

Abstract
Introduction
We previously reported genetic associations of the top Alzheimer's disease (AD) risk alleles with amyloid deposition and neurodegeneration. Here, we report the association of these variants with [18F]flortaucipir standardized uptake value ratio (SUVR).
Methods
We analyzed the [18F]flortaucipir scans of 352 cognitively normal (CN), 160 mild cognitive impairment (MCI), and 54 dementia (DEM) participants from Alzheimer's Disease Neuroimaging Initiative (ADNI)2 and 3. We ran step‐wise regression with log‐transformed [18F]flortaucipir meta—region of interest SUVR as the outcome measure and genetic variants, age, sex, and apolipoprotein E (APOE) ε4 as predictors. The results were visualized using parametric mapping at familywise error cluster‐level–corrected P < .05.
Results
APOE ε4 showed significant (P < .05) associations with tau deposition across all disease stages. Other significantly associated genes include variants in ABCA7 in CN, CR1 in MCI, BIN1 and CASS4 in MCI and dementia participants.
Discussion
We found significant associations to tau deposition for ABCA7, BIN1, CASS4, and CR1, in addition to APOE ε4. These four variants have been previously associated with tau metabolism through model systems.
Keywords: Alzheimer's disease, Alzheimer's Disease Neuroimaging Initiative, flortaucipir, imaging genetics, risk genes
1. INTRODUCTION
An estimated 5.8 million people in the United States are currently living with Alzheimer's disease (AD), of which 99% of cases are considered sporadic in origin. 1 In contrast to the Mendelian autosomal dominant inheritance of single gene mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) in familial early‐onset AD, the risk for sporadic AD is conferred by a large number of genetic and environmental risk factors. 2 , 3 , 4
Understanding the complex network of genetic risk factors contributing to sporadic AD has been challenging. For many years, the only risk gene associated with sporadic AD was the apolipoprotein E (APOE) gene. APOE has a dose‐dependent risk, where one copy of the ε4 allele triples AD risk while two copies increase risk by as much as 12‐fold. 1 Although this variant confers significant risk, it has been estimated that APOE ε4 accounts for less than half of the total genetic hazard. 5 , 6
To explain the remaining genetic risk, large‐scale genome‐wide association studies (GWAS) and meta‐analysis of GWAS studies have identified risk variants in or near 20 genes involved in pathways such as cholesterol metabolism, immune response, and endocytosis. 7 , 8 , 9 , 10 , 11 , 12 Though each of these variants contributes a relatively small amount of the overall risk, inheriting multiple risk variants results in a risk summation. While many of the genes have been attributed to known dysfunctional pathways in AD, the precise mechanisms of action of a significant number of them are still yet to be uncovered.
Through the use of imaging genetics, our group previously associated the top AD GWAS‐identified variants with imaging biomarkers of amyloidosis, brain hypometabolism, and atrophy using data from the Alzheimer's Disease Neuroimaging Initiative (ADNI). 13 , 14 We used a multivariable model to find variant associations with AD‐specific disease biomarker endophenotypes, across the three diagnostic categories—cognitively normal (CN), mild cognitive impairment (MCI), and dementia (DEM)—as well as in a pooled sample, to capture disease‐specific associations across the AD spectrum. We reported stage‐dependent associations of FERMT2 rs17125944 with amyloid deposition; 13 EPHA1 rs11771145 and SLC24A4/RIN3 rs10498633 with brain atrophy; 14 and CD2AP rs9349407, CLU rs9331949, and NME8 rs2718058 with brain metabolic activity 14 indicating that the genetic impact on AD evolves as the disease progresses.
RESEARCH IN CONTEXT
Systematic Review: Relevant literature was reviewed using standard scholarly databases (PubMed, Google Scholar, etc.). Previously our group and others have identified genetic risk variants associated with amyloidosis and neurodegeneration using imaging genetics of Alzheimer's disease (AD).
Interpretation: Ten of the top 20 AD risk genes showed an association with tau deposition in an AD‐like pattern. This, in conjunction with our previous work, reveals biomarker‐specific associations within the amyloid/tau/neurodegeneration framework and across the disease spectrum.
Future Directions: Increasing the sample size and including a longitudinal component may further clarify the genetic variant–biomarker associations in AD. Discovering the precise mechanisms of action of these genes on tau deposition requires further investigation.
Ten genes from the top 20 Alzheimer's disease (AD) risk genes were associated with tau deposition.
We previously associated these 20 risk genes with amyloid positron emission tomography (PET), fluorodeoxyglucose PET, and magnetic resonance imaging.
These associations underscore tau's important role in AD pathophysiology.
With the addition of tau positron emission tomography (PET) neuroimaging to the ADNI protocol, we were able to expand on the previous work to conduct a multivariable analysis of the associations of GWAS‐validated AD risk variants with cortical tau burden, which was measured via [18F]flortaucipir PET standardized uptake value ratio (SUVR). The goal of this study was to establish the relative genetic contribution to tau burden using stepwise multivariable regression as we have done previously for brain amyloidosis and neurodegeneration. 13 , 14
2. METHODS
2.1. Participants
Data used in the preparation of this article were obtained from the ADNI database (http://adni.loni.usc.edu/). ADNI was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. For up‐to‐date information, see www.adni‐info.org.
