

Abstract
To examine whether insertion-duplication mutagenesis with chimeric DNA as a transformation donor could be valuable as a gene knockout tool for genomic analysis in Streptococcus pneumoniae, we studied the transformation efficiency and targeting specificity of the process by using a nonreplicative vector with homologous targeting inserts of various sizes. Insertional recombination was very specific in targeting homologous sites. While the recombination rate did not depend on which site or region was targeted, it did depend strongly on the size of the targeting insert in the donor plasmid, in proportion to the fifth power of its length for inserts of 100 to 500 bp. The dependence of insertion-duplication events on the length of the targeting homology was quite different from that for linear allele replacement and places certain limits on the design of mutagenesis experiments. The number of independent pneumococcal targeting fragments of uniform size required to knock out any desired fraction of the genes in a model genome with a defined probability was calculated from these data by using a combinatorial theory with simplifying assumptions. The results show that efficient and thorough mutagenesis of a large part of the pneumococcal genome should be practical when using insertion-duplication mutagenesis.
Streptococcus pneumoniae (pneumococcus) is a naturally competent species that takes up DNA and inserts it into its genome by homologous recombination after competence for DNA uptake is induced by an intercellular signaling peptide. With a linear homologous DNA donor, the result of recombination is replacement of a linear chromosomal region by a segment of the donor DNA, but if the donor is a chimeric circle the result can be insertion of the entire circle bounded by a duplication (Fig. 1) (24, 31). In the latter case, if the homologous “targeting” portion of the chimeric circle is internal to a gene and the heterologous region has a selectable marker, then the gene may be inactivated and the resulting mutant can be isolated by selection. Such insertion-duplication mutagenesis (IDM) has been used routinely for two decades to knock out genes in pneumococcus (15, 19, 23, 27). Mutagenesis involving circular integration or so-called single-crossover (SCO) recombination has also been commonly used in a variety of other bacterial species and genetic contexts. Usually, one of the molecules participating in the SCO is a chromosome containing the targeted gene. The other participant can be provided in different forms, such as a replicating plasmid present in the cell for multiple generations or a nonreplicating plasmid presented transiently. Some device is then used to withdraw the donor plasmid before the target chromosome is examined for integrants. Natural genetic transformation offers another efficient route for delivery of a nonreplicative vector, without a need for a temperature-sensitive vector or for high-efficiency electroporation. In this case, the apparent SCO recombination involves linear single-stranded DNA formed during uptake by competent cells and presented in a highly recombinogenic but nonreplicating state (14). IDM, like all insertion mutagenic methods, makes mutations that may be polar. However, it is valuable as a general mutagen because of the balancing advantages of a potential for highly random insertions and the generation of a sequence-tagged site, but it can also be used for specific targeting of selected genes.
FIG. 1.

Contrasting recombination products from linear homologous DNA versus circular chimeric DNA donors. The top object represents donor DNA; the middle object represents the recipient chromosome; and the bottom object represents the recombinant chromosome. (A) Linear homologous donors cause simple allele replacement within a homologous portion of the resident chromosome; the hatched box of donor DNA with a marker (+) contains a sequence homologous to the open box of the recipient chromosome. + and −, donor and recipient alleles. (B) Circular chimeric donor allows integration of heterologous DNA (cat) between duplicate, recombinant copies of a homologous target sequence; the vertically hatched box of donor DNA is homologous to the horizontally hatched target box of recipient chromosome.
For pneumococcal transformation, integrative recombination of plasmids carrying large (2-kb) targeting inserts yields about 10,000 to 100,000 recombinants per μg of DNA (13, 15, 18, 31). While this is sufficient for convenient production of complex mutant libraries, the potential of kilobase-sized inserts to disrupt individual genes is so limited that smaller targeting fragments are required for effective mutagenesis. The significance of the size of the targeting insert in a disruption vector for the rate of recombination during natural transformation is difficult to predict from the available precedents. If the principal class of donor molecules is monomeric circles, targeting homologous bases lying at the ends of linear single strands after uptake might be subject to a severe end exclusion effect of the type demonstrated for bases near the end of larger linear donor DNA molecules in allele replacement recombination (16). If, however, a large proportion of donor activity resides in plasmid dimers, the protection afforded such terminal markers by adjacent heterologous sequences (7) might render insertion vectors largely immune to “end exclusion.”
The dependence of recombinant yield on the length (I) of the targeting homology provided by an insert carried by an integration vector has been investigated for each of the delivery strategies mentioned above in at least one bacterial species. In all cases, shorter targeting segments correspond to lower recombination yields. However, the shape of the relation reported varies, and none of the published reports provides a direct basis for the design of IDM in S. pneumoniae. For SCO between double-stranded DNA substrates, the yield generally declines slowly with decreasing length (in proportion to I or I2 (I3 in one case [1]) in the range of 1,000 to 100 bp, and then it falls more precipitously toward undetectable levels at a “minimum effective length” of 20 to 70 bp. Fitting this description are various genetic systems for delivering and assaying the recombining chromosomes, including Escherichia coli phage-plasmid recombination (30, 32), chromosome-plasmid recombination in Bacillus sp. (11), plasmid-plasmid recombination in Lactococcus lactis (1), and direct electroporation in Lactobacillus sake (17). In only one case was a decrease of more than 10-fold observed between 1,000 and 100 bp; nor did any of the data suggest an extrapolated decrease of more than 200-fold over that range.
