Biotin, Cat. No.: ab 79962, Abcam), anti-DNA-POD Ab (cell death detection kit, Cat. No.: 11 544 675 001, Roche Diagnostics, GmbH, Mannheim, Germany), FITC-mouse anti-MPO Ab (clone: 2D4; Abcam), rabbit anti-histone H3 (CitH3) Ab (Cat. No 5103: Abcam), PE-donkey anti-rabbit IgG (clone: Poly4064; Biolegend), rabbit polyclonal IgG (clone: Poly29108, Biolegend), and FITC-mouse IgG1 (B11/5, ab91356, Abcam).
2.3 |. Isolation and enrichment of bone marrow-derived neutrophils (BMDN)
Mice were euthanized and the femurs and the tibias were isolated and transected. Marrow contents were removed by flushing with RPMI 1640 medium and filtered through a 70-μm cell strainer. Cells were enriched by negative selection using EasySep mouse neutrophil enrichment kit. The purity of the sorted neutrophils was assessed by labeling the cells with Ly6G Ab using a BD LSRII flow cytometer (BD Biosciences, San Jose, CA).
2.4 |. Isolation of cells from lung tissues
Lungs were perfused with 30 mL normal saline through right ventricle of the heart under 2% of isoflurane inhalation anesthesia. Lung tissues were harvested from mice and filled with Ca++ and Mg++ free HBSS. Lungs were sliced into small pieces by sterile surgical blade and were digested in HBSS supplemented with 100 U/mL of collagenase I for 30 minutes at 37°C with periodic shaking. The resultant fragments were meshed by a 70 μm cell strainer and a 10 mL syringe plunger and washed with HBSS. The remaining RBCs were lysed with lysing buffer. The numbers of isolated lung cells were counted using a microscope (Eclipse TS100; Nikon, Tokyo, Japan).
2.5 |. Assessment of TREM-1 expression of BMDN
Recombinant murine (rm) CIRP was prepared in-house as described previously.20 A total of 106 BMDN were stimulated with rmCIRP (1 μg/mL) in RPMI medium for different lengths of time. Cells were then stained with antibodies (Ab) to TREM-1 and Ly6G and assessed by flow cytometry.
2.6 |. Assessment of the phosphorylation of DAP12 and Syk
The phosphorylation of DAP12 in BMDN was assessed by anti-phosphotyrosine Ab after immunoprecipitation with anti-DAP12 Ab. Briefly, 5 × 106 BMDN were stimulated with rmCIRP (1 μg/mL) for 5 minutes and the proteins were extracted using a buffer containing 25 mM Tris, 0.15 M NaCl, 1 mM EDTA, 1% NP-40, 5% glycerol, 2 mM Na3VO4, and protease inhibitor cocktail, pH 7.4. The extracted proteins were immunoprecipitated with anti-DAP12 Ab and protein A/G plus agarose beads, and the eluted samples were subjected to Western blotting. Finally, the blots were detected with anti-phosphotyrosine and anti-DAP12 Abs. For the assessment of the phosphorylation of Syk, 106 BMDN were stimulated with rmCIRP (1 μg/mL) for 5 minutes and the proteins were extracted using the preceding buffer. Extracted proteins were subjected to Western blotting and the blots were reacted with pSyk, Syk, and β-actin primary Abs. The blots were reacted with fluorescent-labeled secondary Abs (Li-Cor Biosciences, Lincoln, NE) and detection was done using an Odyssey FC Dual-Mode Imaging system (Li-Cor Biosciences). The densitometry intensities of the bands were measured using Image Studio Ver 5.2 (Li-Cor Biosciences).
2.7 |. Assessment of ICAM-1 expression of BMDN with LP17 treatment
A total of 106 BMDN were pretreated with LP17 (80 μg/mL) or scramble peptide (80 μg/mL) for 30 minutes followed by the stimulation of rmCIRP (1 μg/mL) for 4 hours. Cells were stained with anti-ICAM-1 Ab and assessed by flow cytometry after gating with anti-Ly6G Ab.
2.8 |. In vivo rmCIRP and LP17 injection
Mice were subjected to intraperitoneal injection (ip) of rmCIRP (5 mg/kg) with/without concomitant ip administration of LP17 (5 mg/kg). After 4 hours, the lungs and blood were harvested. The ICAM-1 expression of the neutrophils were assessed using flow cytometry after gating with anti-Ly6G Ab.
2.9 |. Mouse model of sepsis
Cecal ligation and puncture (CLP) was performed to induce sepsis.31 The cecum was exposed through a midline incision, ligated with a 4-0 silk suture 1 cm proximal from its distal extremity, and punctured twice with a 22-gauge needle. The wound was closed in layers. Mice were allocated to treatment or vehicle group. Treatment mice received an intraperitoneal injection of 5 mg/kg body weight (BW) LP17 at the time of abdominal closure. Vehicle groups received an equivalent volume of phosphate-buffered saline (PBS). Sham animals underwent a laparotomy without cecal ligation or puncture. After closure, the mice received a subcutaneous injection of 1 mL of normal saline to avoid surgery-induced dehydration. After 4 hours, the lungs and blood were harvested. The ICAM-1 expression of the neutrophils were assessed using flow cytometry after gating with anti-Ly6G Ab. Similarly, CLP was used to induce sepsis in ICAM-1−/− mice and after 4 hours of CLP, lungs were harvested for single cell staining with anti-Ly6G, -MPO, and -citH3 Abs to detect NETs in the neutrophils.
2.10 |. Coculture of neutrophils with macrophages
BMDN (106/mL) were stimulated with rmCIRP (1 μg/mL) for 4 hours and stained with anti-ICAM-1 Ab. ICAM-1+ and ICAM-1− BMDN were sorted by using a BD FACSAria cell sorter (BD Biosciences). A total of 5 × 104 ICAM-1+ or ICAM-1− neutrophils were cocultured with 1.5 × 105 peritoneal (PerC) macrophages isolated by magnetic sorting (STEMCELL) and, after 20 hours of coculture, TNF-α, and IL-6 levels in the culture supernatants were assessed by ELISA (BD Biosciences). BMDN (5 × 104) isolated from WT and ICAM-1−/− mice were cultured separately with PerC macrophages (1.5 × 105). After 20 hours of stimulation with rmCIRP (1 μg/mL) TNF-α and IL-6 levels in the culture supernatants were assessed by ELISA. WT BMDN (5 × 104) cocultured with PerC macrophages (1.5 × 105) were simultaneously treated with PAD4 inhibitor CI-amidine at a dose of 5 μM and rmCIRP (1 μg/mL). After 20 hours of stimulation with rmCIRP, TNF-α, and IL-6 levels in the culture supernatants were assessed by ELISA.
2.11 |. NETs assay by ELISA and flow cytometry
A total of 106 BMDN isolated from WT or ICAM-1−/− mice were stimulated with rmCIRP (5 μg/mL) for 4 hours at 37°C in a 6-well culture plate in 5% CO2 humidified incubator. After 4 hours of stimulation with rmCIRP, media containing the floating BMDN was gently aspirated and discarded, leaving only an adherent layer of NETs and neutrophils. 1.5 mL of cold PBS without Ca++ and Mg++ was used to wash the bottom of each well in order to lift off all adherent materials. The suspension containing neutrophils and NETs was then collected in a 1.5 mL micro-centrifuge tube and centrifuged for 10 minutes at 300 g at 4°C. After centrifugation, neutrophils and any remaining cells were pelleted at the bottom, leaving a cell-free NET-rich supernatant. This NET-rich supernatant was then collected into a 1.5 mL micro-centrifuge tubes, and centrifuged at 18 000 g for an additional 10 minutes at 4°C. The supernatant was discarded and the resulting pellet containing crude NETs extract was suspended in 100 μL of cold PBS. Crude NETs extracts isolated above were sonicated, diluted in PBS, and 100 μL of each sample was loaded into anti-MPO Ab or anti-NE Ab coated plates. For generating standard curve DNA concentration in one of the rmCIRP-treated samples was measured, serially diluted and loaded in the anti-MPO Ab- or anti-NE Ab-coated ELISA plates. Plate was incubated at room temperature for 2 hours. After that the plate was washed with PBS, reacted with anti-DNA-POD Ab and subsequently the optical density was recorded at 405 nm after adding TMB substrate to each well. Besides these in-house NETs ELISA tests, NETs in context of its NEDNA contents were assessed using NETs assay kit (Cayman Chemicals) following the manufacturer’s protocol. To assess the effect of TREM-1 inhibition on NET formation, WT BMDN were pretreated with LP17 (80 μg/mL) or scramble peptide (80 μg/mL) for 30 minutes and stimulated with rmCIRP (5 μg/mL) for 4 hours. NETs (NE-DNA) contents were assessed using NETosis assay kit.
NETs were detected by flow cytometry as described previously.32,33 A total of 106 BMDN isolated from WT or ICAM-1−/− mice were stimulated with rmCIRP (5 μg/mL) for 4 hours at 37°C in a 5% CO2 humidified incubator. After 4 hours of stimulation with rmCIRP, the neutrophils were collected in flow cytometry tubes and surface-stained with APC-rat anti-mouse Ly-6G Ab, FITC-mouse MPO Ab, and rabbit anti-histone H3 (CitH3) Ab followed by staining with PE-donkey anti-rabbit IgG. To avoid staining of intracellular chromatin and enzymes, cells were not permeabilized. More than 10 000 events were acquired using a BD LSR Fortessa Flow Cytometry Analyzer (BD Biosciences) and the data were analyzed by FlowJo software (Tree Star, Ashland, OR).
2.12 |. Assessment of NETs in lung neutrophils by flow cytometry
NET contents in the lungs were assessed by flow cytometry.33 Briefly, single-cell suspensions were fixed with 2% of paraformaldehyde for 20 minutes at room temperature, blocked for 30 minutes with 2% of bovine serum albumin (BSA) in PBS at 37°C. Cells were surface-stained with APC-rat anti-mouse Ly-6G Ab, FITC-mouse anti-MPO Ab, and rabbit anti-histone H3 (CitH3) Ab followed by staining with PE-donkey anti-rabbit IgG. To avoid staining of intracellular chromatin and enzymes, cells were not permeabilized. More than 10 000 events were acquired using a BD LSR Fortessa Flow Cytometry Analyzer (BD Biosciences) and the data were analyzed by FlowJo software (Tree Star, Ashland, OR). In the flow cytometry dot plots, at first the cells were gated based on their size and intracellular granularity by forward and side scatters, respectively. Next, neutrophils were gated based on Ly6G positive staining. In the Ly6G positive population, we then gated the cells based on surface/extracellular MPO and citH3 staining. We chose quadra/rectangular gate to select MPO+citH3+ in Ly6G+ neutrophils to define the NETs+ neutrophils. Purified rabbit polyclonal IgG and FITC-mouse IgG1 were used as the isotype controls.