The clinical description of the ADNI cohort has been previously published. 15 , 16 , 17 Diagnosis of AD was based on the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria. 18 , 19 , 20 Individuals with AD dementia were required to have Mini‐Mental State Examination (MMSE) scores between 20 and 26 and a Clinical Dementia Rating (CDR) score of 0.5 to 1 at baseline. Qualifying individuals with MCI had memory concerns but no significant functional impairment, scored between 24 and 30 on the MMSE, had a global CDR score of 0.5, had a CDR memory score of 0.5 or greater, and had objective memory impairment on the Wechsler Memory Scale–Logical Memory II test. The controls had MMSE scores between 24 and 30, had a global CDR score of 0, and did not meet criteria for MCI and AD. Individuals were excluded if they refused or were unable to undergo MRI, had other neurologic disorders, active depression, a history of psychiatric diagnosis, a history of alcohol or other substance dependence within the past 2 years, had less than 6 years of education, or were not fluent in English or Spanish. The full list of inclusion and exclusion criteria are listed in the online ADNI protocol ( http://adni.loni.usc.edu/wp‐content/uploads/2010/09/ADNI_GeneralProceduresManual.pdf ). Written informed consent was obtained from all participants. Only de‐identified data were used in these analyses.
All non‐Hispanic White ADNI subjects who enrolled or rolled over into ADNI2/3 were included if they had both genetic data and [18F]flortaucipir scans. This resulted in a sample containing 352 CN, 160 MCI, and 54 DEM subjects.
2.2. Gene variant selection and imputation
Participants were genotyped using either the Illumina Human610‐Quad BeadChip (ADNI1), Illumina HumanOmniExpress BeadChip (ADNIGO/2), or Illumina Infinium Global Screening v2 (ADNI3) arrays. Intensity data were processed using either GenomeStudio v2009.1 (ADNI1/GO/2) or GenomeStudio v2.0.4 (ADNI3) according to Illumina Inc. protocols. More information on the genetics core can be found at http://adni.loni.usc.edu/data‐samples/data‐types/genetic‐data/.
Similar to our previous studies, we narrowed our focus to variants in the top 20 well‐established AD risk genes identified and validated in the largest AD GWAS to date. 7 , 8 , 9 , 10 , 11 , 12 We also included all other variants contained within these genes that have previously been associated with brain amyloidosis, a key pathological hallmark of AD. The full list of the 36 variants considered in our analysis can be seen in Table S1 in supporting information.
Missing genotypes (Table S2 in supporting information) were imputed using MaCH and minimac, relying on the 1000 Genomes project data as a reference panel. We applied a low genotype threshold of 0.05, meaning subjects missing >5% of the genotypes were removed. Posterior probabilities of the imputed genotypes for each individual were determined in minimac. We used an imputation threshold of r 2 = 0.30 to accept the imputed genotype per MaCH recommendations (https://genome.sph.umich.edu/wiki/MaCH_FAQ).
For the nine genes containing more than one single nucleotide polymorphism (SNP), we performed linkage disequilibrium (LD) analysis, followed by Cohen κ statistics to guard against collinearity bias (Table S3 and Figure S1 in supporting information). In situations in which variants had significant overlap (D’ ≥ 0.4), we retained the variant with the least missing data. A total of 27 variants were included in our final regression models including several variants from the ABCA7, BIN1, CLU, CR1, EPHA1, and SORL1 genes that were not in LD.
Genotypes were coded by the number of minor alleles (0/1/2 copies); however, when minor allele homozygote frequency was less than 2% the genotype was collapsed into the presence or absence of minor allele (coded as 1 or 0). This was the case for five of the variants: ABCA7 rs3764650, CASS4 rs7274581, CLU rs9331949, DSG2 rs8093731, and SORL1 rs11218343.
2.3. [18F]flortaucipir PET data acquisition protocol and analyses
Standardized [18F]flortaucipir acquisition and preprocessing protocols can be found at www.adni‐info.org. In our main analysis, we downloaded the ADNI University of California (UC) Berkeley [18F]flortaucipir partial volume corrected (PVC) SUVR FreeSurfer 6.0 data from ADNI's website (http://adni.loni.usc.edu). This website also houses the detailed description of the processing methods for UC Berkeley. We computed a size‐weighted AD‐specific meta‐region of interest (ROI) using the a priori regions defined by Jack et al., containing entorhinal, amygdala, parahippocampal, fusiform, inferior temporal, and middle temporal regions. 21 The meta‐ROI was then intensity‐normalized to the inferior cerebellar gray‐matter reference region (defined by UC Berkeley).
2.4. Statistical analysis
Clinical, demographic, and biomarker values of interest (age, sex, number of APOE ε4 alleles, MMSE, and [18F]flortaucipir meta‐ROI SUVR) for each diagnostic group (CN, MCI, and DEM) were compared using analysis of variance (ANOVA) or χ 2 tests with 2‐sided P‐values as appropriate. Because the meta‐ROI SUVR data were highly skewed, we normalized the data using a natural log‐transformed meta‐ROI SUVR as the outcome measure. Variant association with the [18F]flortaucipir meta‐ROI SUVR was determined using multivariable stepwise linear regression models in SAS 9.4, with all 27 AD risk variants included as predictors, adjusting for age, sex, and APOE ε4 genotype. For the pooled sample we also controlled for diagnosis. Model selection was based on the Akaike information criterion (AIC) critical P‐value threshold of 0.157. 22 We used false discovery rate (FDR)‐corrected P‐values to protect against type I errors using the Yekutieli and Benjamini 23 method. We report gene variant selections in Table 2, though it is worth noting that we also included APOE ε4 outputs (despite being a covariant in all models) as it plays a crucial role in AD and in general had a very large effect. In addition, we report mean SUVR by copy number of each risk variant retained in each model.