In contrast, recombination during transformation of naturally competent Bacillus subtilis cells by a nonreplicating insertion vector is reported to depend more strongly on the length of DNA available for pairing (20) in a manner depending on its molecular form. Monomers have a low activity, even for a 1,000-bp insert (1% of that of unfractionated plasmid DNA), and this falls steeply (∼I5) for smaller inserts. Dimers have a much higher activity, which varies in proportion to I2. These curve shapes may reflect the obligatory scission and strand separation steps of the uptake process and subsequent processing steps of the natural transformation pathway. While the B. subtilis data include only one insert smaller than 500 bp, the properties of plasmids with inserts in the 500- to 50-bp range can be estimated by extrapolation. The only datum point below 560 bp (for I = 260 bp) suggests that the quadratic relation may extend to shorter lengths and, if so, that 1,000 transformants could be obtained per μg donor DNA with 100-bp targeting fragments.
Despite the variety of the contexts of these precedents, they suggest by analogy that targeting fragments as small as 50 to 100 bp might be used to construct libraries of useful size in pneumococcus (i.e., affording yields of at least 100 recombinants per ml). Successful gene inactivation in pneumococcus by using fragments as small as 225 and 154 bp has been reported (25, 28); yet, while IDM has long been used in this species, its length dependence has not been systematically examined.
While the projection of estimates from other species to pneumococcus is uncertain, the data for B. subtilis competent cells are probably the closest available for comparison. However, although the broad outline of DNA processing in naturally competent pneumococcus cells parallels that in competent bacillus cells, such a projection must be considered provisional, as these two species are known to treat plasmid monomers differently (3, 29) and are also known to integrate point markers on small fragments at very different rates (see, for example, references 4 and 21), suggesting a difference in the details of processing of transforming DNA between the species. Since there are so few data for either species on targeting fragments shorter than 500 bp and since a fifth-power dependence would severely restrict the use of the small targeting fragments that are required for the disruption of small genes, we examined the case of transformation in pneumococcus cells experimentally. Here we present evidence on the efficiency and specificity of the recombination of insertion plasmids bearing targeting fragments of various lengths with a view to exploring its utility for genomic analysis, and we incorporate the results in a framework of practical parameters for the design of random mutagenic libraries of such plasmids. The practical conclusion from our data is that while 80-bp fragments can be used for targeting the disruption of individual genes, 300 bp is the approximate lower limit for use in creating complex mutant libraries in pneumococcus cells.
MATERIALS AND METHODS
Bacterial strains and donor DNA.
Strain CP1250 (Mal− Strr Novs Cms Com+) (25) was used for all pneumococcal transformation reactions. Novr DNA was prepared from strain 5MC (4). Plasmids were derivatives of pEVP3 (6), an E. coli plasmid which does not replicate in S. pneumoniae but which carries a cat (chloramphenicol acetyltransferase) marker that confers Cmr (chloramphenicol resistance) to either host. Fragments of the comCDE locus were in plasmids described previously (25) and in pXF530. pXF512 and pXF529 targeted the cel locus (26).
Construction of new plasmids.
pXF530 was derived from pXF517 by self-ligating the 5,288-bp HpaI fragment. pXF529 was derived from pXF512 by trimming a BsrDI-EcoRV fragment of its 3′ protruding end with T4 polymerase before self-ligation. Ligations for pXF530 and pXF529 were done at 19°C with 30 ng of DNA in a 100-μl volume with 10 U of ligase. Plasmids with random 300- to 400-bp inserts were made by cloning sonicated fragments of 5MC DNA at the SmaI site of pEVP3; the insert size was determined by DNA sequencing. The new clones’ insert sequences were mapped onto the partial genome sequence contigs (Institute for Genomic Research, December 1997 release, type 4 strain) to assign the following apparent gene targets: 300-1, contig 4102, bp 10776 to 11079 (rpoB, RNA polymerase β subunit homologue); 300-2, contig 4167, bp 5212 to 5494 (kdgK, 2-keto-3-deoxyglucuronate kinase homologue); 300-9, contig 4251, bp 5458 to 5757 (trkA, K+ transport protein homologue); 300-10, contig 4113, bp 6008 to 6290 (rf2, peptide chain release factor 2 homologue); 400-4, contig 4188, bp 11767 to 12043 (axe1, xylan esterase 1 homologue); 400-5, contig 4130, bp 6303 to 5925 (leuA, 2-isopropylmalate synthase homologue); 400-6, contig 4218, bp 437 to 123 (pbp1b, penicillin-binding protein 1B); and 400-9, contig 4179, bp 23885 to 24257 (ffh, signal recognition particle protein homologue). All were mapped as internal gene fragments except for 400-9, which included the end of the ffh homologue gene. T4 DNA ligase was obtained from Gibco-BRL, and the restriction enzymes were from New England Biolabs.