2.13 |. Active Rho assay
Active Rho (GTP-bound) in PBS- or rmCIRP-treated BMDN was detected by GTP-bound GTPase pull-down process using the active Rho detection kit. BMDN (2.5 × 106) were stimulated with PBS or rmCIRP (5 μg/mL) for 120 minutes. After stimulating the BMDN with rmCIRP, cells were lysed in lysis buffer and ~500 μg of protein was loaded in spin cup containing GST-Rhotekin-RBD fusion protein captured by the glutathione resin. Spin cups were centrifuged at 6000 g for 30 seconds, followed by the washing step using wash buffer. Samples were eluted in 50 μL elusion buffer. Western blot analysis of the eluted sample was performed using a rabbit anti-Rho Ab (1:667 dilution) in 10 mL primary antibody dilution buffer. Anti-rabbit IgG was used as a secondary Ab. A fraction of each lysate was retained to determine the relative level of Rho (total Rho) by Western blot using rabbit anti-Rho Ab as primary and anti-rabbit IgG as secondary Abs, followed by the detection by Odyssey FC Dual-Mode Imaging system (Li-Cor Biosciences). The densitometry intensities of the bands we measured by using Image Studio Ver 5.2 (Li-Cor Biosciences).
2.14 |. Statistical analysis
Data represented in the figures are expressed as mean ± SE or SD. ANOVA was used for one-way comparison among multiple groups and the significance was determined by the Student-Newman-Keuls (SNK) test or the Tukey method, as appropriate. The paired Student t test was applied for two-group comparisons. Significance was considered for P ≤ .05 between study groups. Data analyses were carried out using GraphPad Prism graphing and statistical software (GraphPad Software, San Diego, CA).
3 |. RESULTS
3.1 |. eCIRP induces the surface expression of TREM-1 and activates TREM-1 downstream effectors in neutrophils
We first interrogated whether eCIRP could induce the expression of TREM-1 on the surface of neutrophils. At baseline, 18% of the BMDN expressed TREM-1 on their surface. After exposure to rmCIRP, BMDN significantly increased the expression of TREM-1 in a time-dependent manner, up to 46% at 4 hours after exposure to rmCIRP (Figure 1A,B). Next, we determined whether rmCIRP can activate TREM-1’s downstream molecules DAP12 and Syk in neutrophils. We found that BMDN exposed to rmCIRP had increased tyrosine phosphorylation of both DAP12 and Syk (Figure 1C,D). These data indicate that eCIRP induces TREM-1 surface expression and activates TREM-1’s downstream molecules in neutrophils.
FIGURE 1.
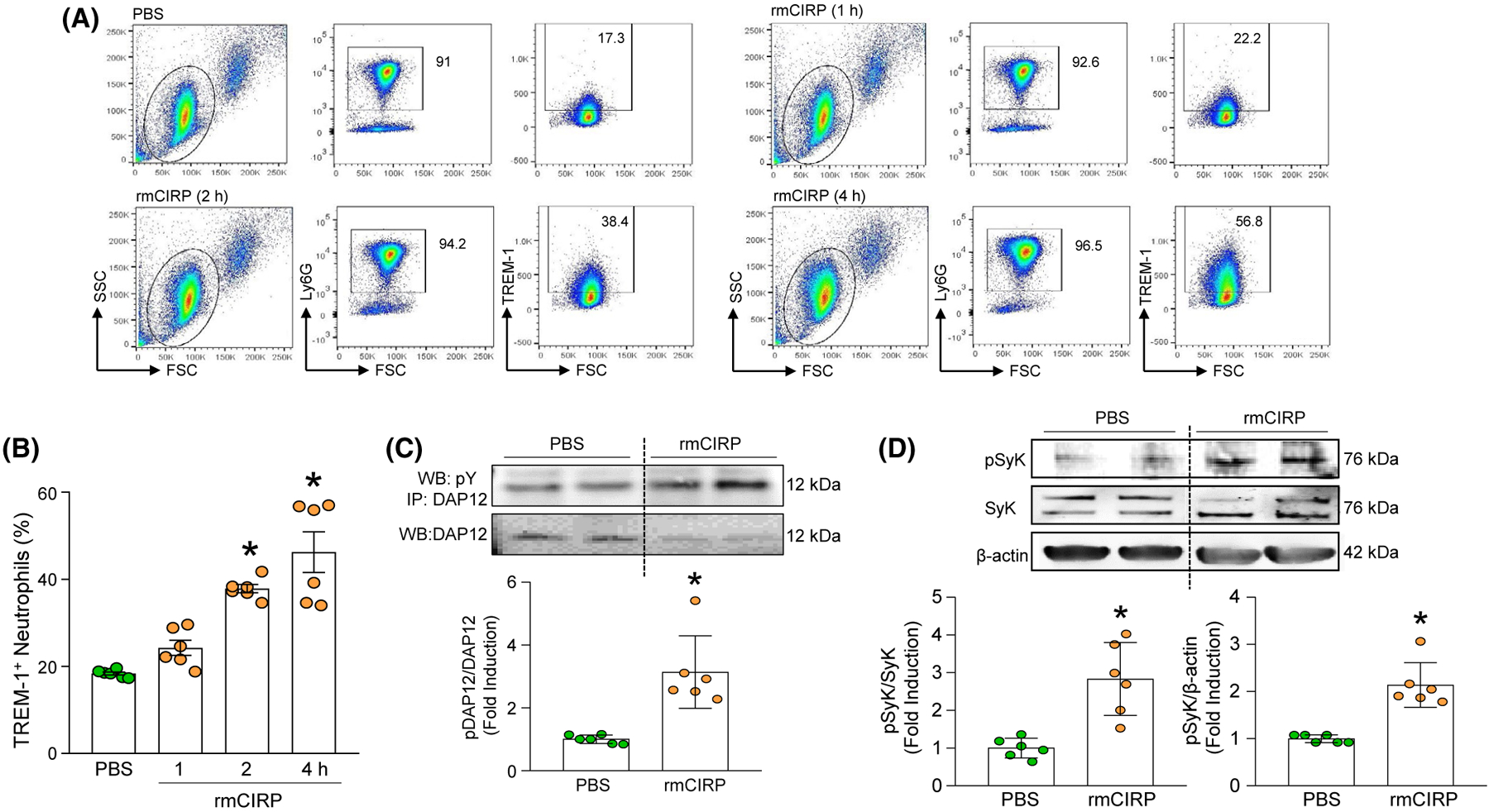
eCIRP induces TREM-1 expression and DAP12 and Syk phosphorylation in neutrophils. A, B, A total of 1 × 106 BMDN/mL were stimulated with PBS or rmCIRP (1 μg/mL) at various time points. After rmCIRP stimulation, the cells were washed with PBS and stained with Alexa Fluor 488-Ly6G and APC-TREM-1 Ab. The frequencies of TREM-1 expressing cells in LY6G gated population were assessed by flow cytometry. Experiments were repeated at least three times. Data are expressed as means ± SE (n = 6 samples/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs PBS-treated group). C, BMDN (5 × 106 cells/mL) were stimulated with PBS or 1 μg/mL of rmCIRP and after 5 minutes of rmCIRP stimulation, the neutrophils were lysed and total proteins were extracted. Equal amount (~200 μg) of protein of each sample was immunoprecipitated with anti-DAP12 Ab, followed by western blotting using anti-phospho Tyr (4G10) Ab. Representative western blots for phosphotyrosine (4G10) and DAP12 are shown. Each blot was quantified by densitometric analysis. Phosphotyrosine (pDAP12) expression in each sample was normalized to total DAP12 expression and the mean values of PBS-treated groups were standardized as one for comparison. Experiments were repeated at least three times. Data are expressed as means ± SD (n = 6 samples/group). The groups were compared by paired t test (*P < .05 vs PBS-treated group). D, BMDN (1 × 106 cells/mL) were stimulated with PBS or 1 μg/mL of rmCIRP and after 20 minutes of rmCIRP stimulation, neutrophils were lysed and total proteins were extracted to perform western blotting using anti-phospho Syk Ab. The blot was stripped and incubated with anti-Syk or anti-β-actin Abs to serve as control. Representative western blots for pSyk, Syk, and β-actin are shown. Each blot was quantified by densitometric analysis. pSyk expression in each sample was normalized to Syk or β-actin expression and the mean values of PBS-treated groups were standardized as one for comparison. Experiments were repeated at least three times. Data are expressed as means ± SD (n = 6 samples/group). The groups were compared by paired t test (*P < .05 vs PBS-treated group)
3.2 |. TREM-1 mediates the eCIRP induction of ICAM-1+ neutrophils in vitro
To determine whether eCIRP induces ICAM-1+ neutrophils via TREM-1, we evaluated the induction of surface ICAM-1 in BMDN from WT and TREM-1−/− mice exposed to rmCIRP. In BMDN from WT mice, rmCIRP significantly increased the surface expression of ICAM-1 by 6.5-fold. In BMDN from TREM-1−/− mice, however, rmCIRP induction of ICAM-1 significantly decreased by relative 27.6%, compared to rmCIRP-treated WT BDMN (Figure 2A,C). To further determine whether eCIRP induces ICAM-1+ neutrophils via TREM-1, we exposed BMDN from WT mice to rmCIRP in the presence or absence of LP17, a competitive inhibitor of TREM-1.30 As expected, exposure to rmCIRP significantly induced BMDN’s surface expression of ICAM-1. The addition of LP17, however, significantly attenuated eCIRP induction of ICAM-1+ BMDN by relative 31% (Figure 2B,D). Of note, cells treated with LP17’s scramble peptide did not show altered induction of ICAM-1+ BMDN (Figure 2B,D), underlining LP17’s specificity for TREM-1 inhibition. These results confirm that eCIRP induces ICAM-1+ neutrophils partially via TREM-1. The possible factors, such as not knowing the optimum dose of LP17 and the time point of rmCIRP’s stimulation, might influence the amount of decrease of ICAM-1+ BMDN in rmCIRP-treated TREM-1−/− BMDN or in LP17-treated WT BMDN.
FIGURE 2.
