

ABSTRACT
Background
Progressive supranuclear palsy (PSP)–pallido‐nigro‐luysian atrophy (PNLA) is a neuropathological entity thought to be a variant of classic PSP. Clinical features and pathologic hallmarks are the same in both conditions; however, age and order of symptom onset, disease duration and prognosis, and distribution and density of pathology differentiate the 2 entities.
Objectives
This study presents a PSP‐PNLA case confirmed pathologically with a clinical presentation of hemichorea/ballism, spasticity, progressive hemiparesis, and a frontal behavioral syndrome with relative cognitive sparing early in the disease course.
Methods
We describe the clinical progression in this unique case supplemented with video and imaging findings in the form of magnetic resonance imaging and brain single photon emission computed tomography. Final diagnosis is via pathological analysis at autopsy.
Results
We present an elderly gentleman who manifested a clinical syndrome consisting of subacute onset of chorea that at presentation was distinctly unilateral and a frontal behavioral syndrome in the setting of mild thrombocytopenia and elevated anticardiolipin antibodies. Positive antiphospholipid antibodies resulted in an initial antemortem diagnosis of primary antiphospholipid syndrome as a cause of his chorea. Longitudinal follow‐up over 5 years demonstrated a progression of clinical features with hemi‐motor impersistence/chorea, disinhibition and impulsivity, and eventually corticospinal distribution weakness on the initially affected side. He required nursing home care and falls necessitated wheelchair use. Postmortem neuropathological study revealed a diagnosis of frontotemporal lobar degeneration‐tau, PSP‐PNLA.
Conclusions
This case broadens the phenotype of PSP‐PNLA and to our knowledge is the only case presenting with unilateral chorea.
Keywords: progressive supranuclear palsy, chorea
Pathologically, progressive supranuclear palsy (PSP) is considered a tauopathy because of the presence of abnormally phosphorylated tau in the neurons and glia in subcortical and cortical areas. 1 The substantia nigra, subthalamic nucleus, and globus pallidus consistently contain tau pathology. PSP–pallido‐nigro‐luysian atrophy (PNLA) is a rare neurodegenerative disease defined by bilateral degeneration of these same structures. Although PNLA and PSP share prominent degeneration of the pallidonigroluysian region, PNLA has less widespread degeneration, few or absent neurofibrillary changes, absent or rare tufted astrocytes, and fewer/shorter argyrophilic threads compared with PSP. Thus, important pathological differences exist.
There have been cases described that fulfill the criteria for both PSP and PNLA, and these are called PSP‐PNLA. Pathologically the same types of lesions are observed in PSP and PSP‐PNLA but with differences in the distribution and density of tau pathology, with less tau in the motor cortex, striatum, pontine nuclei, and cerebellum in PSP‐PNLA. 2
Clinical presentation of PSP‐PNLA includes a younger onset age and longer disease duration than patients with PSP (57.1 ± 3.1 vs. 65.5 ± 1.9 years of age at onset, respectively, and disease durations of 11.5 ± 1.6 vs. 7.6 ± 0.5 years, respectively), 2 and with early handwriting and gait abnormalities, such as freezing. Falls, dysphagia, and vertical gaze palsy were late features of PSP‐PNLA in contrast to PSP‐Richardson syndrome in which these clinical findings are pathognomonic and occur early on. Although cases of PSP‐PNLA are extremely rare and there is limited clinical information available, uniformly all cases are clinically characterized by akinesia and parkinsonism.
We describe a case of PSP‐PNLA confirmed pathologically with a clinical presentation of hemichorea/ballism, spasticity, progressive hemiparesis, and a frontal behavioral syndrome with relative cognitive sparing early on. This clinical presentation in the setting of laboratory findings resulted in an antemortem diagnosis of primary antiphospholipid syndrome (aPS). Longitudinal clinical evaluation and laboratory, neuropsychological, and neuroimaging investigations serve to document this rare presentation of PSP‐PNLA.
Case Report
A 79‐year‐old right‐handed Palestinian man developed 1 year of right‐hand clumsiness and involuntary movements. He noted difficulties including writing and dropping objects from his right hand. He experienced falls, tripping over his cane with the right leg. Involuntary, low‐amplitude movements of his right arm and leg presented insidiously. There were word‐finding difficulties, but he continued to work as an international consultant.
