

ABSTRACT
Background
Contrary to pure cases, the influence of comorbid argyrophilic grain disease (AGD) in progressive supranuclear palsy (PSP) has not been sufficiently evaluated.
Objectives
We compared the clinicoradiological features of 12 patients with PSP with (PSPw/AG) and 8 patients without AGD (PSPw/oAG).
Methods
Medical records and magnetic resonance imaging were checked retrospectively from a single brain bank database.
Results
Other than AGD, no differences were observed in any other neurodegenerative pathologies between the 2 groups. Ages at onset and deaths of patients with PSPw/AG were higher than those of patients with PSPw/oAG (77.9 ± 4.9 vs. 68.9 ± 5.9, and 87.0 ± 5.7 vs. 78.1 ± 5.0; P = 0.003 and P = 0.002, respectively). In addition to the later onset of motor symptoms, initial amnestic presentations were limited to 5 patients with PSPw/AG. Both characteristic midbrain atrophy and severe ambient gyrus atrophy were detected exclusively in 8 patients with PSPw/AG.
Conclusions
Initial amnestic presentations and ambient gyrus atrophy may be characteristic of PSPw/AG.
Keywords: ambient gyrus, argyrophilic grain disease, comorbidity, magnetic resonance imaging, progressive supranuclear palsy
Progressive supranuclear palsy (PSP) is a well‐known 4‐repeat (4R) tauopathy characterized by tau‐positive intracellular lesions, including tuft‐shaped astrocytes, pretangles and neurofibrillary tangles (NFT), neuropil threads, and oligodendroglial coiled bodies. 1 Typical clinicoradiological features of PSP are progressive vertical gaze palsy, prominent postural instability, axial rigidity, frontal cognitive dysfunction, and variable degrees of midbrain atrophy. 2 In addition to the pathognomonic neuropathologies, comorbid neurodegenerative pathologies are frequently observed in patients with PSP (18.8%–80%). 3 Unlike the usual mild degree of other neuropathologies present, argyrophilic grain disease (AGD) may show more widespread extensions. 4
AGD is a sporadic 4R tauopathy estimated to be the second most common neurodegenerative pathology after Alzheimer's disease (AD) in very elderly patients with dementia. 5 It has been reported that pure AGD causes amnestic and psychiatric symptoms and asymmetric atrophy relatively localized in the anterior temporal and limbic lobes. 6 , 7 Contrary to pure cases, the influence of comorbid AGD on other neurodegenerative pathologies including PSP remains arguable. 8 , 9 Furthermore, the neuroradiological features of PSP comorbid with AGD remain unclear. This prompted us to explore the clinicoradiological features of pathologically confirmed cases with concomitant PSP and AGD. This retrospective study was designed to compare the clinicoradiological features of PSP with (PSPw/AG) and without AGD (PSPw/oAG) in the expectation that evaluation of pathologically confirmed cases would provide new insights into the clinicoradiological aspects of PSP.
Methods
Subjects and Assessments
The study population was selected by searching the available medical records at our institution between January 2003 and December 2021. Patient backgrounds were standardized by applying the following inclusion criteria: (1) neuropathological diagnoses according to the published criteria of PSP and AGD and (2) sufficient medical records to evaluate the initial symptoms and clinical course. 10 , 11 Other neurodegenerative pathologies such as corticobasal degeneration or large destructive lesions (eg, cerebrovascular diseases, trauma, or neoplasms) were excluded. 12 Slight accumulation of Lewy bodies below the level needed to fulfill the diagnostic criteria of dementia with Lewy bodies was accepted as reflecting minimal senile neuropathologic changes. 13 Medical records such as admission summaries were retrospectively checked to evaluate the clinical course including the initial symptoms and clinical course. In patients undergoing brain magnetic resonance imaging (MRI), the neuroradiological features were evaluated using semiquantitative and qualitative markers. The degree of the ambient gyrus atrophy (AGA) was semiquantitatively assessed using the 4‐point scale (0 = normal, 1 = mild, 2 = moderate, 3 = severe). In addition, the presence/absence of the humming bird sign indicating midbrain atrophy was evaluated visually. 14 Detailed information about the clinical, MRI, and neuropathological analyses is provided in the Supplementary file.