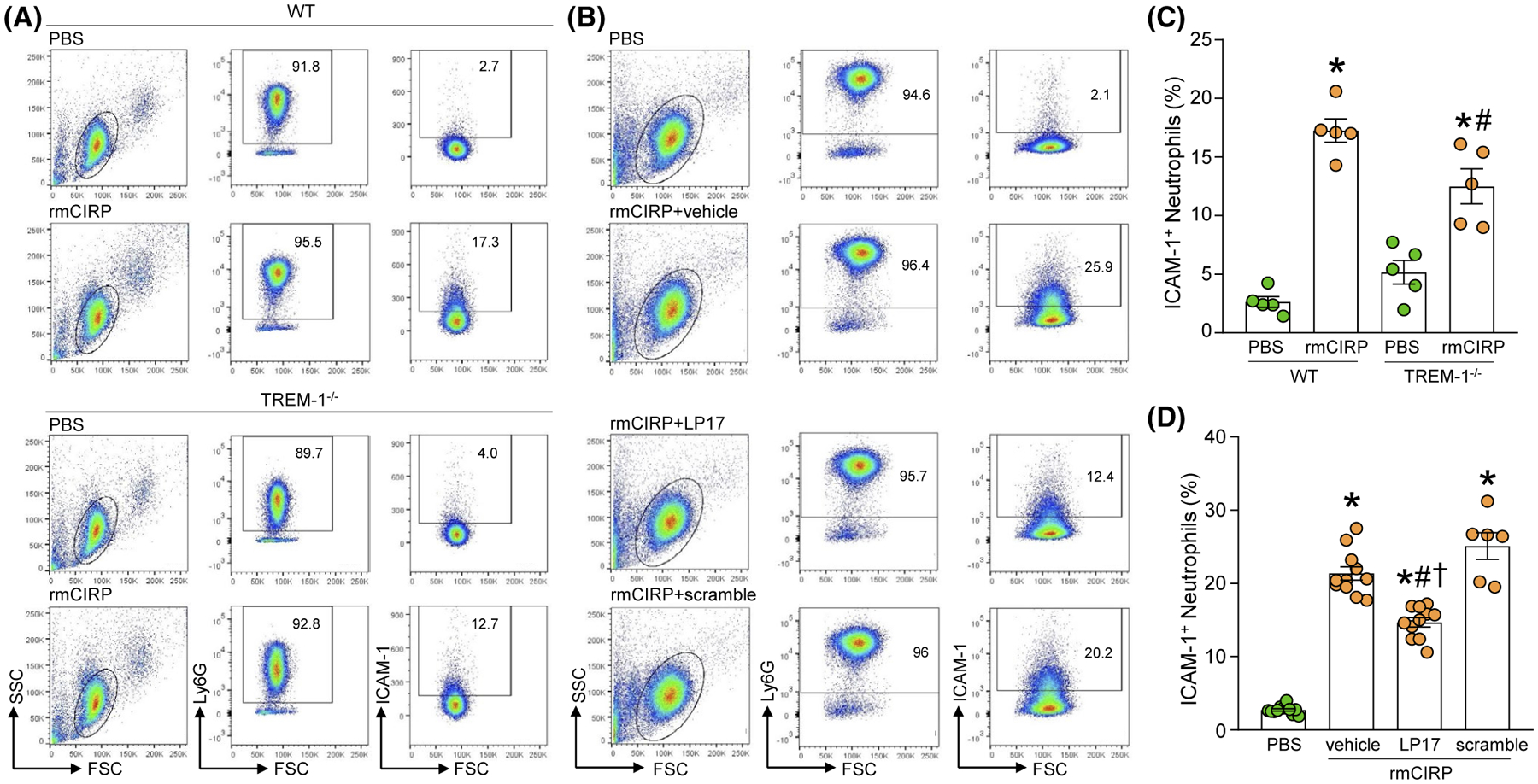
Inhibition of TREM-1 attenuates eCIRP-induced ICAM-1 expression on neutrophils in vitro. A, B, A total of 1 × 106 BMDN isolated from WT and TREM-1−/− mice were stimulated with PBS or rmCIRP at a dose of 1 μg/mL for 4 hours. After rmCIRP stimulation, BMDN were washed with PBS and stained with APC-Ly6G and FITC-ICAM-1 Ab. The frequencies of ICAM-1+ cells in LY6G gated population were assessed by flow cytometry. Experiments were repeated at least three times. Data are expressed as means ± SE (n = 5 samples/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs PBS-treated WT BMDN; #P < .05 vs rmCIRP-treated WT BMDN). C, D, BMDN (1 × 106 cells/ml) were treated with PBS, vehicle (PBS as LP17’s solvent), LP17 (80 μg/mL), and scramble peptide (80 μg/mL). After 30 minutes of the pretreatment of the cells with vehicle, LP17, and scramble, the cells were then stimulated with rmCIRP at a dose of 1 μg/mL for 4 hours. After stimulation with rmCIRP, the cells were washed with PBS and stained with APC-Ly6G and FITC-ICAM-1 Abs. ICAM-1 expression in LY6G+ cells were determined by flow cytometry. Experiments were repeated at least three times. Data are expressed as mean ± SE (n = 11 samples in PBS, vehicle, and LP17 groups; n = 6 samples in scramble group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs PBS-treated BMDN; #P < .05 vs vehicle + rmCIRP-treated BMDN; †P < .05 vs scramble + rmCIRP-treated BMDN)
3.3 |. TREM-1 promotes in vivo rmCIRP injected or sepsis-induced ICAM-1+ neutrophil formation
To demonstrate that TREM-1 also mediates the eCIRP-induction of ICAM-1+ neutrophils in vivo, we intraperitoneally (i.p.) injected mice with rmCIRP plus either vehicle (saline) or LP17 simultaneously. At 4 hours after injection, we collected the blood and lungs to assess the presence of ICAM-1+ neutrophils. Compared with saline, administration of rmCIRP resulted in an increase of relative 73% in the frequency of ICAM-1+ neutrophils in the blood and of relative 76% in the lungs (Figure 3A–D). By contrast, concomitant administration of rmCIRP and LP17 resulted in a significantly decreased frequency of ICAM-1+ neutrophils in the blood and lungs (Figure 3A–D). Thus, eCIRP induces ICAM-1+ neutrophils via TREM-1 not only in vitro, but also in vivo.
FIGURE 3.
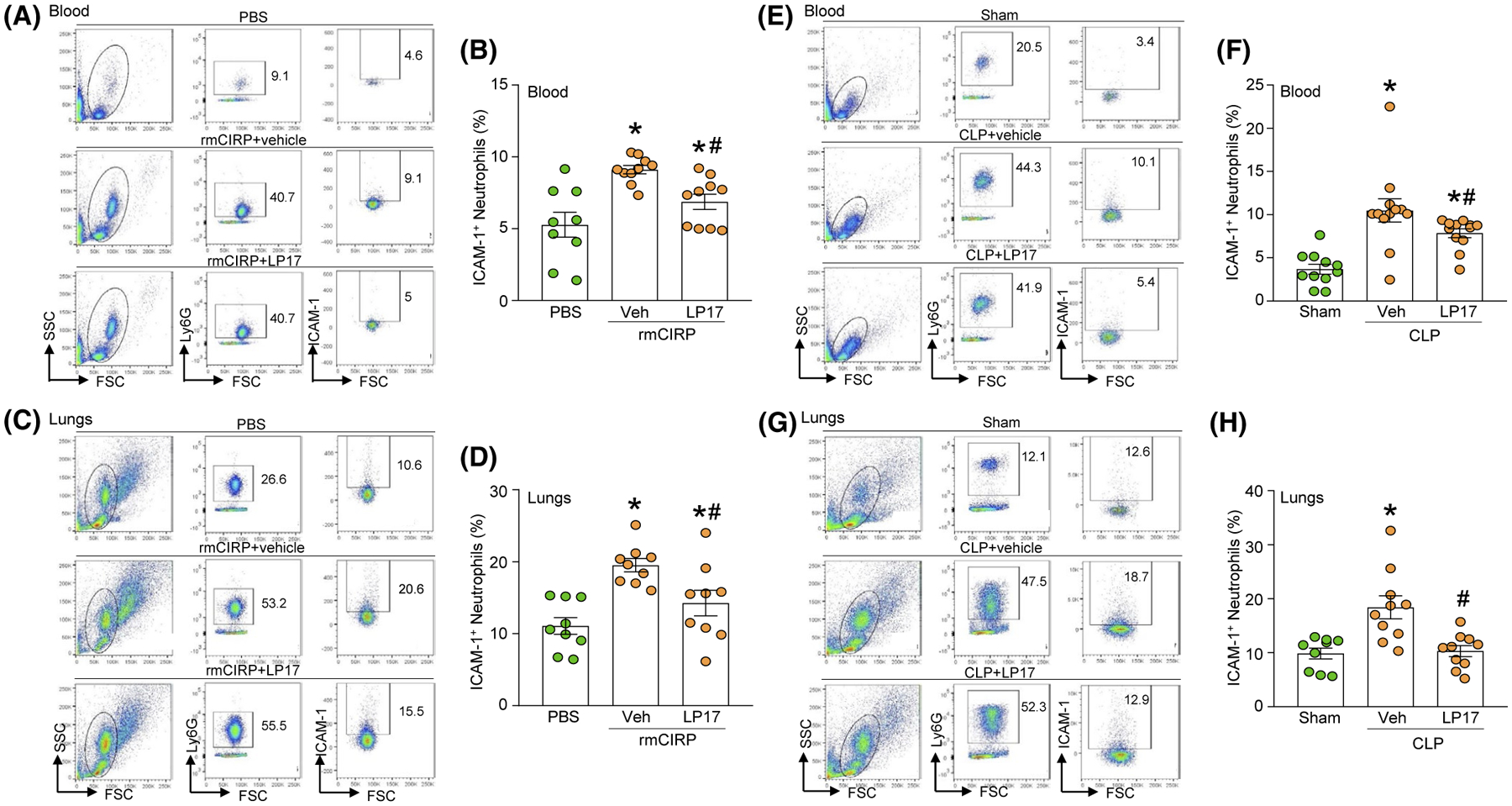
LP17 inhibits ICAM-1 expression in blood and lung neutrophils in rmCIRP-treated or septic mice. Mice were injected with PBS or rmCIRP (5 mg/kg BW) intraperitoneally (i.p.). At the same time mice were i.p. injected with vehicle (PBS) or LP17 (5 mg/kg BW). After 4 hours of injecting PBS, rmCIRP, vehicle, and LP17 mice were sacrificed to collect (A, B) blood and (C, D) lungs to determine ICAM-1 expression in neutrophils. Single cell suspension of blood and lungs were stained with APC-Ly6G and FITC-ICAM-1 Abs. ICAM-1 expression in LY6G+ cells were determined by flow cytometry. Experiments were repeated at least three times. Data are expressed as mean ± SE (n = 9 mice/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs PBS-treated mice (without rmCIRP treatment); #P < .05 vs vehicle + rmCIRP-treated mice). E-H, Sepsis was induced by CLP in WT mice. Mice were instilled with vehicle (PBS) or LP17 (5 mg/kg BW) in 100 μL volume in the peritoneal cavity at the time of CLP operation before closing the abdomen. Mice undergoing only laparotomy without cecal ligation or puncture served as sham group. Four hours after sham or CLP operation, mice were sacrificed to collect blood and lungs. Single cell suspensions of (E, F) blood and (G, H) lungs were stained with APC-Ly6G and FITC-ICAM-1 Abs. ICAM-1 expression in LY6G+ cells were determined by flow cytometry. Experiments were repeated at least three times. Data are expressed as mean ± SE (n = 11 mice/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs sham mice; #P < .05 vs vehicle-treated CLP mice)
We have shown that TREM-1 is important for eCIRP induction of ICAM-1+ neutrophils. Since eCIRP is significantly elevated during sepsis,20 we reasoned that TREM-1 inhibition should decrease the number of ICAM-1+ neutrophils in septic mice. Therefore, we subjected mice to CLP, a model of severe polymicrobial sepsis of abdominal origin.31 We administered LP17 or vehicle immediately after CLP and, at 4 hours after CLP, we collected the blood and lungs to assess the presence of ICAM-1+ neutrophils. Compared with sham, CLP resulted in significant increase of relative 187% in the frequency of ICAM-1+ neutrophils in the blood and of relative 88.5% in lungs (Figure 3E–H). By contrast, CLP with LP17 treatment resulted in a significant decrease in the frequency of ICAM-1+ neutrophils in the blood lungs (Figure 3E–H). These results clearly demonstrate the potential link between eCIRP, TREM-1, and ICAM-1+ PMN in sepsis.
