

SUMMARY
Pediatric cancers often mimic fetal tissues and express proteins normally silenced postnatally that could serve as immune targets. We developed T cells expressing Chimeric Antigen Receptors (CAR) targeting glypican-2 (GPC2), a fetal antigen expressed on neuroblastoma (NB) and several other solid tumors. CARs engineered using standard designs control NBs with transgenic GPC2 overexpression but not those expressing clinically relevant GPC2 site densities (~5000 molecules/cell, range 1–6 ×103). Iterative engineering of transmembrane (TM) and costimulatory domains plus overexpression of c-Jun lowered the GPC2-CAR antigen density threshold, enabling potent and durable eradication of NBs expressing clinically relevant GPC2 antigen densities, without toxicity. These studies highlight the critical interplay between CAR design and antigen density threshold, demonstrate potent efficacy and safety of a lead GPC2-CAR candidate suitable for clinical testing and credential oncofetal antigens as a promising class of targets for CAR T cell therapy of solid tumors.
Keywords: immunotherapy, chimeric antigen receptor, CAR T cell, neuroblastoma, GPC2, Glypican-2, oncofetal antigen, antigen density
Graphical Abstract
eTOC Blurb
Heitzeneder et. al develop potent Chimeric Antigen Receptor (CAR) T cells targeting glypican-2 (GPC2), an oncofetal antigen expressed on neuroblastoma. Accurate quantification of antigen density on clinical samples uncovers a profound impact limiting CAR potency. Iterative engineering of CARs overcomes this limit, achieving profound anti-tumor efficacy without compromising safety.
INTRODUCTION
T cells expressing chimeric antigen receptors (CAR) targeting high and homogenously expressed B lineage antigens mediate impressive clinical benefit with acceptable toxicity. Similar success is not demonstrated in solid tumors (June and Sadelain, 2018; Majzner and Mackall, 2019) due to limited trafficking, a hostile tumor microenvironment (Labanieh et al., 2018), and a need to target antigens expressed at lower and more heterogeneous densities, since lineage antigens on solid tumors usually pose unacceptable risks to vital tissues. CAR receptors engineered using traditional designs manifest high antigen density requirements for full activation due to limited Zap70 recruitment to the immune synapse, but the antigen density thresholds can be tuned by modifying the CAR design (Majzner et al., 2020). Designs that enhance signal strength, modifications to the transmembrane domain (Majzner et al., 2020) and c-Jun overexpression (Lynn et al., 2019) lower the antigen density thresholds, but it remains unknown whether CAR T cells tuned for low antigen density can effectively target antigen densities present on human solid tumors and would avoid targeting of normal tissues.
Pediatric solid tumors and brain tumors often arise in the context of stalled developmental programs (Jessa et al., 2019; Marshall et al., 2014) and express fetal antigens with limited expression on postnatal, non-malignant tissues. Cerebroglycan or glypican-2 (GPC2), a prototypic oncofetal target, is highly expressed in the developing nervous system, where it serves as a signaling co-receptor regulating cell growth (Filmus et al., 2008; Kaur and Cummings, 2019). GPC2 is overexpressed on neuroblastoma (NB) and other solid tumors (Bosse et al., 2017; Li et al., 2017), but shows limited postnatal expression. GPC2-CARs engineered using standard designs controlled NB cell lines engineered to overexpress GPC2 but not non-engineered cell lines due to subthreshold antigen density. Using flow cytometry to measure GPC2 antigen density on metastatic NBs in pediatric bone marrow (BM) samples, we demonstrate that GPC2 antigen density in clinical specimens is below the threshold required to control tumor growth for traditionally designed GPC2-CAR T cells. Sequential GPC2-CAR design modifications significantly lowered the antigen density threshold, yielding lead candidates capable of impressive NB regression and sustained disease control in several tumor models expressing clinically relevant GPC2 levels, without toxicity. This work validates the hypothesis that next generation CAR engineering can tune antigen density thresholds and deliver therapeutics capable of targeting of non-mutant, non-lineage cell surface antigens expressed on solid tumors while sparing normal tissues.
RESULTS
Efficacy of traditionally designed GPC2-CAR T cells against NB is limited by target antigen density.
We identified three novel anti-GPC2 single-chain variable fragments (scFvs) (GPC2.19, GPC2.27, GPC2.D4) within a human naïve antigen-binding fragment (Fab) phage library and a fourth previously reported (GPC2.D3) (Bosse et al., 2017). Fabs incorporating each of the 4 scFvs were thermostable (Figure S1A,B), exhibited high binding affinities towards human GPC2 and GPC2.19, GPC2.D3 and GPC2.D4 also bound murine GPC2 (Figure S1C). GPC2.D4 and GPC2.27 require C-terminal residues (493–553) for binding and GPC2.19 and GPC2.D3 share overlapping epitopes (Figure 1A, Figure S1D,E) including residues 396–400 (Figure 1B), known components for GPC2.D3 (Raman et al., 2021). Using a CD8α hinge/transmembrane (TM) and 4–1BB costimulatory domain backbone that mimics tisagenlecleucel, an FDA approved CD19-CAR active against B-ALL, we engineered T cells to express GPC2-CARs incorporating each scFv, both in VH/VL and VL/VH orientations (Figure S1F). Based upon favorable biophysical properties, robust expression in primary, activated T cells (Figure S1G), killing capacity (Figure S1H-J), low baseline IFNγ and high antigen induced cytokine production, GPC2.19- and GPC2.D3-CAR T cells with VL/VH orientations (Figure S2A-C) in the absence of Fc-mutated spacer domains (Hudecek et al., 2015) were prioritized for further testing (Figure 1C, Figure S2D-I).
Figure 1. GPC2-CAR T cell efficacy is limited by GPC2 antigen density on NB.
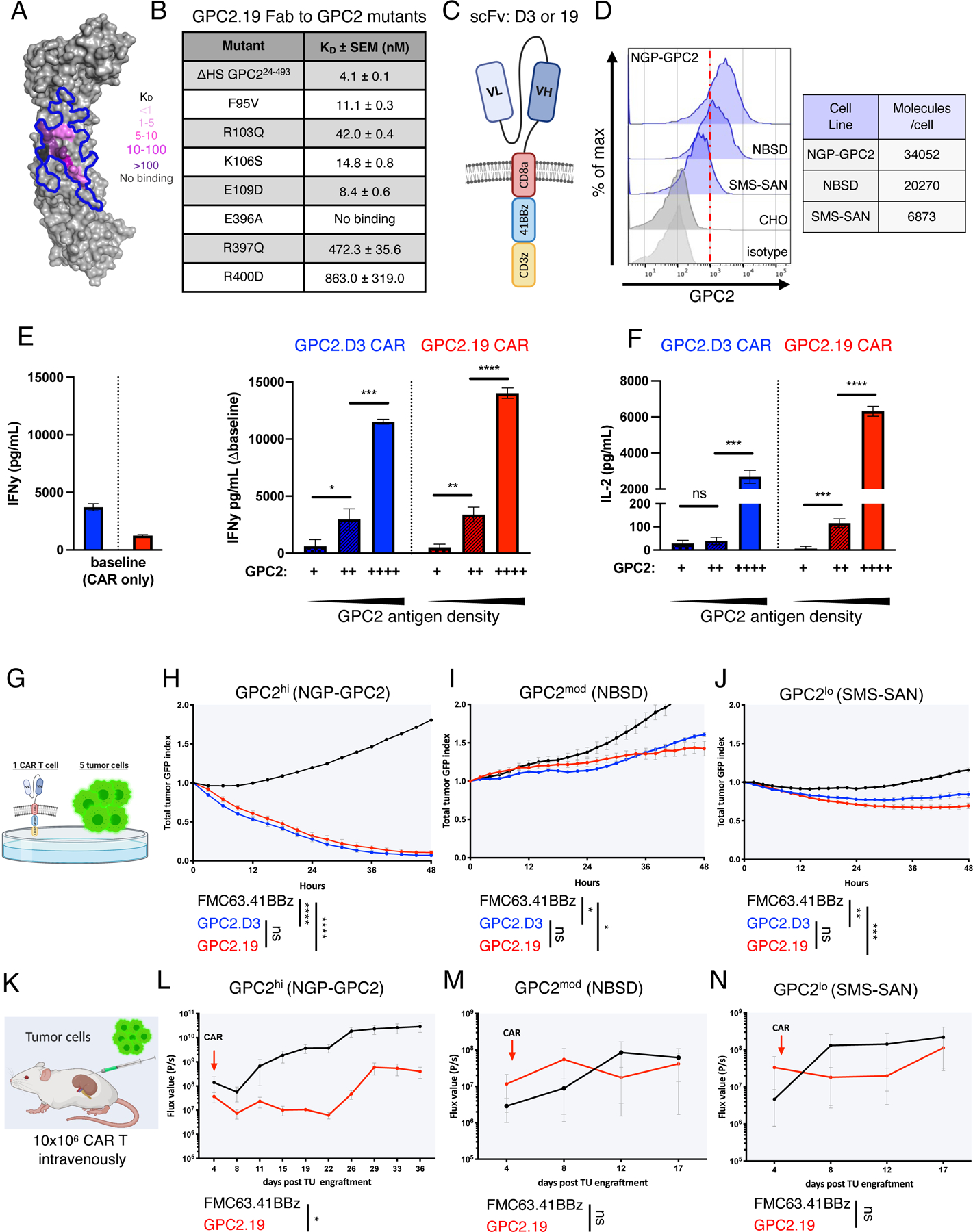
A) Human GPC2 crystal structure (PDB ID: 6WJL) and effect of point mutations on GPC2.19 Fab binding affinity. Blue line delineates previously defined GPC2.D3 epitope.
B) Binding dissociation constants for GPC2.19 Fab binding to human GPC2 mutants.
C) Schematic of GPC2.8aTM.41BBz CAR constructs in VLVH orientation of scFv’s.
D) Flow cytometric cell surface expression of GPC2 on indicated NB cell lines and negative CTRL (CHO) and respective isotype CTRL. Representative histogram of n=4 independent experiments. Table shows molecules/cell of GPC2 as determined by QuantiBRITE PE assay.
E-F) Baseline levels of (E) IFNγ (left) and secretion of IFNγ (∆baseline, right) and (F) IL2 by GPC2-CAR T cells in response to NB cell lines shown in (D), with increasing antigen levels of GPC2. Representative of n=4 independent experiments (mean ± SD).
G) Schematic of in vitro killing assays at 1:5 effector:target ratio.
H-J) Cytolytic activity of GPC2.D3.8TM.41BBz- and GPC2.19.8TM.41BBz-CAR T cells in vitro against (H) NGP-GPC2 (I) NBSD and (J) SMS-SAN NB cell lines at 1:5 E:T ratio. Values represent mean ± SEM, representative of n=4 independent experiments.
K) Schematic of experimental outline testing GPC2.19.8TM.41BBz-CAR CAR T cells in para-orthotopic NB renal capsule xenograft models.
L-N) NGP-GPC2 post engraftment of 0.75 MIO tumor cells on day 0 and treated on day 4 (n=6 mice for CTRL.FMC63 and n=5 mice for GPC2.19 CAR T cells). M) NBSD or N) SMS-SAN post engraftment of 1 MIO tumor cells on day 0 and treated with CAR on day 5 (n=3 mice/group). For L-N): n=1 experiment each. Tumor burden assessed by IVIS imaging. Values represent FLUX [P/s] mean ± SEM.
Statistics: (E,F) represent Student’s t-test, (H-J) and (L-N) represent two-way RM-ANOVA (**** = p<0.0001, *** = p<0.001, ** = p<0.01, * = p<0.05), ns = p > 0.05).
See also Figures S1-2.
We measured potency against NB cell lines with variable GPC2 site density, including NGP-GPC2, an isogenic line with transgenic GPC2 at high levels (GPC2hi, 34052 molecules/cell), NBSD, a cell line expressing moderate GPC2 levels (GPC2mod, 20270 molecules/cell) and SMS-SAN, which expresses GPC2 at low levels (GPC2lo, 6873 molecules/cell) (Figure 1D) and observed that cytokine production by GPC2.D3- and GPC2.19-CARs directly correlated with GPC2 antigen density (Figure 1E-F), a dependency that held true across a larger panel of NB cell lines (Figure S2J). At low effector to target ratios in vitro (1:5 E:T), GPC.D3- and GPC2.19-CARs killed GPC2hi, but not GPC2mod or GPClo targets (Figure 1G-J) and the requirement for high antigen density was confirmed in vivo since GPC.19-CARs controlled GPC2hi xenografts, but not GPC2mod and GPC2lo xenografts (Figure 1K-N).
We next compared antigen density thresholds for GPC2.19-CARs vs. a CD19-CAR mimicking tisagenlecleucel (FMC63-CAR) using tumor lines expressing antigen densities similar to GPC2 and CD19 respectively (Figure S2K) (Majzner et al., 2020). Both CARs demonstrated a similar dependence on antigen density, as both killed antigenlo targets at 1:1 ratios but neither produced significant cytokines in responses to antigenlo targets (Figure S2M-O). These results demonstrate that GPC2-CARs incorporating a standard CAR design utilized in the FDA approved tisagenlecleucel show significant and specific GPC2-directed activity, but potency varied greatly depending upon antigen density, highlighting a critical need to quantitate GPC2 antigen density on human NBs to predict the potential for clinical efficacy.
Quantification of immunotherapy-relevant NB cell surface antigens.
To precisely measure GPC2 antigen density on the surface of NB cells in patients, we developed a multicolor antigen density quantification assay for 6 immunotherapy-relevant antigens. We applied this assay on nine bone marrow (BM) samples infiltrated with metastatic NB (Figure S3A), a panel of cell lines and several PDX tumors. BM was selected since it is a common site for NB metastasis and allowed analysis without enzymatic tissue digestion. Hematopoietic cells were excluded based upon CD45 expression, and BM stromal cells were excluded based upon CD13 expression (Theodorakos et al., 2019), which is not expressed by NB (Figure 2A,B). Gated CD45–CD13– cells co-expressed NCAM and GD2, known NB associated molecules (Figure 2C) (Warzynski et al., 2002). On the tumor population, we measured fluorophore signal emanating from target-specific antibodies alongside the signal from pre-calibrated fluorescent beads, allowing calculation of target molecules/cell. Metastatic NB in the BM demonstrated 4262±469 GPC2 molecules/cell (range 1425–6041) with little interpatient variation, which is remarkably lower than that present on most NB cell lines and PDX samples assessed simultaneously (Figure 2D, Figure S3B) and approximating the level present on SMS-SAN, our GPC2lo line.
Figure 2. Antigen density of candidate immunotherapy targets on clinical samples of NB cells metastatic to the bone marrow.
