

Abstract
Novel and environmentally benign ionic liquids (ILs) comprised of DABCO were successfully synthesized. These ILs were used as robust catalysts for the sonochemical one pot multi-component synthetic route for the functionalized annulated imidazoles in water with excellent yields. The present protocol scored well in terms of yield economy as compared with the conventional procedures. The merits of the current green method include simplicity, applicability, broad functional groups tolerance, clean reaction profiles, and no tedious work-up and they give high-to-quantitative yields. From the environmental viewpoint, these eco-friendly green catalysts can be effortlessly retrieved and reused at least seven successive times without substantial loss in catalytic activity.
Novel and environmentally benign ionic liquids are synthesized and used as robust catalysts for a sonochemical one pot multi-component synthetic route to functionalized annulated imidazoles in water with excellent yields.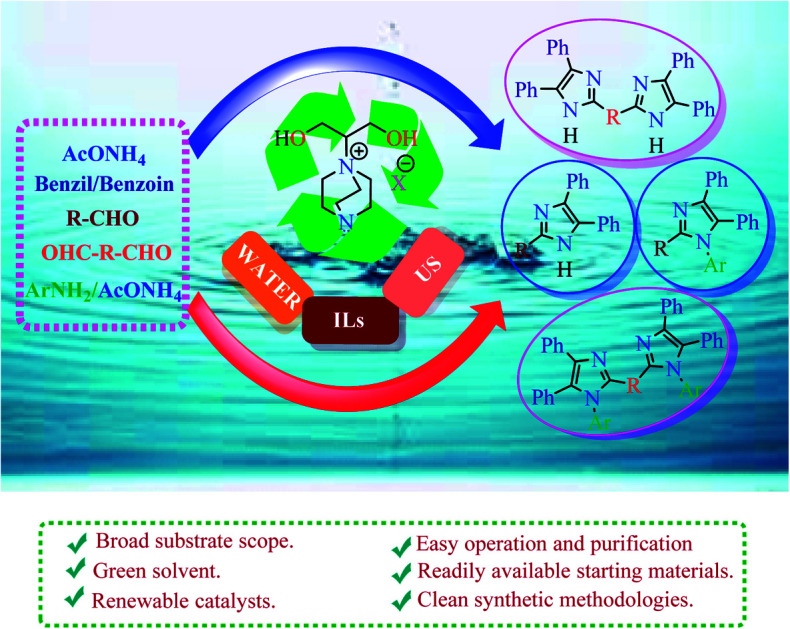
Introduction
Multicomponent reactions (MCRs) have been demonstrated to be significantly successful in producing the desired complex products from readily obtainable materials in a single synthetic reaction that comprises three or more substrates.1 The creating of novel MCRs and improving of published MCRs are presently under intensive focus due to their broad scope of applications in modern drug design.2 One of these applications is the assembly of multisubstituted imidazoles. The heterocyclic molecules comprised of an imidazole moiety whether synthetic or naturally occurring exhibit an array of pharmacological activities and play substantial roles in biochemical processes. They are well-known as anti-inflammatory agents, P38 MAP kinase inhibitors, fungicides, herbicides, therapeutic and antithrombotic agents.3 As well, imidazole derivatives are employed in photography as photosensitive molecules.4 Moreover, several imidazole derivatives are utilized as IL-1 biosynthesis inhibitors and eclectic antagonists for the glucagon receptors.5 In addition, the skeleton of many drug molecules such as eprosartan and trifenagrel, as well numerous biological compounds such as histamine and histidine, comprise imidazole moieties.6 Applications such as N-heterocyclic carbenes in catalysts design7 and ILs in green chemistry8 are some other important merits of the imidazoles derivatives. Recently, the evolution of the synthetic protocols for imidazoles preparation has attracted continued interest among medicinal and organic chemists.9 Among these protocols, the multicomponent cyclo-condensation reaction of diverse aldehydes, 1,2-dicarbonyl compounds (benzils or benzoins), and a nitrogen exporter that is widely utilized for the design of multisubstituted imidazole derivatives with alteration of catalysts and parameters.10 Nevertheless, some of these synthetic procedures associated with more than one serious drawback such as acidic media, low yield, long reaction time, formation of by-products and intricate work up and purification. Furthermore, the construction of such heterocyclic molecules has been commonly performed using polar solvents, for example, acetic acid (AcOH) and N,N-dimethylformamide (DMF) resulting in recovery procedure and complex isolation. Thus, presenting novel and green catalysts having excellent recyclability is an essential task for synthetic organic chemists to gain control over these deficiencies and satisfy the standards of an environmentally benign, effective and mild procedures for the assembly of imidazoles. As of late, from a green chemistry standpoint, the evolution of sustainable protocols requires the utilization of sustainable catalysts and benign solvents. From this aspect, ILs displayed good catalytic activities as a result of their negligible vapor pressure, recovery from reaction mixture, selectivity, reusability and capability to dissolve plentiful of substrates.11 ILs were successfully utilized as catalyst and solvent for assortment of reactions that have many applications in organic preparations and industry.12 Solvents, the most essential parameter of the ecological performance, are another factor, which ought to be thought about in green procedures especially in chemical manufacture processes. Usage of environmentally preferable solvents, such as water, found to be diminished the environmental influences that resulting from the utilization of other organic solvents in chemical production.13 Ultrasound-accelerated organic chemical reactions are acquiring a great interest in several chemistry research branches.14 Ultrasound irradiation displayed an alternative source of energy for organic synthesis, which proceed by the generation, growth and collapse of acoustic bubbles in the reaction mixture. These straightforwardly help in lessening the reaction duration and enhancing the reaction outcome.15 In the current work, an endeavor has been made for the construction of mono- and bis-multisubstituted imidazoles in water under sonication within few minutes using new ILs as an eco-friendly, reusable and sustainable catalysts. After carefully literature survey, we can believe that this is the first report of design, preparation, and characterization of ionic liquids 3a–c (Scheme 1) and their application as catalyst in organic syntheses (Schemes 2–5).