3.4 |. ICAM-1+ neutrophils induce macrophage inflammation by NETs
To reveal the pro-inflammatory role of ICAM-1+ neutrophils, we cocultured FACS sorted ICAM-1+ or ICAM-1− neutrophils of WT mice with peritoneal macrophages. We found that although the peritoneal macrophages cocultured with ICAM-1− neutrophils released significantly higher levels of TNF-α and IL-6 in the culture supernatants compared to the peritoneal macrophages when they were cultured alone, intriguingly, the macrophages cultured with ICAM-1+ neutrophils produced significantly higher levels of TNF-α and IL-6 in the culture supernatants compared to culture with ICAM-1− neutrophils by 62% and 132%, respectively (Figure 4A,B). We next cocultured WT macrophages with neutrophils isolated from either WT or ICAM-1−/− mice, stimulated with rmCIRP, and assessed for TNF-α and IL-6 levels in the culture supernatants. We found that after rmCIRP stimulation WT neutrophils cocultured with WT macrophages produced significantly increased levels of TNF-α and IL-6 in the culture supernatants compared to WT macrophages cultured alone. However, we found that under rmCIRP-treated condition, WT macrophages cocultured with neutrophils from ICAM-1−/− mice released significantly decreased levels of TNF-α and IL-6 in the culture supernatants by 42%, and 52%, respectively, compared to WT macrophages cocultured with WT neutrophils (Figure 4C,D). Collectively, these data suggest that ICAM-1+ neutrophils induce macrophages to produce more pro-inflammatory cytokines. We previously identified that NETs were mainly produced by the ICAM-1+ neutrophils,10 we therefore aimed to determine whether or not by targeting NETs the pro-inflammatory effects of ICAM-1+ neutrophils on macrophages can be controlled. PAD4 is expressed in granulocytes and catalyzes citrullination of histones, leading to chromatin decondensation which is essential for NETosis.34 PAD4 not only induces chromatin decondition, but also helps release chromosomal DNA into the extracellular space.34 Cl-amidine, is a pharmaceutical inhibitor of PAD4.35 It irreversibly inactivates PAD4 by covalently modifying an active site cysteine that is important for its catalytic activity.36 We cocultured neutrophils with macrophages with or without the PAD4 inhibitor CI-Amidine, and then, after stimulating them with rmCIRP, we assessed TNF-α and IL-6 in the culture supernatants. We found that macrophages cocultured with neutrophils significantly increased the release of TNF-α and IL-6 in the culture supernatants after treatment with rmCIRP compared to PBS-treated control. However, when macrophages and neutrophils were cocultured in the presence of PAD4 inhibitor, after treatment with rmCIRP they released significantly decreased levels of TNF-α and IL-6 in the culture supernatants by 39%, and 48%, respectively, compared to vehicle-treated coculture of macrophages and neutrophils (Figure 4E,F). Taken together, these data suggest that eCIRP-induced NET-forming ICAM-1+ neutrophils induce macrophages to produce increased levels of pro-inflammatory cytokines.
FIGURE 4.
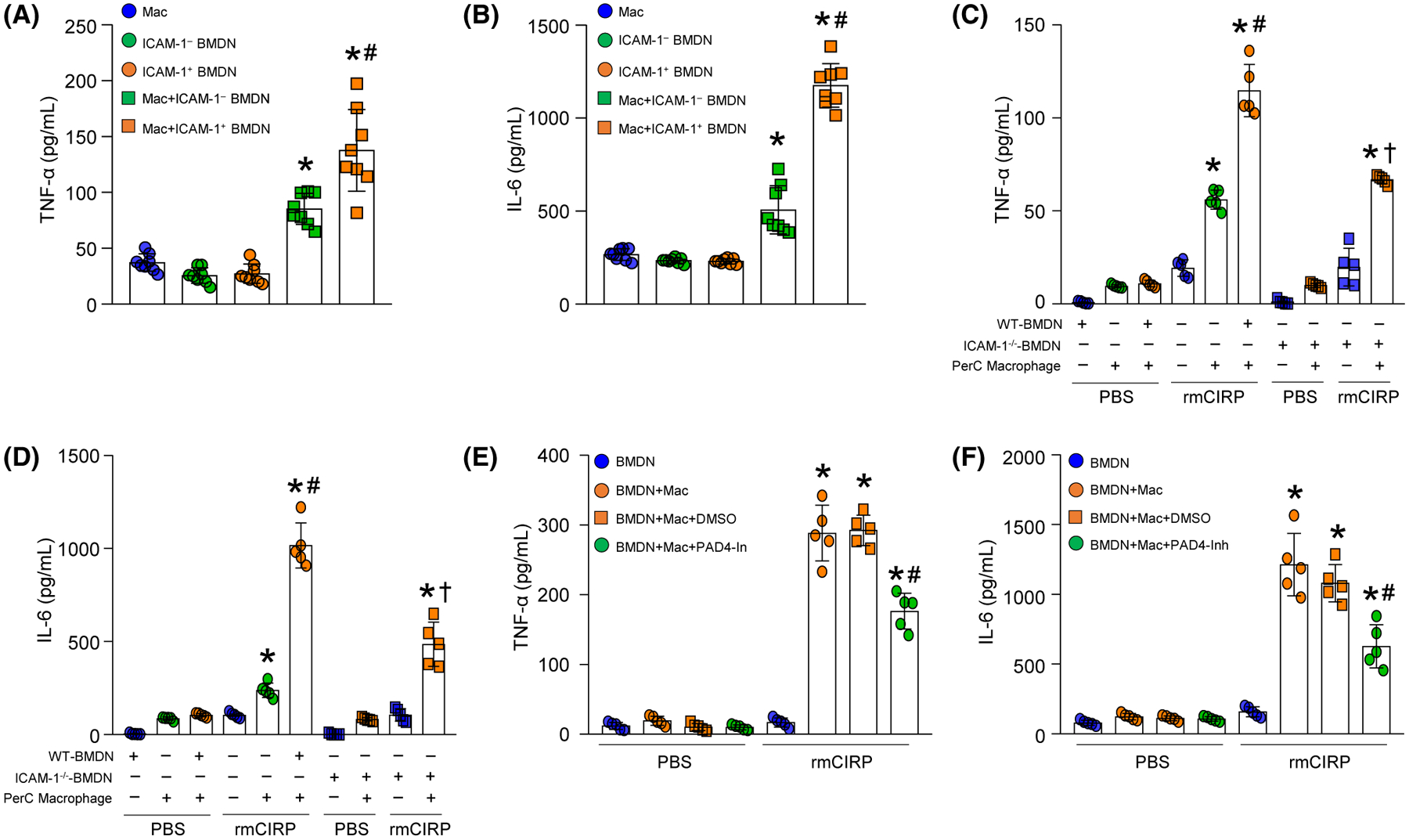
ICAM-1+ neutrophils induce macrophages to produce pro-inflammatory cytokines through increased NET formation. A total of 5 × 104 ICAM-1+ or ICAM-1− neutrophils sorted from rmCIRP-treated BMDN were cocultured with 1.5 × 105 peritoneal (PerC) macrophages, and 20 hours of coculture (A) TNF-α and (B) IL-6 levels in the culture supernatants were assessed by ELISA. Experiments were repeated at least three times. Data are expressed as mean ± SD (n = 8 samples/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs macrophage only; #P < .05 vs macrophage + ICAM-1− PMN). C, D, BMDN (5 × 104) isolated from WT and ICAM-1−/− mice were cultured separately with PerC macrophages (1.5 × 105). After 20 hours of stimulation with rmCIRP (1 μg/mL) (C) TNF-α and (D) IL-6 levels in the culture supernatants were assessed by ELISA. Experiments were repeated at least three times. Data are expressed as mean ± SD (n = 5 samples/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs PBS macrophages; #P < .05 vs rmCIRP macrophages; †P < .05 vs rmCIRP macrophage + WT-PMN). E, F, WT BMDN (5 × 104) cocultured with PerC macrophages (1.5 × 105) were simultaneously treated with PAD4 inhibitor CI-amidine at a dose of 5 μM and rmCIRP (1 μg/mL). After 20 hours of stimulation with rmCIRP, (E) TNF-α and (F) IL-6 levels in the culture supernatants were assessed by ELISA. Experiments were repeated at least three times. Data are expressed as mean ± SD (n = 5 samples/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs PBS macrophages; #P < .05 vs rmCIRP macrophages + DMSO)
3.5 |. ICAM-1 deficient neutrophils produce less NETs in vitro and in vivo
We treated neutrophils isolated from WT and ICAM-1−/− mice bone marrow with rmCIRP and assessed NETs using various tools. We found that following rmCIRP stimulation in WT BMDN, NET formation was significantly increased. However, in ICAM-1−/− BMDN stimulation with rmCIRP dramatically decreased their NET formation compared to WT neutrophils by relative 45%, 64%, and 52%, as revealed by MPO:DNA and NE:DNA ELISAs, and flow cytometry, respectively (Figure 5A–C). To determine the impact of ICAM-1 deficiency on eCIRP-induced NET formation in vivo, we injected WT and ICAM-1−/− mice with rmCIRP intratracheally (i.t.), and 4 hours after rmCIRP instillation into lungs we assessed NETs in lungs. We found that in ICAM-1−/− mice, administration with rmCIRP i.t. into lungs produced significantly decreased frequency and number of NET-forming neutrophils compared to WT mice instilled with rmCIRP by 61% and 60%, respectively (Figure 5D,E).