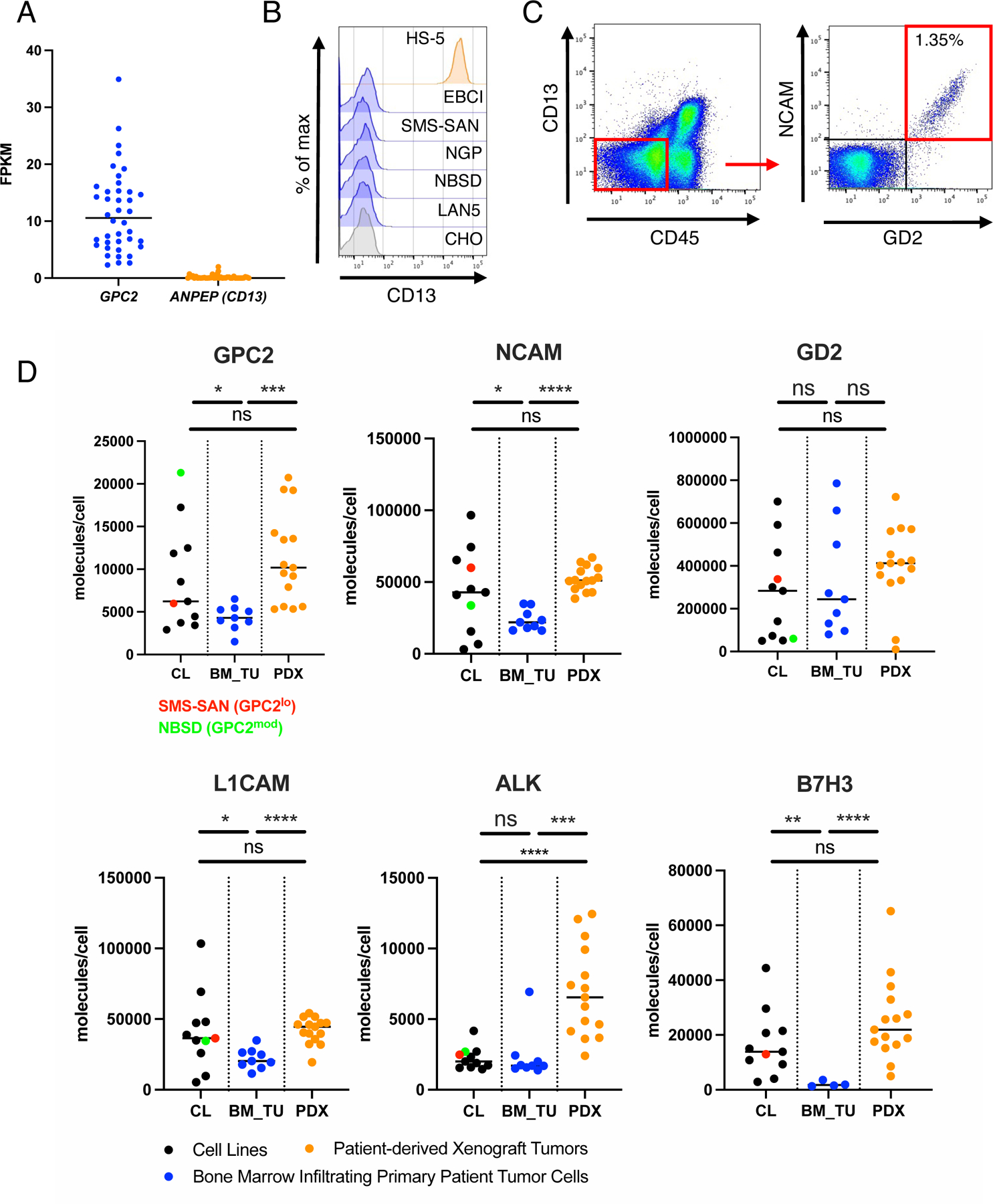
A) Gene expression of GPC2 and ANPEP (CD13) in a panel of NB cell lines (dataset: GSE89413)
B) Flow cytometric cell surface expression of CD13 on negative CTRL cells (CHO), NB cell lines and HS-5 BM stroma cells.
C) Representative CD45-CD13- and NCAM+GD2+ gating strategy to identify NB tumor cell populations in clinical BM samples.
D) Cell surface antigen quantification (molecules/cell) of GPC2, NCAM, GD2, L1CAM, ALK and B7-H3 on BM infiltrating NB cells (BM_TU), Cell Lines (CL) and patient derived xenograft tumors (PDX) assessed by multicolor flow cytometry antigen density quantification assay. Horizontal lines indicate median. Statistics represent Welch’s t-test (**** = p<0.0001, *** = p<0.001, ** = p<0.01, * = p<0.05), ns = p > 0.05). Statistical parameters of the samples are shown in Figure S3B.
See also Figure S3.
We also compared expression levels of GPC2 to several other immunotherapy targets and observed a wide range in antigen density between antigens. GPC2 levels are lower than GD2, NCAM and L1CAM, but higher than anaplastic lymphoma kinase (ALK) and B7-H3. The highest antigen density was observed for the disialoganglioside GD2, demonstrating almost 30x more molecules than GPC2 (Figure S3C). Similar to GPC2, NCAM, L1CAM and B7-H3 antigen density showed little interpatient variability and was significantly lower in metastatic NB compared to cell lines and PDX samples (Figure 2D, Figure S3B). Paired diagnostic/relapse samples from one patient demonstrated a drop in antigen density for each target at relapse, except for GPC2, which was modestly increased on the relapsed sample (Figure S3D,E). Together, these data highlight a critical role for antigen density in modulating CAR T cell potency, wide ranges in antigen density across targets with most antigens showing little interpatient variability, and poor concordance between levels on cell lines and PDX samples compared to that present on metastatic NB.
GPC2-CARs incorporating a CD28 TM domain are efficacious against GPC2 densities expressed on metastatic NB tumor cells.
We previously demonstrated that a CD28 TM domain lowered the CAR T cell antigen density threshold (Majzner et al., 2020). Hence, we compared GPC2.D3- and GPC2.19-CAR T cells with a CD28TM± a CD28 costimulatory endodomain substituted into original CD8 TM constructs (Figure 3A, Table S1). These structural changes did not impact CAR surface expression (Figure S4A,B), exhaustion profiles (Figure S4C) or T cell differentiation phenotypes (Figure S4D), but GPC2-CARs incorporating CD28TM domains, with either 41BB or CD28 costimulatory endodomains, demonstrated augmented killing of GPC2mod and GPC2lo targets (Figure 3B, Figure S4E,F) and increased IL-2 and IFNy production in response to GPC2mod tumor cells (Figure S4G,H). These in vitro results were mirrored in vivo against GPC2mod lines, where standard design GPC2.19.8TM.41BBz-CARs were ineffective, but both GPC2.19.28TM.41BBz- and GPC2.19.28TM.28z-CARs mediated complete responses (CRs) with significant survival benefit (Figure 3D-G). Similar anti-tumor effects were achieved in vivo in a metastatic model against GPC2lo (SMS-SAN) NB (Figure 3H-K).
Figure 3. GPC2-CAR T cells incorporating a CD28 TM eradicate GPC2mod and GPC2lo NBs.
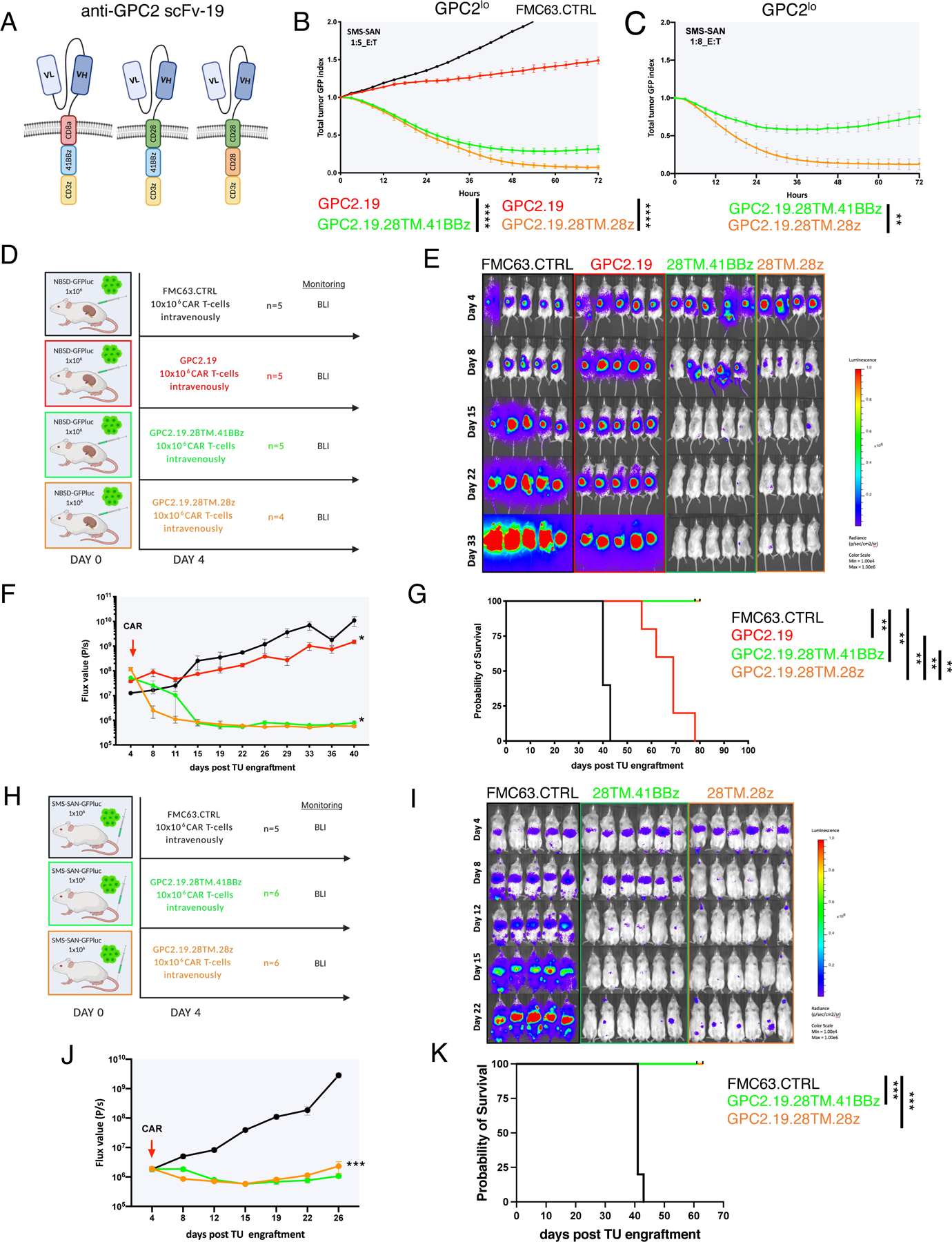
A) Schematic of GPC2.19.8TM.41BBz (left), GPC2.19.28TM.41BBz (center) and GPC2.19.28TM.28z (right) CAR constructs.
B-C) Cytolytic activity of these CAR T cells against SMS-SAN (GPC2lo) at B) 1:5 and C) 1:8 effector:target ratio. Representative of n=3 independent experiments with n=3 individual donors.
D) Experimental in vivo setup testing GPC2.19-CAR T cell constructs shown in (A) in a para-orthotopic GPC2mod (NBSD) NB renal capsule model.
E-G) Bioluminescence images (E) (BLI) and F) FLUX [P/s] values of tumor burden assessed by IVIS imaging, and G) Kaplan-Meier survival analysis of treatment arms shown in (D). Statistical analysis for survival curves represents log-rank test. One mouse in the FMC63.CTRL group died during imaging on day 40 and was censored from the analysis. Representative of n=3 independent experiments with n=3 individual donors.
H) Experimental in vivo setup testing constructs shown (A) in GPC2lo (SMS-SAN) metastatic xenograft model.
I-K) BLI images (I) and J) FLUX [P/s] values of tumor burden assessed by IVIS imaging, and K) Kaplan-Meier survival analysis of treatment arms shown in H. Representative of n=1 experiment. Statistical analysis for survival curves represents log-rank test. Values in B, C, F, J represent mean ± SEM. Statistical test in B, C, F, J represents two-way RM-ANOVA (**** = p<0.0001, *** = p<0.001, ** = p<0.01, * = p<0.05), ns = p > 0.05).
To further assess effects of the costimulatory endodomain, we compared killing of GPC2.19.CD28TM-CARs incorporating a CD28 vs. 41BB endodomains in stress tests in vitro. Using 1:8 E:T ratios against GPC2mod and GPC2lo NB lines, we observed superior killing by GPC2-CARs incorporating a CD28 costimulatory endodomain (Figure 3C, Figure S4I). While both mediated significant tumor regression in vivo against patient-derived xenografts with GPC2mod antigen density (Figure 2D) in animals with moderate (MOD: range mean TU VOL 0.22–0.24 cm3) or high (HI: range mean TU VOL 0.65–0.78 cm3) tumor burden, those containing a CD28 endodomain were superior (Figure 4A-E). Together, these results demonstrate that iterative CAR engineering can tune the antigen density threshold to enable effective targeting of cells expressing GPC2 at approximately 5×103 molecules/cell, well below the level a standard CD8TM and 41BB-CAR configuration can target and approximating levels present on clinical samples of metastatic NB.
Figure 4. GPC2-CAR T cells with CD28 TM domains control patient derived xenografts and CD28 outperforms 41BB costimulatory endodomains.
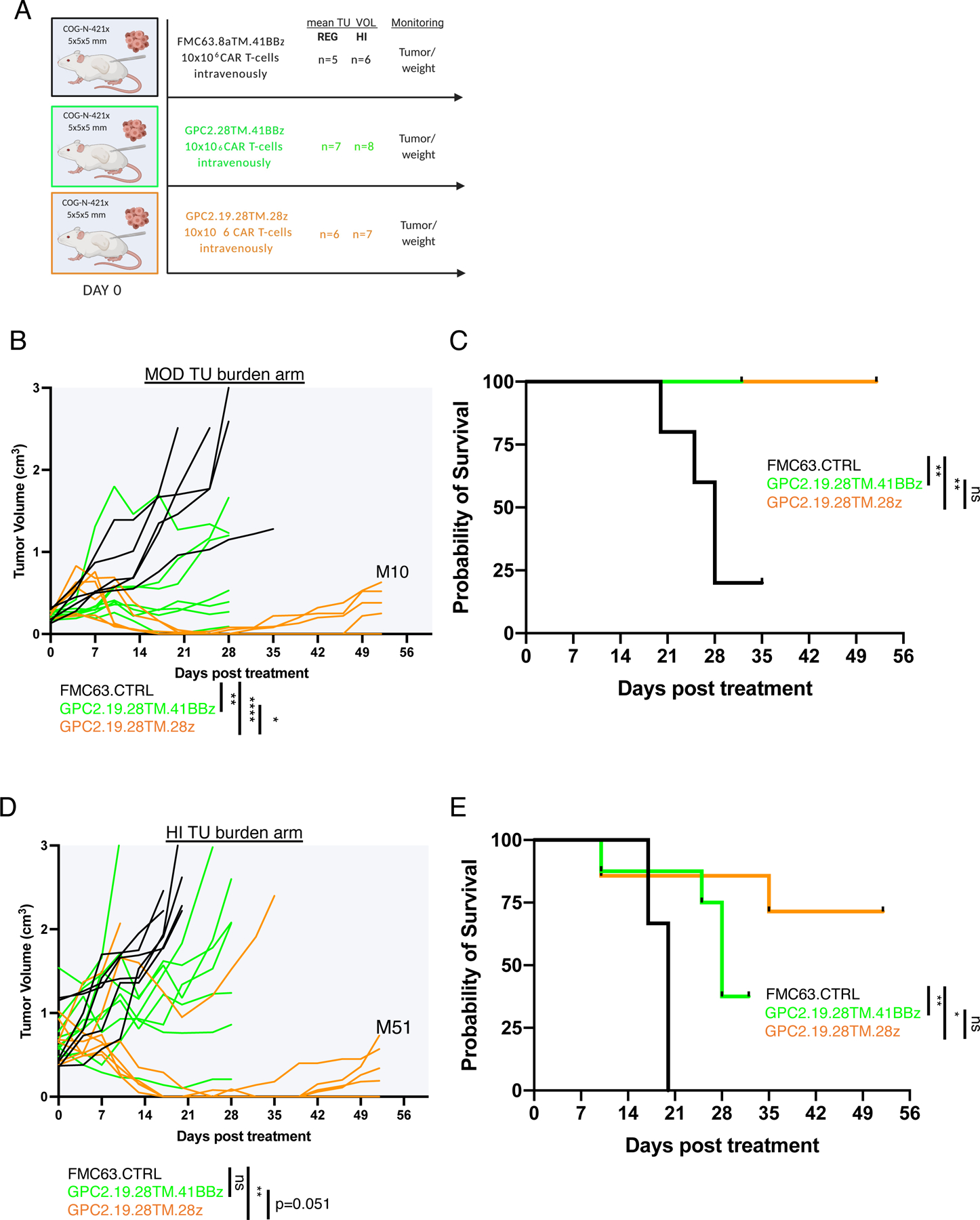
A) Experimental in vivo setup testing GPC2.19-CAR T cells in COG-N-421x PDX models bearing moderate size tumor burden (MOD: range mean TU vol 0.22–0.24 cm3) or high size tumor burden (HI: range mean TU vol 0.65–0.78 cm3).