Scheme 1. Synthesis of diol functionalized ILs and exchange of anion.

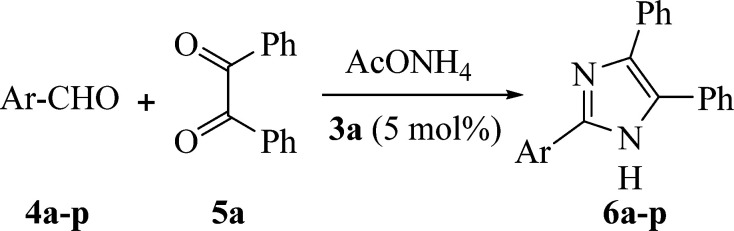
Scheme 2. Synthesis of 2,4,5-triphenyl-1H-imidazole 6a.

Scheme 3. Synthesis of bis-2,4,5-trisubstituted imidazoles 10a–f.

Scheme 4. Synthesis of bis-1,2,4,5-tetrasubstituted imidazoles 11a–f.

Scheme 5. Plausible mechanism for the catalytic synthesis of 2,4,5-trisubstituted imidazoles (6) over [DABCO-DOL][OAc].

Results and discussion
In the context of our search for the evolution of eco-friendly and efficacious protocols for the construction of biologically significance heterocyclic compounds,16 we studied the preparation of substituted imidazoles as potential drug derivatives, utilizing novel ILs (3a–c) as catalysts (Scheme 1). Initially, ILs (3a–c) were prepared from commercially available 1,4-diazobicyclo[2.2.2]octane (DABCO) and 2-chloro-1,3-propandiol (1), followed by the reaction with different salts for anion exchange (Scheme 1). The structure of the prepared ILs was elucidated from their FT-IR, NMR, and elemental analyses. Further, these ILs can be employed effectively in water that is compatible with “green chemistry”.17
In order to assess the catalytic efficacy of the newly synthesized ILs and to determine the most convenient reaction conditions; the three component reaction (3-MCR) of benzaldehyde (4a, 1 mmol), benzil (5a, 1 mmol), and NH4OAc (2.5 mmol, ammonia source) in water (5 mL) at 120 °C (oil bath temperature) was selected as the model study (Scheme 2) and the outcomes are summed up in Table 1. From these results (Table 1), we observed that this cyclo-condensation reaction could be effectively catalyzed by any one of the ILs (3a–c) and afforded the required product (6a) in moderate yields (Table 1, entries 1–4).
Synthesis of 6a using DABCO derived catalyst systems (3a–c)a.
| Entry | Catalyst | Method | Time (min) | Yield (%) |
|---|---|---|---|---|
| 1 | [DABCO-DOL][OAc] | Stirring | 20 | 94 |
| 2 | [DABCO-DOL][BF4] | Stirring | 25 | 88 |
| 3 | [DABCO-DOL][PF6] | Stirring | 27 | 86 |
| 4 | [DABCO-DOL][Cl] | Stirring | 30 | 77 |
| 5 | DABCO | Stirring | 60 | 23 |
| 6 | None | Stirring | 120 | NRb |
| 7 | [DABCO-DOL][OAc] | US | 20 | 99 |
| 8 | [DABCO-DOL][OAc] | US | 10 | 99 |
| 9 | [DABCO-DOL][OAc] | US | 5 | 99 |
| 10 | [DABCO-DOL][OAc] | US | 3 | 97 |
4a (1 mmol), 5a (1 mmol), NH4OAc (2.5 mmol) and 3a–c (5 mol%) in water (5 mL) at 120 °C (60 °C in case of US).
NR no reaction.
The template reaction (Scheme 2) was proceeded well under the catalytic influence of diversified DABCO based catalysts. The preferable outcome was acquired with acetate catalyst (3a), furnished 6a in 94% yield after 20 min (Table 1, entry 1). The other catalysts afforded lesser yields in a longer reaction times (Table 1, entries 2–4) with the relative catalytic potential following the order AcO > BF4 ≈ PF6 > Cl. To recognize the function of hydroxyl groups in the current catalytic system, two separately reactions with 5 mol% of each 3a and DABCO were employed under the optimized reaction conditions. As obvious from Table 1, lower reaction rate and hence lower yield are detected with DABCO (Table 1, entry 5), which distinctly indicating that hydroxyl groups are the active centre in catalyst core and responsible for promoting this reaction. When carried out the template reaction without using catalyst, no product was observed even after stirring for 2 h (Table 1, entry 6). So, in the next studies, catalyst [DABCO-DOL][OAc] (3a) will be utilized as the optimal choice for further investigation. Further improvements in both reaction yield and reaction rate were observed on running the aforementioned reaction (Scheme 2) under ultrasonic irradiation (40 kHz, Table 1, entries 7–10).