His previous medical history included coronary artery disease, hypertension, hypercholesterolemia, and lumbar degenerative disc disease. He had no history of autoimmune conditions, cancer, or nonatherosclerotic thrombotic events. There was no exposure to antipsychotics or antiemetics. There was no relevant family history.
Vital signs and general examination were normal. Mini‐Mental State Examination was 28/30. He behaved inappropriately, talking loudly and seeming overly familiar; he touched the examiner's shoes because he “liked them.” He was short‐tempered with his wife. This was out of character according to the family. Extraocular movements were full with saccadic pursuit. There was occasional blinking with horizontal saccades. His face was symmetric, and the tongue was midline with no motor impersistence or orobuccal dyskinesia. There was a positive glabellar tap and bilateral palmomental reflexes. Muscle bulk, tone, and power were normal. Reflexes were exaggerated in the right arm and leg (3+) and normal on the left (2+), with bilateral flexor plantar responses. There was no ataxia. Primary and secondary sensory modalities were intact. Chorea was present at rest and increased with action in the right arm and leg, sometimes with a ballistic quality. He easily stood up from a chair and had a narrow gait with chorea in his right hand and leg while walking. The chorea in his leg sometimes impaired his walking and balance (Video 1, segment S1).
Video 1.
Video demonstrating the clinical phenomenology at presentation and 2 years later.
The following year, chorea increased in amplitude but remained unilateral. He became more irritable, impulsive, and unpredictable in his behavior, for example, tipping a musician excessively and increased interest in sexual activity with his wife discordant with her wishes. There was no paranoia, hallucinations, or delusions. Cognitively, his family noted distractibility and short‐term memory problems. However, he remained intact in his instrumental activities of daily living. Efforts to reduce chorea were unsuccessful: tetrabenazine caused sedation, and risperidone did not help.
He scored within normal limits on the Mattis Dementia Rating Scale, although a formal neuropsychological assessment revealed mild to moderate impairment in areas of speeded information processing, verbal working memory, complex visuospatial construction, effortful encoding, and retrieval of verbal information, suggesting frontosubcortical and mild left temporal dysfunction. Frontal Behavioral Inventory score was 33 (cutoff score ≥ 30).
Initial investigations including brain magnetic resonance imaging revealed mild generalized atrophy and microangiopathic changes consistent with age, but with no strokes or other pathology in the subthalamic nuclei or basal ganglia to account for the hemichorea (Fig. 1). Blood work, including electrolytes, ESR, AST/ALT, B12, ANA, CPK, C3/C4, INR/PTT, and thyroid functions, was normal. Further investigations were pursued, including brain single photon emission computed tomography showing increased perfusion in the basal ganglia bilaterally compared with matched normal controls (patient vs. control average percentage increase: left, +12%; right, +18%; see Fig 1E). Serum paraneoplastic antibody panel was negative. Huntington's disease and SCA 1, 2, 3, 6, 7, 8, and 17 genetic testing were negative. CBC was normal except for thrombocytopenia (platelets 120). Lupus anticoagulant was negative; however, anticardiolipin immunoglobulin G (IgG) antibody level was 54 (repeated 3 months later = 66; normal, 0–18), with an immunoglobulin M (IgM) value of 33 (repeated 3 months later = 18; normal, 0–10). Because of these results in conjunction with his clinical presentation, he was reviewed by a rheumatologist who suggested a diagnosis of aPS.
FIG. 1.