TABLE 2.
Regression results in each diagnostic group
| Pooled sample, model R 2 = 0.3970/adjusted R 2 = 0.3861, P < .0001 | |||||
|---|---|---|---|---|---|
| Variants selected | Parameter estimate | Standard error | Parameter P‐value/FDR‐corrected P‐value | [18F]flortaucipir SUVR by risk allele | SUVR P‐value |
| APOE ε4 | 0.052 | 0.011 | <.0001 | 1.54/1.64/2.01 | <.001 |
| ABCA7 rs3752246 | 0.024 | 0.013 | .0567/.1395 | 1.59/1.62/1.81 | .097 |
| BIN1 rs6733839 | 0.014 | 0.010 | .1395/.1395 | 1.56/1.64/1.62 | .100 |
| CR1 rs3818361 | 0.019 | 0.012 | .1211/.1395 | 1.59/1.64/1.61 | .325 |
| NME8 rs2718058 | –0.018 | 0.010 | .0785/.1395 | 1.66/1.57/1.56 | .037 |
| ZCWPW1 rs1476679 | –0.017 | 0.011 | .0991/.1395 | 1.66/1.54/1.58 | .007 |
| CN sample, model R 2 = 0.0971/adjusted R 2 = 0.0840, P < .0001 | |||||
|---|---|---|---|---|---|
| Variants selected | Parameter estimate | Standard error | Parameter P‐value/FDR‐corrected P‐value | [18F]flortaucipir SUVR by risk allele | SUVR P‐value |
| APOE ε4 | 0.0255 | 0.011 | .0195 | 1.47/1.48/1.65 | .014 |
| ABCA7 rs3764650 | 0.044 | 0.014 | .0028 /.0057 | 1.46/1.54 | .003 |
| CR1 rs12034383 | –0.015 | 0.008 | .0799/.0799 | 1.50/1.49/1.46 | .365 |
| MCI sample, model R 2 = 0.2108/adjusted R 2 = 0.1744, P < .0001 | |||||
|---|---|---|---|---|---|
| Variants selected | Parameter estimate | Standard error | Parameter P‐value/FDR‐corrected P‐value | [18F]flortaucipir SUVR by risk allele | SUVR P‐value |
| APOE ε4 | 0.076 | 0.020 | .0002 | 1.55/1.76/1.78 | <.001 |
| BIN1 rs6733839 | 0.045 | 0.020 | .0272 /.0464 | 1.54/1.68/1.68 | .037 |
| CASS4 rs7274581 | 0.074 | 0.035 | .0348 /.0464 | 1.61/1.74 | .065 |
| CLU rs9331949 | 0.113 | 0.070 | .1091/.1091 | 1.63/1.81 | .206 |
| CR1 rs3818361 | 0.051 | 0.024 | .0324 /.0464 | 1.59/1.70/1.80 | .099 |
| DEM sample, model R 2 = 0.4240/adjusted R 2 = 0.3216, P = .0009 | |||||
|---|---|---|---|---|---|
| Variants selected | Parameter estimate | Standard error | Parameter P‐value/FDR‐corrected P‐value | [18F]flortaucipir SUVR by risk allele | SUVR P‐value |
| APOE ε4 | 0.095 | 0.046 | .0455 | 2.21/2.29/2.60 | .332 |
| BIN1 rs744373 | –0.177 | 0.064 | .0085 /.0423 | 2.38/2.37/1.72 | .271 |
| CASS4 rs7274581 | 0.257 | 0.104 | .0176 /.0441 | 2.27/2.63 | .206 |
| DSG2 rs8093731 | –0.426 | 0.276 | .1296/.1296 | 2.35/1.49 | .278 |
| EPHA1 rs11767557 | 0.110 | 0.071 | .1264/.1296 | 2.23/2.47/2.99 | .406 |
| SORL1 rs11218343 | –0.208 | 0.116 | .0794/.1296 | 2.36/2.05 | .350 |
Note: Final results from stepwise linear regression models using 27 AD risk variants, including covariates of age, sex, and APOE ε4 (and diagnosis for pooled model) and log‐transformed outcome measure. Model selection for the risk variants was P = .157.
Abbreviations: AD, Alzheimer's disease; ANOVA, analysis of variance; APOE, apolipoprotein E; CN, cognitively normal; DEM, dementia; FDR, false discovery rate; MCI, mild cognitive impairment; SUVR, standardized uptake value ratio.
2.5. Analysis in imaging space
Next, we visualized the selected variant spatial effects in SPM12. Preprocessed [18F]flortaucipir scans were downloaded from ADNI ( http://adni.loni.usc.edu/ ), where PET frames were coregistered, averaged, and image and voxel sizes were standardized and smoothed to a uniform resolution. Using SPM12, each subject's preprocessed [18F]flortaucipir scan was then coregistered to that subject's closest‐visit MRI, spatially normalized into Montreal Neurological Institute space, and intensity normalized to the cerebellar crus to generate SUVR images. To visualize the spatial distribution of the genetic associations, we reproduced the regression models using voxelwise regression in SPM12. For display purposed our figures only include variants that were significant (uncorrected P < .05) or trending (uncorrected P < .10) in the models for meta‐ROI SUVR. As with our regression modeling in SAS, we covaried for age, sex, and APOE ε4 genotype, including diagnosis as a covariate in the pooled sample. The variant associations were displayed at a cluster‐level familywise error (FWE)–corrected P < .05, which was used instead of FDR as it is more easily integrated into an SPM analysis.