To assess the impact of catalyst loading on this reaction, the template reaction (Scheme 2) was performed using different amounts of IL 3a (Table 2). From the results displayed in Table 2, it was established that, 5 mol% is the optimal molar ratio of 3a that afforded the desired product in excellent yield (Table 2, entry 4). When the molar ratio of 3a was decreased to 4 and 3 mol%, the yields were also high, whilst the rates were diminished (Table 2, entries 2 and 3). Moreover, when the catalyst molar ratio was further diminished to 1 mol%, the required product was acquired in moderate yield with notable decrease of the rate (Table 2, entry 1). Whereas, increasing the amount of 3a over 8 mol% and 10 mol%, neither the reaction rate nor the yield was enhanced (Table 2, entries 5 and 6).
Effect of catalyst loading during the assembly of 6aa.
| Entry | Catalyst (mol%) | Time (min) | Yield (%) |
|---|---|---|---|
| 1 | 1 | 30 | 73 |
| 2 | 3 | 15 | 89 |
| 3 | 4 | 7 | 97 |
| 4 | 5 | 5 | 99 |
| 5 | 8 | 5 | 99 |
| 6 | 10 | 5 | 99 |
The mixture of 4a (1 mmol), 5a (1 mmol), NH4OAc (2.5 mmol) and 3a in water (5 mL) was sonicated at 60 °C.
The operating reaction temperature was also assessed and optimized. The best temperature was demonstrated to be 60 °C (Table 3, entry 2). An elevation of the operating temperature (70 °C) did not demonstrate further improvement of the yield of 6a (Table 3, entry 3). While, on employing the reaction at room temperature, the desirable product (6a) was acquired in moderate yield (Table 3, entry 1). Next, to evaluate any beneficial/detrimental impact of solvents on the [DABCO-DIOL][OAc] (3a) catalyzed substituted imidazole formation, the template reaction (Scheme 2) was employed in protic polar, aprotic polar, halogenated hydrocarbon, hydrocarbon solvents and ethereal (Table 3, entries 3–10). As showed in Table 3, water is obviously the best choice, as well as EtOH, for this reaction. While the yield of 6a diminished drastically with other solvents, denoting the detrimental influence of these solvents.
The influence of solvents and temperature on preparation of 6aa.
| Entry | Solvent | Temperature (°C) | Yield (%) |
|---|---|---|---|
| 1 | H2O | 25 | 68 |
| 2 | H2O | 60 | 99 |
| 3 | H2O | 70 | 99 |
| 4 | Non | 80 | 82 |
| 5 | EtOH | 40 | 95 |
| 6 | DCM | 60 | 21 |
| 7 | MeCN | 60 | 26 |
| 8 | THF | 60 | 33 |
| 9 | Dioxane | 60 | 15 |
| 10 | Toluene | 60 | Trace |
The mixture of 4a (1 mmol), 5a (1 mmol), NH4OAc (2.5 mmol) and 3a (5 mol%) in solvent (5 mL) was sonicated for 5 min at different temperatures.
The impact of the nitrogen sources was also investigated by performing the model reaction (Scheme 2) using different ammonium salts (Table 4) instead of NH4OAc and the optimum outcome was acquired with NH4OAc (Table 4, entry 2) as the most effective nitrogen source.
The effect of diverse ammonium salts as the nitrogen source on preparation of 6aa.
| Entry | Nitrogen source | Equiv. | Time (min) | Yield (%) |
|---|---|---|---|---|
| 1 | NH4OAc | 1 | 5 | 91 |
| 2 | NH4OAc | 2.5 | 5 | 99 |
| 3 | NH4Cl | 3 | 10 | 88 |
| 4 | NH4HCO3 | 3 | 10 | 82 |
| 5 | (NH4)2CO3 | 3 | 10 | 85 |
| 6 | HCOONH4 | 3 | 15 | 55 |
The mixture of 4a (1 mmol), 5a (1 mmol) and 3a (5 mol%) in water (5 mL) was sonicated at 60 °C using different ammonium salts.
Moreover, in order to study the influence of the power of ultrasonic intensity, the model reaction (Scheme 2) was employed at 100%, 80%, 60%, 40% and 20% of the power rate of the ultrasonic bath (200, 160, 120, 80 and 40 W). The outcomes are outlined in Table 5. The ultrasonic intensity is directly proportionate to the vibration amplitude of the ultrasound exporter and, as such, a rise in the vibration amplitude will result in a rise in both the vibration intensity and the sonochemical influences. As outlined in Table 5, increasing the intensity of the power up to 80% affords excellent yield in short reaction time; beyond this value, the yield diminishes somewhat. Next, the same template reaction (Scheme 2) was also performed using benzoin (5b) instead of benzil. The desirable product was obtained in acceptable yield (96%) but in a longer reaction time (15 min), indicating benzil is more convenient, affording the required product (6a) in shorter reaction time with higher yield.