Statistical Analysis
Data were analyzed using IBM SPSS statistics 24 (IBM, Armonk, NY). The demographic and clinicoradiological variables of PSPw/AG were compared with those of PSPw/oAG. Welch's t test and the Mann–Whitney U test were used for normally distributed and skewed data, respectively. Fisher's exact test for dichotomous variables was used for other group comparisons that did not involve skewed data. Significance level was set at P < 0.05. A receiver operating characteristic (ROC) curve analysis was performed to evaluate the utility of the AGA score for the differentiation of PSPw/AG and PSPw/oAG.
Results
Patient characteristics are summarized in Table 1. The PSPw/AG group consisted of 12 patients with various degrees of argyrophilic grain neuropathologies (AGs), mainly Saito stages I and II. 11 A total of 2 patients with PSPw/AG showed a moderate Braak NFT stage (ie, IV) despite which no difference was observed in the severity of NFT stage between the PSPw/AG and PSPw/oAG groups. Severe Braak NFT stage was not detected in either patient group. Similarly, no significant differences in brain weight or coexistence rate of trans activation responsive region (TAR) DNA‐binding protein of 43 kDa (TDP‐43) or Lewy body pathologies were found. Despite the lack of difference in disease duration, age at onset and death were both higher in the patients with PSPw/AG (77.9 ± 4.9 vs. 68.9 ± 5.9 and 87.0 ± 5.7 vs. 78.1 ± 5.0; P = 0.003 and P = 0.002, respectively). Detailed results of the neurological and neuropathological analyses are outlined in Table S1.
Table 1.
Patient characteristics
| Variable | PSPw/AG | PSPw/oAG | P Value |
|---|---|---|---|
| N | 12 | 8 | NA |
| Male/female | 7/5 | 4/4 | 1.0 a |
| Neuropathological features | |||
| Brain weight (g) | 1105 ± 105 | 1155 ± 104 | 0.37 b |
| Argyrophilic grain (Saito) (1–3) | 1.6 ± 0.7 | 0.0 ± 0.0 | NA |
| Braak NFT stage (1–6) | 2.2 ± 0.9 | 1.5 ± 0.5 | 0.14 b |
| CERAD 0/A/B/C | 1/6/2/3 | 5/1/1/1 | 0.08 a |
| Lewy‐body pathology | 8.3 (1/12) | 0.0 (0/8) | 1.0 a |
| TDP‐43 pathology c | 25.0 (1/4) | 0.0 (0/2) | 1.0 a |
| Age at onset (y) | 77.9 ± 4.9 | 68.9 ± 5.9 | 0.003 d |
| Age at death (y) | 87.0 ± 5.7 | 78.1 ± 5.0 | 0.002 d |
| Disease duration (m) | 104.5 ± 32.7 | 104.4 ± 58.1 | 0.46 d |
| Type of initial predominant symptoms | |||
| Fgd/Mem/Beh/Lmb | 4/5/2/1 | 4/0/3/1 | 0.24 a |
| Onset of MoS (m) | 44.6 ± 44.6 | 14.2 ± 13.8 | 0.046 d |
| Onset of MPS (m) | 16.6 ± 34.8 | 20.7 ± 26.9 | 0.79b |
| Clinical phenotype | |||
| RS/A/F/CBS/PI/P | 4/4/2/1/0/1 | 3/0/3/1/1/0 | 0.33 d |
| MRI features e | |||
| AGA score (1–3) | 2.6 ± 0.6 | 1.3 ± 0.6 | 0.024 b |
| Humming bird sign | 100 (8/8) | 100 (3/3) | 1.0 a |
Data are shown as absolute number, percentage (number), or mean ± standard deviation.
Mann–Whitney U test.
Fisher′s exact test.
TDP‐43 pathologies were not fully evaluated in all patients with PSP.
Welch′s t test.
MRI was performed in 8 patients with PSPw/AG and 3 patients with PSPw/oAG.