Preparation and quantitation of donor DNA.
Plasmids were prepared from transformed DH10B (Gibco-BRL) by using the Wizard Plus MaxiPrep kit (Promega). Donor DNA samples were quantitated by measuring the fluorescent intensities in ethidium bromide-stained agarose gels relative to that of a standard DNA of known concentration.
Transformation protocols.
To determine the plasmid transforming activities, a frozen CP1250 stock (optical density at 550 nm [OD550] = 0.1) was diluted 1:100 in CTM plus 3 mM HCl (25) and cultured to an OD550 of 0.025 at 38°C. After the addition of competence-stimulating peptide and NaOH to 200 ng/ml and 6 mM, respectively, and a 1/100 volume of donor DNA, incubation was continued for 45 min. The transformation reactions (up to 0.5 ml per 100-mm-diameter plate) were plated as described earlier (22) with a top layer containing 10 μg of chloramphenicol or 10 μg of novobiocin per ml or no drug, and the colonies were counted after 20 h at 37°C. A typical 1-ml transformation reaction contained 108 CFU/ml. To determine competence phenotypes of recombinants, the microtiter well competence test was done as described earlier (22) but with competence-stimulating peptide added to the growth medium.
Combinatorial description of mutagenic hits in an insertion-duplication library.
Let Mi be the probability of mutating a certain set of i genes within a genome containing N genes. Pi = NCi × Mi is then the probability of mutating any set of i genes out of N genes, where NCi = N!/[(N − i)!¡!]. Since (G − I) is the effective length of the mutatable region within a gene, Q = [L − (G − I) (N − i)]/L = 1 − (N − i)q is the fraction of the genome containing the mutatable regions of a specific set of i genes plus all of the nonmutatable regions, where q = (G − I)/L, G is the size of a gene, I is the size of an insert or targeting fragment, L is the genome size (L = N × G), and n is the number of random clones (library size). Thus, Q is the probability for an insert to target one of the i genes or one of the other nonmutatable regions. So Qn = [1 − (N − i)q]n is the probability that all of n random clones target one of the mutatable regions of a specific set of i genes or one of nonmutatable regions. Since it is also equal to the sum of the probabilities to mutate any set of j genes (j = 0, 1, …, i) out of i genes (iCjMj), Σj=0i iCjMj = [1 − (N − i)q]n (i = 0, 1, …, N). Pi can now be obtained by calculating Mi from the above equation as follows:
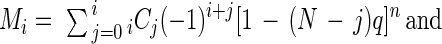 |
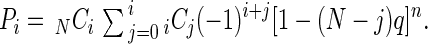 |
Note that the average number of mutated genes can be calculated by Σi=0N i × Pi, and is the same as N times the probability for a certain gene to be mutated, which is 1 − (1 − q)n (3). Therefore, the average fraction of mutated genes is <F> = 1 − (1 − q)n, and (1 − q)n can be approximated by P(0) of the Poisson distribution.
RESULTS
To investigate the potential of IDM for use as a mutagenic tool, we prepared a series of plasmids with pneumococcal inserts ranging in size from 96 to 2.4 kb (Table 1). The targeted genes were chosen, with two exceptions, because they were nonessential, and several were at characterized loci with known mutant phenotypes affecting genetic competence (25). Transformation reactions were carried out with cells induced to competence with synthetic pheromone peptide (10) to determine the importance of the three parameters: DNA dose, target location, and length of target homology.
TABLE 1.
Donor plasmids used in this study
| Plasmid (size [kb]) | Targeting fragment
|
Phenotypeb after integration | Source or referencec | |
|---|---|---|---|---|
| Size (bp) | Gene(s)a | |||
| pEVP3 (6.3) | 0 | 6 | ||
| pXF512 (8.7) | 2,364 | ‘celB, orfr1’ | Com+ | 26 |
| pXF517 (6.7) | 390 | ‘comD’ | Com− | 25 |
| pXF518 (6.5) | 154 | ‘comE’ | Com− | 25 |
| pXF519 (6.5) | 172 | ‘orfL | Com+ | 25 |
| 300-1 (6.6) | 300 | ‘hrpoB’ | Lethal | pEVP3 plus sonicated DNA |
| 300-2 (6.6) | 283 | ‘hkdgK’ | NT | pEVP3 plus sonicated DNA |
| 300-9 (6.6) | 330 | ‘htrkA’ | NT | pEVP3 plus sonicated DNA |
| 300-10 (6.6) | 283 | ‘hrf2’ | Lethal | pEVP3 plus sonicated DNA |
| 400-4 (6.6) | 279 | ‘haxel’ | NT | pEVP3 plus sonicated DNA |
| 400-5 (6.7) | 379 | ‘hleuA’ | NT | pEVP3 plus sonicated DNA |
| 400-6 (6.7) | 315 | ‘pbp1b’ | NT | pEVP3 plus sonicated DNA |
| 400-9 (6.6) | 373 | ‘hffh | NT | pEVP3 plus sonicated DNA |
| pXF521 (6.8) | 513 | comD, comC, orfL | Com+ | 25 |
| pXF529 (6.2) | 1,017 | ‘celB’ | Com− | pXF512 |
| pXF530 (5.3) | 96 | ‘comD’ | Com− | pXF517 |
For fragments containing less than a whole gene, truncation of the gene is indicated by an apostrophe. Genes identified by sequence homology are indicated by an “h” prefix.