Influence of US intensity power on the assembly of 6aa.
| Entry | Power intensity (%) | Watt (W) | Time (min) | Yield (%) |
|---|---|---|---|---|
| 1 | 20 | 40 | 25 | 83 |
| 2 | 40 | 80 | 20 | 88 |
| 3 | 60 | 120 | 10 | 93 |
| 4 | 80 | 160 | 5 | 99 |
| 5 | 100 | 200 | 7 | 96 |
The mixture of 4a (1 mmol), 5a (1 mmol), NH4OAc (2.5 mmol) and 3a (5 mol%) in water (5 mL) was sonicated at 60 °C.
Following these examinations, the best outcome was acquired at 60 °C, using 5 mol% of 3a as promoting catalyst, which yielded the desirable product (6a) in 99% on sonication for 5 min. In order to assess the scope and efficiency of this protocol and to represent pharmacologically pertinent substitution designs; diverse examples clarifying the current methodology for the construction of 2,4,5-trisubstituted imidazole derivatives 6a–p was investigated. The results are summed up in Table 6. These outcomes manifested that the electronic influence of the substituents on the aldehydes had almost no impact on the yield and the reaction rate: that is, the reactions are equally activated with both electron-withdrawing and electron-donating substituents on the aldehyde moieties. Additionally, the current methodology has been utilized successfully for both heteroaromatic and aliphatic aldehydes, and desired imidazole derivatives were acquired in considerable yields (Table 6, entries 14–16).
Synthesis of 2,4,5-trisubstituted imidazoles (6a–p) via the reaction of benzil (5a), aldehydes (4a–p) and NH4OAc catalyzed by 3aa.
| |||||
|---|---|---|---|---|---|
| Entry | Ar | Time (min) | Yield (%) | mp | |
| Found | Reported | ||||
| 1 | C6H5 | 5 | 99 | 272–273 | 272–274 (ref. 18) |
| 2 | 4-CH3O–C6H4 | 4 | 99 | 230–232 | 229–231 (ref. 18) |
| 3 | 4-HO–C6H4 | 4 | 98 | 234–236 | 232–234 (ref. 18) |
| 4 | 4-Me2N–C6H4 | 7 | 97 | 255–227 | 255–256 (ref. 18) |
| 5 | 2-HO–C6H4 | 6 | 95 | 202–204 | 201–203 (ref. 18) |
| 6 | 3,4-(MeO)2C6H3 | 6 | 98 | 214–216 | 213–216 (ref. 19) |
| 7 | 4-CH3–C6H4 | 8 | 95 | 230–232 | 226–227 (ref. 18) |
| 8 | 4-F–C6H4 | 8 | 96 | 260–262 | 262–264 (ref. 20) |
| 9 | 4-Br–C6H4 | 5 | 98 | 265–266 | 261–263 (ref. 21) |
| 10 | 2-Cl–C6H4 | 5 | 98 | 192–193 | 199–201 (ref. 21) |
| 11 | 4-Cl–C6H4 | 7 | 97 | 263–265 | 262–263 (ref. 22) |
| 12 | 2,4-Cl2C6H3 | 6 | 96 | 171–173 | 172–173 (ref. 20) |
| 13 | 4-NO2–C6H4 | 7 | 96 | 242–243 | 239–242 (ref. 21) |
| 14 | 2-Thienyl | 5 | 99 | 255–257 | 254–256 (ref. 18) |
| 15 | 2-Furyl | 5 | 98 | 201–202 | 201–203 (ref. 21) |
| 16 | CH3 | 6 | 99 | 244–246 | 243–245 (ref. 23) |
The mixture of 4a–p (1 mmol), 5a (1 mmol), NH4OAc (2.5 mmol) and 3a (5 mol%) in water (5 mL) was sonicated at 60 °C.
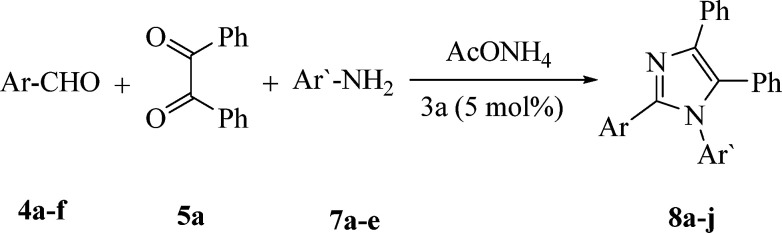
The promising results obtained in case of the three-component cyclo-condensation assembly of imidazoles (6a–p) promoted us to investigate the efficiency of catalyst 3a for the construction of 1,2,4,5-tetrasubstituted imidazoles (8a–j) via the four component reaction comprising 4a–f (1 mmol), 5a (1 mmol), NH4OAc (1 mmol) and amines 7a–e (1 mmol) (Table 7). The corresponding 1,2,4,5-tetrasubstituted imidazoles (8a–j) were acquired in excellent yields without the isolation of any by-products like the generation of 2,4,5-trisubstituted imidazoles, oxidized products of aldehydes and anilines that are commonly obtained under the impact of strong acids.