Abbreviations: PSPw/AG, progressive supranuclear palsy with argyrophilic grain; PSPw/oAG, progressive supranuclear palsy without argyrophilic grain; NFT, neurofibrillary tangle; CERAD, Consortium to Establish a Registry for Alzheimer Disease; TDP‐43, TAR DNA‐binding protein‐43; Fgd, fall and gait disorder; Mem, memory; Beh, behavior; Lmb, limb; MoS, motor symptoms; MPS, memory and psychiatric symptoms; RS, Richardson′s syndrome; A, initial amnestic presentation; F, predominant frontal presentation; CBS, corticobasal syndrome; PI, postural instability; P, predominant parkinsonism; MRI, magnetic resonance imaging; AGA, ambient gyrus atrophy; NA, not applicable; TAR, trans activation responsive.
Falls and gait disorder were the most prevalent initial signs irrespective of the presence of AGs. Intriguingly, initial amnestic predominant presentations were reported solely in 5 patients with PSPw/AG (5/12 [41.6%] vs. 0/8 [0%]; P = 0.055). Compared with PSPw/oAG, the onset of motor symptoms was later in patients with PSPw/AG (44.6 ± 44.6 vs. 14.2 ± 13.8; P = 0.046). Using the Movement Disorder Society (MDS) PSP diagnostic criteria, patients were classified into clinical phenotypes including PSP–Richardson′s syndrome (PSP‐RS), PSP–predominant frontal presentation, PSP–corticobasal syndrome, and PSP–postural instability based on the combination of initial symptoms and clinical course (Table 1). 15 In addition, we termed 4 patients with PSPw/AG presenting with initial amnestic predominant symptoms, preceding motor or psychiatric symptoms by more than 3 years, as PSP with initial amnestic predominant presentation (PSP‐A). Even in the clinical phenotype of PSP‐RS, 1 patient with severe AGs (ie, Saito III) initially presented with memory loss and depression. The period between the onset of memory and motor symptoms was longer in patients with PSP‐A than other phenotypes (94.4 ± 29.3 vs. 29.0 ± 19.6 months; P = 0.001).
MRI was performed in 8 patients with PSPw/AG and 3 patients with PSPw/oAG. There was no difference in disease duration at MRI between PSPw/AG and PSPw/oAG (101.3 ± 41.4 vs. 79.7 ± 68.9; P = 0.41). Representative MRI images are presented in Figure 1. Compared with PSPw/oAG, patients with PSPw/AG showed higher AGA scores, reflecting more severe atrophy of the ambient gyrus (2.6 ± 0.6 vs. 1.3 ± 0.6; P = 0.024). The AGA score exhibited areas under the curve of 0.94 for differentiating between PSPw/AG and PSPw/oAG on ROC analysis (Figure S1).
FIG 1.
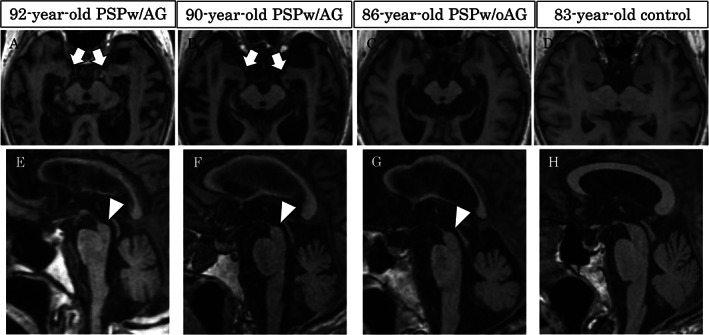
Representative axial and sagittal magnetic resonance images of PSPw/AG, PSPw/oAG and healthy control. In contrast to midbrain atrophy in all PSP (E‐G; arrowheads), severe atrophy of the bilateral ambient gyri was detected exclusively in PSPw/AG (A and B; arrows). PSP, progressive supranuclear palsy; w/AG, with argyrophilic grains; w/oAG, without argyrophilic grains. No atrophy of the midbrain and ambient gyri was detected in a healthy control (D, H).
Discussion
This is the first study to investigate the clinicoradiological features of patients with PSPw/AG. A novel aspect was that it compared the MRI findings between pathologically confirmed patients with PSPw/AG and PSPw/oAG. In addition to the older age at onset and death, some patients with PSPw/AG presented with initial amnestic predominant symptoms preceding motor symptoms by several years (ie, PSP‐A). MRI exhibited atrophy not only of the midbrain but also of the ambient gyrus in patients with PSPw/AG, suggesting that the finding of ambient gyrus atrophy could raise the possibility of comorbid AGs in elderly patients with PSP.