For certain fragments from known genes, the competence phenotypes of mutants can be predicted. NT, not tested.
pEVP3 plus sonicated DNA, random clones from a library of 20,000 independent clones (unpublished data).
DNA dose response.
The DNA dose-response curves for a point marker, nov-1, in linear chromosomal donor DNA (20 kb) and for an insertion plasmid with a large targeting insert were compared (Fig. 2). nov-1 exhibited the expected linear relation between transformants and a donor concentration below 50 ng/ml and reached a plateau at below 1,000 ng/ml, a finding consistent with the results of Cato and Guild (4). For pXF512, a representative circular DNA with a 2,364-bp pneumococcal insert, similarly extensive data also fit a linear relation at concentrations up to about 10 ng/ml but then approached a plateau more slowly than the nov-1 marker without reaching a plateau in the concentration range studied. In the linear range of the transformation curves, the relative transformation yields for pXF512 and nov-1 were 1:60 on a mass basis but 1:15,000 on a molar basis (for a chromosome of 2.25 Mb).
FIG. 2.

Transformation dose dependence for various insertion plasmids. Transformation by insertion plasmids (circular chimeric plasmid donor, Cmr) was compared to the standard allele replacement reaction (linear chromosomal DNA donor, Novr) in competent cultures of 108 cells/ml. The yield for different donors is indicated by the symbol shapes as follows: linear chromosomal DNA (■), 2,634-bp-insert plasmid (•), 172-bp-insert plasmid (□), and 300- to 400-bp-insert plasmids marked with the homology identifications presented in Table 1 (×). Data interpolated or extrapolated to 100 ng of DNA/ml by using the curve shape shown for pXF512 (2,634-bp insert) are presented: from the bottom, the diamonds (⧫) represent 96-, 154-, 390-, 300-, 513-, and 1,013-bp inserts, respectively. Curves were fit manually to the chromosomal data ( ) and to that for the 2,634-bp-insert plasmid (
) and to that for the 2,634-bp-insert plasmid ( ). The curve shape for the latter was used to represent the dose dependence of the other plasmids, as illustrated for the 172-bp-insert plasmid pXF519. The background level of pEVP3 insertion was ≤0.2 Cmr transformants/ml at 200 ng/ml.
). The curve shape for the latter was used to represent the dose dependence of the other plasmids, as illustrated for the 172-bp-insert plasmid pXF519. The background level of pEVP3 insertion was ≤0.2 Cmr transformants/ml at 200 ng/ml.
The transforming activity of pXF519, a plasmid with a smaller (172-bp) insert, was much lower but had a dose-response pattern similar to that of pXF512 within the range studied, as shown in Fig. 2. For plasmids with even smaller inserts, the DNA concentrations used were above 100 ng/ml because the transformant number for those plasmids was too low to give statistically reliable data at lower doses (not shown). In this high-dose range, the patterns of dose-response for pXF530 (96-bp insert) and pXF518 (154-bp insert) were also quite similar to that of pXF512. As the dose-response patterns for all other plasmids were also similar to that of pXF512 (not shown), a standard curve fit to the data for pXF512 was used for interpolating or extrapolating to the activity at 100 ng/ml for subsequent comparisons. Transforming activity estimated in this way was a strong function of insert size, varying 25,000-fold between 2,364 and 96 bp.
Specificity of gene targeting.
Transformation yields for plasmids with very small inserts were so low that nonspecific insertion by those plasmids was considered possible (see reference 11) even though the recombination of small-insert plasmids appeared to depend on their pneumococcal homology. To determine whether these rare recombination events occurred at the relevant chromosomal site, we transformed CP1250 (wild type in competence) with pXF530 and pXF518, plasmids with 96- and 154-bp inserts, respectively, targeting internal regions of comD and comE (both required for competence), and with pXF519, a plasmid with a 172-bp insert targeting orfL (not required for competence). On determining the competence phenotype of the resulting transformants as described in Materials and Methods, all clones of the recipient strain CP1250 and of the pXF519 transformants (3 and 11 independent clones, respectively) were found to be fully competent (105 transformants/ml), while with all 11 independent pXF518 transformants and with 5 of 6 of the independent pXF530 transformants tested, there were fewer than 5 × 102 transformants/ml, as is typical for the comDE class of transformation-defective mutants. Table 2, in which these results on site specificity of insertion events are combined with data from several previous reports, shows that the targeted gene disruption achieved by this method is highly specific, with the targeted gene being mutated in 65 of 66 recombinants overall.
TABLE 2.