Preparation of 1,2,4,5-tetrasubstituted imidazoles (8a–j) via the reaction of benzil (5a), aldehydes (4a–f), amines (7a–e) and NH4OAc catalyzed by 3aa.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Ar | Ar′ | Time (min) | Yield (%) | mp | |
| Found | Reported | |||||
| 1 | C6H5 | C6H5 | 5 | 98 | 220–222 | 219–221 (ref. 18) |
| 2 | 4-CH3–C6H4 | C6H5 | 7 | 96 | 188–189 | 185–187 (ref. 18) |
| 3 | 4-CH3O–C6H4 | C6H5 | 5 | 96 | 183–185 | 183–185 (ref. 18) |
| 4 | 4-Cl–C6H4 | C6H5 | 6 | 95 | 161–162 | 160–163 (ref. 24) |
| 5 | 4-Br–C6H4 | C6H5 | 7 | 96 | 209–211 | 207–211 (ref. 18) |
| 6 | C6H5 | C6H4–CH2 | 5 | 98 | 160–164 | 160–164 (ref. 24) |
| 7 | C6H5 | 4-CH3O–C6H4–CH2 | 5 | 98 | 150–153 | 150–154 (ref. 24) |
| 8 | 4-CH3–C6H4 | 4-CH3–C6H4–CH2 | 5 | 98 | 132–135 | 132–135 (ref. 24) |
| 9 | 4-F–C6H4 | 4-F–C6H4–CH2 | 6 | 96 | 158–161 | 158–162 (ref. 24) |
| 10 | 4-CH3O–C6H4 | 4-CH3O–C6H4–CH2 | 6 | 96 | 152–154 | 152–154 (ref. 24) |
The mixture of 4a–f (1 mmol), 7a–e (1 mmol), 5a (1 mmol), NH4OAc (1 mmol) and 3a (5 mol%) in water (5 mL) was sonicated at 60 °C.
A comparative study of the current methodology with the reported protocols for the assembly of 6a is compiled in Table 8, to show the efficiency and the merit of the current work. As shown in Table 8, using of 3a is obviously the best choice for this reaction that could be used for the construction of a series of multisubstituted imidazoles under ultrasonic conditions with respect to reaction time and yield of the products.
Comparison of reaction yields and times for the assembly of derivative 6a under different methodologies.
| Entry | Method | Catalyst | Time (min) | Yield (%) | Yield economy |
|---|---|---|---|---|---|
| 1 | US – 60 °C | 3a | 5 | 99 (present) | 19.8 |
| 2 | Grinding | I2 | 30 | 42 (ref. 25) | 1.40 |
| 3 | Heating | NH4OAc | 360 | 71 (ref. 26) | 0.197 |
| 4 | Heating | Phosphomolybdic acid | 55 | 93 (ref. 27) | 1.69 |
| 5 | Heating – 65 °C | CANa | 80 | 82 (ref. 28) | 1.02 |
| 6 | MW | [HeMIM]BF4b | 4 | 65 (ref. 29) | 16.25 |
| 7 | Heating – 120 °C | HBF4–SiO2 | 15 | 92 (ref. 30) | 6.13 |
| 8 | Heating – 80 °C | Cu(NO3)2–zeolite | 30 | 96 (ref. 31) | 3.20 |
CAN: ceric ammonium nitrate.
[HeMIM]BF4: 1-methyl-3-heptylimidazolium tetrafluoroborate.
All the prepared derivatives (6 and 8) were confirmed by IR, NMR, and MS and also by comparing their spectral data with those of authentic samples. In order to explore more of the applicability of the current protocol, the same reaction conditions were applied for the construction of the bis-2,4,5-trisubstituted imidazoles (10a–f) via a similar one-pot, 3-MCR condensation of benzil (2 mmol), dialdehydes (9a–f) (1 mmol), and NH4OAc (4.5 mmol) as outline in Scheme 3 and the four component reaction (4-MCR) comprising benzil (2 mmol), dialdehydes (9a–f) (1 mmol), aniline (2 mmol) and NH4OAc (2 mmol) to form bis-1,2,4,5-tetrasubstituted imidazoles (11a–f) as displayed in Scheme 4. The reaction was completed within 7 min, and considerable yields of products were obtained (≈95%). The structures of hitherto unreported derivatives (10b, 10c, 10e, 10f and 11a–f) were interpreted from their IR, NMR and MS (see ESI†).
A reasonable mechanism for the preparation of multisubstituted imidazoles mediated by the [DABCO-DOL][OAc] catalyst is displayed in Scheme 5. The hydroxyl groups of 3a probably induce the polarization of the carbonyl groups in both aldehyde and benzil under sonication. The catalyst stimulates the aldehydic carbonyl group, in the presence of NH3, to form the non-isolable intermediate 12 (hydroxylamine) that is dehydrated to imine 13. Nucleophilic attack of another NH3 molecule yields intermediate 14, which attacks the activated carbonyl group of benzil 5a, affording 15. Intermediate 15 undergoes dehydration reaction forming 16. Intramolecular nucleophilic attack on 16 leads to the cyclization of 17. Subsequently, dehydration of 17 affords the trisubstituted imidazole product 6 with the recovery of 3a. The suggested mechanism may be belongs to the “on water” class,32 where water molecules at the organic–aqueous interface has OH group that acts as catalytic site for the reaction. These OH groups could be acted as both acceptors and hydrogen-bond donors, and thence they are partaken in trans-phase hydrogen bonding with the transition state that diminishes the reaction activation energy.17,33
Recyclability is another favorably merit of the catalyst under investigation. After accomplishment of the reaction, the product obtained was filtered off and washed with deionized water. By removal of the water, the retrieved catalyst was washed with ethyl acetate and reused for seven consecutive runs of reactions without any considerable diminish in reaction yields (Fig. 1).