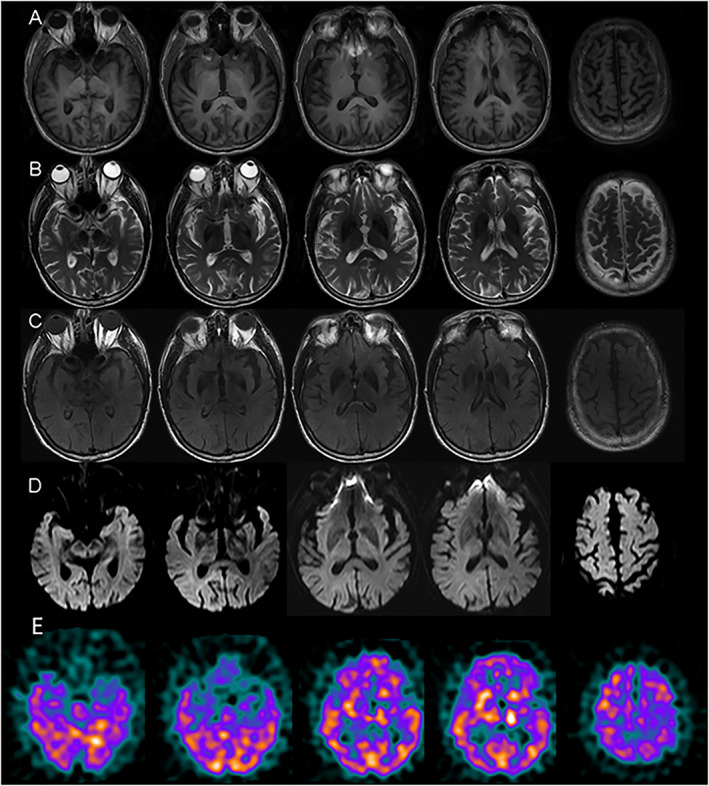
(A–D) Axial T1, T2, fluid‐attenuated inversion recovery (FLAIR), and diffusion magnetic resonance imaging without gadolinium showing mild generalized atrophy. (E) Brain Technetium‐99m ethyl cysteinate dimer (99mTc‐ECD) single photon emission computed tomography showing increased perfusion of the bilateral basal ganglia, z scores >2 (normalized to 8 matched controls from the Sunnybrook Dementia Study).
He received a 3‐day course of methylprednisolone 1000 mg intravenously daily followed by longer term immunosuppression with methotrexate. Despite improvement in his antibody parameters, he continued to progress clinically. He had motor impersistence of the right arm resulting in difficulty maintaining any posture in addition to choreic/ballistic movements exacerbated with action. There was truncal chorea and subtle left leg chorea with action (Video 1, segment S1). Reflexes were exaggerated bilaterally (right > left), and gait was unsteady with lunging and ballistic movements. A neurodegenerative disorder was suspected. Given the asymmetry at presentation, atypical corticobasal syndrome was considered, but he did not meet the clinical criteria. Apraxia was hard to assess given the chorea, but he lacked any cortical sensory loss, and his neuropsychological assessment was also not consistent with that diagnosis. Symptomatic treatment with flupenthixol depot at 10 mg intramuscularly every 3 weeks was added to both dampen movements and improve behavior. This reduced the chorea; however, he continued to have progressive loss of use of his right arm with associated 4+ corticospinal distribution weakness and superimposed motor impersistence. Treatment with intravenous immunoglobulin at a dose of 1 g/kg/day for 2 days was instituted without benefit.
During the next 3 years he became progressively disabled with spastic right hemiplegia and increasing chorea/ballism on his left. He was admitted to a nursing home, had frequent falls, and became wheelchair bound. He became dysarthric to the point of incomprehensibility, and in the last months of life he had vertical gaze palsy and severe pseudobulbar dysfunction. He died of sepsis secondary to an infected sacral ulcer, approximately 5 years after symptom onset. His brain was submitted for postmortem examination after informed consent was obtained.
Pathological Findings
The brain was examined and sampled for microscopy according to a standard neurodegenerative neuropathology blocking protocol. Formalin‐fixed, paraffin‐embedded blocks were cut into 4‐μm thick sections, mounted on glass slides, and stained with hematoxylin–eosin/luxol fast blue, Gallyas, and the immunohistochemistry antibodies listed in Table 1.
TABLE 1.
Immunohistochemical methods employed
| Antibody | Manufacturer | Dilution | Detection system |
|---|---|---|---|
| Tau | Invitrogen (Canada) | 1/200 | Mach 4 |
| β‐amyloid | Dako (U.S.A) | 1/200 | Mach 4 |
| p62 | Becton, Dickinson and Company (Canada) | 1/200 | Dako Omnis instrument using Envision Flex detection System |
| α‐synuclein | Invitrogen (Canada) | 1/2000 | Dako Omnis instrument using Envision Flex detection System |
| GFAP | Cell Marque (U.S.A) | Prediluted | Ultraview‐Ventana |
| TDP‐43 | Proteintech Group (U.S.A) | 1/2000 | Dako Omnis instrument using Envision Flex detection system |
Abbreviations: GFAP, glial fibrillary acidic protein; TDP‐43, transactive response DNA binding protein 43 kDa.