Target specificity of gene disruption insertion vectors
| Target gene | Length of targeting fragment (bp)a | No. of mutants/no. of recombinantsb | Reference |
|---|---|---|---|
| comD | 96 | 5/6 | This study |
| comE | 154 | 11/11 | This study |
| recP | 190 | 6/6 | 28 |
| coiA | 301 | 5/5 | 25 |
| recP | 360 | 6/6 | 28 |
| comD | 390 | 12/12 | 25 |
| recP | 450 | 6/6 | 28 |
| comE | 510 | 8/8 | 25 |
| celB | 1,283 | 6/6 | 25 |
Chimeric donor plasmids carrying an internal region of each target gene in vectors pJDC9 (5) or pEVP3.
Number of transformation-defective recombinants per total number of recombinants tested.
Dependence of recombination yield on targeting homology length.
Since the plasmids we studied targeted various sites on the chromosome, a strong site dependence of recombination frequency could confound any effect of targeting fragment length on the transformation yield. To check for such a site preference, eight additional plasmids with similar-sized inserts were chosen randomly for assay. The results distinguished two classes of plasmid. One class produced no recombinants, apparently because they targeted essential genes (rpoB and rf2). The other class, plasmids making nondisruptive insertions or targeting dispensible genes, had transformation yields (Fig. 2) that were all quite similar (400 to 600 transformants/ml at 160 ng of DNA/ml). Thus, within the limitation of these statistics, the recombination efficiency of insertion-duplication was not very sensitive to the site targeted, although the actual recovery of mutants produced naturally depended on their individual phenotypes.
In contrast, the recombination rate for insertion-duplication did depend strongly on the targeting fragment’s size. Figure 3 shows the relation of the transformation rate to insert length, incorporating the intercept at 100 ng/ml presented in Fig. 2. The results stand in striking contrast to the results of an extensive study of the rate of allele replacement by linear donor DNA of various lengths by Cato and Guild (4), which was fit well by a theoretical curve derived by Lacks (12) predicting zero transformation activity at and below 450 bp. Here we report that circular DNA with a 390-bp targeting fragment recombined at a significant rate (600 Cmr transformants/ml, ∼5% of that for 2,400-bp inserts) and that targeting fragments as small as 96 bp yielded detectable numbers of transformants, yet with high target specificity. Thus, for a given length of homologous DNA available for pairing and for a constant mass of donor DNA, the transformation yield for insertion-duplication was quite low compared to linear DNA for lengths above 1 kb but was dramatically higher below 500 bp. Nonetheless, the data also show that as the insert size decreased transformation dropped rapidly. Over the range of 96 to 500 bp, the actual yield for plasmid integration at 100 ng of DNA/ml is approximated by a fifth-power relation (Fig. 3).
FIG. 3.

Size dependence of plasmid insertion and allele replacement transformation in pneumococcus cells. Symbols: •, transformation yield for insertion-duplication with circular donor plasmids (Cmr) with various-sized inserts (96 to 2,364 bp) at a constant DNA concentration (100 ng/ml) as presented in Fig. 2; ○, allele replacement yield for linear donor DNA (Novr) duplexes of various sizes (470 to 25,000 bp) as given by Cato and Guild (4). The curve shows the relation: y = 6.0 × 10−11 x5.0 (Equation 1), where y is the number of transformants per milliliter and x is the available pairing length in base pairs, for the range of 70 to 500 bp.
DISCUSSION
The data presented here show that fragments so small as to have zero transforming activity as free linear donors do, when present in an insertion vector, direct recombination to the homologous site of the resident chromosome at an appreciable rate. Yet despite the implied protection from “end effects,” the rate of such recombination is still a strong (fifth-power) function of the length of the targeting homology. The pattern presented here for natural transformation in pneumococcus may be contrasted with another natural competence system, that of B. subtilis. For purified monomeric insertion plasmids in bacillus, as for bulk plasmid with pneumococcus, the yield follows an I5 relation. In B. subtilis, transformation with purified dimeric insertion plasmids displayed a lower dependence on insert length (∼I2). As suggested by Michel et al. (20), this is consistent with the idea that several required size-dependent steps are involved in insertion-duplication. For example, for dimers, these might be the pairing of two copies of the target with the chromosome. For monomers, additional steps could be cutting within the target segment for uptake (1) and escape by these targeting end-pieces from possible end-degrading activities in the competent cell after uptake (2, 3). In pneumococcus cells, unfractionated plasmid was approximately as efficient as dimers or unfractionated plasmids in bacillus cells and much more efficient than were monomers in bacillus cells, but they still followed a fifth-power relation like that for monomers in the bacillus system. The contrast may reflect differences between the two DNA-processing pathways, which is also suggested by the fact that plasmid monomers transform pneumococcus but not bacillus (3, 29). Alternatively, the difference may merely reflect the lack of data below 500 bp in bacillus cells.