Fig. 1. Recyclability of 3a in the preparation of 6a.

In order to demonstrate the potential for scale-up of the protocol, the MCR was performed on the gram scale under the optimal conditions, which in turn afforded the required products (6a, 8a, 10a and 11a) in quantitative yields (99, 99, 99 and 97%, respectively, Scheme 6).
Scheme 6. Scaled-up syntheses of derivatives 6a, 8a, 10a and 11a.

Conclusion
In the current work, an environmentally benign, efficient, straightforward and atom-economic green protocol for preparation of structurally diverse mono- and bis-multisubstituted imidazoles using renewable [DABCO-DOL][OAc] as promoter under sonication was described. Compared to the traditional methods, the current procedure can avert the utilization of harmful organic solvents and extremely enhances the reaction rate and thereby the excellent yields of the reactions that would greatly participate to the manufacturing of drugs. This methodology is bestowed with several unique merits including an easy work up, simple experimental process and no side reactions. Moreover, the catalyst can be facilely recycled and reused at least seven runs with negligible loss of its activity or alter in the structure, consequently, considerably contributes to the practice of green chemistry.
Experimental
Chemical reagents in high purity were obtained commercially and were used without further purification. Melting points were determined in open capillaries using an Electrothermal IA9100 melting point apparatus (UK). 1H and 13C NMR spectra were recorded using a Bruker UltraShield spectrometer at 400 and 100 MHz, respectively. High resolution mass spectra (HRMS) were performed utilizing a Bruker Daltonics microTOF spectrometer. FT-IR spectra were obtained with potassium bromide pellets in the range of 400–4000 cm−1 using a Perkin-Elmer Spectrum One spectrometer. Ultrasonication was done in a SY5200DH-T ultrasound cleaner.
Synthesis of the catalysts [DABCO-DOL][X] (3a–c)
A mixture of 1,4-diazobicyclo[2.2.2]octane (DABCO) (0.224 g, 2 mmol) and 2-chloro-1,3-propandiol (0.22 g, 2 mmol) in EtOH (15 mL) was sonicated for 3 h (TLC). Then, the solvent was removed under reduced pressure to afford compound (2). Then, NaOAc (0.164 g, 2 mmol), KPF6 (0.368 g, 2 mmol) and/or NaBF4 (0.22 g, 2 mmol) was appended to the residue 2 (0.444 g, 2 mmol) in EtOH (20 mL). The resulting solution was sonicated for 2 h and then, the solvent was evaporated in vacuo to afford the corresponding ILs (3a–c) in quantitative yields.
General procedure for the preparation of imidazoles 6a–p and bis-imidazoles 10a–f
A 25 mL Erlenmeyer flask was charged with aldehydes, 4a–p, (1 mmol) and/or dialdehyde, 9a–f, (0.5 mmol), benzil (1 mmol in case of 4a–p, and 2 mmol in case of 9a–f), NH4OAc (2.5 mmol in case of 4a–p, and 4.5 mmol in case of 9a–f), and 3a (5% mol in case of 4a–p, and 8% mol in case of 9a–f) in 5 mL water. The reaction vessel was placed in the ultrasonic bath, where the surface of solution is slightly lower than the water level, the reaction mixture was exposed to ultrasound irradiation at 60 °C for appropriated time. After completion of reaction (confirmed by TLC, eluent: MeOH/DCM = 1 : 4 vol), the solid that separated out was filtered washed with H2O, dried and recrystallized from a proper solvent to afford the analytically pure product. The catalyst was recovered from the aqueous layer under vacuum, washed with ethyl acetate and reused for the next reactions. All known products were confirmed by comparing their melting points and NMR spectra (see ESI†).