B-C) Tumor volume (B) after treatment and C) Kaplan-Meier survival analysis of the MOD tumor burden arm. Mice in the GPC2.19.28TM.41BBz group were euthanized due to clinical signs of GvHD by day 32, as was one mouse in the FMC63.CTRL group on d35 and were censored from the analysis.
D-E) Tumor volume (B) after treatment and E) Kaplan-Meier survival analysis of the HI tumor burden arm. Three mice in the GPC2.19.28TM.41BBz group were euthanized due to clinical signs of GvHD by d32 and were censored from the analysis. Endpoints were defined maximum tumor burden >2 cm3 volume or signs of GVHD.
Statistics in B and D represent two-way RM-ANOVA (**** = p<0.0001, *** = p<0.001, ** = p<0.01, * = p<0.05), ns = p > 0.05).
Late relapse is associated with emergence of GPC2ultralo NBs and can be abrogated by c-JUN overexpression in GPC2.28TM.28z-CAR T cells.
The significant tumor regression observed in GPC2.19.28TM.28z CAR T cells tuned to target antigen density thresholds to levels found on human NBs bodes well for clinical activity, but such constructs are associated with shorter persistence due to the development of CAR T cell exhaustion (Lee et al., 2015; Long et al., 2015) and could predispose to acquired resistance. We followed animals long-term after CRs induced by GPC2.19.28TM.28z CAR T cells and studied relapses occurring in a fraction of animals (Figure 5A, S5A,B). Immunohistochemistry (IHC) and flow cytometry of recurrent tumors demonstrated decreased GPC2 expression, accompanied by substantial CAR T cell infiltration (Figure 5B-D), whereas expression of other antigens such as NCAM, L1CAM, or GD2 were not changed (Figure 5C,D, Figure S5C,D). Upon re-engraftment into non-CAR T cell bearing mice, GPC2 expression on relapsed tumors returned to baseline, indicating high tumor cell plasticity in regulation of GPC2 expression (Figure 5D) mediated by CAR T cell immune pressure.
Figure 5. Acquired resistance following GPC2-CAR therapy is associated with reversible downregulation of GPC2 cell surface expression.
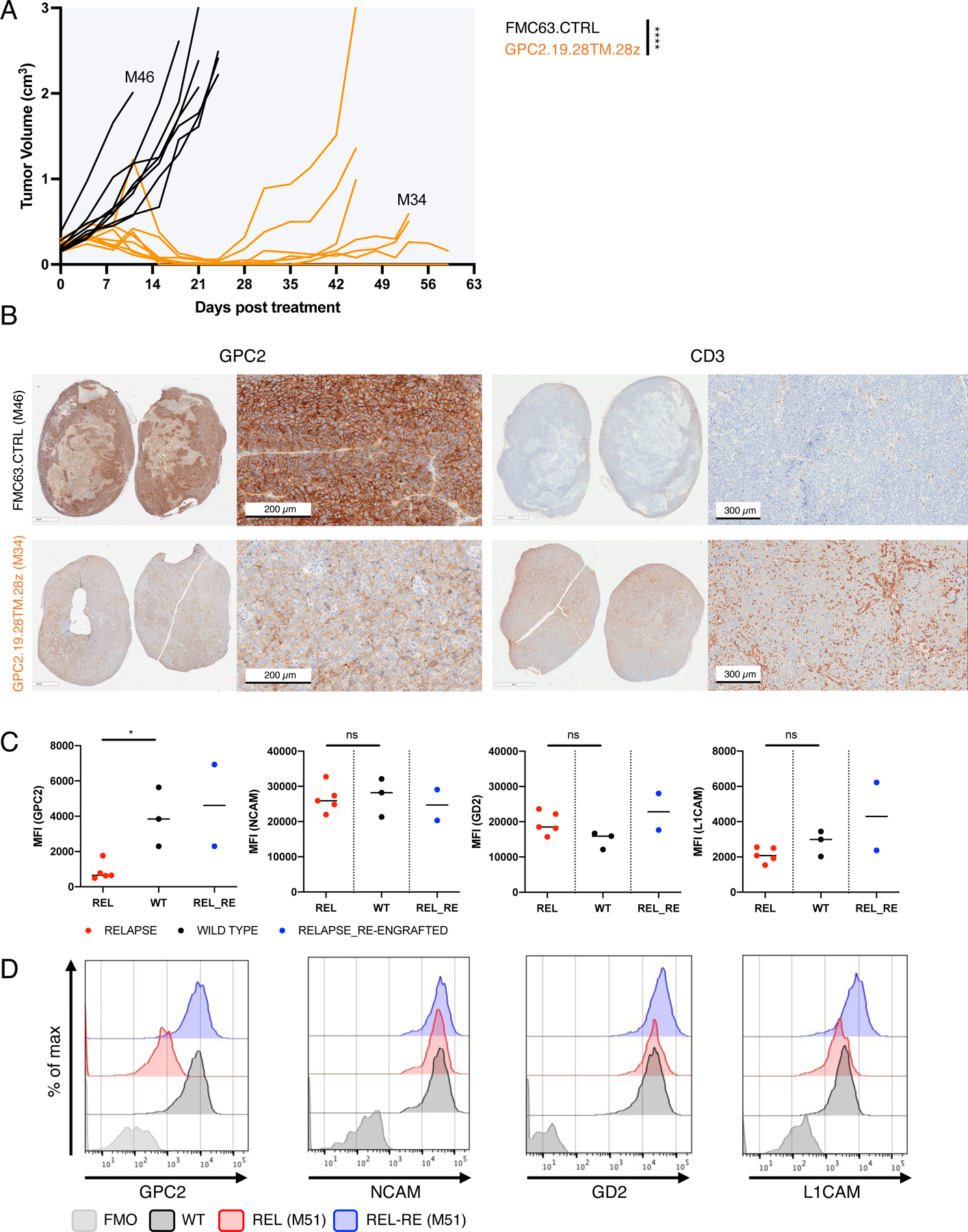
A) Tumor volumes of PDX (COG-N-421x) flank tumors (MOD: range mean TU vol 0.22–0.24 cm3 tumor volume) in mice treated with GPC2.19.28TM.28z- or FMC63.CTRL-CAR T cells. Primary (M46) and recurrent (M34) tumors were harvested for further analysis.
B) Representative IHC images of GPC2 and CD3 expression in post-GPC2.19.28TM.28z recurrent (M34, harvested d56) or CTRL (M46, harvested d12) tumors.
C) Mean Fluorescent Intensity (MFI) of NB cell surface markers in tumors harvested from untreated (WT) and GPC2.19.28TM.28z CAR treated, relapsed samples harvested at the endpoint of experiment shown in Figure 4. Two relapsed samples (M10 and M51), were re-engrafted as single cell suspensions into non-CAR T cell bearing mice and harvested 120 days later and analyzed simultaneously (REL_RE). Horizontal lines indicate median. Statistic represents Mann-Whitney test (* = p<0.05).
D) Representative histograms of one untreated (WT), one relapsed (REL) and one matched relapsed/re-engrafted (REL-RE) tumor.
See also Figure S5.
We recently showed that c-Jun overexpression (OE) enhances potency and persistence of CAR T cells towards antigen-low tumor cells (Lynn et al., 2019), thus we generated a bicistronic c-Jun.GPC2.19.28TM.28z-CAR construct (cJun.GPC2) and tested whether it enhanced in vivo anti-tumor activity against GPC2lo NBs and/or diminished recurrence. cJun.GPC2 demonstrated similar cell surface CAR expression as GPC2.19.28TM.28z (Figure 6A,B) and outperformed all other constructs in killing capacity across varying levels of antigen (Figure S5E). To study functionality in a clinically relevant model, we engrafted patient-derived NB cells isolated from the BM of patient ST16 post treatment at tumor relapse (ST16-BM4224) and transduced with GFP-Luc, into the renal capsule in a para-orthotopic manner (Figure 6C). Interestingly, GPC2 molecules/cell increased 2x during in vitro expansion (5450 on primary NB vs 10843 post expansion) and both CAR constructs effectively cleared tumor (Figure 6D). Yet, in a GPC2lo metastatic model (6873 m/cell) in which both GPC2.19.28TM.28z and GPC2.19.28TM.41BBz were unable to mediate and maintain CRs, cJun.GPC2-CAR T cells exhibited markedly increased anti-tumor activity, CAR expansion and persistence (Figure 6E-G) and also significantly outperformed original constructs against GPC2ultralo tumors expressing even lower antigen levels (3425 molecules/cell) (Figure S5F,G). Moreover, cJun.GPC2 CARs mediated increased durable disease control and CAR-persistence in animals para-orthotopically engrafted with a high-burden of GPC2hi NB (Figure 6H-J) and cleared GPC2hi and GPC2lo tumors (4460 m/cell) equally well when they were co-engrafted, while GPC2.19.8TM.41BBz constructs selectively targeted GPC2hi tumors, but completely spared GPC2lo tumors (Figure 6K-O). Together these data demonstrate that c-Jun OE improves GPC2-CAR T cell potency and mediates durable antitumor activity against NBs expressing clinically relevant GPC2 antigen density, and therefore emerged as the lead candidate for clinical testing.
Figure 6. Overexpression of c-Jun enhances GPC2-CAR potency.
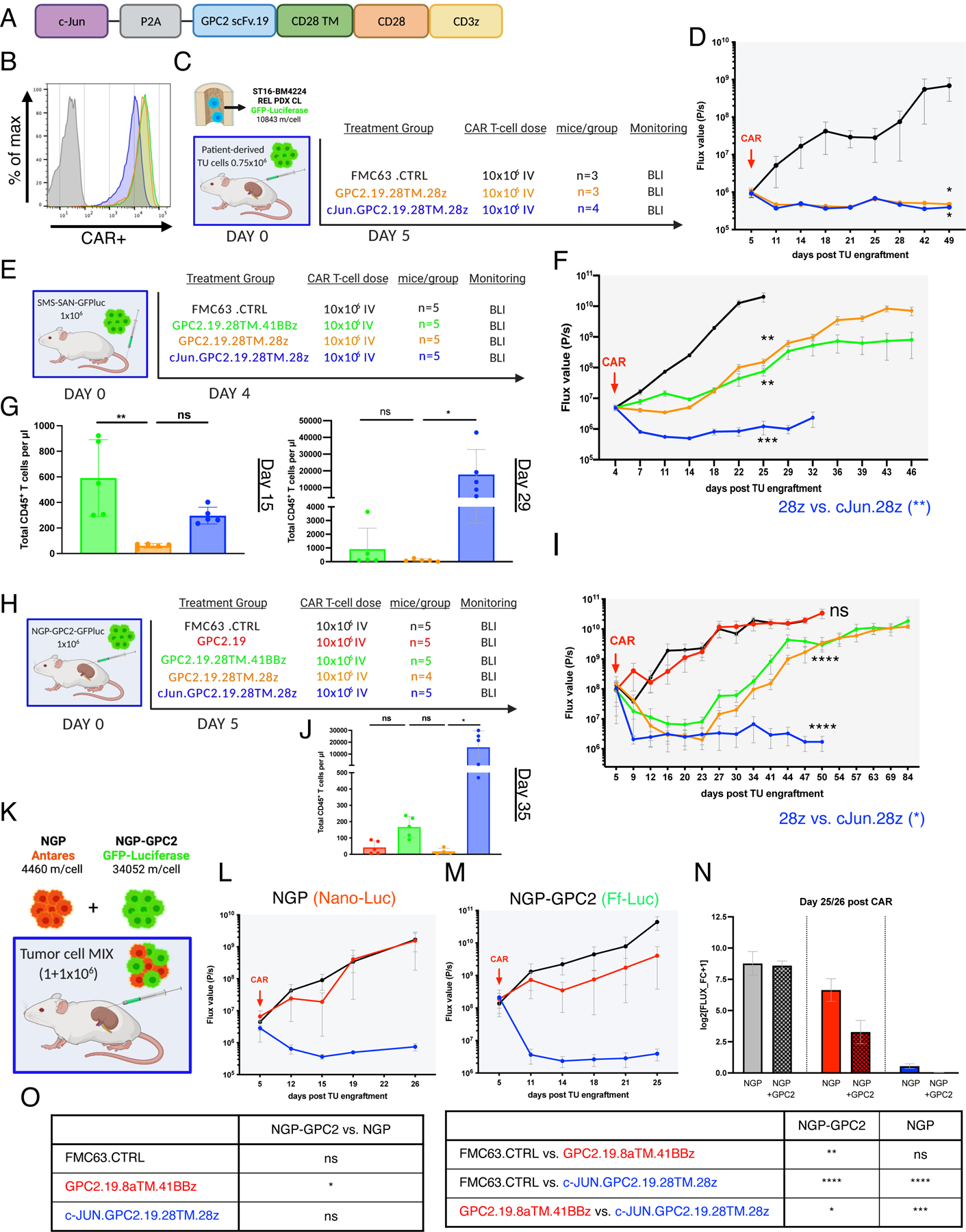
A) Schematic of the c-Jun.GPC2.19.28TM.28z expression vector.
B) Flow cytometry cell surface analysis of GPC2.19-CAR T cell constructs used in vivo as compared to Mock T cells. Color legend of B, D, F and G shown in E.
C) Experimental in vivo setup testing GPC2.19.28TM.28z CAR T cells +/− c-Jun in para-orthotopic renal capsule xenograft model of tumor cells isolated from the BM of patient ST16 at tumor relapse.
D) Corresponding FLUX [P/s] values of tumor burden assessed by IVIS imaging. Statistic represents one-tailed Mann Whitney test at experimental endpoint (d49).
E) Experimental in vivo setup testing c-Jun overexpressing GPC2.19-CAR T cell constructs in comparison to 28TM.41BBz and 28TM.28z in GPC2lo (SMS-SAN) metastatic xenograft model. Representative of n=1 experiment.
F) Corresponding FLUX [P/s] values of tumor burden assessed by IVIS imaging. Statistic represents 2-Way RM-ANOVA by day 25. Endpoint of c-Jun.GPC2.19.28TM.28z group was onset of GvHD on d32.
G) Persistence of CD45+ CAR T cells/µl blood on mice shown in (F) on d15 and d29. Statistic represents one-way multiple comparisons ANOVA.