Although predominant, otherwise unexplained impairment of episodic memory suggestive of AD is regarded as 1 exclusion criterion in the MDS PSP diagnostic criteria, and previous studies have reported memory impairment in patients with pathologically confirmed PSP. 15 , 16 , 17 , 18 In addition, comorbid AD neuropathologies were observed in 2 studies. 16 , 18 In contrast, not only 2 patients with PSPw/AG with intermediate‐level AD neuropathologies but also 3 patients with no/low‐level comorbid AD neuropathologies initially presented with amnestic predominant symptoms in this study. As well as AD neuropathologies, comorbid AGs might contribute to the amnestic symptoms in some patients with PSPw/AG.
Despite the relatively high frequency of comorbid AGs found in various neurodegenerative disorders, their clinical impact is unclear. Previous studies reported that comorbid AGs may aggravate the psychiatric symptoms in patients with Parkinson's disease and poststroke depression. 8 , 9 In addition, this study showed that some patients with PSPw/AG presented with a peculiar clinical course with antecedent amnestic symptoms followed by motor and psychiatric symptoms. This raises the possibility that comorbid AGs may cause an atypical clinical presentation other than typical PSP‐RS. Older onset and atypical clinical presentation of PSP‐A were reminiscent of previously reported elderly patients with PSP with mild or absent motor symptoms. 19 , 20 However, a conflicting result, indicating no clinical impact of comorbid neurodegenerative disorders or vascular copathologies in PSP, has also been reported. 4 Considering these inconsistent results, further studies will be necessary to confirm the significance of comorbid AGs in PSP.
Head MRI is useful to detect minute structures such as the midbrain and ambient gyrus, which are frequently damaged in PSP and AGD, respectively. It is well known that midbrain atrophy indicative of the “humming bird sign” is a characteristic MRI finding of PSP. 2 , 14 A recent neuroradiological study clarified that MRI can detect atrophy of the ambient gyrus in patients with pathologically confirmed AGD. 6 In addition to the typical “humming bird sign,” MRI exhibited more significant atrophy of the ambient gyrus in PSPw/AG than PSPw/oAG. Notably, severe atrophy such as AGD was exclusively detected in patients with PSPw/AG. Because the limbic system is less frequently impaired in PSP, this finding could be a noninvasive biomarker of concomitant AGs in PSP. 21 In the context of devising future anti‐tau strategies against PSP, which will require a precise diagnosis of pure PSP, such MRI findings may provide clues to diagnose comorbid AGs in PSP. 22
A strength of the study is that because all cases were referred to a single brain bank, neuropathologic procedures were systematic and standardized. However, limitations related to its retrospective nature should be mentioned because the patients were recruited from a referral brain bank in a medical long‐term care sanatorium. As a result, autopsies were performed mainly in long‐standing severely demented patients. Therefore, it was difficult to obtain the standardized medical documentation and detailed neurological findings at the early disease stage. Despite the inclusion criteria with sufficient clinical documentation checked by an experienced neurologist, not all patients were directly examined by neurologists. This might have caused a classification bias, particularly in cases with atypical presentations. Similarly, it was difficult to evaluate the MRI findings including the presence or absence of midbrain atrophy at the early disease stage. Even with the rarity of severe AD neuropathologies in PSP, some degree of NFT was detected in both PSP groups. 16 , 18 Considering the possibility of superimposed effect of NFT to the medial temporal atrophy, comparison between comorbid AGD and AD may be necessary to evaluate the impact of these neuropathologies to the mesiotemporal structures in PSP. Despite the utility of other biomarkers such as amyloid‐42 and phosphorylated‐tau 217 of cerebrospinal fluid and plasma to differentiate between PSP and AD, there are no sufficient data for the diagnosis of PSP‐A. 23 , 24 , 25 A prospective long‐term observational study including other biomarkers will be required to comprehend the clinicoradiological impact of comorbid AGs in patients with PSP.