General procedure for the synthesis of imidazoles 8a–j and bis-imidazoles 11a–f
A 25 mL Erlenmeyer flask was charged with aldehydes (4a–f, 1 mmol) and/or dialdehyde (9a–f, 0.5 mmol), benzil (1 mmol in case of 4a–f, and 2 mmol in case of 9a–f), ammonium acetate (1 mmol in case of 4a–f, and 2 mmol in case of 9a–f), amines (7a–e, 1 mmol, in case of 4a–f) and/or aniline (1 mmol, in case of 9a–f) and 3a (5% mol in case of 4a–f and 8 mmol in case of 9a–f) in 5 mL water. The reaction flask was located in the ultrasonic bath, where the surface of reactants is slightly lower than the level of the water, the reaction mixture was exposed to ultrasound irradiation at 60 °C for appropriated time. After completion of reaction (confirmed by TLC, eluent: MeOH/DCM = 1 : 3 vol), the solid that separated out was filtered washed with water, dried and recrystallized from dioxane containing few drops of DMF to afford the analytically pure product.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
I am very thankful to the Department of Organic Chemistry, Arrhenius Lab., Stockholm University for providing laboratory facilities.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02755b
Notes and references
- Ibarra I. A. Jácome A. I. Zamora E. G. Org. Biomol. Chem. 2018;16:1402–1418. doi: 10.1021/cr400615v. [DOI] [PubMed] [Google Scholar]; Rotstein B. H. Zaretsky S. Rai V. Yudin A. K. Chem. Rev. 2014;114:8323–8359. doi: 10.1021/cr400615v. [DOI] [PubMed] [Google Scholar]
- Yang J. Mei F. Fu S. Gu Y. Green Chem. 2018;20:1367–1374. doi: 10.1039/C7GC03644B. [DOI] [Google Scholar]; Xiao F. Liu C. Wang D. Huang H. Deng G. J. Green Chem. 2018;20:973–977. doi: 10.1039/C7GC03644B. [DOI] [Google Scholar]
- Gee D. I. M. Bahramnejad M. Dabiri M. Tetrahedron Lett. 2013;54:2591–2594. doi: 10.1016/j.tetlet.2013.03.008. [DOI] [Google Scholar]; Keivanloo1 A. Bakherad M. Imanifar E. Mirzaee M. Appl. Catal., A. 2013;467:291–300. doi: 10.1016/j.tetlet.2013.03.008. [DOI] [Google Scholar]
- Schmierer R., Mildenberger H. and Buerstell H., German Patent 361464, 1987Chem. Abstr., 1988, 108, 37838; Maier T., Schmierer R., Bauer K., Bieringer H., Buerstell H. and Sachse B., US Pat. 4820335, 1989Chem. Abstr., 1989, 111, 19494
- Gadekar L. S. Mane S. R. Katkar S. S. Arbad B. R. Lande M. K. Cent. Eur. J. Chem. 2009;75:50–554. [Google Scholar]
- Koike H. Konse T. Sada T. Ikeda T. Hyogo A. Hinman D. Saito H. Yanagisawa H. Annu. Rep. Sankyo Res. Lab. 2003;55:1–91. doi: 10.1021/ol049682b. [DOI] [PubMed] [Google Scholar]; Abrahams S. L. Hazen R. J. Batson A. G. Phillips A. P. J. Pharmacol. Exp. Ther. 1989;249:359–365. doi: 10.1021/ol049682b. [DOI] [PubMed] [Google Scholar]; Leister C. Wang Y. Zhao Z. Lindsley C. W. Org. Lett. 2004;6:1453–1456. doi: 10.1021/ol049682b. [DOI] [PubMed] [Google Scholar]
- Beillard A. Bantreil X. Métro T. X. Martineza J. Lamaty F. Green Chem. 2018;20:964–968. doi: 10.1039/C7GC03539J. [DOI] [Google Scholar]; Chiarotto I. Feeney M. M. M. Feroci M. Inesi A. Electrochim. Acta. 2009;54:1638–1644. doi: 10.1039/C7GC03539J. [DOI] [Google Scholar]
- Chowdhury S. Mohan R. S. Scott J. L. Tetrahedron. 2007;63:2363–2389. doi: 10.1016/j.tet.2006.11.001. [DOI] [Google Scholar]
- Marzouk A. A. Abu-Dief A. M. Abdelhamid A. A. Appl. Organomet. Chem. 2018;32:3794. doi: 10.1039/C7GC02484C. [DOI] [Google Scholar]; Yang D. Shan L. Xu Z. F. Li C. Y. Org. Biomol. Chem. 2018;16:1461–1464. doi: 10.1039/C7GC02484C. [DOI] [PubMed] [Google Scholar]; Jayrama J. Jeena V. Green Chem. 2017;19:5841–5845. doi: 10.1039/C7GC02484C. [DOI] [Google Scholar]
- Chawla A. Sharma A. Kumar A. Pharma Chem. 2012;4:116–140. doi: 10.1002/jhet.924. [DOI] [Google Scholar]; Rahman M. Bagdi A. K. Kundu D. Majee A. Hajra A. J. Heterocycl. Chem. 2012;49:1224–1228. doi: 10.1002/jhet.924. [DOI] [Google Scholar]; Ramesh K. Murthy S. N. Karnakar K. Nageswar Y. V. D. Vijayalakhshmi K. Prabhavathi Devi B. L. A. Prasad R. B. N. Tetrahedron Lett. 2012;53:1126–1129. doi: 10.1002/jhet.924. [DOI] [Google Scholar]; Niknam K. Deris A. Naeimi F. Majleci F. Tetrahedron Lett. 2011;52:4642–4645. doi: 10.1002/jhet.924. [DOI] [Google Scholar]; Hasaninejad A. Zare A. Shekouhy M. Rad J. A. J. Comb. Chem. 2010;12:844–849. doi: 10.1002/jhet.924. [DOI] [PubMed] [Google Scholar]; Murthy N. S. Madhav B. Nageswar Y. V. D. Tetrahedron Lett. 2010;51:5252–5257. doi: 10.1002/jhet.924. [DOI] [Google Scholar]; Mukhopadhyay C. Tapaswi P. K. Drew M. G. B. Tetrahedron Lett. 2010;51:3944–3950. doi: 10.1002/jhet.924. [DOI] [Google Scholar]; Zhang P. F. Chen Z. C. Z. Synthesis. 2001:2075–2077. doi: 10.1002/jhet.924. [DOI] [Google Scholar]