H) Schematic of experimental setup testing c-Jun overexpressing GPC2.19-CAR T cell constructs in comparison to GPC2.19.28TM.41BBz and GPC2.19.28TM.28z in para-orthotopic NB renal capsule xenograft model engrafted with NGP-GPC2 (GPC2hi) cells. Representative of n=1 experiment.
I) Corresponding FLUX [P/s] values of tumor burden assessed by IVIS imaging. Endpoint of c-Jun.28TM.28z group was onset of GvHD on day 50. Statistic represents 2-Way RM-ANOVA by day 50.
J) Persistence of CD45+ CAR T cells/µl blood on day 35. Statistic represents one-way multiple comparisons ANOVA.
K) Experimental in vivo setup for dual-imaging in para-orthotopic NB renal capsule xenograft model engrafted with GPC2lo/GPC2hi-MIX.
L-M) Corresponding FLUX [P/s] values of NGP tumor burden assessed by IVIS imaging using (L) Nano-Luciferase substrate and M) NGP-GPC2 tumor burden using Firefly-Luciferase substrate post treatment with 10×106 FMC63.CTRL (n=3), GPC2.19.8TM.41BBz (n=3) or c-Jun.GPC2.19.28TM.28z (n=3) CAR T cells N) Tumor burden of GPC2lo and GPC2hi normalized to pre-treatment. Data represents log2 transformed FLUX fold-change+1 at the endpoint (day 26 for NGP, day 25 for NGP-GPC2). Values in D, F, I, L, M, N represent mean ± SEM. Values in G, J represent mean ± SD.
O) Statistical analysis of data shown in N), representing one-way multiple comparison ANOVA (**** = p<0.0001, *** = p<0.001, ** = p<0.01, * = p<0.05), ns = p > 0.05).
See also Figure S5.
GPC2 is an embryonal antigen with expression restricted to fetal brain development.
Next, we sought to evaluate the potential for on-target/off-tumor toxicity of our low antigen density tuned GPC2-CAR T cells. Using IHC, protein expression of GPC2 has been reported to be very limited in normal adult (Li et al., 2017) and pediatric tissues (Bosse et al., 2017). Consistent with this, published databases of GPC2 gene expression during human organ development (Cardoso-Moreira et al., 2019) demonstrate expression restricted to the developing brain, peaking around 9 weeks post-conception, followed by a gradual decrease and complete absence after birth, a pattern mirrored in murine developing tissues (Figure S6A,B). Protein expression in a mass-spectrometry database confirmed this pattern, demonstrating expression of GPC2 limited to the fetal brain with low levels present postnatally only in adult testes (Figure S6C). We further confirmed an inverse correlation between GPC2 expression and gestational age by IHC staining of prenatal, infant and pediatric brain tissues (Figure 7A), and found significantly higher levels of GPC2 on NBs compared to pediatric and infant brain (Figure 7B,C), where H-scores were consistently <100.
Figure 7. GPC2 expression is restricted to fetal brain.
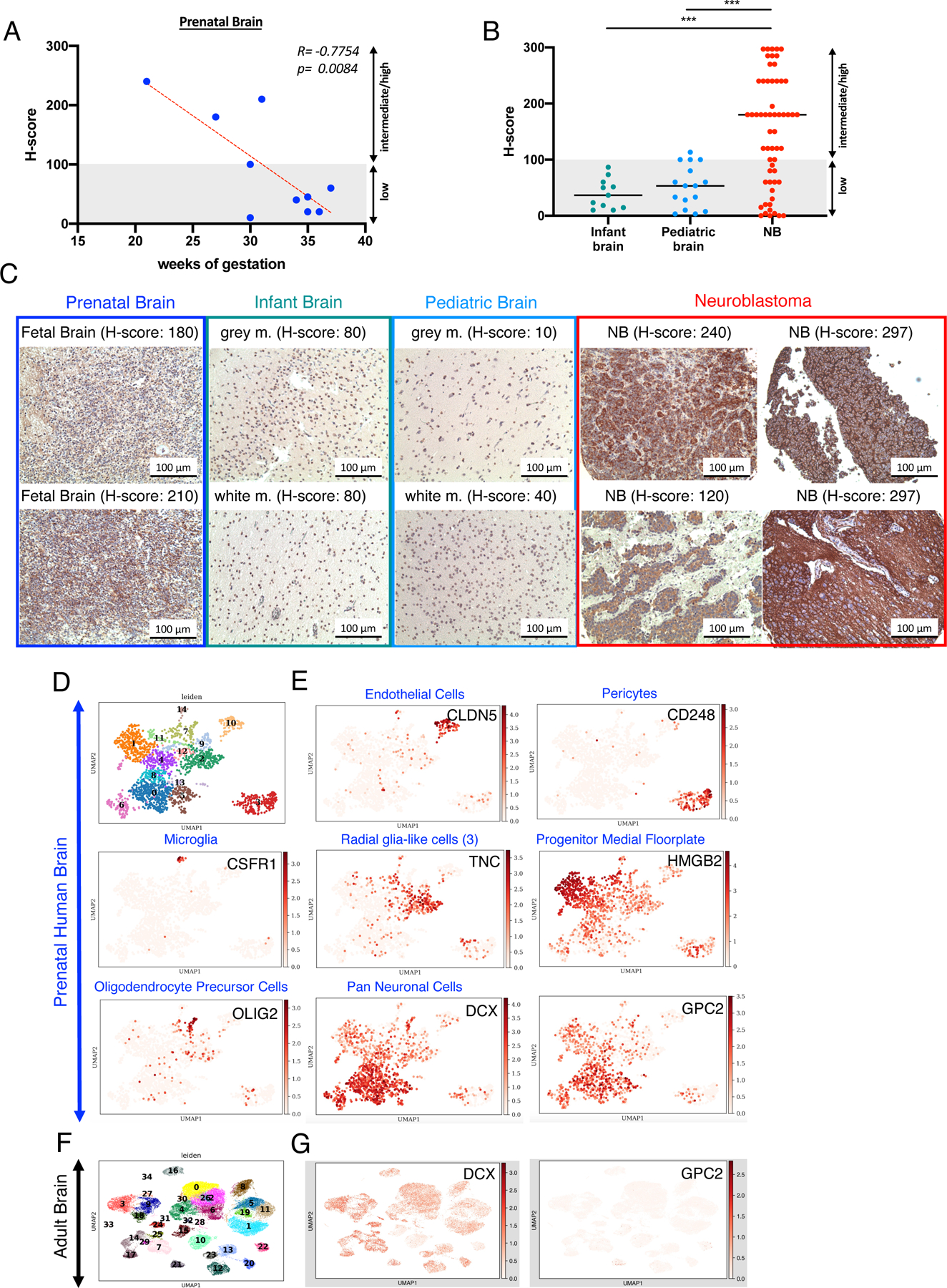
A) Pearson Coefficient Correlation between GPC2 immunostaining (H-score) and gestational age in prenatal brain (n=10) tissues.
B) Immunostaining (H-scores) of GPC2 in infant (n=11) and pediatric brain (n=16) and NB tumor samples (n=58). Statistical analysis represents one-way multiple comparison ANOVA (*** = p<0.001). Prenatal brain vs. NB =ns. Horizontal lines indicate median.
C) Representative IHC images of GPC2 staining in prenatal brain, infant brain, pediatric brain and NB tumors.
D) UMAP projections showing distinct cell populations from scRNAseq data in fetal, human ventral midbrain (from La Manno et. al, Cell 2016), colored by cluster ID.
E) UMAP projection of scRNAseq data from adult human brain, colored by expression of the indicated genes, which mark various brain cell populations. Color indicates log-transformed, depth-normalized counts per cell. Data sourced via GEO accession GSE76381 (La Manno et al., 2016) for human prenatal brain and via the Allen Brain Atlas for human adult brain (https://portal.brain-map.org/atlases-and-data/rnaseq/human-multiple-cortical-areas-smart-seq).
F) Distinct cell populations in single cells of adult human brain (Allen Brain Atlas), colored by cluster ID G) Expression of GPC2 in DCX+ neuronal population in the adult human brain. As in (F), color indicates log-transformed depth-normalized counts per cell.
See also Figure S6.
Analysis of bulk RNAseq datasets can mask gene expression in rare cell types. We therefore analyzed single-cell RNAseq datasets of fetal and adult brain (La Manno et al., 2016), Allen Brain Atlas: http://www.brain-map.org). Of 14 distinct cell populations defined by UMAP clustering in fetal human brain samples, GPC2 was primarily expressed in the neuronal progenitor compartment, in a cluster driven by Doublecortin (DCX) expression (Figure 7D-G), a pattern also observed in the murine developing midbrain (Figure S6D,E). DCX, a microtubule-associated protein found in immature, migrating, neuronal progenitors (Ayanlaja et al., 2017) is downregulated as neurons mature, but remains expressed at reduced levels in single cells of the adult brain, where it is no longer accompanied by GPC2 (Figure 6F,G). Further, flow cytometric quantification of GPC2 on primary neurons and astrocytes revealed levels that were below the threshold of activation for our most potent GPC2 CAR constructs GPC2.19.28HTM.28z +/− c-Jun (Figure S7G-K). Together, these data confirm that GPC2 is a neuronal developmental antigen with insignificant expression in tissues after birth and therefore likely to be a safe target for CAR T cell therapy.
GPC2-CAR T cells eradicate tumor in the absence of toxicity.
We next assessed the specificity of GPC2.19 via a membrane proteome array, testing the GPC2.19-IgG1 mAB for reactivity against a library of >5,300 human membrane proteins, including 94% of all single-pass, multi-pass and GPI-anchored proteins. No off-target binding at mAB concentrations of up to 1.25 µg/mL was observed (Figure S7A). At >5µg/mL, we observed low level off-target binding to voltage-gated hydrogen channel 1 (HVCN1), which is primarily expressed on B cells (Capasso et al., 2010) and granulocytes (Morgan et al., 2009). However, GPC2.19-IgG1 demonstrated only minimal binding to B cells and granulocytes (Figure S7B) and CAR T cells did not mediate cytotoxicity against these cell types, indicating no biologically significant cross-reactivity (Figure S7C,D).
Since gene expression profiles across organ development are comparable between human and murine GPC2 (Figure S6A,B), we used mouse models to assess potential off-target and on-target/off-tumor related toxicities. GPC2.19 exhibits high affinity binding towards human (KD: 11.0±3) and murine GPC2 (77.2±10.2) and GPC2.19 CAR T cells showed effective cytokine production against plate-bound human and murine GPC2 (Figure S7E,F). Using our previously established para-orthotopic renal capsule xenograft model, we tested toxicity of our most potent GPC2.19.28TM.28z CAR T cells +/− c-Jun compared to FMC63.CTRL and performed necropsy and blood analysis 14 days post treatment (Figure 8A, Figure S8A). Animals exhibited no weight loss or clinical sign of toxicity (Figure 8B, Figure S8B) despite potent anti-tumor effects (Figure 8C,D, Figure S8C,D) and no significant changes in blood cell counts or liver function parameters (Figure 8E, Figure S8E). Cellular infiltrates were observed in the liver (2/3) and lung (3/3) of animals treated with cJun.GPC2-CAR T cells, yet all tissues including the liver, testis and CNS were grossly and histologically within normal limits for both GPC2 CARs and FMC63.CTRL treated animals, as evaluated by a blinded pathologist (Figure 8F, Figure S8F). We conclude that GPC2.19.28TM.28z CAR T cells +/− c-JUN OE effectively control tumor in representative disease models without evidence of toxicity.
Figure 8. c-Jun overexpressing GPC2-CAR T cells eradicate tumor in the absence of toxicity.
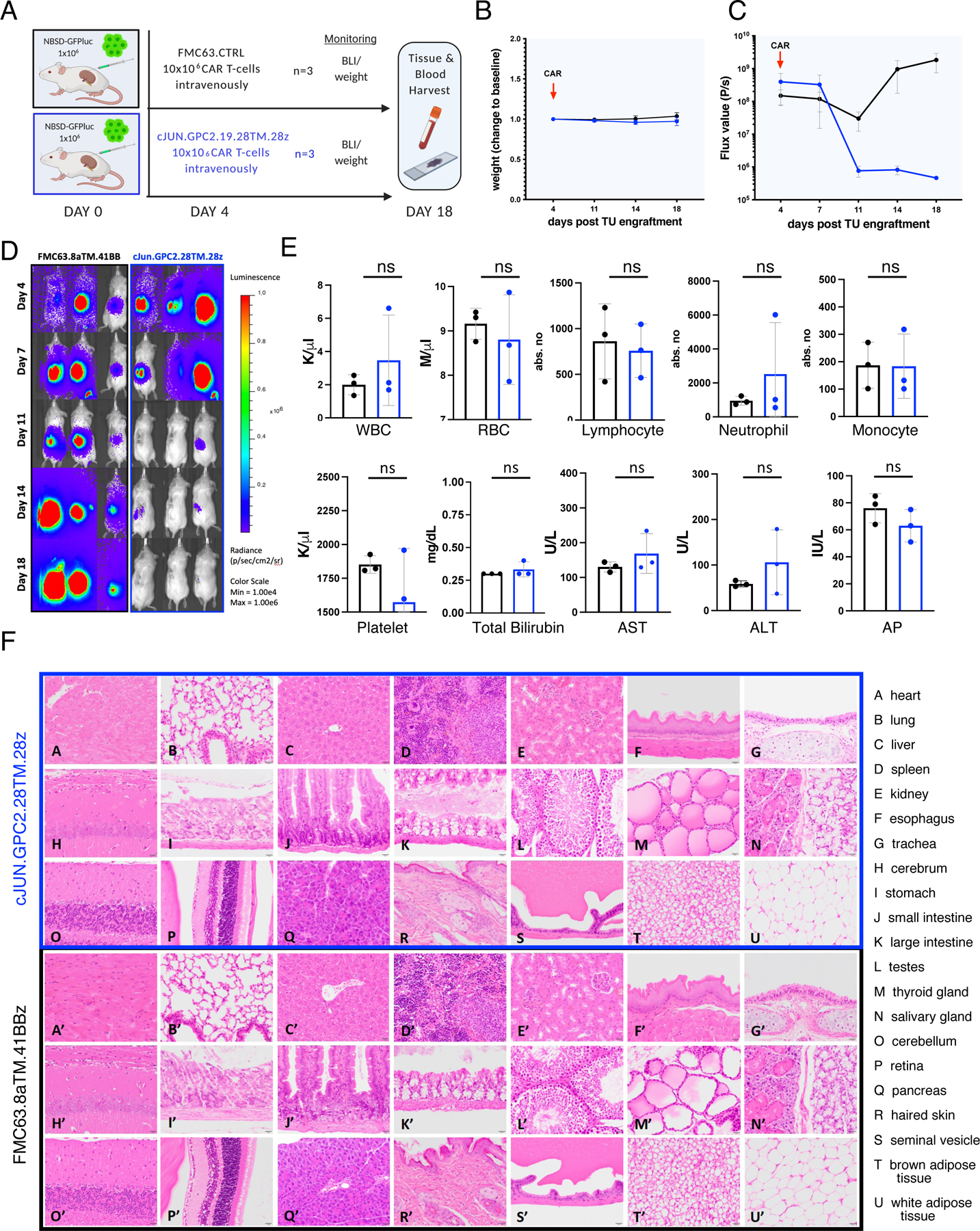
A) Schematic of experimental setup (n=1): NSG mice were engrafted with 1×106 NBSD tumor cells beneath the left renal capsule and treated with 10×106 c-Jun.GPC2.19.28TM.28z or control FMC63 CAR T cells on d4 via IV tail vein injection. Tumor burden and weight was followed until the endpoint on day 18.