3. RESULTS
Group comparisons of clinical, demographic, biomarker, and carrier distribution of variants retained in the regression models are seen in Table 1. Our sample included 352 CN, 160 MCI, and 54 DEM subjects who had available GWAS and [18F]flortaucipir data. There were significant differences in age, sex, APOE ε4 carrier percentage, MMSE, and meta‐ROI SUVR among diagnostic groups (P < .001 for all). All gene variant selection models were corrected for age, sex, and APOE ε4. There were no significant diagnostic group differences in minor allele distribution for any of the gene variants selected in our regression models.
TABLE 1.
Demographic, biomarker data, and minor allele distribution for variants retained in the regression models in each diagnostic group
| Variables | CN (N = 352) | MCI (N = 160) | DEM (N = 54) | P ‐value |
|---|---|---|---|---|
| Age, years, mean (SD) | 73.6 (7.1) | 75.7 (8.2) | 77.4 (9.1) | <.001 |
| Sex, % male | 43.5 | 65.0 | 53.7 | <.001 |
| APOE ε4 alleles, % 0/1/2 | 66/31/3 | 61/28/11 | 43/33/24 | <.001 |
| MMSE, mean (SD) | 29.1 (1.3) | 27.4 (2.5) | 20.2 (6.0) | <.001 |
|
Meta‐ROI flortaucipir SUVR, mean (SD) |
1.48 (0.19) | 1.63 (0.35) | 2.33 (0.77) | <.001 |
| ABCA7 rs3752246, % 0/1/2 | 63/34/4b | 73/26/1 | 63/33/4 | .098 |
| ABCA7 rs3764650, % 0/1a | 80/20 | 85/15 | 85/15 | .334 |
| BIN1 rs6733839, % 0/1/2 | 40/43/17 | 34/52/14 | 35/46/19 | .394 |
| BIN1 rs744373, % 0/1/2 | 49/42/9 | 48/42/10 | 46/46/7b | .968 |
| CASS4 rs7274581, % 0/1a | 85/15 | 81/19 | 83/17 | .627 |
| CLU rs9331949, % 0/1a | 94/6 | 96/4 | 96/4 | .433 |
| CR1 rs12034383, % 0/1/2 | 15/49/36 | 16/48/36 | 9/52/39 | .807 |
| CR1 rs3818361, % 0/1/2 | 68/29/4b | 64/32/4 | 63/35/2 | .757 |
| DSG2 rs8093731, % 0/1a | 97/3 | 98/2 | 98/2 | .869 |
| EPHA1 rs11767557, % 0/1/2 | 66/30/4 | 66/31/3 | 63/35/2 | .913 |
| NME8 rs2718058, % 0/1/2 | 38/48/15b | 41/48/12b | 46/43/11 | .734 |
| SORL1 rs11218343, % 0/1a | 90/11b | 92/8 | 89/11 | .670 |
| ZCWPW1 rs1476679, % 0/1/2 | 51/41/8 | 46/43/11 | 67/32/2b | .050 |
Note: This table shows a demographic, neuropsychological, and imaging measure comparison across each of the diagnostic groups. We also display the percentage of subjects who had each copy number for the genes retained in any of the four regression models. P‐values were generated using ANOVA and Chi‐square where necessary.
aCollapsed because minor allele homozygote frequency was <2%.
bDoes not add up to 100% due to rounding.
Abbreviations: ANOVA, analysis of variance; APOE, apolipoprotein E; CN, cognitively normal; DEM, dementia; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; ROI, region of interest; SD, standard deviation; SUVR, standardized uptake value ratio.
In addition, we investigated demographic differences between risk variant minor allele carriers and noncarriers among the variants retained in our regression (not shown). We observed no significant differences in age, sex, or APOE ε4 carrier percentage.
3.1. Pooled sample
In the pooled sample, our stepwise regression model achieved an R 2 value of 0.397 (Table 2). APOE ε4 was significantly associated with the meta‐ROI SUVR (P < .0001). While ABCA7 rs3752246 (P FDR = 0.1395), NME8 rs2718058 (P FDR = 0.1395), and ZCWPW1 rs1476679 (P FDR = 0.1395) were associated with tau meta‐ROI SUVR at an uncorrected trend level, they did not survive FDR correction. Visual assessment of voxelwise analysis displayed significant large clusters of association to tau deposition with APOE ε4 and ABCA7 rs3752246 in an AD‐like pattern including temporoparietal and frontal regions (Figure 1). ZWPW1 rs1476679 showed a more restricted right lateralized association pattern primarily in the inferior and temporal polar regions.
FIGURE 1.
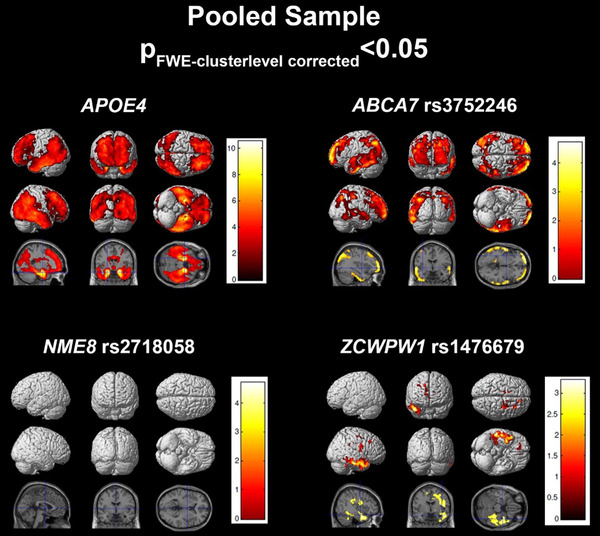
Regression results in the pooled sample. SPM visualization of association patterns for the regression selected significant or trending genes (P < .10). Results are displayed at a familywise error (few)‐cluster–level correction of P < .05
3.2. CN sample
In CN subjects, the regression model achieved an R 2 of 0.0971 (Table 2). APOE ε4 (P = .0195) and ABCA7 rs3764650 (P FDR = 0.0057) were significantly associated with tau meta‐ROI SUVR, while CR1 rs12034383 (P FDR = 0.0799) was associated at a trend level. Voxelwise analysis shows that all three variants have significant associations with tau deposition in the temporal, parietal, and frontal lobes. APOE ε4 and CR1 rs12034383 showed additional associations in the occipital region (Figure 2).