- Silva S. S. Mano J. F. Reis R. L. Green Chem. 2017;19:1208–1220. doi: 10.1039/C6GC02827F. [DOI] [Google Scholar]
- Yokota Y. Miyamoto H. Imanishi A. Inagaki K. Morikawa Y. Fukui K. Phys. Chem. Chem. Phys. 2018;20:6668–6676. doi: 10.1039/C7CP07313E. [DOI] [PubMed] [Google Scholar]
- Capello C. Fischer U. Hungerbühler K. Green Chem. 2007;9:927–934. doi: 10.1039/B617536H. [DOI] [Google Scholar]
- Arafa W. A. A. Abdel-Magied A. F. ARKIVOC. 2017;(v):327–340. doi: 10.1039/C7QI00389G. [DOI] [Google Scholar]; Dalodière E. Virot M. Dumas T. Guillaumont D. Illy M. C. Berthon C. Guerin L. Rossberg A. Venault L. Moisy P. Nikitenkoa S. I. Inorg. Chem. Front. 2018;5:100–111. doi: 10.1039/C7QI00389G. [DOI] [Google Scholar]; Letort S. Lejeune M. Kardos N. Métay E. Popowycz F. Lemaire M. Draye M. Green Chem. 2017;19:4583–4590. doi: 10.1039/C7QI00389G. [DOI] [Google Scholar]
- Mason T. J. Chem. Soc. Rev. 1997;26:443–451. doi: 10.1039/CS9972600443. [DOI] [Google Scholar]; Banerjee B. Ultrason. Sonochem. 2017;35:15–35. doi: 10.1039/CS9972600443. [DOI] [PubMed] [Google Scholar]
- Arafa W. A. A. Ibrahim H. M. RSC Adv. 2018;8:10516–10521. doi: 10.1039/C8RA01253A. [DOI] [PMC free article] [PubMed] [Google Scholar]; Arafa W. A. A. J. Heterocycl. Chem. 2018;55:456–464. doi: 10.1039/C8RA01253A. [DOI] [Google Scholar]
- Arafa W. A. A. Mohamed A. M. Abdel-Magied A. F. Heterocycles. 2017;94:1439–1455. doi: 10.3987/COM-17-13726. [DOI] [Google Scholar]
- Esfahani M. N. Montazerozohori M. Abdizadeh T. Chem. Pap. 2015;69:1491–1499. [Google Scholar]
- Safari J. Khalili S. D. Rezaei M. Banitaba S. H. J. Chem. Sci. 2010;122:437–441. doi: 10.1007/s12039-010-0051-6. [DOI] [Google Scholar]
- Zang H. Su Q. Mo Y. Cheng B. W. Jun S. Ultrason. Sonochem. 2010;17:749–751. doi: 10.1016/j.ultsonch.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Heravi M. R. P. Vessally E. Behbehani G. R. R. C. R. Chim. 2014;17:146–150. doi: 10.1016/j.crci.2012.12.007. [DOI] [Google Scholar]
- Kidwai M. Mothsra P. Bansal V. Somvanshi R. K. Ethayathulla A. S. Dey S. Singh T. P. J. Mol. Catal. A: Chem. 2007;265:177. doi: 10.1016/j.molcata.2006.10.009. [DOI] [Google Scholar]
- Waheed M. Ahmed N. Alsharif M. A. Alahmdi M. I. Mukhtar S. ChemistrySelect. 2017;2:7946–7950. doi: 10.1002/slct.201701299. [DOI] [Google Scholar]
- Samanta S. Roy D. Khamarui S. Maiti D. K. Chem. Commun. 2014;50:2477–2480. doi: 10.1039/C3CC48437H. [DOI] [PubMed] [Google Scholar]
- Parveen A. Ahmed M. d. R. S. Shaikh K. A. Deshmukh S. P. Pawar R. P. ARKIVOC. 2007;xvi:12–18. [Google Scholar]
- Tian M. Wang C. Wang L. G. Luo K. Zhao A. Guo C. C. Luminescence. 2014;29:540–548. doi: 10.1002/bio.2580. [DOI] [PubMed] [Google Scholar]
- Jadhav S. D. Kokare N. D. Jadhav S. D. J. Heterocycl. Chem. 2008;45:1461–1464. doi: 10.1002/jhet.5570450533. [DOI] [Google Scholar]
- Sangshetti J. N. Kokare N. D. Kotharkara S. A. Shinde D. B. J. Chem. Sci. 2008;120:463–467. doi: 10.1007/s12039-008-0072-6. [DOI] [Google Scholar]
- Xia M. Lu Y. J. Mol. Catal. A: Chem. 2007;265:205–208. doi: 10.1016/j.molcata.2006.10.004. [DOI] [Google Scholar]
- Kumar D. Kommi D. N. Bollineni N. Patel A. R. Chakraborti A. K. Green Chem. 2012;14:2038–2049. doi: 10.1039/C2GC35277J. [DOI] [Google Scholar]
- Sivakumar K. Kathirvel A. Lalitha A. Tetrahedron Lett. 2010;51:3018–3021. doi: 10.1016/j.tetlet.2010.04.013. [DOI] [Google Scholar]
- Jung Y. Marcus R. A. J. Am. Chem. Soc. 2007;129:5492–5502. doi: 10.1021/ja068120f. [DOI] [PubMed] [Google Scholar]
- Wadhwa P. Kaur T. Singh N. Singh U. P. Sharma A. Asian J. Org. Chem. 2016;5:120–126. doi: 10.1002/ajoc.201500397. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.