B) Weight of treated mice as change to baseline over the course of the experiment. Values represent mean ± SD.
C) FLUX [P/s] values of tumor burden assessed by IVIS imaging and D) BLI images. Values in C) represent mean ± SEM.
E) Assessment of blood cell populations and liver function parameters (transaminases AST, ALT and alkaline phosphatase). Values represent mean ± SD. Statistic represents Student’s t-test (**** = p<0.0001, *** = p<0.001, ** = p<0.01, * = p<0.05), ns = p > 0.05).
F) Hematoxylin and eosin [H&E] stained tissues from mice either treated with GPC2.19 28TM.28z CAR T cells (A-U) or FMC63 control CAR T cells (A’-U’). Magnification: 40x. Scale bar: 20μm.
See also Figures S7-8.
DISCUSSION
Given the rarity of tumor-specific cell surface molecules, expanding the reach of CAR T cells to solid tumors will require the field to develop approaches to exploit quantitative differences in antigen expression between tumors and normal tissues. In contrast to T cell receptors, which recognize cells expressing <10 peptides, CARs require thousands of molecules for full T cell activation (Dong et al., 2020; Gudipati et al., 2020; Majzner et al., 2020). Current understanding holds that CAR T cells require high antigen density due to inefficient immune synapse formation (Dong et al., 2020) and weak proximal signaling associated with inefficient Zap70 recruitment (Gudipati et al., 2020; Majzner et al., 2020), which is unlikely to be significantly impacted by further affinity enhancement of the scFvs. This property has profound implications for clinical translation, since even low-level target antigen expression on vital tissues can prove fatal when using TCRs (Cameron et al., 2013; Linette et al., 2013; Morgan et al., 2013), while antigens expressed at low levels on vital tissues may be viable targets for CAR T cells, as suggested by the safety of CARs targeting GD2, a target expressed at low levels on neural tissues (Heczey et al., 2017; Straathof et al., 2020). The requirement for high antigen density, however, also increases the risk of relapse with antigen low variants following CAR T cell therapy, which has been observed following BCMA (Cohen et al., 2019), EGFRvIII (O’Rourke et al., 2017), IL13Ra2 (Brown et al., 2016), CD22 (Fry et al., 2018) and CD19-targeted CARs (Spiegel et al., 2021).
The convergence of data highlighting antigen density as a core feature regulating CAR T cell activity emphasizes a need for approaches to accurately quantify cell surface antigen density on clinical samples. We previously used quantitative flow cytometry to measure antigen density on B-ALL which was informative for CD22 (Fry et al., 2018; Haso et al., 2013) and CD19 (Lee et al., 2015) and recently demonstrated that quantitative flow cytometry outperformed IHC in predicting risk for disease progression following CD19-CAR for lymphoma, identifying <3000 CD19 molecules/cell as associated with a greater risk for relapse (Spiegel et al., 2021). Here, we measured antigen density on metastatic neuroblastoma cells in BM samples and obtained remarkably similar results despite conducting the analyses at two separate institutions. We observed limited interpatient variability in the expression density of GPC2, NCAM, L1CAM, ALK and B7-H3, which contrasted with highly variable expression of GD2 in clinical samples and variable expression of GD2, GPC2, NCAM, L1CAM and B7-H3 on cell lines. PDX tumors demonstrated higher antigen density levels than primary tumors for each target, except GD2. While we acknowledge that the generalizability of these findings are limited due to the small sample size of our clinical specimens, and the fact that primary tumors and recurrent tumors were not analyzed, the data clearly demonstrate that overexpressed tumor antigens can vary widely in the level and variation of expression, and therefore disease models used for CAR T cell development, must be validated to express clinically relevant levels of target antigen if they are to inform predicted potency against disease in humans.
NB is the most common extracranial solid tumor of childhood, and long-term survival for high-risk cases remains less than 50% despite dose intensive, toxic treatment regimens (Bosse and Maris, 2016; Richards et al., 2018). CAR T cells targeting GD2 have shown some promise, although the objective responses rates are low and a clear impact on survival has not been demonstrated (Louis et al., 2011; Pule et al., 2008; Straathof et al., 2020). Additional immunotherapy targets are needed, since tumor heterogeneity is a fundamental feature of cancer and patients who have received GD2-targeted immunotherapies (CAR T cells or dinutuximab) are likely to be at risk for GD2 loss or downregulation. GPC2 is overexpressed on NB compared to normal tissues and immunotoxins (Li et al., 2017) and an antibody-drug conjugate targeting GPC2 has shown promising anti-tumor effects in vivo in preclinical models (Bosse et al., 2017; Raman et al., 2021). We thus sought to develop GPC2-CAR T cells targeting NB but our initial designs were wholly inadequate to control NBs expressing clinically relevant levels of GPC2. Similarly, a previous report using single-domain binding moieties led to only modest anti-tumor effects in in vivo models (Li et al., 2017).
Numerous factors, including scFv affinity, epitope specificity, TM domain, costimulatory signals and levels of CAR T cell exhaustion modulate the antigen density threshold for CAR T cell activation (Caruso et al., 2015; Haso et al., 2013; Majzner et al., 2019; Watanabe et al., 2018). Informed by data regarding the antigen density level required for effective GPC2 targeting by CAR T cells in metastatic NB, we iteratively optimized GPC2-CARs to lower the antigen density threshold. Substitution of the CD28 TM for a CD8 TM domain was remarkably effective, and although 41BB costimulatory domains have been emphasized due to their enhanced persistence (Zhao et al., 2015) and decreased propensity for exhaustion (Long et al., 2015), we found that GPC2-CAR T cells with CD28 costimulatory endodomains emerged as the lead candidate. Despite these design modifications, we observed recurrence in some animals with GPC2ultralo NB, which was ultimately prevented by incorporating c-Jun expression, further demonstrating the capacity for c-Jun to enhance the potency of CAR T cells (Lynn et al., 2019). Together, these iterations lowered the antigen density threshold approximately 10-fold. Tuning CAR T cells to recognize low antigen density potentially increases the risk for on-target or off-target toxicity, however our data demonstrate that GPC2 expression is very limited in postnatal human and murine tissues, and we did not observe evidence for on-target or off-target toxicity with our most potent GPC2-CAR T cells, which bind to murine GPC2, albeit with slightly lower affinity than to human GPC2.
In summary, this work highlights a profound impact of antigen density on the potency of CAR T cells and illustrates an essential role for both accurate quantitation of antigen density on clinical samples and iterative engineering of CAR T cells focusing on antigen density thresholds. We demonstrate that numerous accessible solutions are available to tune CAR T cells based upon a desired antigen density threshold. Using stringent efficacy and toxicity models, we have credentialed potent lead GPC2-CAR candidates that are ready for clinical testing in NB, as well as a variety of other currently lethal cancers for which standard therapies are inadequate (Bosse et al., 2017; Raman et al., 2021). Finally, these data also credential oncofetal antigens as a class of nonmutant targets that are ripe for targeting with CAR T cells.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Crystal L. Mackall (cmackall@stanford.edu).
Materials Availability
Plasmids generated in this study will be made available upon request and completion of a Material Transfer Agreement.
Data and Code Availability
This study did not generate any novel datasets. Sources of publicly available scRNAseq datasets are La Manno et al. 2016 via GEO (accession GSE76381) for human and murine prenatal brain, the Allen Brain Atlas for human adult brain data (https://portal.brain-map.org/atlases-and-data/rnaseq/human-multiple-cortical-areas-smart-seq).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines, cell culture and PDX tumors and specimens
The NB cell lines SMS-SAN, NBSD, NGP, NGP-GPC2, EBC1, NB1643, CHLA255, SMS-KCNR, CHLA-90, Kelly, SKNAS, Lan5 and the CHO-K1 cells were cultured in RPMI-1640, supplemented with 10% heat-inactivated FBS (Gibco, Life Technologies), 10mM HEPES, 100U/mL penicillin, 100 μg/ml streptomycin and 2mM L-glutamine (Gibco, Life technologies). The HS-5 cell line was cultured in DMEM, supplemented with 10% heat-inactivated FBS (Gibco, Life Technologies), 10 mM HEPES, 100 U/mL penicillin, 100 μg/ml streptomycin and 2m M L-glutamine (Gibco, Life technologies). All cell lines used were fingerprinted every 6 months and regularly tested negative for Mycoplasm using LabCorp Genetica Cell Line Testing. The NB cell lines SMS-SAN, NBSD, NGP-GPC2 were stably transduced with a retroviral vector encoding the GFP-Firefly-Luciferase gene for the monitoring tumor burden by IVIS imaging in in vivo models. Patient-derived cell lines established from bone marrows of a NB patient at diagnosis and relapse (ST16–4045 and ST16–4224) were cultured in a final media containing 1 part of medium A (EBM2 medium with the following EGM2 SingleQuote supplements: FBS, Hydrocortisone, hFGF-B, R3-IGF-1, Ascorbic acid, hEGF, GA-1000 and Heparin (Lonza, Cat# CC-3162) and 3 parts of medium B (Corning™ cellgro™ RPMI 1640 Medium, Cat# MT15041CV) without L-Glutamine supplemented with 5% FBS, 1X glutamine and ITS (500x) Insulin, Transferrin and Selenium (Lonza, Cat# 17–838Z), 100 U /ml penicillin, and 100 μg/ml streptomycin (Gibco, Life technologies). Patient-derived xenograft tumors for multicolor antigen density quantification were obtained from Dr. John Maris. After thawing, tumors were processed into homogenous single cell suspensions via mechanical dissociation using a gentleMACS dissociator (Miltenyi), passage through 70 micron filters and ACK lysis followed by 2 wash steps with PBS. The sex of cell lines/PDX samples used in this study is female for CHO, SMS-SAN, NBSD, SKNAS, Kelly, COG-N-440x, COG-N-471x, COG-N-496x, COG-N-561x, COG-N-624x, CHLA-79 xenograft and male for HS-5, NGP, EBC1, NB1643, SMS-KCNR, CHLA-90, Lan5, ST16 and COG-N-421x, COG-426x-Felix, COG-N-453x, COG-N-519x, COG-N-557x, COG-N-549x, COG-N-590x, COG-N-603x, NB-1643 xenograft and unspecified for CHLA255.
Human Samples
Bone marrow of pediatric NB patients was collected and utilized following the guidelines of Stanford University Institutional Review Board (IRB)-approved protocols (#45458 and #56619) and stored at the Stanford Pediatric Bass Center Tissue bank until use (n=5). ST16-BM4045 (at diagnosis) and ST16-BM4224 (at relapse) were derived from a 2-yrs 9 mo old male HR-NBL patient with MYCN amplification. All other samples were derived from patients without MYCN amplification, ST36 (at diagnosis) from a female 2-yrs 10 mo old HR-NBL, ST77 (at diagnosis) from a 7-yrs 5 mo old male HR-NBL patient and ST5 (at relapse) from a 6-yrs 1 mo old patient with composite pheochromocytoma. The matched patient-derived cell lines from patient ST16 were established by either injection into NSG mice followed by harvest/dissociation via collagenase digestion for BM4045 (diagnostic timepoint) or Ficoll-purification for BM4224 (relapse timepoint) and subsequent cell culture to generate stable cell lines. ST16-BM4224 was transduced with GFP-Luciferase prior to engraftment in para-orthotopic in vivo models. Additional bone marrow samples of pediatric NB patients (n=4) were obtained at the St. Anna Children’s Hospital (Vienna, Austria)/Children’s Cancer Research Institute under the ethics committee of the Medical University of Vienna (EK)-approved protocols (EK#115/2006, EK#1853/2016 and EK#1216/2018). All 4 samples were taken at diagnosis of HR-NBL patients, three with MYCN amplification (AT1: female/1 yrs 9 mo, AT3: male/2 yrs 5 mo and AT4: female/7 yrs 2 mo) and one (AT2: female/7 years) without. All specimens were obtained with written informed consent in accordance with the Declaration of Helsinki from either Lucile Packard Children’s Hospital (Stanford, CA) or St. Anna Children’s Hospital (Vienna, Austria). For immunohistochemistry, tissue sections of fetal and infant brain were obtained as previously described (Theruvath et al., 2020). A tissue microarray containing pediatric CNS and PNS tissue (normal tissue from children diagnosed with non-cancer-related pathologies) was obtained from the BC Children’s Hospital (Vancouver, BC, Canada). A tissue microarray containing 58 NB cases was obtained from the Children’s Oncology Group (COG). Primary human CNS cells were purchased from Neuromics (Edina, MN), Primary Human Neurons (#HNC001) and Human Brain Astrocytes (#HMP200) were grown in T25 flasks pre-coated with AlphaBiocoat Solution (#AC001) in Neuron Growth Media (#HNM001) or Astrocyte-Growth Media (#PGB003) respectively and media was exchanged every 2–3 days.
Murine Studies
Animals were housed in cages of up to 5 mice in pathogen-free conditions at a barrier facility at the Lokey Stem Cell Building (SIM1) at Stanford School of Medicine (Stanford, California) or at the Children’s Hospital of Philadelphia Animal Facility, fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). All animal handling, surveillance, and experimentation was performed in accordance with and approval from the Stanford University Administrative Panel on Laboratory Animal Care (Protocol #APLAC-31287) or approved by the CHOP Institutional Animal Care and Use Committee (IACUC; Approved IACUC Protocol #643). For para-orthotopic and metastatic xenograft models, 6–10 weeks old male or female in-house bred NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ: The Jackson Laboratory) were used. Animals bearing engrafted tumors were randomized into cohorts to ensure a similar mean tumor burden/group based on bioluminescent FLUX [P/s] values at study enrollment.
For patient derived xenograft models, 6–7 weeks old female (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, purchased from: The Jackson Laboratory, #005557) mice were used. Animals bearing engrafted tumors were randomized assigned into cohorts, to ensure a similar mean tumor volume/group based on caliper measurements at study enrollment.
METHOD DETAILS
Generation of binder sequences targeting GPC2
A naive human Fab and a scFv phage display libraries constructed from peripheral blood B cells of 50 healthy donors (Zhu and Dimitrov, 2009) was used for selection of Fabs and scFvs respectively against purified recombinant GPC2 ectodomain (R&D Systems, Cat#2304). Briefly, the isolated Fabs and scFvs were expressed, purified and tested for binding to the GPC2 ectodomain through ELISA and to GPC2 positive cells through FACS, and two scFv binders, designated as GPC2.19 and GPC2.27 were selected from the human scFv phage display library for further characterization, two Fabs binders were isolated from the human Fab phage display library, designated as GPC2.D3 and GPC2.D4, were converted to scFvs as CAR for further characterizations. Binders GPC2.19 and GPC2.D3 were also converted to full-length human IgG1. The scFvs and Fabs were expressed in HB2151 cells and purified as previously described (Zhu and Dimitrov, 2009). Briefly, plasmids were transformed into HB2151 cells. A single colony was picked from the plate containing freshly transformed cells, inoculated into 200 mL 2YT medium broth containing 100 μg/ml ampicillin and 0.2% glucose, and incubated at 37°C with shaking at 250 rpm. When the culture OD at 600 nm reached 0.90, isopropyl-β-d-thiogalactopyranoside at a 0.5 mM final concentration was added, and the culture was further incubated overnight at 30°C. The bacterial pellet was collected after centrifugation at 8,000 × g for 20 minutes and resuspended in PBS buffer containing 0.5 mil polymyxin B (Sigma-Aldrich, Cat# P4119). After 30 minutes incubation with rotation at 50 rpm at room temperature, the mixture was centrifuged at 25,000 × g for 25 minutes at 4°C, and the supernatant was used for 6× His-tagged scFv and Fab purification using Nickel charged resin (Ni-NTA Agarose, QIAGEN, #30210). The full length lgG1 DNA constructs were transiently transfected into Freestyle™ 293-F cells (Thermo Fisher Scientific, Cat#R79007) for antibody production and the GPC2.19 and GPC2.D3 lgG1 were purified on a protein A column.