The results presented here show that the targeting of IDM is very specific and that its efficiency varies little with chromosomal site but does drop steeply as the length of homologous DNA presented for recombination decreases. Thus, a mixture of plasmids with random inserts of uniform size can be expected to yield a population of insertion mutants distributed quite randomly among dispensible genes. The strong dependency of the relative transformation efficiency on the length of the DNA available for pairing implies that the insert sizes in plasmid mixtures used for such a transformation should be uniform, since most transformants from a mixture of plasmids with heterogeneously sized inserts would arise primarily from those with the larger inserts. Thus, for example, targeting fragments created with a restriction enzyme would be likely to produce a nonrandom set of mutations. The actual transformation yield reported here also implies that making a large collection of mutants from plasmids with very small inserts can require inconveniently large amounts of DNA and competent culture, as discussed below. Nonetheless, the detectable transforming activity of plasmids with small inserts would readily allow disruption of a single chosen gene by using a plasmid carrying an insert as small as 100 bp (10 transformants from 2 μg of plasmid DNA and 20 ml of culture). Consistent with the higher specific activity of dimeric plasmid forms (31), DNA preparations from the host LE392, which are enriched in dimers, have been reported to give higher yields for IDM (5a). In our study plasmids with targeting fragments of 172 or 390 bp gave 10-fold-higher insertion rates if prepared from LE392 instead of DH10B (data not shown). This improvement corresponds to a reduction in the minimum targeting size for library preparation of about 30 to 40% and should be considered if the smallest possible duplications are desired.
Implications for design of random mutagenic libraries.
The low rate of insertion-duplication recombination and its steep dependence on homology length place certain limits on the design of libraries for mutagenesis of the pneumococcal genome: the yield limits the number of mutants it is practical to make, while the size of the insert used affects the chance that an insertion will be mutagenic (i.e., disrupt a gene). To illustrate the interrelation of these parameters and to explore the library size required to approach saturation of the pneumococcal genome, we estimated the number of plasmids needed to hit with at least one mutation nearly all of the genes in a model genome as a function of insert (targeting-fragment) size. To simplify the calculation, we made assumptions that (i) all genes have the same size (denoted by G) and that (ii) only fragments targeting the internal region of a gene (of size [G − I], where I is the insert size) produce a mutation. (The theory could be readily extended to accommodate any explicit distribution of gene sizes by integrating the results over the size distribution, since G is an explicit parameter of the function. However, since the shape of the gene size distribution is not known, we have not done that here.) We derived Pi (see Materials and Methods), the probability that a collection of n random clones of plasmids with inserts of size l will mutate any i genes among all N genes. The probability to mutate at least k genes is then Σi=kN Pi. We define the minimum knockout fraction F = k/N as the proportion of genes that will be hit at least once with 99% probability, where k is determined by Σi=kN Pi = 0.99 to obtain an explicit statistical estimate of genome coverage. In Table 3, values of F are shown for several insert and library sizes. (One could use the average knockout fraction <F> = 1 − (1 − q)n (see Materials and Methods), but F is a more strict criterion because the probability to mutate more than N × <F> genes is only about 50%). Table 3 illustrates the asymptomatic nature of the approach to saturation of the genome with mutations as the number of clones is increased. (For example, for 300-bp inserts, a library size of 10 × N can mutate at least 99.6% of the genes, while a 5 × N library can mutate at least 95.2%; note that at this high coverage a 4% increase in coverage requires doubling the library.)
TABLE 3.
Effect of insert length on library size and competent culture volume required to approach saturation mutagenesis by using an insertion vector
| Targeting insert length I (bp) | Transformation yield (no. of Cmr transfor-mants/ml)a | Minimum knockout fraction, F (%)b, for a library size (n) of:
|
Cell culturec vol (ml) | |||
|---|---|---|---|---|---|---|
| 1 × N | 3 × N | 5 × N | 10 × N | |||
| 0d | 60.1 | 93.0 | 98.5 | 99.90 | ||
| 100 | 0.6 | 56.1 | 91.0 | 98.4 | 99.85 | 15,750 |
| 200 | 19.2 | 51.6 | 88.3 | 96.8 | 99.80 | 530 |
| 300 | 146 | 46.8 | 84.8 | 95.2 | 99.6 | 80 |
| 400 | 614 | 41.5 | 80.2 | 90.2 | 99.2 | 30 |
| 500 | 1875 | 35.5 | 74.0 | 86.0 | 98.5 | 10 |
Data for 100 ng of plasmid DNA per ml are from Fig. 3.
F values were obtained by calculating Σi=kN Pi with expression as described for Pi in Materials and Methods by using the mathematical program Mathematica. Assuming that L = 2.25 × 106 bp (genome size of pneumococcus [9]) and G = 1,000 bp (average gene size from sequenced bacteria such as H. influenzae [8] and Methanococcus jannaschii [2]), then N = L/G = 2,250.
Approximate culture volume necessary to knock out 95% of the genes in the genome with 99% probability.
The case of a transposon targeting randomly throughout the genome is included for comparison.
To explore the effect of insert length on required library size, we calculated conditions for at least 95% of the genes to be mutated with 99% probability. Roughly 9,000 plasmids are required with randomly chosen 100-bp inserts, 11,000 plasmids are required with 300-bp inserts, 17,000 plasmids are required with 400-bp inserts, and 19,000 plasmids are required with 500-bp inserts.