FIGURE 2.
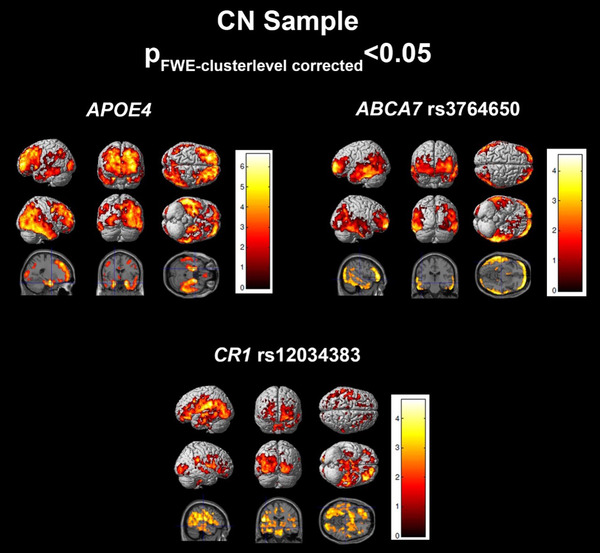
Regression results in the cognitively normal (CN) sample. SPM visualization of association patterns for the regression selected significant or trending genes (P < .10). Results are displayed at a familywise error (few)‐cluster–level correction of P < .05
3.3. MCI sample
In MCI subjects, the regression model achieved an R 2 of 0.2108 (Table 2). APOE ε4 (P = .0002), BIN1 rs6733839 (P FDR = 0.0464), CASS4 rs7274581 (P FDR = 0.0464), and CR1 rs3818361 (P FDR = 0.0464) were significantly associated with tau deposition. Voxelwise analysis displayed right greater than left significant associations to tau burden for APOE ε4 and CR1 rs3818361 in temporal, parietal, and occipital cortices (Figure 3). CASS4 rs7274581 showed significant association in temporoparietal regions. Interestingly, voxelwise analysis of BIN1 rs6733839 did not show any FWE cluster‐level corrected associations to tau deposition.
FIGURE 3.
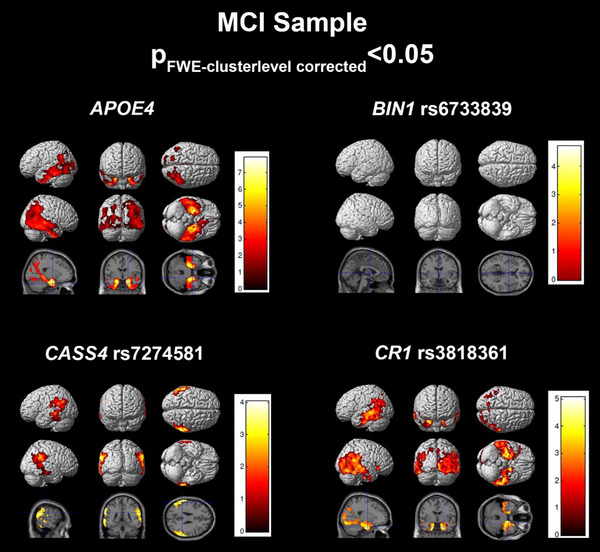
Regression results in the mild cognitive impairment (MCI) sample. SPM visualization of association patterns for the regression selected significant or trending genes (P < .10). Results are displayed at a familywise error (few)‐cluster–level correction of P < .05
3.4. DEM sample
In DEM subjects, the regression model achieved an R 2 of 0.4240 (Table 2). APOE ε4 (P = .0455), BIN1 rs744373 (P FDR = 0.0423), and CASS4 7274581 (P FDR = 0.0464) were significantly associated with [18F]flortaucipir meta‐ROI SUVR. SORL1 rs11218343 (P FDR = 0.1296) was associated to tau at an uncorrected trend level. Voxelwise analysis of BIN1 rs744373 showed significant association with tau in left temporal and parietal and bilateral occipital lobes (Figure 4). CASS4 rs7274581 showed association to tau burden in the bilateral occipital and temporo‐occipital, right inferior temporal regions, and sensorimotor cortices. APOE ε4 was significantly associated to tau in left medial temporal and temporoparietal cortices.
FIGURE 4.