Molecular Cloning and DNA plasmids
Genes encoding for GPC2 scFv’s were synthesized as either gene fragments (gBlock, IDT DNA) or gene-encoding plasmids synthesized by GeneArt (LifeTechnologies) and then cloned into a MSGV1 retroviral expression vector containing a granulocyte-macrophage colony-stimulating factor leader sequence using restriction cloning (Rapid DNA Ligation Kit, Roche) or using the In-fusion HD cloning system (Clontech, Takara) according to manufacturer instructions. The wild type c-JUN and P2A sequence was derived as previously used in our lab (Lynn et al., 2019). Final constructs were expressed in Stellar Competent Cells (Clontech, Takara) and bacterial cultures were grown overnight at 37°C in 8% CO2 while shaking, subsequently pelleted and DNA was extracted using ZymoPURE II Plasmid Maxiprep Kit (Zymo Research).
Protein expression and purification
Full-length human GPC2 and mouse GPC2 ectodomains with C-terminal His6x tags were purchased from R&D Systems (catalogue #2304-GP and 2355-GP, respectively). The gene for the human GPC2 construct GPC224−493 containing a C-terminal His6x tag was codon-optimized for expression in human cells and cloned into the pHLsec vector (GeneArt), as previously described (Raman et al., 2021). GPC2 point mutants were generated using a KOD-Plus-Mutagenesis Kit following the manufacturer’s instructions. All constructs were transiently transfected and expressed in HEK293F cells and purified using Ni-NTA affinity chromatography, followed by size exclusion chromatography in 20 mM Tris, pH 8.0 and 150 mM NaCl. Genes for the heavy and light chains of the four Fabs were codon-optimized for expression in human cells and cloned into the pcDNA3.4 vector (GeneArt). Fabs were transiently expressed in HEK293F cells and purified using Kappa-Select affinity chromatography (GE Healthcare), followed by size exclusion chromatography in phosphate buffer saline (PBS).
Biolayer interferometry binding and competition assays
The binding affinity of Fabs to human and mouse GPC2 was measured by BLI on an Octet Red96 instrument (FortéBio) at 25°C. All proteins were diluted in 1X kinetics buffer (PBS, pH 7.4, 0.01% [w/v] BSA, 0.002% [v/v] Tween-20). GPC2 was immobilized on Ni-NTA biosensors, which were subsequently dipped into wells containing serial dilutions of Fabs to determine the rate of association. Sensors were then dipped back into kinetics buffer to monitor the dissociation rate. Curves were fitted to a 1:1 binding model and the kinetic parameters (kon and koff) and dissociation constant (KD) were determined using FortéBio’s data analysis software (Version 9.0). For tandem competition assays, GPC2 was immobilized on Ni-NTA sensors followed by binding to a first Fab and followed by dipping the biosensor into a second Fab at 500 nM concentration.
Thermostability assay
The melting temperature (Tm) and aggregation temperature (Tagg) of the different Fabs were determined using a UNit system (Unchained Labs). Samples at a concentration of 1 mg/mL in PBS were subjected to a thermal gradient from 25 to 95° C with 1° C increments. Tm values were calculated from the barycentric mean fluorescence and Tagg values were determined from the static light scattering at a 266 nm wavelength.
Retroviral vector production and transduction of human T cells
Retroviral supernatant was produced via transient transfection of the 293GP packaging cell line as previously described (Haso et al., 2013). Briefly, 70% confluent cells were co-transfected via Lipofectamine 2000 (Life Technologies) in 150mm Poly-D-Lysine culture dishes with the plasmids encoding the CARs and the RD114 envelope protein. Media was replaced at 24 and 48 hours post transfection. Viral supernatant was harvested 48- and 72-hours post-transfection and centrifuged to remove cell debris and stored at −80°C until use. Primary human T cells were isolated from healthy donors derived from the Stanford Blood Bank using the RosetteSep Human T cell Enrichment kit (Stem Cell Technologies) using buffy coats derived from the Stanford Blood Center and processed according to the manufacturer’s protocol using Lymphoprep density gradient medium and SepMate-50 tubes. Isolated T cells were cryopreserved in CryoStor CS10 cryopreservation medium (Stem Cell Technologies). Cryopreserved T cells were thawed and activated with Dynabeads Human T-Expander CD3/CD28 (Thermo Fischer Scientific, Gibco) at a 3:1 beads:cell ratio in AIM-V media supplemented with 5% FBS, 10mM HEPES, 2 mM l-glutamine, 100 U/mL penicillin, and 100ug/mL streptomycin (Thermo Fischer Scientific, Gibco) and with 100 IU/ml of recombinant human IL-2 (Preprotech). T cells were transduced with retroviral vector on Retronectin-coated (Takara) non-tissue culture treated plates on days 3 and 4 post activation and anti-CD3/CD28 beads were removed on day 5. CAR T cells were maintained at 0.3–1×106 cells per mL in T cell medium supplemented with 100 IU/ml IL2. CAR T cells were used for in vitro assays or transferred into mice on day 10 or 11 post activation.
Flow Cytometry and Cell Phenotyping
Data was collected with an LSR Fortessa X-20 (BD Bioscience) and analyzed using FlowJo software. Cells were harvested, washed twice with FACS buffer (PBS supplemented with 2% FBS and 0.4% 0.5M EDTA) and stained for 30 min in the dark on ice. Cells were washed 3 times with FACS buffer after each incubation step. Cells were gated on viable cells (fixable viability dye eF780, and singlet discrimination (FSC-A/FSC-H) was performed before assessment of antigen expression. The antibodies used in this study were specific to human CD4, CD8, PD1, Tim3, Lag3, CD62L, CD45RA, CD3, CD45, CD13, CD19, CD15, CD66b, HVCN1, GD2, NCAM, L1CAM, B7-H3, ALK and murine CD45, TER-119, H-2Kb and CD31 conjugated with PE, FITC, APC, PE-Cy7, Per-CP-Cy5.5, BUV395, BUV495, BUV805, BUV737, BV510, BV421, BV605, BV711 and PacBlue flurochromes. The GPC2-specific antibodies D3-IgG1 (provided by Dimiter S. Dimitrov, University of Pittsburgh) and 19-IgG1 were labelled with Dylight650 Microscale Antibody Labelling Kits (Fisher Scientific, Cat# 84536). Details on usage in Multicolor Antigen Density Quantification Assay are specified in the next section. CAR expression was assessed by Flow Cytometry after incubation with soluble, recombinant, human GPC2 (R&D systems) for GPC2-CAR T cells or FMC63 anti-idiotype antibody (provided by Laurence Cooper, MD Anderson Cancer Center) for CTRL CD19 CAR T cells, labelled using Dylight650 Microscale Antibody Labelling Kits (Fisher Scientific) respectively. Target binding to murine GPC2 was assessed by Flow Cytometry after incubation with soluble, recombinant, murine GPC2 (R&D systems) labelled using Dylight488 Microscale Antibody Labelling Kits (Fisher Scientific).
Multicolor Quantibrite Antigen Density Quantification Assay
The cell surface quantification of immunotherapy-relevant NB markers on NB infiltrated bone-marrow samples from patients was enumerated by Flow Cytometry using BD QuantiBRITE beads and Custom Quantitation Beads (BD Biosciences, San Jose, CA, USA). Ficoll-purified samples were thawed and incubated with 1mL of Cellstripper (Corning, #MT25056CI) non-enzymatic Dissociation Reagent to break up tumor cell spheres and washed with FACS buffer. FC Block (BD Human FC Block #564219) was added to prevent non-specific staining by FC receptor expressing cells and incubated with the antibody cocktail in Brilliant Stain buffer (BD Biosciences, San Jose, CA, USA) without washing and incubated on ice for 30 minutes. Post incubation, samples were washed twice with FACS buffer and passed through Cell Strainer Cap Falcon Round-Bottom Polystyrene tubes. Analysis gates were drawn based on FSC/SSC, viable cells (eBioscience™ Fixable Viability Dye eFluor™ 780), and single cells (FSC-A/FSC-H). Non-tumor cell populations were gated out based on CD45 for hematopoietic cells and CD13 for bone marrow stromal cells. The tumor-cell population was identified based on NCAM (BV605, Biolegend, clone 5.1H11) and GD2 (BV510, Biolegend, clone 14g2a) and MRD negative bone marrow samples from ALL patients with and without spike-in of NB ST16-BM4045 patient-derived cell lines were used as controls. In addition, GPC2 (PE-conjugated using Abcam PE Conjugation kit Lightning-Link, clone D3), L1CAM (BV421, BD Biosciences, clone 5G3), ALK (APC-conjugated using Abcam APC Conjugation kit Lightning-Link ab201807, clone ALK-48) and B7-H3 (PE-Cy7, Biolegend, clone MIH42) were included in the panel and gated based on background staining on fluorescent minus one (FMO) and/or CHO negative control cells. All antibodies were titrated prior to use to ensure optimal saturating conditions. In conjunction, BD QuantiBRITE-PE beads and BD Custom Quantitation Beads in BV421, BV510, BV605 and APC were run. These are pre-calibrated standard bead sets containing known numbers of fluorophore molecules bound per bead to calibrate and convert flow cytometry fluorescence signal into number of fluorophores. They allow for calculation of antigens per cell when using antibodies at saturating conditions and accounting for the corresponding Fluorophore to Protein Ratio (F:P) of each antibody. Information on respective F:P Ratios of each antibody was provided by Biolegend for NCAM-BV605 (Lot#B280386=3.4:1), B7-H3-PE-Cy7 (Lot#B293754=0.92:1) and GD2-BV510 (Lot#B280958=3.1:1 or Lot#B305929=4.2:1) and experimentally determined for L1CAM-BV421 (Lot#9241038=2.21:1 and Lot#246703=1.83:1) and ALK-APC (3.34:1) by using PE-conjugated versions of the same clones (F:P =1:1) and extrapolating F:P ratios from BD QuantiBRITE-PE measurements. For PE-conjugated antibodies (GPC2.D3-IgG1) using Abcam PE Conjugation kit Lightning-Link, a resulting F:P ratio of 1:1 was taken into consideration, according to manufacturer’s instructions. For quantification of GPC2 on primary human Astrocytes and Neurons the same PE-conjugated GPC2.D3-IgG1 antibody was used in conjunction with QuantiBRITE-PE beads. Molecules/cell were calculated post subtracting background signal emanating from a respective isotype control antibody (PE human IgG1 antibody, Biolegend, Clone QA16A12).
ELISA
Cytokine release was assayed by co-incubating 0.1×106 CAR+ T cells and 0.1×106 tumor cells in complete RPMI-1640 in triplicates. At 24 hours, culture media were collected stored at −20 °C until analyzed. Cytokines levels for IFNγ and IL-2 were measured in supernatants after thawing (Biolegend). Baseline cytokine production of CAR only conditions was subtracted for IFNγ as indicated in the figures.
IncuCyte killing assays
For IncuCyte killing assays 0.05×106 GPF-positive tumor cells were plated in triplicates in 96-well flat-bottom plates and co-incubated with GPC2 CAR-positive T cells or an equivalent number of FMC63.CTRL CAR T cells targeting CD19 at either 1:1, 1:5 or 1:8 effector to target ratios in 200 μl RPMI-1640. Plates were imaged every 2–3 hours using the IncuCyte ZOOM Live-Cell analysis system (Essen Bioscience) and 4 images per well at 10X zoom were collected at each time point. Total integrated GFP intensity per well was assessed as a quantitative measure of viable, GFP-positive tumor cells. Values were normalized to the starting measurement and plotted over time.
Xenograft Mouse Models
For para-orthotopic NB models, NB tumor cells co-expressing GFP-Luciferase (0.75 or 1×106, as indicated in the figure legend) were surgically implanted beneath the left renal capsule as previously described (Patterson et al., 2011). Tumor cells were harvested, washed twice with PBS and resuspended as 1×106 cells per 100 μl and stored on ice until injection. For metastatic NB models, 1×106 SMS-SAN-GFP-Luciferase or 0.75 ×106 SKNAS-GFP-Luciferase tumor cells were washed twice with PBS and injected intravenously into the tail vein in a total volume of 200 μl PBS. Tumor engraftment and growth was followed via bioluminescence imaging on an IVIS spectrum instrument (Caliper Life Science, Hopkinton, MA, USA) and quantified with Living Image software (PerkinElmer, Waltham, MA, USA). Isofluorane-anesthetized mice were imaged 4 minutes after 3mg D-luciferin (Perkin-Elmer) was injected intraperitoneally at an exposure time of 30 seconds. For dual-imaging of GPC2lo/GPC2hi-MIX, 1×106 GPC2lo NB cells (NGP) transduced with Antares were mixed at a 1:1 ratio with 1×106 GPC2hi cells (NGP-GPC2) transduced with GFP-Firefly-Luciferase and surgically implanted beneath the left renal capsule. The GPC2hi (NGP-GPC2-GFP-Firefly-Luciferase) population was imaged using D-luciferin (Perkin-Elmer) as described above and the GPC2lo population was imaged using Nano-Glo Luciferase substrate (Promega, #N1110). On the initial timepoint before CAR T treatment, populations were imaged 8 hours apart to rule out signal overlap and on 2 consecutive days for all other imaging timepoints.
Four to five days after tumor implantation (as indicated in the figure legend and/or schematic of experimental outline), mice received 10×106 CAR-positive GPC2-CAR T cells or an equivalent number of FMC63.CTRL-CAR T cells in 200 μl PBS intravenously via tailvein injections. Endpoints for both para-orthotopic renal capsule and metastatic models were defined as max. tumor of FLUX [P/s] values >1010 or displayed sign of morbidity due to tumor burden or GvHD. For patient-derived xenograft models, CHOP-N-421x PDX tumors were implanted into the flank of mice. Tumor engraftment and growth was followed using caliper measurements at least twice weekly. Tumor volumes were calculated as volume = ((diameter1/2 + diameter2/2)3*0.5236)/1000. All mice weights were also measured at least twice weekly and mice were monitored daily for signs of clinical toxicity. Mice were euthanized when tumor volumes reached/exceeded 2 cm3 or an animal displayed signs of clinical toxicity including excessive weight loss due to tumor burden or GvHD.
Peripheral blood sampling of mice was conducted via retro-orbital blood collection under isoflurane anesthesia at the indicated time point. 50µL blood was labeled for CD45, CD4, and CD8, lysed using BD FACS Lysing Solution (10x Concentrate, BD Biosciences) and quantified using CountBright Absolute Counting beads (Thermo Fisher Scientific) on an LSR Fortessa X-20. Analysis of xenograft tumor tissue was performed after harvesting, optional mechanical dissociation using a gentleMACS dissociator (Miltenyi), passage through 70 micron filters and ACK lysis followed by 2 wash steps with PBS to obtain a homogenous single cell suspension and either processed immediately or cryopreserved until further analysis. Tumor cell population was identified using viable, murine tissue neg. cells based on murine CD45, TER-119, H-2Kb and CD31 and further gated on human CD45 neg., human NCAM+ populations.