FIGURE 5.
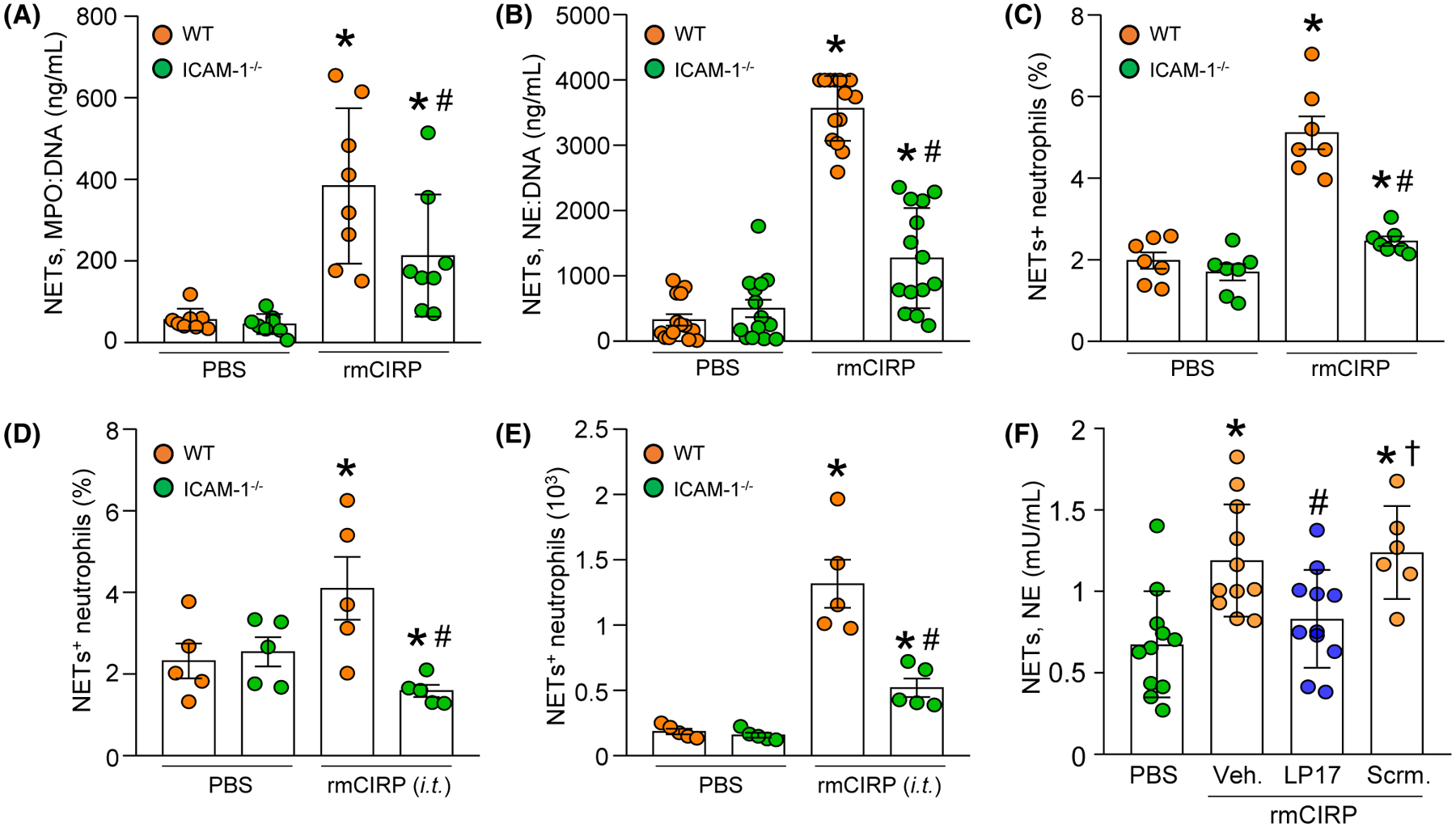
ICAM-1 deficient neutrophils or blockade of TREM-1 in WT neutrophils with LP17 produce less NETs following rmCIRP stimulation. A total of 106 BMDN isolated from WT or ICAM-1−/− mice were stimulated with rmCIRP for 4 hours. After stimulation with rmCIRP, isolated NETs were loaded in (A) MPO:DNA and (B) NE:DNA coated plates and assessed by ELISA. C, A total of 106 BMDN isolated from WT or ICAM-1−/− mice were stimulated with rmCIRP for 4 hours. After stimulation with rmCIRP, the neutrophils were surface-stained with APC-rat anti-mouse Ly-6G Ab, FITC-mouse MPO Ab, and rabbit anti-histone H3 (CitH3) Ab followed by staining with PE-donkey anti-rabbit IgG. NETs (MPO+citH3+ neutrophils) were assessed by flow cytometry. D, E, After 4 hours of injection of rmCIRP intratracheally (i.t.) in WT and ICAM-1−/− mice lungs were perfused and harvested. Single cell suspensions of lung tissues were surface-stained with APC-rat anti-mouse Ly-6G Ab, FITC-mouse anti-MPO Ab, and rabbit anti-histone H3 (CitH3) Ab followed by staining with PE-donkey anti-rabbit IgG and NETs were determined by flow cytometry. D, The frequencies and (E) numbers of NET forming neutrophils in lungs are shown. Experiments were repeated at least three times. Data are expressed as mean ± SE or SD (n = 5–14 samples/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs WT PBS; #P < .05 vs WT rmCIRP). F, A total of 106 BMDN/mL in 24-well plate were treated with PBS or rmCIRP in the presence of vehicle (PBS), LP17 or scramble. After 4 hours of rmCIRP stimulation, NETs were collected from the cells and assessed NE contents by ELISA. Experiments were repeated at least three times. Data are expressed as mean ± SD (n = 6–11 samples/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs PBS-treated group; #P < .05 vs vehicle (PBS) + rmCIRP-treated group; †P < .05 vs scramble + rmCIRP-treated group)
Since, eCIRP induces ICAM-1+ neutrophils via TREM-1 (Figure 2) and eCIRP-induced ICAM-1+ neutrophils produces more NETs,10 we hypothesized that inhibition of TREM-1 should limit eCIRP-induced NET formation. We exposed BMDN to rmCIRP plus vehicle (PBS), LP17, or LP17 scramble peptide. Four hours after stimulation, we assessed changes in NET formation. Compared with PBS-treated cells, BMDN exposed to rmCIRP plus vehicle had a significant increase of 76% in the frequency of the NET+ cells (Figure 5F). By contrast, BMDN exposed to rmCIRP and LP17 displayed a 30% reduction in NET formation, compared to BMDN rmCIRP with vehicle (Figure 5F). NET formation after exposure to rmCIRP and the scramble peptide was comparable to NET formation after BMDN exposure to rmCIRP plus vehicle (Figure 5F). These data clearly indicate that TREM-1-mediated eCIRP induction of ICAM-1 promotes NET formation by the neutrophils.
3.6 |. ICAM-1 deficient mice produce less NETs in the lungs during sepsis
We induced sepsis in WT and ICAM-1−/− mice and assessed NETs in the lungs. We found that after sepsis WT mice significantly increased the frequency and numbers of NET-forming neutrophils in the lungs compared to sham-operated mice. By contrast, in ICAM-1−/− mice the frequency and numbers of NET-forming neutrophils in the lungs were significantly decreased compared to WT mice in sepsis by 59.6%, and 57.3%, respectively (Figure 6A–C). Therefore, ICAM-1 can control eCIRP-induced NET formation in sepsis.
FIGURE 6.