In conclusion, morphological changes reflecting PSP comorbid with AGs can be detected in patients with PSPw/AG. Even with presentation with initial amnestic predominant symptoms, clinicians should observe both the midbrain and ambient gyrus in elderly patients.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
K.S.: 1A, 1C, 2A, 2B, 3A
D.K.: 1A, 1B, 1C, 3B
S.M.: 1A, 2C, 3B
Y.U.: 2A, 2C, 3B
S.I.: 2A, 2C, 3B
Y.K.: 1A, 2C, 3B
T.K.: 1A, 3B
K.I.: 1B, 3B
Y.H.: 1A, 1B, 1C
Disclosures
Ethical Compliance Statement: The authors declare that the present study was approved by the institutional review board of Fukushimura Hospital (Research ID: 433, 2022), and was therefore performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Postmortem consent was obtained from the bereaved of all patients for use of their data for academic purposes. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: A part of this work was supported by JSPS KAKENHI Grant Number JP 16H06277 (CoBiA). The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months: None of the authors have any disclosures to report.
Supporting information
Data S1 Definitions of each progressive supranuclear palsy subtype and procedures of neuropathological and neuroradiological analyses.
Figure S1 Receiver operating characteristic (ROC) curves using the ambient gyrus atrophy score for the differentiation between progressive supranuclear palsy with (PSPw/AG) and without argyrophilic grain disease (PSPw/oAG) and a visual rating scale of the ambient gyrus atrophy.
Figure S2 Visual rating scale of the ambient gyrus atrophy. The ambient gyrus atrophy (AGA) scores were defined as 0 for the bulgy medial temporal protuberance indicative of normal volume (A), 1 for the mildly decreased medial temporal protuberance indicative of mild atrophy (B), 2 for the slightly prickly medial temporal protuberance indicative of moderate atrophy (C), and 3 for the small acicular or invisible medial temporal protuberance indicative of severe atrophy (D, E).
Table S1 Detailed results of the neurological, neuropathological, and neuroradiological analyses in all patients.
Acknowledgments
We thank John Gelblum for his English proofreading.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Dickson DW, Rademakers R, Hutton ML. Progressive supranuclear palsy: pathology and genetics. Brain Pathol 2007;17(1):74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sakurai K, Tokumaru AM, Shimoji K, et al. Beyond the midbrain atrophy: wide spectrum of structural MRI finding in cases of pathologically proven progressive supranuclear palsy. Neuroradiology 2017;59(5):431–443. [DOI] [PubMed] [Google Scholar]
- 3. Yokota O, Miki T, Ikeda C, et al. Neuropathological comorbidity associated with argyrophilic grain disease. Neuropathology 2018;38(1):82–97. [DOI] [PubMed] [Google Scholar]
- 4. Jecmenica Lukic M, Kurz C, Respondek G, et al. Copathology in progressive supranuclear palsy: does it matter? Mov Disord 2020;35(6):984–993. [DOI] [PubMed] [Google Scholar]
- 5. Esteban de Antonio E, López‐Álvarez J, Rábano A, et al. Pathological correlations of neuropsychiatric symptoms in institutionalized people with dementia. J Alzheimer's Dis 2020;78(4):1731–1741. [DOI] [PubMed] [Google Scholar]
- 6. Sakurai K, Tokumaru AM, Ikeda T, et al. Characteristic asymmetric limbic and anterior temporal atrophy in demented patients with pathologically confirmed argyrophilic grain disease. Neuroradiology 2019;61(11):1239–1249. [DOI] [PubMed] [Google Scholar]
- 7. Sakurai K, Kaneda D, Inui S, et al. Simple quantitative indices for the differentiation of advanced‐stage Alzheimer's disease and other limbic tauopathies. J Alzheimer's Dis 2021;81(3):1093–1102. [DOI] [PubMed] [Google Scholar]
- 8. Grau‐Rivera O, Gelpi E, Rey MJ, et al. Prominent psychiatric symptoms in patients with Parkinson's disease and concomitant argyrophilic grain disease. J Neurol 2013;260(12):3002–3009. [DOI] [PubMed] [Google Scholar]
- 9. Nishida N, Hata Y, Yoshida K, Kinoshita K. Neuropathologic features of suicide victims who presented with acute poststroke depression: significance of association with neurodegenerative disorders. J Neuropathol Exp Neurol 2015;74(5):401–410. [DOI] [PubMed] [Google Scholar]