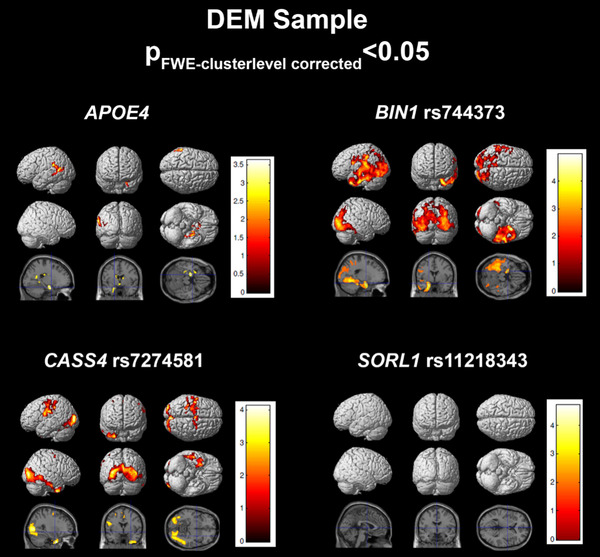
Regression results in the dementia (DEM) sample. SPM visualization of association patterns for the regression selected significant or trending genes (P < .10). Results are displayed at a familywise error (few)‐cluster–level correction of P < .05
4. DISCUSSION
To our knowledge, this is the first comprehensive analysis of the association of the top 20 AD risk genes with tau burden. This work highlights the importance of modeling genetic associations in AD, as well as other complex diseases, in a polygenic fashion. Associations with tau burden were seen in each diagnostic group, for some variants across multiple diagnostic groups. The reason behind this likely is very complex and involves direct and indirect relationships to tau burden with regional associations that complicate our understanding further. We replicated previously reported associations to tau for ABCA7, BIN1, CASS4, CLU, CR1, EPHA1, NME8, and SORL1, while identifying novel associations for DSG2 and ZCWPW1. 24 , 25 , 26 , 27 , 28 , 29 Like with our previous work, 13 , 14 we also visualized the spatial associations using voxelwise regression maps (Figures 1, 2, 3, 4).
In total, we found 10 of the top 20 genes implicated in late‐onset AD to have an association with tau burden. To better understand the potential role each gene may play in tau pathology, we provide a brief literature review that includes probable functions as well as a previous association to tau (if applicable) and any associations to amyloidosis or neurodegeneration from our earlier publications 13 , 14 using a similar technique and sample.
4.1. APOE
Apolipoprotein E encodes a 317 amino acid apolipoprotein involved in a variety of pathways from lipoprotein metabolism to neuronal maintenance and repair. 30 , 31 APOE has three major alleles, ε2, ε3 (the most common), and ε4 (the risk allele for sporadic AD). Though the APOE ε4 variant has long been associated with the amyloid pathway, 32 recent work has also linked APOE ε4 to tau pathology and neurodegeneration. 33 We previously identified a strong effect of APOE ε4 on amyloid deposition across all diagnostic stages and with atrophy and hypometabolism in dementia and MCI, respectively. 13 , 14 APOE ε4 was identified to have a significant association with tau burden across all disease stages, with the strongest association in CN and MCI subjects.
4.2. ABCA7
ATP‐binding cassette subfamily A member 7 (ABCA7) encodes a 2146 amino acid member of the ABC transporter family comprising proteins involved in lipid transport and plays a role in macrophage‐mediated phagocytosis. 34 ABCA7 loss of function was associated with increased production of amyloid beta. 35 ABCA7 has also been linked to cerebrospinal fluid (CSF) and [18F]florbetapir measured amyloidosis, 36 while expression of ABCA7 has been associated with tangle density. 24 In our earlier work, ABCA7 was associated with amyloidosis early in the disease course (i.e., in CN and MCI) and with atrophy in the later disease stages (i.e., DEM). 13 , 14 Here, ABCA7 association to the AD meta‐ROI was found in our pooled sample for ABCA7 rs3752246 and in the CN sample for ABCA7 rs3764650 variant. This staged effect (early association with amyloid and tau and late association with brain atrophy) is consistent with the notion that tau deposition precedes, and potentially drives, neurodegeneration in AD.
4.3. BIN1
Bridging integrator 1 (BIN1) encodes a 593 amino acid protein that may be involved with endocytosis of synaptic vesicles and trafficking as well as control of amyloid production. 37 Additionally, the gene product of BIN1 has been shown to directly interact with tau and further impact phosphorylation of tau. 25 BIN1 was recently associated with increased binding of [18F]flortaucipir. 26 We failed to find BIN1 associations with amyloidosis or atrophy/hypometabolism in our prior work. 13 , 14 Here we found associations with tau of BIN1 rs6733839 variant in the prodromal and BIN1 rs744373 in the dementia stages.
4.4. CASS4
Cas Scaffold Protein Family Member 4 (CASS4) encodes a 786 amino acid scaffolding protein that regulates focal adhesion kinase (FAK) signaling and activation, ultimately impacting cellular adhesion and spreading. 38 , 39 A recent study linked the CASS4 Drosophila ortholog, p130CAS, as a modulator of tau toxicity. 27 CASS4 was not associated with either amyloidosis or neurodegeneration in our previous work. 13 , 14 Here, we found a significant association of CASS4 rs7274581 variant with the [18F]flortaucipir meta‐ROI SUVR in our MCI and DEM samples.