Finally, to select a practicable insert size, we calculated the volume of cell culture (and, by implication, the amount of plasmid DNA) necessary to obtain libraries of pneumococcal insertion mutant cells of a given transformant number produced by 95% mutagenic libraries of plasmids with different inserts (Table 3) by using Equation 1 (see Fig. 3). If a library of plasmids with 100-bp inserts is used, the work necessary for knocking out 95% of genes with 99% probability is quite large. (In addition to culturing 15,750 ml of cells, 2 × 15,750 plates would be required to select the transformants.) For 200-bp inserts, the work necessary for the same level of mutagenesis is still rather large but is not prohibitive. As insert size increases further, the work necessary to achieve the same insertion rate is reduced further, but the fraction of nonmutable genes increases strongly as I approaches 1,000. Overall, we think that the smallest insert requiring a practical amount of work for mutating almost all genes (95 or 99%) is 200 to 300 bp. Of course, genes smaller than 1,000 bp would be hit less efficiently, and larger ones would be hit more efficiently, while genes smaller than 300 bp would not be mutated at all; however, these values offer a valuable rough guide for planning experiments.
The lower limit to the size of targeting fragments for library construction means that very small genes will escape mutation by this method unless revealed by polar insertion in upstream genes of the same operon. For these very small genes, an attractive alternative would be transposon mutagenesis because transposons typically disrupt target genes with negligible duplication of target sequences. If a transposon with essentially no target preference were identified, then the statistical function Σi=kN Pi could be used to predict the degree of saturation expected by setting I = 0. The corresponding predictions for a model 1-kb-gene genome and a random transposon are included in Table 3.
ACKNOWLEDGMENTS
This work was supported in part by the U.S. National Science Foundation (award MCB-9722821).
We thank Brian Dougherty for the sequence data on several new plasmids and David Engstrom for technical help.
REFERENCES
- 1.Biswas I, Gruss A, Ehrlich S D, Maguin E. High-efficiency gene inactivation and replacement system for gram-positive bacteria. J Bacteriol. 1993;175:3628–3635. doi: 10.1128/jb.175.11.3628-3635.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bult C J, White O, Olsen G J, et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 3.Canosi U, Morelli G, Trautner T A. The relationship between molecular structure and transformation efficiency of some S. aureus plasmids isolated from B. subtilis. Mol Gen Genet. 1978;166:259–267. doi: 10.1007/BF00267617. [DOI] [PubMed] [Google Scholar]
- 4.Cato A, Guild W R. Transformation and DNA size. J Mol Biol. 1968;37:157–178. doi: 10.1016/0022-2836(68)90080-6. [DOI] [PubMed] [Google Scholar]
- 5.Chen J D, Morrison D A. Cloning of Streptococcus pneumoniae DNA fragments in E. coli requires vectors protected by strong transcriptional terminators. Gene. 1987;55:179–187. doi: 10.1016/0378-1119(87)90278-2. [DOI] [PubMed] [Google Scholar]
- 5a.Claverys, J.-P. Personal communication.
- 6.Claverys J-P, Dintilhac A, Pestova E V, Martin B, Morrison D A. Construction and evaluation of new drug-resistance cassettes for gene disruption mutagenesis in Streptococcus pneumoniae, using an ami test platform. Gene. 1995;164:123–128. doi: 10.1016/0378-1119(95)00485-o. [DOI] [PubMed] [Google Scholar]
- 7.Claverys J-P, Louarn J M, Sicard A M. Cloning of Streptococcus pneumoniae DNA: its use in pneumococcal transformation and in studies of mismatch repair. Gene. 1981;13:65–73. doi: 10.1016/0378-1119(81)90044-5. [DOI] [PubMed] [Google Scholar]
- 8.Fleischmann R D, Adams M D, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 9.Gasc A M, Kauc L, Barraillé P, Sicard M, Goodgal S. Gene localization, size, and physical map of the chromosome of Streptococcus pneumoniae. J Bacteriol. 1991;173:7361–7367. doi: 10.1128/jb.173.22.7361-7367.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Håvarstein L S, Coomaraswamy G, Morrison D A. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc Natl Acad Sci USA. 1995;92:11140–11144. doi: 10.1073/pnas.92.24.11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khasanov F K, Zvingila D J, Zainullin A A, Prozorov A A, Bashkirov V I. Homologous recombination between plasmid and chromosomal DNA in Bacillus subtilis requires approximately 70 bp of homology. Mol Gen Genet. 1992;234:494–497. doi: 10.1007/BF00538711. [DOI] [PubMed] [Google Scholar]
- 12.Lacks S A. Theoretical relationship between probability of marker integration and length of donor DNA in pneumococcal transformation. J Mol Biol. 1968;37:179. doi: 10.1016/0022-2836(68)90081-8. [DOI] [PubMed] [Google Scholar]
- 13.Lacks S A. Modes of DNA interaction in bacterial transformation. In: Chopra V L, editor. Genetics: new frontiers. Vol. 1. New Delhi, India: Oxford and IBH; 1984. pp. 149–158. [Google Scholar]