Macroscopic
The brain weighed 1040 grams. There was mild symmetrical atrophy of the frontal and temporal lobes bilaterally. The midbrain and pons were small in caliber. There was mild dark discoloration of the putamen and nucleus of Luys bilaterally, and the substantia nigra appeared pale (Fig. 2A).
FIG. 2.
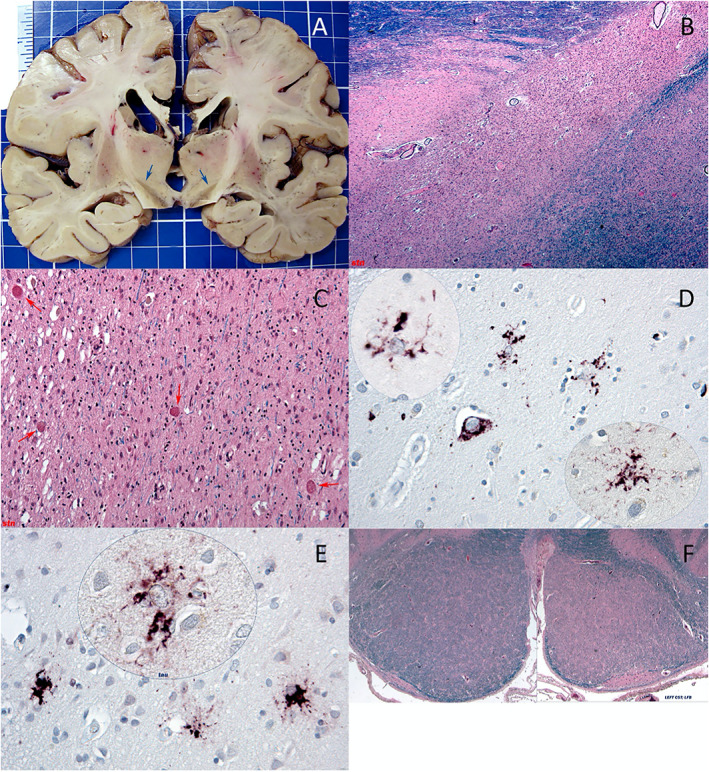
See main text. (A) Coronal; blue arrows highlight subthalamic nuclei. (B,C) Low/high magnification; subthalamic nuclei; hematoxylin–eosin (H&E)/luxol fast blue (LFB). (D) High magnification; putamen; tau (AT8). (E) High magnification; frontal cortex; p62 immunolabeling and tau (AT8; inset). (F) Low magnification; left medullary pyramid; H&E/LFB.
Microscopic
Microscopic examination revealed a unique glioneuronal tauopathy with atrophy of the corticospinal tracts (left greater than right; Fig. 2F) and subthalamic nuclei and sparing of the inferior olivary and dentate nuclei. The tau‐positive inclusions strongly stained with Gallyas and were predominantly glial and cytoplasmic; the oligodendroglial inclusions were mostly coiled bodies, and the astrocytic inclusions often had a “bushy” morphology. Neurofibrillary tangles were a relatively minor component.
Examination of the deep gray structures showed numerous oligodendroglial tau‐positive inclusions, less frequent bushy astrocytic tau‐positive inclusions, no tufted astrocytes, some neuronal cytoplasmic tau‐positive inclusions in the putamen (Fig. 2D), a normal anterior striatum, neuronal dropout and gliosis of the pallidum, and severe neuronal loss, vacuolation, glial inclusions, axonal spheroids (Fig. 2C, red arrows) and iron deposition in the subthalamic nuclei (Fig. 2B,C). The substantia nigra showed neuronal loss, gliosis, and coiled bodies on tau immunolabeling. The remainder of the brainstem and cerebellum appeared normal without tau inclusions, including the olivary and dentate nuclei. Neocortical pathology was mild and included slight cortical neuronal loss with scattered bushy tau‐immunopositive astrocytic inclusions (Fig. 2E). Only rare cortical ballooned neurons were present, and no glial plaques were noted. In the amygdala and hippocampus, there were neuronal pretangles and glial tau‐positive inclusions. There was no evidence of hippocampal sclerosis. Transactive response DNA binding protein 43 kDa (TDP‐43), α‐synuclein, and β‐amyloid staining were negative, and p62 highlighted the tau pathology only. There was no significant cerebrovascular pathology including lacunes or thrombotic microangiopathy.