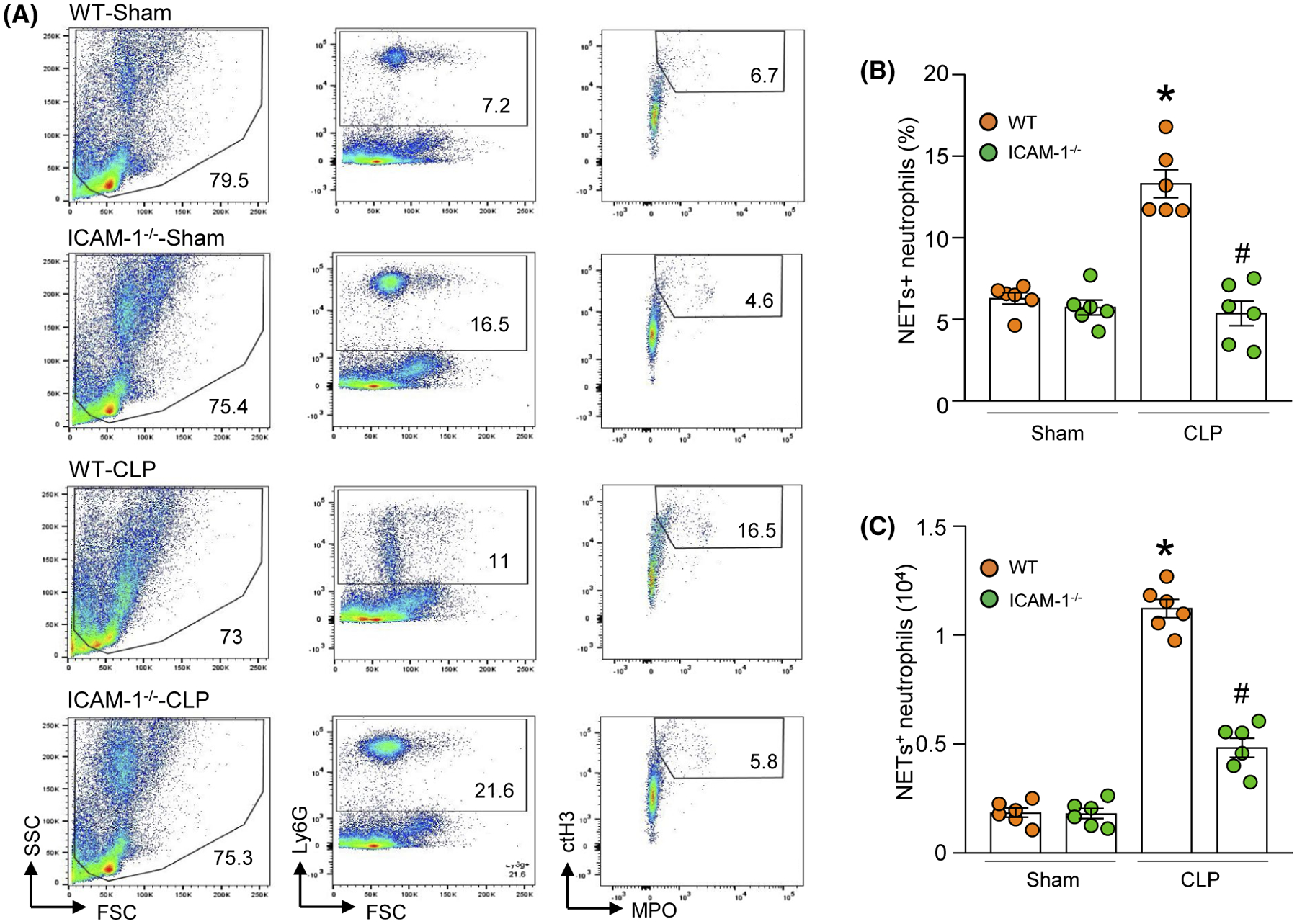
ICAM-1−/− mice produce less NETs in the lungs in sepsis. Sepsis was induced in WT and ICAM-1−/− mice by cecal ligation and puncture (CLP). After 4 hours of CLP, lung tissues were harvested and single cell suspensions were prepared. Cells were surface-stained with APC-rat anti-mouse Ly-6G Ab, FITC-mouse anti-MPO Ab, and rabbit anti-histone H3 (CitH3) Ab followed by staining with PE-donkey anti-rabbit IgG and NETs were determined by flow cytometry. A, Representative dot plots of the NETs+ cells in the lungs are shown. B, The frequencies and (C) numbers of NET forming neutrophils in lungs are shown. Experiments were repeated at least three times. Data are expressed as mean ± SE (n = 6 mice/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs WT sham; #P < .05 vs WT CLP)
3.7 |. eCIRP-induced ICAM-1-mediated Rho activation leads to increased NET formation
We stimulated BMDN with rmCIRP and assessed Rho activation at various time points. We found that stimulation of BMDN with rmCIRP significantly increased Rho activation, where the highest increase in Rho activity occurred at 120 minutes of rmCIRP treatment (Figure S1). We next treated BMDN with ICAM-1 neutralizing Ab and rmCIRP simultaneously for 120 minutes, and found that although rmCIRP treatment increased Rho activity in the BMDN by 2.4-fold compared to PBS-treated cells, blocking ICAM-1 by its neutralizing Ab significantly decreased Rho activation by 64% compared to only rmCIRP-treated BMDN (Figure 7A). We then treated BMDN with a Rho kinase inhibitor Y-27632 and rmCIRP and assessed NET formation. We found that BMDN stimulated with rmCIRP increased NET formation; however, when the cells were treated with Rho kinase inhibitor these cells produced significantly decreased amount of NETs by relative 28% compared to only rmCIRP-treated BMDN (Figure 7B,C). These data suggest that eCIRP-induced ICAM-1 pathway increases Rho activation in neutrophils, which leads to NET formation.
FIGURE 7.
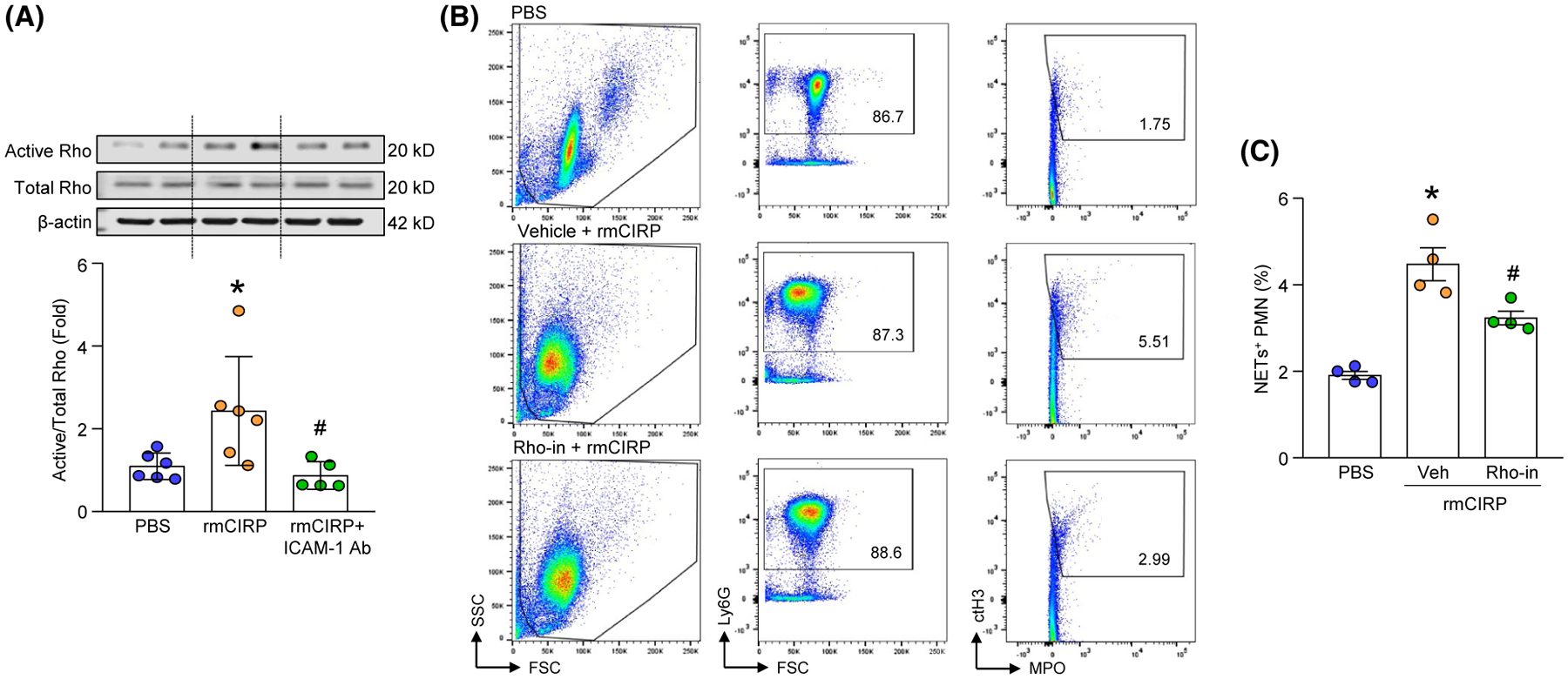
Inhibition of Rho activation reduces NET formation in BMDN. A, BMDN (2.5 × 106) were stimulated with PBS or rmCIRP in presence of ICAM-1 neutralizing Ab for 120 minutes. After stimulating the BMDN with rmCIRP, cells were lysed in lysis buffer and Rho activity was determined by GTP pull down process, followed by western blot assays using anti-Rho Ab. A fraction of each lysate was retained to determine the total Rho and β-actin by western blot using rabbit anti-Rho and β-actin Abs. Representative western blots for active Rho, total Rho, and β-actin are shown. Active Rho in each sample was normalized to total Rho expression and the mean values of PBS-treated group was standardized as one for comparison. Experiment was repeated twice. Data are expressed as mean ± SD (n = 5–6 samples/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs PBS; #P < .05 vs rmCIRP). B, C, A total of 106 BMDN were stimulated with rmCIRP for 4 hours in presence of vehicle control (PBS) or Rho inhibitor. After stimulation with rmCIRP, the neutrophils were surface-stained with APC-rat anti-mouse Ly-6G Ab, FITC-mouse MPO Ab, and rabbit anti-histone H3 (CitH3) Ab followed by staining with PE-donkey anti-rabbit IgG. NETs (MPO+citH3+ neutrophils) were assessed by flow cytometry. B, Representative dot plots of the NETs+ BMDN are shown. C, Bar diagram showing the frequencies of NETs+ neutrophils in the lungs are shown. Data are expressed as mean ± SE (n = 4 samples/group). The groups were compared by one-way ANOVA and SNK method (*P < .05 vs PBS; #P < .05 vs vehicle + rmCIRP)
4 |. DISCUSSION
In the current study, we identified TREM-1 as a novel mechanism through which eCIRP is able to induce the formation of ICAM-1+ neutrophils and NETs (Figure 8). During sepsis and sterile inflammation, certain intracellular contents are released and act as DAMPs to fuel further inflammation and ultimately cause organ injury.2 eCIRP is a DAMP found to be released by cells undergoing ischemic and thermal stress as well as during sepsis, hemorrhagic shock, and ischemia-reperfusion (I/R) injury.20,21 Elevated plasma levels of eCIRP have been independently correlated with increased disease severity in patients with sepsis.20,37,38 Once in the extracellular space, eCIRP can bind the TLR4-myeloid differentiation factor 2 (MD2) receptor complex on the surface of macrophages, leading to activation of the TLR4/MyD88/NF-κB pathway and culminating in the release of TNF-α, IL-6, and IL-1β.20,21 Besides macrophages, eCIRP also affects other cells. We have discovered that eCIRP is a strong inducer of NET formation by neutrophils.33 Moreover, we identified that eCIRP also induces a neutrophil subpopulation identifiable by ICAM-1+ surface expression and characterized by increased production of ROS, iNOS, and NETs.10,39 But the exact receptor through which eCIRP is able to induce ICAM-1+ neutrophils and the impact of ICAM-1 receptor on NET formation had never been investigated. We have previously assessed eCIRP levels in the blood and lungs after CLP and found significant increases in the levels of eCIRP as compared to sham-operated mice.10,20 Since we used the same model of creating sepsis, we expect increased levels of eCIRP in the current study.
FIGURE 8.