4.5. CR1
Complement Receptor Type 1 (CR1) encodes a 2039 amino acid membrane glycoprotein that has been linked to mediation of the binding between immune cells and their targets. 40 , 41 Deletion of the murine ortholog of CR1, Crry, was previously shown to significantly reduce tau phosphorylation. 42 Here, we found that CR1 rs3818361 was associated with the AD tau meta‐ROI in a pooled and MCI sample, while CR1 rs12034383 showed associations in the CN sample. In our earlier work, CR1 was associated with hypometabolism in preclinical subjects 14 while having no associations with amyloidosis. 13
4.6. Trend‐level associations
It is worth briefly mentioning the trend‐level associations (P < .10) here as some of these genes have been previously associated with tau in AD in the pre‐existing literature. Carrying the Clusterin (CLU) rs11136000 minor allele was associated with CSF tau levels in AD patients and intracellular CLU was found to interact with tau in cell culture. 29 The Drosophila ortholog of EPH receptor A1 (EPHA1), Eph, has been identified as a tau toxicity modulator. 27 NME/NM23 Family Member 8 (NME8) rs2718058 genotype was shown to be significantly associated to CSF tau levels. 43 Sortilin related receptor 1 (SORL1) has previously been associated with CSF total and phospho‐tau protein levels. 44 , 45 Zinc finger CW‐type and PPWP domain containing 1 (ZCWPW1) has no previous reported associations with tau. To date, there have been no reported associations of Desmoglein 2 (DSG2) to tau. All five genes were associated with neurodegeneration (atrophy and/or hypometabolism) in our previous work 14 and all but NME8 were also associated with brain amyloidosis. 13
5. STRENGTHS AND LIMITATIONS
This study has several strengths and limitations that merit discussion. One of the main strengths lies in the rigorous clinical, biomarker, and genetic characterization of all individuals enrolled in ADNI. ADNI uses standardized subject assessment, data collection, and quality control practices as well as an imaging normalization to bring images from different scanner types and locations to their closest alignment. Another strength of ADNI is that it is a well‐characterized cohort. A major limitation of our study is that we report only cross‐sectional analyses, making it difficult to determine with certainty that specific genetic variants exert stage‐specific effects. However, leveraging the full dementia continuum, we suggest that there are differential early and late genetic influences on cortical tau burden. We also have not included any analysis for combined risk of carrying a variant within a loci, which possibly may identify additional associations that may otherwise have been too small at a SNP level. However, this assumes that variants within a loci associate with AD in a similar manner (conferring risk or offering protection), which was not the case in our analysis. Another limitation of our work is the sample size limiting our power to detect smaller associations, a product of the relative newness of [18F]flortaucipir scanning in ADNI. This is an emerging field of research and warrants future studies as more subjects with genotyping and imaging are added. Finally, ADNI is a clinic‐based research cohort that is not necessarily representative of the general population; thus, the continuation of our study will aim to validate our findings in a large, independent, longitudinal cohort.
6. CONCLUSIONS
Using a multivariable regression modeling approach across the cognitive continuum, we found associations of 10 of the top 20 AD risk genes to tau deposition in an AD‐specific meta‐ROI. Some of the genes showed stage‐dependent association. These observations may inform future studies on potential gene targets for further in vivo research.
CONFLICTS OF INTEREST
ES, KL, SG, and KN have no relationships/activities/interests to disclose related to the content of this submission. SR serves as an associate editor of the Diagnosis, Assessment & Disease Monitoring journal. AS has received research support from Eli Lilly and AVID Radiopharmaceuticals and serves on the editorial board of the Diagnosis, Assessment & Disease Monitoring journal. LA serves on an Advisory Board for Eli Lilly and on the Speakers Bureau for Piramal and Eli Lilly. LA has also received research support from GE Healthcare. LA serves as editor‐in‐chief of the Diagnosis, Assessment & Disease Monitoring journal.
Supporting information
SUPPORTING INFORMATION
SUPPORTING INFORMATION
{kind=link}
SUPPORTING INFORMATION
SUPPORTING INFORMATION
ACKNOWLEDGMENTS
Data used in preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wpcontent/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. The analyses reported in this manuscript were funded in part by NIA grants R01 AG040770, K02 AG048240, P30 AG010133, K01 AG049050, R56 AG057195, R01 AG019771 and U01 AG068057, NLM grant R01 LM013463, and the Easton Consortium for Alzheimer's Drug Discovery and Biomarker Development. Data collection and sharing for this project was funded by the ADNI (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer's Association; Alzheimer's Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol‐Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Stage E, Risacher SL, Lane KA, et al. Association of the top 20 Alzheimer's disease risk genes with [F]flortaucipir PET. Alzheimer's Dement. 2022;14:e12308. 10.1002/dad2.12308
REFERENCES
- 1. Alzheimer's Association . Alzheimer's disease facts and figures. Alzheimers Dement. 2019;15(3):321‐387. [Google Scholar]
- 2. Goldman JS, Hahn SE, Catania JW, et al. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med. 2011;13:597‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168‐174. [DOI] [PubMed] [Google Scholar]
- 4. Gatz M, Pedersen NL, Berg S, et al. Heritability for Alzheimer's disease: the study of dementia in Swedish twins. J Gerontol A Biol Sci Med Sci. 1997;52:M117‐M125. [DOI] [PubMed] [Google Scholar]
- 5. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921‐923. [DOI] [PubMed] [Google Scholar]
- 6. Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18:421‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harold D, Abraham R, Hollingworth P, et al. Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lambert JC, Heath S, Even G, et al. Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094‐1099. [DOI] [PubMed] [Google Scholar]