For re-engraftment of relapsed tumors into non-CAR T cell bearing mice in COG-N-421x patient-derived xenograft models, homogenous single cell suspensions harvested from tumors at predetermined tumor volume endpoint and 1 million cells were resuspended in 100 uL of Matrigel and injected subcutaneously into the flanks of 3 and 2 unique CAR naïve SCID mice for the tumors derived from mouse M10 and M51, respectively. Re-engrafted tumors were harvested after 120 days (at a volume of 1–2 cm3) and made into single cell suspensions and viably cryopreserved as described above.
Immunohistochemistry
Formalin-fixed, paraffin-embedded TMA sections were analyzed for GPC2 expression. In brief, tissue sections were incubated in Tris EDTA buffer (cell conditioning 1; CC1 standard) at 95°C for 1 hour to retrieve antigenicity, followed by incubation with GPC2 antibody (Santa Cruz sc-393824) at 1:100 for 1 hour. Slides were then incubated with the respective secondary antibody (Jackson Laboratories) with 1:500 dilution, followed by Ultramap HRP and Chromomap DAB detection. Intensity scoring was done on a common four-point scale. Descriptively, 0 represents no staining, 1 represents low but detectable degree of staining, 2 represents clearly positive staining, and 3 represents strong expression. Expression was quantified as H-Score, the product of staining intensity, and % of stained cells using the following standard formula: [1 × (% cells 1+) + 2 × (% cells 2+) + 3 × (% cells 3+)].
Analysis of single cell RNA-seq datasets
Single-cell RNA-sequencing data was download as processed cell-by-gene counts as follows: for human and mouse prenatal data, from La Manno et al. 2016, via GEO (accession GSE76381); for human adult data, from the Allen Brain Atlas (https://portal.brain-map.org/atlases-and-data/rnaseq/human-multiple-cortical-areas-smart-seq). The data were processed using Scanpy v1.6.0. For each dataset, the data were first imported, then filtered to remove low-quality cells containing fewer than 250 detected genes, fewer than 500 detected counts, or greater than 25% mitochondrial reads. The data was then depth-normalized to a total of 10,000 reads per cell, and log-transformed with a pseudocount of 1. The top 2000 variable genes were identified and used as inputs for downstream processes: batch effects due to the number of genes detected per cell were regressed out, gene expression values were scaled across cells, PCA was performed, and the top 50 PCs were used as inputs for nearest neighbors calculations. UMAP clustering was performed with default settings, and leiden clustering was performed with a resolution of 0.2–1, depending on the dataset. Unless otherwise noted, gene expression was visualized as log-transformed, depth-normalized counts.
Antibody generation of GPC2.19-IgG1
Fully human GPC2.19-IgG1 antibody was transiently expressed in 293 FreeStyle cells. Antibody was isolated from culture supernatant using protein A beads (Tucker et al.). Isolated antibody was washed using PBS and Amicon 10,000 kDa filter columns.
Membrane Proteome Array
The Membrane Proteome Array (MPA) testing GPC2.19-IgG1 was conducted at Integral Molecular, Inc. (Philadelphia, PA). A protein library composed of >5,300 distinct human membrane protein clones, each overexpressed in HEK293T cells from expression plasmids was individually transfected in separate wells of a 384-well plate (Tucker et al., 2018) followed by a 36-hour incubation. Cells expressing each individual MPA protein clone were arrayed in duplicate in a matrix format for high-throughput screening. Prior, the GPC2.19-IgG1 antibody concentration for screening was determined on cells expressing positive (membrane-tethered Protein A and GPC2) and negative (mock-transfected) binding controls, followed by detection by flow cytometry using a fluorescently labeled secondary antibody. The GPC2.19-IgG1 antibody was then added to the MPA at the predetermined concentration, and binding across the protein library was measured on an IntellicytiQue using a fluorescently labeled secondary antibody. Each array plate contains both positive (Fc-binding) and negative (empty vector) controls to ensure plate-by-plate reproducibility. GPC2.19-GPC2-IgG1 antibody interactions with any targets identified by this MPA screening were validated in a second flow cytometry experiment using serial dilutions of the test antibody, and the target identity was re-verified by sequencing.
HVCN1 cross-reactivity
Whole blood was derived from the Stanford Blood Bank and either used directly for Flow Cytometric analysis following ACK lysis and FC-blocking (BD Biosciences) or for isolation of distinct cell populations. T cells were isolated as described above. Granulocytes and B-cells were isolated using a whole blood column kit (Miltenyi) and magnetic beads for granulocytes (StraightFrom Whole Blood CD66b MicroBeads, Miltenyi) and for B-cells (StraightFrom Whole Blood CD19 MicroBeads, Miltenyi). Autologous T cells were activated as described above. Granulocytes were viably cryopreserved in CryoStor, CS10 cryopreservation medium (Stem Cell Technologies) and until use. B-cells were cultured and expanded for 10 days in ExCellerate B Cell Media, Xeno-Free (R&D Systems) supplemented with the CellXVivo Human B Cell Expansion Kit (R&D Systems) and a bone marrow stroma feeder-layer using HS-5 cells. CAR T cells were transduced as described above. Co-cultures between 0.1×106 autologous CAR T cells and NB tumor cells (NBSD), freshly harvested B-cells and thawed granulocytes were performed on day 10 after T cell activation at 1:1 ratios between effector and target cells. After 24 hours, supernatant culture media was collected stored at −20 °C until analyzed by ELISA and remaining cells were analyzed by Flow Cytometry.
Toxicity evaluation of GPC2-CAR T cells in NSG xenograft model
Para-orthotopic tumors (1×106 NBSD-GFP-Luciferase cells) were engrafted beneath the left renal capsule of 8 weeks old NSG mice as described above and treated with 10×106 GPC2.19.28TM.28z CAR T cells (n=3) or an equivalent number of FMC63.CTRL CAR T cells (n=3) four days after tumor implantation. Tumor growth via bioluminescence imaging and body weight of animals was measured every 2–4 days. Mice were euthanized 14 days after CAR T cell injection. Tissues were collected and processed by the Comparative Medicine Animal Histology Department at Stanford University and analyzed blindly by a board-certified veterinary pathologist. Tissues were collected in 10% neutral buffered formalin (NBF), and routinely processed for paraffin embedding, sectioned at 5.0μm, and stained with hematoxylin and eosin (H&E). Tissues were visualized using an Olympus BX43 upright brightfield microscope, and images captured using an Olympus DP27 camera and cellSens software. Blood was collected at the endpoint and processed and analyzed by the Animal Diagnostic Laboratory at the Stanford Veterinary Service Center.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis and visualization was performed using Prism 8.4 (GraphPad Software). Graphs represent either group mean values ± SEM or SD as indicated in the figure legends. All statistical tests were two-sided, except for data shown in Figure 6D, statistics represents one-tailed Mann Whitney test at experimental endpoint (day 49). A P<.05 was considered statistically significant and P-values are denoted with asterisks as follows (**** = p<0.0001, *** = p<0.001, ** = p<0.01, * = p<0.05), ns = p > 0.05). Number of repeats performed and statistical tests used are described in the relevant figure legend. Briefly, molecules/cell on cell lines vs. bone marrow infiltrating NB cells were assessed using the Welch’s t-test. Effects on tumor growth curves in vitro and in vivo were calculated using two-way repeated measures analysis of variance (RM-ANOVA). One-way multiple comparisons ANOVA with Tukey’s multiple comparison test was used when more than 2 groups/conditions were compared. Kaplan–Meier survival curves of vivo experiments were analyzed using log-rank. Mice that died due to reasons other than defined MAX TU endpoint were censored from the analysis. For all other experiments, a Student’s t-test was used.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| human GPC2 Clone D3, Dylight650 labelled) | Dimiter S. Dimitrov (university of Pittsburgh) | N/A |
| human GPC2 Clone 19, Dylight650 labelled) | This manuscript | N/A |
| human isotype CTRL (Dylight650 labelled) | Crown Bioscience | Cat# C0001 |
| human CD4-BUV395 (Clone SK3) | BD Biosciences | Cat# 563550 |
| human CD8-BUV805 (clone SK1) | BD Biosciences | Cat# 564912 |
| human PD1-PE-Cy7 | Thermo Fisher Scientific (ebioscience) | Cat# 25–2799-41 |
| human Tim3-BV510 (Clone F38–2E2) | Biolegend | Cat# 345030 |
| human Lag3-PE | Thermo Fisher Scientific (ebioscience) | Cat# 12–2239-42 |
| human CD62L-BV650 (Clone DREG-56) | BD Biosciences | Cat# 562719 |
| human CD45RA-FITC (Clone L48) | BD Biosciences | Cat# 649458 |
| FMC63-anti-idiotype (Dylight650 labelled) | Dr. Laurence Cooper, MD Anderson Cancer Center | N/A |
| human CD13-BUV737 (L138) | BD Biosciences | Cat# 749265 |
| human CD45-PerCp-Cy5.5 | Thermo Fisher Scientific | Cat# 45–0459-42 |
| human GPC2 (Clone D3, PE labelled) | Dr. Dimiter S. Dimitrov (university of Pittsburgh) | N/A |
| human GD2-BV510 (Clone 14g2a) | Biolegend | Cat# 357316 |
| human NCAM-BV605 (Clone 5.1H11) | Biolegend | Cat# 362538 |
| human L1CAM-BV421 (Clone 5G3) | BD Biosciences | Cat# 565732 |
| human ALK (Clone 48, APC labelled) | Mark Vigny | N/A |
| human B7-H3-PE-Cy7 (Clone MIH42) | Biolegend | Cat# 351008 |
| human GD2-PE-Cy7 (Clone 14g2a) | Biolegend | Cat# 357308 |
| human NCAM-BV510 (Clone 5.1H11) | Biolegend | Cat# 362534 |
| human L1CAM-BUV395 (Clone 5G3) | BD Biosciences | Cat# 565736 |
| human B7-H3-BUV805 (Clone 7–517) | BD Biosciences | Cat# 748378 |
| human HVCN1-FITC (polyclonal) | LS BIO | Cat# LS‑C236467 |
| Isotype CTRL FITC (polyclonal) | LS BIO | Cat# LS-C149376 |
| human CD3-BUV737 (Clone UCHT1) | BD Biosciences | Cat# 612750 |
| human CD19-BUV495 (Clone SJ25C1) | BD Biosciences | Cat# 612938 |
| human CD15-BV711 (Clone W6D3) | Biolegend | Cat# 323050 |
| human CD66b-PE-Cy7 (Clone G10F5) | Biolegend | Cat# 305116 |
| murine CD45-PacBlue (Clone ) | Biolegend | Cat# 103126 |
| murine TER-119-PacBlue (Clone ) | Biolegend | Cat# 116232 |
| murine H-2Kb-PacBlue (Clone ) | Biolegend | Cat# 116514 |
| murine CD31-PacBlue (Clone ) | Biolegend | Cat# 102422 |
| Bacterial and Virus Strains | ||
| Stellar Competent Cells | Clontech, Takara | Cat# 636766 |
| Biological Samples | ||
| Buffy Coats | Stanford Blood Bank | N/A |
| Whole Blood Samples of healthy donors | Stanford Blood Bank | N/A |
| Human Neuroblastoma Primary Tumor Microarray (TMA) | Children’s Oncology Group (COG) | N/A |
| Tissue Blocks of human prenatal and infant brain | as described in Theruvath et. al, Nature Medicine 2020 | N/A |
| Tissue Blocks of human pediatric brain | BC Children’s Hospital Vancouver | N/A |
| Human Neuroblastoma infiltrated Bone Marrow Samples | Stanford Pediatric Bass Center Tissue bank (IRB: #45458 and #56619) | N/A |
| Human Neuroblastoma infiltrated Bone Marrow Samples | Children’s Cancer Research Institute; Vienna Austria (EK#115/2006, EK#1853/2016 and EK#1216/2018) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phusion Hot Start Flex 2X Master Mix | New England Biolabs | Cat# M0536L |
| Rapid DNA Ligation Kit | Roche | Cat# 11–635-379–001 |
| In-fusion HD Cloning System | Clontech, Takara | Cat# 639647 |
| ZymoPURE II Plasmid Maxiprep Kit | Zymo Research | Cat# D4203 |
| RosetteSep Human T cell Enrichment kit | Stem Cell Technologies | Cat# 15061 |
| Dynabeads Human T-Expander CD3/CD28 | Thermo Fischer Scientific, Gibco | Cat# 11141D |
| Lipofectamine 2000 Transfection Reagent | Thermo Fischer Scientific | Cat# 11668500 |
| RetroNectin® Recombinant Human Fibronectin Fragment | Takara | Cat# T100B |
| Recombinant Human IL-2 | Preprotech | Cat# 200–02-1mg |
| Recombinant Human Glypican 2 Protein | R&D systems, Inc. | Cat# 2304-GP-050 |
| Recombinant Mouse Glypican 2 Protein | R&D systems, Inc. | Cat# 2355-GP-050 |
| Dylight 650 Micoscale Antibody Labelling Kit | Thermo Fischer Scientific | Cat# 84536 |
| Dylight 488 Micoscale Antibody Labelling Kit | Thermo Fischer Scientific | Cat# 53025 |
| Abcam PE Conjugation kit Lightning-Link | Abcam | Cat# ab102918 |
| Abcam APC Conjugation kit Lightning-Link | Abcam | Cat# ab201807 |
| BD QuantiBRITE PE Beads | BD Biosciences | Cat# 340495 |
| BD Custom Quantitation Beads in PE-Cy7, APC, BV421, BV510, BV605 | BD Biosciences | N/A |
| Critical Commercial Assays | ||
| Human IFN-γ ELISA MAX Deluxe | BioLegend | Cat# 430104 |
| Human IL-2 ELISA MAX Deluxe | BioLegend | Cat# 431804 |
| Whole Blood Column Kit | Miltenyi | Cat# 130–093-545 |
| StraightFrom Whole Blood CD19 MicroBeads, human | Miltenyi | Cat# 130–090-880 |
| StraightFrom Whole Blood CD66b MicroBeads, human | Miltenyi | Cat# 130–104-913 |
| Deposited Data | ||
| scRNAseq data human and murine prenatal brain | La Manno et. al, 2016 | GEO (accession GSE76381) |
| scRNAseq data human adult brain | Allen Brain Atlas | https://portal.brain-map.org/atlases-and-data/rnaseq/human-multiple-cortical-areas-smart-seq |
| Experimental Models: Cell Lines and PDX | ||
| CHO (female) | ATCC | RRID:CVCL_0213 |
| HS-5 (male) | ATCC | RRID:CVCL_3720 |
| NGP (male) | Dr. John Maris (CHOP) | RRID:CVCL_2141 |
| NGP-NPC2 (male) | Dr. John Maris (CHOP) | N/A |
| SMS-SAN (female) | Dr. John Maris (CHOP) | RRID:CVCL_7136 |
| NBSD (female) | Dr. John Maris (CHOP) | RRID:CVCL_LF68 |
| EBC1 (male) | Dr. John Maris (CHOP) | RRID:CVCL_E218 |
| NB1643 (male) | Dr. John Maris (CHOP) | RRID:CVCL_5627 |
| CHLA255 (sex unspecified) | Dr. Robert Seeger (Keck School of Medicine, USC) | RRID:CVCL_AQ27 |
| SMS-KCNR (male) | Dr. Robert Seeger (Keck School of Medicine, USC) | To cite this cell line use: SMS-KCNR (RRID:CVCL_7134 |
| SKNAS (female) | Dr. Bill Chiu (Stanford University) | RRID:CVCL_1700 |
| CHLA-90 (male) | Dr. Bill Chiu (Stanford University) | RRID:CVCL_6610 |
| Kelly (female) | Dr. Kimberly Stegmaier (Harvard Medical School) | RRID:CVCL_2092 |
| Lan5 (male) | Dr. Kimberly Stegmaier (Harvard Medical School) | RRID:CVCL_0389 |
| ST16-BM4045 (male, diagnostic) | This manuscript | N/A |
| ST16-BM4224 (male, relapse) | This manuscript | N/A |
| COG-N-421x PDX (male) | Dr. John Maris (CHOP) | N/A |
| COG-N-426x-Felix PDX (male) | Dr. John Maris (CHOP) | N/A |
| COG-N-440x PDX (female) | Dr. John Maris (CHOP) | N/A |
| COG-N-453x PDX (male) | Dr. John Maris (CHOP) | N/A |
| COG-N-471x PDX (female) | Dr. John Maris (CHOP) | N/A |
| COG-N-496x PDX (female) | Dr. John Maris (CHOP) | N/A |
| COG-N-519x PDX (male) | Dr. John Maris (CHOP) | N/A |
| COG-N-549x PDX (male) | Dr. John Maris (CHOP) | N/A |
| COG-N-557x PDX (male) | Dr. John Maris (CHOP) | N/A |
| COG-N-561x PDX (female) | Dr. John Maris (CHOP) | N/A |
| COG-N-590x PDX (male) | Dr. John Maris (CHOP) | N/A |
| COG-N-603x PDX (male) | Dr. John Maris (CHOP) | N/A |
| COG-N-624x PDX (female) | Dr. John Maris (CHOP) | N/A |
| CHLA-79 PDX (female) | Dr. John Maris (CHOP) | N/A |
| NB-1643 PDX (male) | Dr. John Maris (CHOP) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mice: NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) | The Jackson Laboratory | Cat# JAX:005557; RRID:IMSR_JAX:005557 |
| Oligonucleotides | ||
| N/A | ||
| Recombinant DNA | ||
| RD114 | add | add |
| CAR.GPC2.D3VLVH.8aHTM.BBz | This manuscript | N/A |
| CAR.GPC2.D3VHVL.8aHTM.BBz | This manuscript | N/A |
| CAR.GPC2.D4VLVH.8aHTM.BBz | This manuscript | N/A |
| CAR.GPC2.D4VHVL.8aHTM.BBz | This manuscript | N/A |
| CAR.GPC2.19VLVH.8aHTM.BBz | This manuscript | N/A |
| CAR.GPC2.19VHVL.8aHTM.BBz | This manuscript | N/A |
| CAR.GPC2.27VLVH.8aHTM.BBz | This manuscript | N/A |
| CAR.GPC2.27VHVL.8aHTM.BBz | This manuscript | N/A |
| CAR.GPC2.CH2CH3.D3VLVH.8aHTM.BBz | This manuscript | N/A |
| CAR.GPC2.CH2CH3.19VLVH.8aHTM.BBz | This manuscript | N/A |
| CAR.GPC2.D3VLVH.28HTM.BBz | This manuscript | N/A |
| CAR.GPC2.19VLVH.28HTM.BBz | This manuscript | N/A |
| CAR.GPC2.D3VLVH.28HTM.28z | This manuscript | N/A |
| CAR.GPC2.19VLVH.28HTM.28z | This manuscript | N/A |
| CAR.cJUN-P2A-GPC2.19VLVH.28HTM.28z | This manuscript | N/A |
| CAR.CD19.FMC63.8aHTM.BBz | previously used in our lab | N/A |
| Software and Algorithms | ||
| FlowJo v10.7.1 | FlowJo, LLC | N/A |
| GraphPad Prism v8.4 | GraphPad Software Inc. | N/A |
| Living Image version (IVIS imaging) | Perkin Elmer | N/A |
| Biorender | Biorender | N/A |
| Scanpy v 1.6.0 (ssRNAseq analysis) | Wolf et al., 2018 | N/A |
| Other | ||
| fixable viability dye eFluor780 | Thermo Fisher Scientific (ebioscience) | Cat# 65–0865-14 |
| CountBright Absolute Counting beads | Thermo Fischer Scientific | Cat# C36950 |
| Brilliant Stain buffer | BD Biosciences | Cat# 566349 |
| Human FC Block | BD Biosciences | Cat# 564219 |
| Corning CellStripper Dissociation Reagent | Corning | Cat# MT25056CI |
Add info for ssRNAseq datasets
HIGHLIGHTS.