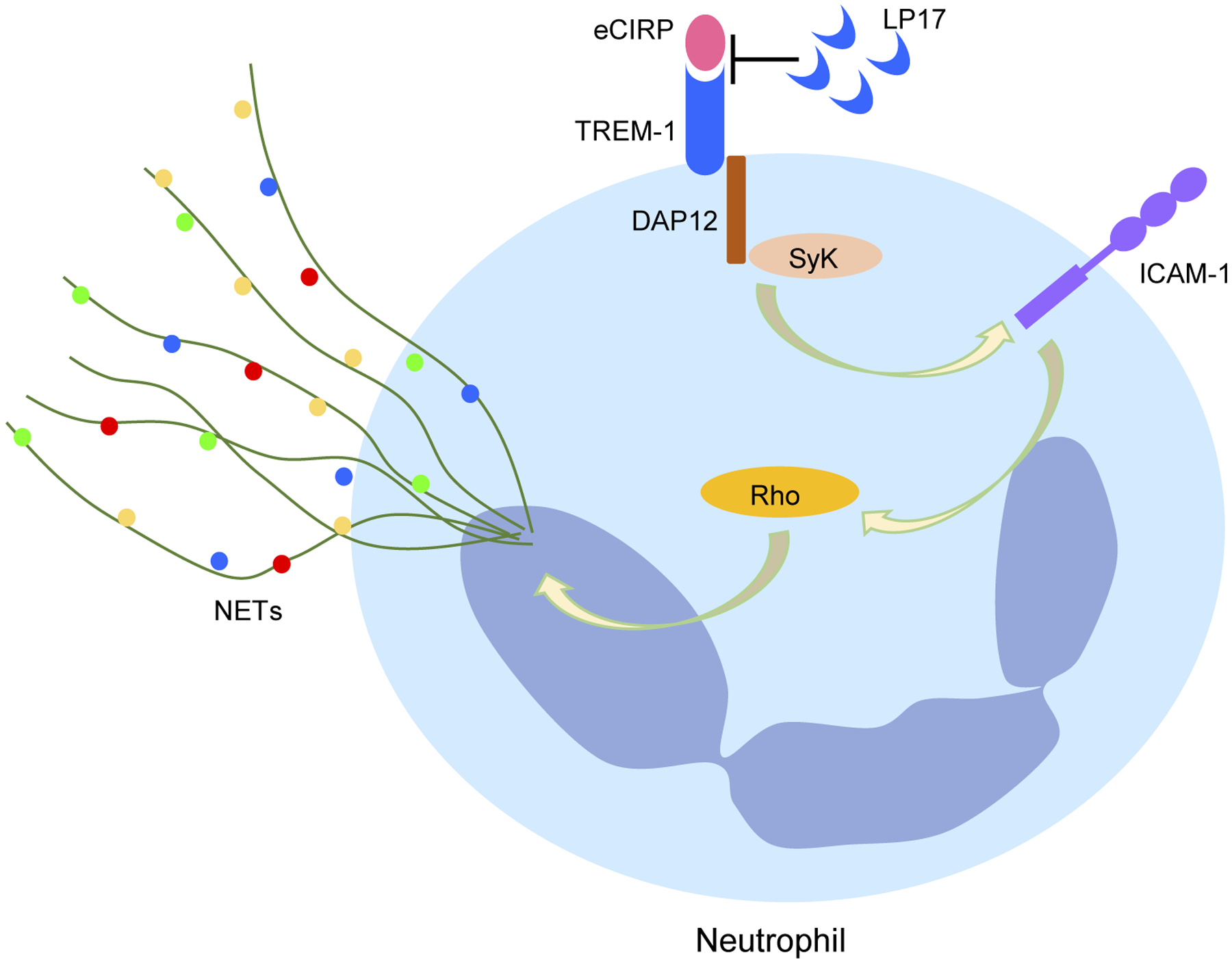
Graphical summary. eCIRP binds to TREM-1. eCIRP increases TREM-1 expression and activates its downstream molecules DAP12 and Syk in neutrophils, which lead to the increased expression of ICAM-1 and activation of Rho. eCIRP-induced Rho activation ultimately leads to increased NET formation. Targeting TREM-1-eCIRP interaction with LP17 which is a decoy peptide of TREM-1 inhibits eCIRP-TREM-1-mediated expression of ICAM-1 in the neutrophils and subsequently attenuating NET formation
A potential neutrophil receptor for eCIRP is TREM-1.23 TREM-1 is an innate immune receptor expressed primarily on neutrophils and macrophages.24 TREM-1 can be activated directly or in conjunction with TLR4.24,29 Once activated, TREM-1 leads to phosphorylation of the signaling adaptor protein DAP12, which in turn promotes phosphorylation of the tyrosine kinase Syk, resulting in the production of cytokines and chemokines.25,29,40 Using surface plasmon resonance (SPR), fluorescence resonance energy transfer (FRET), and immunofluorescence assays, we recently discovered that TREM-1 as a novel receptor of eCIRP.23 Since TREM-1 is known to be expressed on the surface of neutrophils, we decided to investigate whether TREM-1 was required for the eCIRP induction of NET-forming ICAM-1+ neutrophils. First, we identified that eCIRP not only up-regulates neutrophil TREM-1 surface expression, but also activates the neutrophil TREM-1 pathway, as indicated by DAP12- and Syk-phosphorylation. Next, we showed that eCIRP induction of ICAM-1+ neutrophils was dependent on TREM-1 activation. Induction of ICAM-1+ neutrophils was significantly diminished in cells from TREM-1 knockout mice and after treatment with LP17, a TREM-1 pharmacological inhibitor which works a decoy receptor for TREM-1 agonists.30 Here, we unexpectedly noticed the difference of the frequencies of ICAM-1+ BMDN between TREM-1−/− vs WT BMDN at baseline, which could be due to some feedback effects of the absence of TREM-1. Additionally, given the difference in the source of WT and TREM-1−/− mice, their gene expression profile could be slightly different at baseline, which might be reflected by the deviation in the percentages of ICAM-1+ PMN at baseline in WT vs TREM-1−/− mice. Previous report supports the notion that the mice from different vendors, strains, and sources exhibit different gene expression profile at basal level due to difference in their microbiome profile depending on diverse housing environments at different animal facilities.41–43 These results of TREM-1−/− mice are fully supported by our previous studies showing that TREM-1 also mediates the activation of macrophages by eCIRP.23 In those studies, we found that eCIRP increased the expression of pro-inflammatory cytokines and chemokines in macrophages, which was similarly reduced in cells from TREM-1−/− mice and after treatment with LP17. Combined, these results reveal TREM-1’s important role in the eCIRP activation of both macrophages and neutrophils.
It should be noted that our study does not rule out the participation of TL4 pathway.20 Activation of TLR4 leads to TREM-1 activation, which further amplifies TLR4-mediated inflammatory signals.29,44 Therefore, the eCIRP-mediated increased in the surface expression of TREM-1 could be as a result of eCIRP’s interaction with both TLR4 and TREM-1. In addition, as our data demonstrated that treatment of neutrophils with rmCIRP rapidly increased the phosphorylation of DAP12 and Syk, eCIRP-mediated induction of TREM-1 expression could occur through a positive feedback loop by engaging eCIRP-TREM-1 interaction leading to DAP12 and Syk phosphorylation.
One important neutrophil effector function is the release of NETs. We have previously shown that NETs are predominantly produced by activated ICAM-1+ neutrophils.10 Additionally, we have determined that eCIRP not only induces the surface expression of ICAM-1 in neutrophils, but also induces NETosis in these cells.10 Therefore, we decided to verify in the current study whether NETosis was regulated by TREM-1. As expected, inhibition of TREM-1 with LP17 also inhibited eCIRP-induced NET formation. It is possible that LP17 could have an indirect effect on NET formation due to decreased induction of ICAM-1 in neutrophils exposed to LP17 plus eCIRP. We therefore aimed to reveal how neutrophil expression of ICAM-1 results in increased NET formation. One possibility is that ICAM-1 promotes NETosis via the increased production of ROS and MPO, which are both elevated in ICAM-1+ neutrophils10,39 and have been shown to play a vital role in NET formation.2,45 Phagocytosis, for example, is a strong inducer of intracellular ROS and MPO.46 MPO and ROS then translocate from the cytoplasm to the nucleus, where they directly activate PAD4.2,45,47 Increased PAD4 activity causes histone citrullination leading to chromatin decondensation.2,48 Although these events ultimately may result in NET formation, they represent an indirect role of ICAM-1 on NETosis.
ICAM-1 promotes leukocyte transmigration, vascular permeability, and endothelial inflammatory response.12,13 In endothelial cells, ICAM-1-mediated downstream signal transduction leads to Rho GTPases activation, intracellular calcium (Ca2+) influx, actin cytoskeletal rearrangements, and Src family kinases activation.13–17 Rho proteins are activated upon binding to GTP, which play pivotal role in cell cytoskeleton organization, migration, transcription, and proliferation.18 To reveal the involvement of ICAM-1 on Rho activation, followed by NET formation, we first confirmed Rho activation in neutrophils following treatment with rmCIRP. We then blocked ICAM-1 using anti-ICAM-1 Ab which revealed decreased levels of active Rho in rmCIRP-treated neutrophils. We further determined the novel role of Rho in eCIRP-induced NET formation, which showed that inhibition of Rho significantly decreased eCIRP-induced NET formation. Collectively our findings demonstrated that eCIRP-mediated induction of Rho activation requires ICAM-1 and that Rho activation is essential for eCIRP-induced NET formation. Together, these results establish the notion that eCIRP-induced ICAM-1 leads to NET formation through Rho activation. In support of our finding of active Rho-mediated NET formation, a recent study demonstrated the influence of Rho on chromatin swelling and cytoskeletal rearrangement for NET formation.19 Further studies to determine whether or not Rho activation leading to NET formation is PAD4 dependent or independent may provide profound insights on this novel pathway of NET formation driven by eCIRP.
During inflammation, endothelial cells’ ICAM-1 recognizes its counterpart ligand Mac-1 (CD11b/CD18) expressed on leukocytes, such as neutrophils allowing them to adhere to the endothelial layer and finally helps neutrophils extravagate into the lungs.49 Conversely, ICAM-1 is not expressed in neutrophils under normal condition, but upon stimulation of neutrophils with rmCIRP, they express ICAM-1 on their cell surface.10 Since neutrophils also express Mac-1 on their surface, it is likely that the ICAM-1+ neutrophils could be activated by Mac-1 of adjacent neutrophils or same neutrophil through cross-linking. Future studies on crosslinking of ICAM-1 by its ligand(s) Mac-1 or LFA-1 will provide more deep insight into mechanism of activation of ICAM-1+ PMN.