- 10. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat Genet. 2011;43:436‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome‐wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832‐1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Apostolova LG, Risacher SL, Duran T, et al. Associations of the Top 20 Alzheimer disease risk variants with brain amyloidosis. JAMA Neurol. 2018;75:328‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stage E, Duran T, Risacher SL, et al. The effect of the top 20 Alzheimer disease risk genes on gray‐matter density and FDG PET brain metabolism. Alzheimers Dement (Amst). 2016;5:53‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Petersen RC, Aisen PS, Beckett LA, et al. Alzheimer's Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petersen RC, Negash S. Mild cognitive impairment: an overview. CNS Spectr. 2008;13:45‐53. [DOI] [PubMed] [Google Scholar]
- 17. Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56:303‐308. [DOI] [PubMed] [Google Scholar]
- 18. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939‐944. [DOI] [PubMed] [Google Scholar]
- 19. Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS‐ADRDA criteria. Lancet Neurol. 2007;6:734‐746. [DOI] [PubMed] [Google Scholar]
- 20. Jack CR, Jr. , Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jack CR, Jr. , Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 2017;13:205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akaike H. Information Theory and an Extension of the Maximum Likelihood Principle. Selected Papers of Hirotugu Akaike Springer Series in Statistics (Perspectives in Statistics). New York, NY: Springer; 1998. [Google Scholar]
- 23. Yekutieli D, & Benjamini Y. Resampling‐based false discovery rate controlling multiple test procedures for correlated test statistics. JSPI. 1999;82:171‐96. [Google Scholar]
- 24. Yu L, Chibnik LB, Srivastava GP, et al. Association of brain DNA methylation in SORL1, ABCA7, HLA‐DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer Disease. JAMA Neurol. 2015;72:15‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lasorsa A, Malki I, Cantrelle FX, et al. Structural basis of Tau interaction with BIN1 and regulation by Tau phosphorylation. Front Mol Neurosci. 2018;11:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Franzmeier N, Rubinski A, Neitzel J, Ewers M, Alzheimer's Disease Neuroimaging I. The BIN1 rs744373 SNP is associated with increased tau‐PET levels and impaired memory. Nat Commun. 2019;10:1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dourlen P, Fernandez‐Gomez FJ, Dupont C, et al. Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of Tau pathology. Mol Psychiatry. 2017;22:874‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shulman JM, Imboywa S, Giagtzoglou N, et al. Functional screening in Drosophila identifies Alzheimer's disease susceptibility genes and implicates Tau‐mediated mechanisms. Hum Mol Genet. 2014;23:870‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Y, Hayashi I, Wong J, Tugusheva K, Renger JJ, Zerbinatti C. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule‐associated protein Tau in Alzheimer's disease. PLoS One. 2014;9:e103187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622‐630. [DOI] [PubMed] [Google Scholar]
- 31. Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5644‐5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer's disease: accidental encounters or partners? Neuron. 2014;81:740‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mattsson N, Ossenkoppele R, Smith R, et al. Greater tau load and reduced cortical thickness in APOE epsilon4‐negative Alzheimer's disease: a cohort study. Alzheimers Res Ther. 2018;10:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abe‐Dohmae S, Ikeda Y, Matsuo M, et al. Human ABCA7 supports apolipoprotein‐mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J Biol Chem. 2004;279:604‐611. [DOI] [PubMed] [Google Scholar]
- 35. Satoh K, Abe‐Dohmae S, Yokoyama S, St George‐Hyslop P, Fraser PE. ATP‐binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J Biol Chem. 2015;290:24152‐24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao QF, Wan Y, Wang HF, et al. ABCA7 genotypes confer Alzheimer's disease risk by modulating amyloid‐beta pathology. J Alzheimers Dis. 2016;52:693‐703. [DOI] [PubMed] [Google Scholar]
- 37. Miyagawa T, Ebinuma I, Morohashi Y, et al. BIN1 regulates BACE1 intracellular trafficking and amyloid‐beta production. Hum Mol Genet. 2016;25:2948‐2958. [DOI] [PubMed] [Google Scholar]
- 38. Singh MK, Dadke D, Nicolas E, et al. A novel Cas family member, HEPL, regulates FAK and cell spreading. Mol Biol Cell. 2008;19:1627‐1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hsia DA, Mitra SK, Hauck CR, et al. Differential regulation of cell motility and invasion by FAK. J Cell Biol. 2003;160:753‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schifferli JA, Ng YC, Estreicher J, Walport MJ. The clearance of tetanus toxoid/anti‐tetanus toxoid immune complexes from the circulation of humans. Complement‐ and erythrocyte complement receptor 1‐dependent mechanisms. J Immunol. 1988;140:899‐904. [PubMed] [Google Scholar]
- 41. Torok K, Dezso B, Bencsik A, Uzonyi B, Erdei A. Complement receptor type 1 (CR1/CD35) expressed on activated human CD4+ T cells contributes to generation of regulatory T cells. Immunol Lett. 2015;164:117‐124. [DOI] [PubMed] [Google Scholar]
- 42. Killick R, Hughes TR, Morgan BP, Lovestone S. Deletion of Crry, the murine ortholog of the sporadic Alzheimer's disease risk gene CR1, impacts tau phosphorylation and brain CFH. Neurosci Lett. 2013;533:96‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Y, Yu JT, Wang HF, et al. Association between NME8 locus polymorphism and cognitive decline, cerebrospinal fluid and neuroimaging biomarkers in Alzheimer's disease. PLoS One. 2014;9:e114777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alexopoulos P, Guo LH, Tsolakidou A, et al. Interrelations between CSF soluble AbetaPPbeta, amyloid‐beta 1‐42, SORL1, and tau levels in Alzheimer's disease. J Alzheimers Dis. 2012;28:543‐552. [DOI] [PubMed] [Google Scholar]
- 45. Louwersheimer E, Ramirez A, Cruchaga C, et al. Influence of genertic variants in SORL1 gene on the manifestation of Alzheimer's disease. Neurobiol Aging. 2015;36:1605.e1613‐e1620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION
SUPPORTING INFORMATION