- 10. Litvan I, Hauw JJ, Bartko JJ, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 1996;55(1):97–105. [DOI] [PubMed] [Google Scholar]
- 11. Saito Y, Ruberu NN, Sawabe M, et al. Staging of argyrophilic grains: an age‐associated tauopathy. J Neuropathol Exp Neurol 2004;63(9):911–918. [DOI] [PubMed] [Google Scholar]
- 12. Dickson DW, Bergeron C, Chin SS, et al. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 2002;61(11):935–946. [DOI] [PubMed] [Google Scholar]
- 13. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology 2017;89(1):88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kato N, Arai K, Hattori T. Study of the rostral midbrain atrophy in progressive supranuclear palsy. J Neurol Sci 2003;210(1–2):57–60. [DOI] [PubMed] [Google Scholar]
- 15. Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord 2017;32(6):853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gearing M, Olson DA, Watts RL, Mirra SS. Progressive supranuclear palsy: neuropathologic and clinical heterogeneity. Neurology 1994;44(6):1015–1024. [DOI] [PubMed] [Google Scholar]
- 17. Josephs KA, Dickson DW. Diagnostic accuracy of progressive supranuclear palsy in the Society for Progressive Supranuclear Palsy brain bank. Mov Disord 2003;18(9):1018–1026. [DOI] [PubMed] [Google Scholar]
- 18. Sakamoto R, Tsuchiya K, Yoshida R, et al. Progressive supranuclear palsy combined with Alzheimer's disease: a clinicopathological study of two autopsy cases. Neuropathology 2009;29(3):219–229. [DOI] [PubMed] [Google Scholar]
- 19. Daniel SE, de Bruin VM, Lees AJ. The clinical and pathological spectrum of Steele‐Richardson‐Olszewski syndrome (progressive supranuclear palsy): a reappraisal. Brain 1995;118(Pt 3):759–770. [DOI] [PubMed] [Google Scholar]
- 20. Niizato K, Kuroki N, Arai T, Kase K, Iritani S, Ikeda K. An elderly patient with progressive supranuclear palsy without neurological signs. Brain Nerve 1997;49(9):829–833. [PubMed] [Google Scholar]
- 21. Hattori M, Hashizume Y, Yoshida M, et al. Distribution of astrocytic plaques in the corticobasal degeneration brain and comparison with tuft‐shaped astrocytes in the progressive supranuclear palsy brain. Acta Neuropathol 2003;106(2):143–149. [DOI] [PubMed] [Google Scholar]
- 22. Höglinger GU. Does the anti‐tau strategy in progressive supranuclear palsy need to be reconsidered? No. Mov Disord Clin Pract 2021;8(7):1038–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morimoto S, Takao M, Hatsuta H, et al. Homovanillic acid and 5‐hydroxyindole acetic acid as biomarkers for dementia with Lewy bodies and coincident Alzheimer's disease: an autopsy‐confirmed study. PLoS One 2017;12(2):e0171524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abu‐Rumeileh S, Mometto N, Bartoletti‐Stella A, et al. Cerebrospinal fluid biomarkers in patients with frontotemporal dementia spectrum: a single‐center study. J Alzheimer's Dis 2018;66(2):551–563. [DOI] [PubMed] [Google Scholar]
- 25. Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative accuracy of plasma phospho‐tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA 2020;324(8):772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Definitions of each progressive supranuclear palsy subtype and procedures of neuropathological and neuroradiological analyses.
Figure S1 Receiver operating characteristic (ROC) curves using the ambient gyrus atrophy score for the differentiation between progressive supranuclear palsy with (PSPw/AG) and without argyrophilic grain disease (PSPw/oAG) and a visual rating scale of the ambient gyrus atrophy.
Figure S2 Visual rating scale of the ambient gyrus atrophy. The ambient gyrus atrophy (AGA) scores were defined as 0 for the bulgy medial temporal protuberance indicative of normal volume (A), 1 for the mildly decreased medial temporal protuberance indicative of mild atrophy (B), 2 for the slightly prickly medial temporal protuberance indicative of moderate atrophy (C), and 3 for the small acicular or invisible medial temporal protuberance indicative of severe atrophy (D, E).
Table S1 Detailed results of the neurological, neuropathological, and neuroradiological analyses in all patients.