Here, in some results we noticed that the number of NET forming cells was low. This could be due to the possibility that we might have missed the detection of the peak level of NETs by flow cytometry tool following stimulation of BMDN with rmCIRP. The flow cytometry-based detection of NETs, although it provides a quantitative result of the number of NETs, may under report the number of NETs because of the involvement of several experimental steps and mechanical maneuver while processing samples for flow cytometry to detect NETs. Validating the flow cytometry-based NETs data with the results using other tools like microscopy will help generate more solid findings of the decreased amount of NET formation in ICAM-1−/− mice lungs in sepsis. Our previous data using flow cytometry tool showed similar frequencies of NET-forming neutrophils in the lungs in sepsis.33 Even though the low numbers of NET forming cells were detected in lungs after sepsis, each one of NET DNA are decorated with considerable amount of inflammatory molecules like MPO, and H3; thus even a small increase can be deleterious in sepsis.
eCIRP and NETs are both elevated in the sera of patients with sepsis.20,37,50 Since we have previously shown that during sepsis eCIRP induces NET-forming ICAM-1+ neutrophils, we set out to evaluate whether TREM-1 was also relevant to ICAM-1+ neutrophil induction in the context of sepsis. Indeed, while septic mice had increased numbers of ICAM-1+ neutrophils, LP17 caused a reduction in the number of these cells. These results not only confirmed that TREM-1 is needed for maximal induction of ICAM-1+ neutrophils, but also revealed the therapeutic potential of targeting the eCIRP/TREM-1 pathway to treat septic patients. Determining exact role of TREM-1 pathway in human neutrophils in the eCIRP induction of NET-forming ICAM-1+ neutrophils will shed further light on the pathways involved in neutrophil heterogeneity and their functional outcomes in diseases such as sepsis.
In conclusion, the current study revealed TREM-1 as a previously unidentified pathway through which eCIRP induces NET-forming ICAM-1+ neutrophils in the blood and lungs of mice. We also revealed the involvement of Rho in ICAM-1+ neutrophils for NET formation. The discovery of the eCIRP/TREM-1 interaction and ICAM-1-mediated Rho activation are expected to support the development of novel therapeutic molecules able to mitigate inflammation and sepsis by controlling ICAM-1+ neutrophils and NETs.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health (NIH) grants R35GM118337 (PW) and R01GM129633 (MA) and Shock Society’s Faculty Research award grant (MA).
Funding information
National Institutes of Health (NIH), Grant/Award Number: R35GM118337 and R01GM129633; Shock Society’s Faculty Research
Abbreviations:
- BMDN
bone marrow-derived neutrophil(s)
- CIRP
cold-inducible RNA-binding protein
- CLP
cecal ligation and puncture
- DAMPs
damage-associated molecular patterns
- DAP12
DNAX-activating protein of 12 kDa
- eCIRP
extracellular CIRP
- HMGB1
high mobility group box 1
- ICAM-1
intercellular adhesion molecule-1
- LFA-1
lymphocyte function-associated antigen 1
- MPO
myeloperoxidase
- NETs
neutrophil extracellular traps
- PAD4
peptidylarginine deiminase 4
- PGLYRP1
peptidoglycan recognition protein 1
- Syk
spleen tyrosine kinase
- TLR4
Toll-like receptor 4
- TREM-1
triggering receptor expressed on myeloid cells-1
Footnotes
CONFLICT OF INTEREST
The authors declared that they have no competing interests.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denning NL, Aziz M, Gurien SD, Wang P. DAMPs and NETs in sepsis. Front Immunol. 2019;10:2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. 2014;15:602–611. [DOI] [PubMed] [Google Scholar]
- 4.Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. 2019;19:255–265. [DOI] [PubMed] [Google Scholar]
- 5.Silvestre-Roig C, Hidalgo A, Soehnlein O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood. 2016;127:2173–2181. [DOI] [PubMed] [Google Scholar]
- 6.Hidalgo A, Chilvers ER, Summers C, Koenderman L. The neutrophil life cycle. Trends Immunol. 2019;40:584–597. [DOI] [PubMed] [Google Scholar]
- 7.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. 2012;189:2689–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459–489. [DOI] [PubMed] [Google Scholar]
- 10.Ode Y, Aziz M, Wang P. CIRP increases ICAM-1+ phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J Leukoc Biol. 2018;103:693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berendt AR, Simmons DL, Tansey J, Newbold CI, Marsh K. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. Nature. 1989;341:57–59. [DOI] [PubMed] [Google Scholar]
- 12.Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106:584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dragoni S, Hudson N, Kenny BA, et al. Endothelial MAPKs direct ICAM-1 signaling to divergent inflammatory functions. J Immunol. 2017;198:4074–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Etienne S, Adamson P, Greenwood J, Strosberg AD, Cazaubon S, Couraud PO. ICAM-1 signaling pathways associated with Rho activation in microvascular brain endothelial cells. J Immunol. 1998;161:5755–5761. [PubMed] [Google Scholar]
- 15.Adamson P, Etienne S, Couraud PO, Calder V, Greenwood J. Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a rho-dependent pathway. J Immunol. 1999;162:2964–2973. [PubMed] [Google Scholar]
- 16.Etienne-Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J Immunol. 2000;165:3375–3383. [DOI] [PubMed] [Google Scholar]
- 17.Allingham MJ, van Buul JD, Burridge K. ICAM-1-mediated, Srcand Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol. 2007;179:4053–4064. [DOI] [PubMed] [Google Scholar]
- 18.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. [DOI] [PubMed] [Google Scholar]
- 19.Neubert E, Meyer D, Rocca F, et al. Chromatin swelling drives neutrophil extracellular trap release. Nat Commun. 2018;9:3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiang X, Yang WL, Wu R, et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 2013;19:1489–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aziz M, Brenner M, Wang P. Extracellular CIRP (eCIRP) and inflammation. J Leukoc Biol. 2019;106:133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cen C, McGinn J, Aziz M, et al. Deficiency in cold-inducible RNA-binding protein attenuates acute respiratory distress syndrome induced by intestinal ischemia-reperfusion. Surgery. 2017;162:917–927. [DOI] [PubMed] [Google Scholar]
- 23.Denning NL, Aziz M, Murao A, et al. Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI Insight. 2020;5(5):e134172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001;410:1103–1107. [DOI] [PubMed] [Google Scholar]
- 25.Arts RJ, Joosten LA, van der Meer JW, Netea MG. TREM-1: intracellular signaling pathways and interaction with pattern recognition receptors. J Leukoc Biol. 2013;93:209–215. [DOI] [PubMed] [Google Scholar]
- 26.Wu J, Li J, Salcedo R, Mivechi NF, Trinchieri G, Horuzsko A. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012;72:3977–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu L, Han L, Xie C, et al. Identification of extracellular actin as a ligand for triggering receptor expressed on myeloid cells-1 signaling. Front Immunol. 2017;8:917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Read CB, Kuijper JL, Hjorth SA, et al. Cutting edge: identification of neutrophil PGLYRP1 as a ligand for TREM-1. J Immunol. 2015;194:1417–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dower K, Ellis DK, Saraf K, Jelinsky SA, Lin LL. Innate immune responses to TREM-1 activation: overlap, divergence, and positive and negative cross-talk with bacterial lipopolysaccharide. J Immunol. 2008;180:3520–3534. [DOI] [PubMed] [Google Scholar]
- 30.Gibot S, Kolopp-Sarda MN, Béné MC, et al. A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis. J Exp Med. 2004;200:1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aziz M, Holodick NE, Rothstein TL, Wang P. B-1a cells protect mice from sepsis: critical role of CREB. J Immunol. 2017;199:750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gavillet M, Martinod K, Renella R, et al. Flow cytometric assay for direct quantification of neutrophil extracellular traps in blood samples. Am J Hematol. 2015;90:1155–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ode Y, Aziz M, Jin H, Arif A, Nicastro JG, Wang P. Cold-inducible RNA-binding protein induces neutrophil extracellular traps in the lungs during sepsis. Sci Rep. 2019;9:6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Li M, Stadler S, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biron BM, Chung CS, O’Brien XM, Chen Y, Reichner JS, Ayala A. Cl-amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun. 2017;9:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo Y, Arita K, Bhatia M, et al. Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry. 2006;45:11727–11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gurien SD, Aziz M, Jin H, et al. Extracellular microRNA 130b–3p inhibits eCIRP-induced inflammation. EMBO Rep. 2019:e48075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Y, Dong H, Zhong Y, Huang J, Lv J, Li J. The cold-inducible RNA-binding protein (CIRP) level in peripheral blood predicts sepsis outcome. PLoS One. 2015;10:e0137721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woodfin A, Beyrau M, Voisin MB, et al. ICAM-1-expressing neutrophils exhibit enhanced effector functions in murine models of endotoxemia. Blood. 2016;127:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000;164:4991–4995. [DOI] [PubMed] [Google Scholar]
- 41.Keane TM, Goodstadt L, Danecek P, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rasmussen TS, de Vries L, Kot W, Hansen LH, Castro-Mejía JL, Vogensen FK, Hansen AK, Nielsen DS. Mouse vendor influence on the bacterial and viral gut composition exceeds the effect of diet. Viruses. 2019;11(5):435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ericsson AC, Davis JW, Spollen W, et al. Effects of vendor and genetic background on the composition of the fecal microbiota of inbred mice. PLoS One. 2015;10:e0116704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arts RJ, Joosten LA, Dinarello CA, Kullberg BJ, van der Meer JW, Netea MG. TREM-1 interaction with the LPS/TLR4 receptor complex. Eur Cytokine Netw. 2011;22:11–14. [DOI] [PubMed] [Google Scholar]
- 45.Yang H, Biermann MH, Brauner JM, Liu Y, Zhao Y, Herrmann M. New insights into neutrophil extracellular traps: Mechanisms of formation and role in inflammation. Front Immunol. 2016;7:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warnatsch A, Tsourouktsoglou TD, Branzk N, et al. Reactive oxygen species localization programs inflammation to clear microbes of different size. Immunity. 2017;46:421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Claushuis TAM, van der Donk LEH, Luitse AL, et al. Role of peptidylarginine deiminase 4 in neutrophil extracellular trap formation and host defense during Klebsiella pneumoniae—Induced pneumonia-derived sepsis. J Immunol. 2018;201:1241–1252. [DOI] [PubMed] [Google Scholar]
- 48.Wong SL, Wagner DD. Peptidylarginine deiminase 4: a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB. 2018;32(12):6258–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203:2569–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colón DF, Wanderley CW, Franchin M, et al. Neutrophil extracellular traps (NETs) exacerbate severity of infant sepsis. Crit Care. 2019;23:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.