GPC2-CARs with standard 41BB designs require > 3 × 104 molecules for full activation
Metastatic neuroblastomas express ~5 × 103 GPC molecules/cell
CAR antigen density threshold tunable with modular engineering and c-Jun overexpression
Tuned GPC2 CAR T cells mediate potent neuroblastoma regression without toxicity
ACKNOWLEDGEMENTS
This work was supported by the NIH U54 CA232568–01 (CLM, JMM, PHS, KRB), P01CA217959–01 (CLM, JMM, SH) and a St Baldrick’s/Stand Up 2 Cancer (SU2C) Pediatric Dream Team Translational Cancer Research Grant SU2CAACR-DT1113 (CLM, JMM, SH, KRB, RGM, AD, PHS). SU2C is a program of the Entertainment Industry Foundation administered by the AACR. CLM is a member of the Parker Institute for Cancer Immunotherapy (PICI), which supports the Stanford University Cancer Immunotherapy Program. Additional support: CLM: Virginia and D.K. Ludwig Fund for Cancer Research, JMM: R35 grant CA220500, KRB: Damon Runyon Cancer Research Foundation PST-07–16, Alex’s Lemonade Stand Foundation, NCI K08CA230223, EVAN Foundation. RGM: Taube Distinguished Scholar for Pediatric Immunotherapy (Stanford School of Medicine). JPJ: CIFAR Azrieli Global Scholar program, Ontario Early Researcher Awards program, Canada Research Chairs program (JPJ). JT: German Cancer Aid grant P-91650709. ML,BS: Federal Ministry for Digital and Economic Affairs of Austria, National Foundation for Research, Technology and Development of Austria to the Christian Doppler Research Association (Christian Doppler Laboratory for Next Generation CAR T Cells), private donations to the CCRI (Vienna, Austria). ATS: NIH K08CA230188, Burroughs Wellcome Fund, Technology Impact Award from the Cancer Research Institute. HYC: Investigator of the Howard Hughes Medical Institute and PICI.
We thank the staff of the pediatric hematology/oncology department at the Lucile Packard and St. Anna’s Children’s Hospital for collecting BM samples and the patients and their parents for giving their informed consent. We thank Garry L. Coles and Julien Sage for providing the ST16-BM4045 PDX line and BD Biosciences (Scott Bornheimer) for providing proprietary multicolor quantitation beads prior to commercialization. We thank Jean Oak from the Clinical Flow Cytometry Department at Stanford Hospital and Dieter Printz from the FACS Core at the CCRI (Vienna, Austria) for assisting assay setup. We thank JH and PX for mouse colony maintenance and the Stanford Animal Histology Services for preparation of histology slides. All cartoons were created using BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
CLM, SH, JMM, KRB, RGM, DSD and ZZ are coinventors on patents related to this work. CLM (and others) have multiple patents pertinent to CAR T cells. CLM is a cofounder of Lyell Immunopharma and Syncopation Life Sciences, which develop CAR-based therapies and consults for Lyell, NeoImmune Tech, Apricity, Nektar and Immatics. KRB and JMM receive research funding from Tmunity for research on GPC2-directed immunotherapies. DZ, ZZ, DSD, JMM and KRB receive royalties from Tmunity for licensing of GPC2-related IP. RGM and ES are consultants for and hold equity in Lyell Immunopharma. RGM consults for GammaDelta Therapeutics, Aptorum Group, Zai Lab, and Illumina Radiopharmaceuticals and JT for Dorian Therapeutics. SJB is an employee of BD Biosciences. ATS is a founder of Immunai and Cartography Biosciences and receives research funding from Arsenal Biosciences and 10x Genomics. KRP is a cofounder and employee of Cartography Biosciences. HYC is a co-founder of Accent Therapeutics, Boundless Bio and is an advisor to 10x Genomics, Arsenal Bio and Spring Discovery.
INCLUSION AND DIVERSITY
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
REFERENCES
- Ayanlaja AA, Xiong Y, Gao Y, Ji G, Tang C, Abdikani Abdullah Z, and Gao D (2017). Distinct Features of Doublecortin as a Marker of Neuronal Migration and Its Implications in Cancer Cell Mobility. Front Mol Neurosci 10, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse KR, and Maris JM (2016). Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 122, 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse KR, Raman P, Zhu Z, Lane M, Martinez D, Heitzeneder S, Rathi KS, Kendsersky NM, Randall M, Donovan L, et al. (2017). Identification of GPC2 as an Oncoprotein and Candidate Immunotherapeutic Target in High-Risk Neuroblastoma. Cancer Cell 32, 295–309 e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. (2016). Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 375, 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, et al. (2013). Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med 5, 197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso M, Bhamrah MK, Henley T, Boyd RS, Langlais C, Cain K, Dinsdale D, Pulford K, Khan M, Musset B, et al. (2010). HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat Immunol 11, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso-Moreira M, Halbert J, Valloton D, Velten B, Chen C, Shao Y, Liechti A, Ascencao K, Rummel C, Ovchinnikova S, et al. (2019). Gene expression across mammalian organ development. Nature 571, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H, et al. (2015). Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res 75, 3505–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, Vogl DT, Weiss BM, Dengel K, Nelson A, et al. (2019). B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest 129, 2210–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R, Libby KA, Blaeschke F, Fuchs W, Marson A, Vale RD, and Su X (2020). Rewired signaling network in T cells expressing the chimeric antigen receptor (CAR). EMBO J 39, e104730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filmus J, Capurro M, and Rast J (2008). Glypicans. Genome Biol 9, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, Wolters P, Martin S, Delbrook C, Yates B, et al. (2018). CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 24, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudipati V, Rydzek J, Doel-Perez I, Goncalves VDR, Scharf L, Konigsberger S, Lobner E, Kunert R, Einsele H, Stockinger H, et al. (2020). Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat Immunol 21, 848–856. [DOI] [PubMed] [Google Scholar]
- Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM, et al. (2013). Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 121, 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, Liu H, Wu MF, Mei Z, Gee A, et al. (2017). CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther 25, 2214–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, and Riddell SR (2015). The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res 3, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessa S, Blanchet-Cohen A, Krug B, Vladoiu M, Coutelier M, Faury D, Poreau B, De Jay N, Hebert S, Monlong J, et al. (2019). Stalled developmental programs at the root of pediatric brain tumors. Nat Genet 51, 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, and Sadelain M (2018). Chimeric Antigen Receptor Therapy. N Engl J Med 379, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur SP, and Cummings BS (2019). Role of glypicans in regulation of the tumor microenvironment and cancer progression. Biochem Pharmacol 168, 108–118. [DOI] [PubMed] [Google Scholar]
- La Manno G, Gyllborg D, Codeluppi S, Nishimura K, Salto C, Zeisel A, Borm LE, Stott SRW, Toledo EM, Villaescusa JC, et al. (2016). Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells. Cell 167, 566–580 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labanieh L, Majzner RG, and Mackall CL (2018). Programming CAR-T cells to kill cancer. Nat Biomed Eng 2, 377–391. [DOI] [PubMed] [Google Scholar]
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. (2015). T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Fu H, Hewitt SM, Dimitrov DS, and Ho M (2017). Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc Natl Acad Sci U S A 114, E6623–E6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, et al. (2013). Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. (2015). 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 21, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al. (2011). Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118, 6050–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, et al. (2019). c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majzner RG, and Mackall CL (2019). Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med 25, 1341–1355. [DOI] [PubMed] [Google Scholar]
- Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, Myklebust JH, Kadapakkam M, Weber EW, Tousley AM, et al. (2020). Tuning the Antigen Density Requirement for CAR T Cell Activity. Cancer Discov [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, Rietberg SP, Linde MH, Xu P, Rota C, et al. (2019). CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin Cancer Res 25, 2560–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall GM, Carter DR, Cheung BB, Liu T, Mateos MK, Meyerowitz JG, and Weiss WA (2014). The prenatal origins of cancer. Nat Rev Cancer 14, 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Capasso M, Musset B, Cherny VV, Rios E, Dyer MJ, and DeCoursey TE (2009). Voltage-gated proton channels maintain pH in human neutrophils during phagocytosis. Proc Natl Acad Sci U S A 106, 18022–18027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, et al. (2013). Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 36, 133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, et al. (2017). A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson DM, Shohet JM, and Kim ES (2011). Preclinical models of pediatric solid tumors (neuroblastoma) and their use in drug discovery. Curr Protoc Pharmacol Chapter 14, Unit 14 17. [DOI] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al. (2008). Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14, 1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman S, Buongervino SN, Lane MV, Zhelev DV, Zhu Z, Cui H, Martinez B, Martinez D, Wang Y, Upton K, et al. (2021). A GPC2 antibody-drug conjugate is efficacious against neuroblastoma and small-cell lung cancer via binding a conformational epitope. Cell Rep Med 2, 100344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards RM, Sotillo E, and Majzner RG (2018). CAR T Cell Therapy for Neuroblastoma. Front Immunol 9, 2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH, Frank MJ, Shiraz P, Sahaf B, Craig J, et al. (2021). CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med 27, 1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straathof K, Flutter B, Wallace R, Jain N, Loka T, Depani S, Wright G, Thomas S, Cheung GW, Gileadi T, et al. (2020). Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci Transl Med 12. [DOI] [PubMed] [Google Scholar]
- Theodorakos I, Paterakis G, Papadakis V, Vicha A, Topakas G, Jencova P, Karchilaki E, Taparkou A, Tsagarakis NJ, and Polychronopoulou S (2019). Interference of bone marrow CD56(+) mesenchymal stromal cells in minimal residual disease investigation of neuroblastoma and other CD45(−) /CD56(+) pediatric malignancies using flow cytometry. Pediatr Blood Cancer 66, e27799. [DOI] [PubMed] [Google Scholar]
- Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, Labanieh L, Dhingra S, Leruste A, Majzner RG, et al. (2020). Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med 26, 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker DF, Sullivan JT, Mattia KA, Fisher CR, Barnes T, Mabila MN, Wilf R, Sulli C, Pitts M, Payne RJ, et al. (2018). Isolation of state-dependent monoclonal antibodies against the 12-transmembrane domain glucose transporter 4 using virus-like particles. Proc Natl Acad Sci U S A 115, E4990–E4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzynski MJ, Graham DM, Axtell RA, Higgins JV, and Hammers YA (2002). Flow cytometric immunophenotyping test for staging/monitoring neuroblastoma patients. Cytometry 50, 298–304. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Kuramitsu S, Posey AD Jr., and June CH (2018). Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front Immunol 9, 2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, Plotkin J, and Sadelain M (2015). Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 28, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, and Dimitrov DS (2009). Construction of a large naive human phage-displayed Fab library through one-step cloning. Methods Mol Biol 525, 129–142, xv. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any novel datasets. Sources of publicly available scRNAseq datasets are La Manno et al. 2016 via GEO (accession GSE76381) for human and murine prenatal brain, the Allen Brain Atlas for human adult brain data (https://portal.brain-map.org/atlases-and-data/rnaseq/human-multiple-cortical-areas-smart-seq).