- 14.Lacks S A. Mechanisms of genetic recombination in gram-positive bacteria. In: Kucherlapati R, Smith G, editors. Genetic recombination. Washington, D.C: American Society for Microbiology; 1988. pp. 43–86. [Google Scholar]
- 15.Lacks S A. Cloning and expression of pneumococcal genes in Streptococcus pneumoniae. Microb Drug Resist. 1997;3:327–337. doi: 10.1089/mdr.1997.3.327. [DOI] [PubMed] [Google Scholar]
- 16.Lataste H, Claverys J-P, Sicard A M. Relation between the transforming activity of a marker and its proximity to the end of the DNA particle. Mol Gen Genet. 1981;183:199–201. doi: 10.1007/BF00270163. [DOI] [PubMed] [Google Scholar]
- 17.Leloup L, Ehrlich S D, Zagorec M, Moreldeville F. Single-crossover integration in the Lactobacillus sake chromosome and insertional inactivation of the ptsI and lacL genes. Appl Environ Microbiol. 1997;63:2117–2123. doi: 10.1128/aem.63.6.2117-2123.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mannarelli B M, Lacks S A. Ectopic integration of chromosomal genes in Streptococcus pneumoniae. J Bacteriol. 1984;160:867–873. doi: 10.1128/jb.160.3.867-873.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mejean V, Claverys J-P, Vasseghi H, Sicard A M. Rapid cloning of specific DNA fragments of Streptococcus pneumoniae by vector integration into the chromosome followed by endonucleolytic excision. Gene. 1981;15:289–293. doi: 10.1016/0378-1119(81)90139-6. [DOI] [PubMed] [Google Scholar]
- 20.Michel B, Niaudet B, Ehrlich S D. Intermolecular recombination during transformation of Bacillus subtilis competent cells by monomeric and dimeric plasmids. Plasmid. 1983;10:1–10. doi: 10.1016/0147-619x(83)90052-5. [DOI] [PubMed] [Google Scholar]
- 21.Morrison D A, Guild W R. Activity of deoxyribonucleic acid fragments of defined size in Bacillus subtilis transformation. J Bacteriol. 1973;112:220–223. doi: 10.1128/jb.112.1.220-223.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison D A, Lacks S A, Guild W R, Hageman J M. Isolation and characterization of three new classes of transformation-deficient mutants of Streptococcus pneumoniae that are defective in DNA transport and genetic recombination. J Bacteriol. 1983;156:281–290. doi: 10.1128/jb.156.1.281-290.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrison D A, Trombe M-C, Hayden M K, Waszak G, Chen J-D. Isolation of transformation-deficient Streptococcus pneumoniae mutants defective in control of competence using insertion-duplication mutagenesis with the erythromycin resistance determinant of pAMβ1. J Bacteriol. 1984;159:870–876. doi: 10.1128/jb.159.3.870-876.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mortier-Barriere I, Humbert O, Martin B, Prudhomme M, Claverys J-P. Control of recombination rate during transformation of Streptococcus pneumoniae: an overview. Microb Drug Resist. 1997;3:233–242. doi: 10.1089/mdr.1997.3.233. [DOI] [PubMed] [Google Scholar]
- 25.Pestova E V, Håvarstein L S, Morrison D A. Regulation of competence for genetic transformation in Streptococcus pneumoniae by an auto-induced peptide pheromone and a two-component regulatory system. Mol Microbiol. 1996;21:853–862. doi: 10.1046/j.1365-2958.1996.501417.x. [DOI] [PubMed] [Google Scholar]
- 26.Pestova E V, Morrison D A. Isolation and characterization of three Streptococcus pneumoniae transformation-specific loci by use of a lacZ reporter insertion vector. J Bacteriol. 1998;180:2701–2710. doi: 10.1128/jb.180.10.2701-2710.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puyet A, Greenberg B, Lacks S A. Genetic and structural characterization of EndA, a membrane-bound nuclease required for transformation of Streptococcus pneumoniae. J Mol Biol. 1990;213:727–738. doi: 10.1016/S0022-2836(05)80259-1. [DOI] [PubMed] [Google Scholar]
- 28.Rhee D K, Morrison D A. Genetic transformation in Streptococcus pneumoniae: molecular cloning and characterization of recP, a gene required for genetic recombination. J Bacteriol. 1988;170:630–637. doi: 10.1128/jb.170.2.630-637.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saunders C W, Guild W R. Monomer plasmid DNA transforms Streptococcus pneumoniae. Mol Gen Genet. 1981;181:57–62. doi: 10.1007/BF00339005. [DOI] [PubMed] [Google Scholar]
- 30.Shen P, Huang H V. Homologous recombination in Escherichia coli: dependence on substrate length and homology. Genetics. 1986;112:441–457. doi: 10.1093/genetics/112.3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vasseghi H, Claverys J-P, Sicard A M. Mechanism of integrating foreign DNA during transformation of Streptococcus pneumoniae. In: Polsinelli M, Mazza G, editors. Transformation—1980. Oxford, England: Cotswold Press, Ltd.; 1981. pp. 137–153. [Google Scholar]
- 32.Watt V M, Ingles C J, Urdea M S, Rutter W J. Homology requirements for recombination in Escherichia coli. Proc Natl Acad Sci USA. 1985;82:4768–4772. doi: 10.1073/pnas.82.14.4768. [DOI] [PMC free article] [PubMed] [Google Scholar]