Discussion
This case features a man with a frontal behavioral syndrome and hemichorea in the setting of positive antiphospholipid antibodies that resulted in an initial diagnosis of aPS, although subsequent clinical evolution led to the suspicion of an atypical neurodegenerative disorder, ultimately confirmed at postmortem study to be PSP‐PNLA.
Chorea, particularly unilateral, without an obvious explanation is exceedingly rare. 3 The working diagnosis of aPS was reasonable considering the clinical presentation and laboratory findings. aPS is a rare but well‐established cause of chorea, including unilateral presentations. 4 Treatment with steroids, methotrexate, and intravenous immunoglobulin did not improve the chorea despite antiphospholipid antibody normalization. He progressed clinically, ultimately developing chorea on the left, a vertical gaze palsy, and pseudobulbar dysfunction.
Postmortem neuropathological study revealed atrophy and tau immunopathology in the pallidum, substantia nigra, and most severely, the subthalamic nuclei, with unilateral corticospinal tract degeneration. In contrast, the areas usually found to be affected in conventional PSP such as the inferior olives and dentate nuclei were largely spared, and tufted astrocytes, a feature commonly found in PSP, were mostly absent. PSP‐PNLA has pathological findings with a similar distribution to our case. However, of the published cases of PSP‐PNLA, none presented with unilateral chorea to our knowledge. Wong et al 5 describe the case of a 70‐year‐old man who presented with slurred speech and went on to develop gait difficulties and left‐sided chorea. At autopsy, this patient was found to have right‐predominant degeneration in the globus pallidus and subthalamic nucleus. TDP‐43 staining was positive in the hippocampus, amygdala, cerebellum, and frontoparietal cortices. Immunohistochemistry revealed p62‐positive, tau‐negative, thread‐like profiles in glial cells. There were no tau‐positive glial inclusions, and the patient was diagnosed with PNLA. Chorea is described in PNLA and can be seen in up to 20% of cases, but it is a relatively uncommon symptom in adult neurodegenerative diseases and when present should raise the question of PNLA or dentatorubral‐pallidoluysian atrophy. 6
Despite the presence of nigral pathology, our case had no parkinsonian features as described in PSP and PNLA. Supranuclear gaze and pseudobulbar palsies, clues to the PSP phenotype, only appeared near the end when neurological functions were severely impaired. The prominent degeneration and tauopathy affecting the subthalamic nucleus (STN) likely accounted for the chorea/ballism. The relative sparing of the dentate and olives, the dominant “bushy” morphology to the astrocytic inclusions, and corticospinal tract degeneration are atypical of PSP. 7 This correlated poorly with the clinical phenotype marked by hemichorea, hemiparesis, and lack of parkinsonism. Some of the observed pathologic findings are reminiscent of those described in globular glial tauopathies (GGT), 8 including the involvement of the corticospinal tract8, 9 However, the present case contained more abundant coiled bodies then usually encountered in GGT, and some astrocytic inclusions stained positively with Gallyas.
The laboratory findings of aPS were a red herring. Anti‐immunoglobulin‐like cell adhesion molecule 5 (IgLON5) antibodies were not described at the time of this case and so are unavailable, but the neuropathology is not consistent with this condition. 10 Other diagnoses that have a similar tau burden and distribution to our case include some cases of frontotemporal dementia‐parkinsonism linked to chromosome 17 associated with MAPT gene mutations (FTDP‐17T). Microtubule‐associated protein tau (MAPT) mutation screening was negative in our patient, although he had the MAPT H1/H1 genotype, a common genetic risk factor for PSP and corticobasal degeneration. 11 This case broadens the phenotype of PSP‐PNLA and is the only case to our knowledge presenting with unilateral chorea.
Author Roles
(1) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
G.K.: 1A, 1B
S.A.R.: 1A, 1B
J.B.: 1B
J.K.: 1B
E.R.: 1B
S.E.B.: 1B
A.E.L.: 1B
M.M.: 1B
Disclosures
Ethical Compliance Statement
This work was embedded within the Sunnybrook Dementia Study (SDS) (ClinicalTrials.gov: NCT01800214) ‐ a prospective observational study of patients with neurodegenerative dementia and movement disorders. It is approved by the local Research Ethics Board at Sunnybrook Health Sciences Centre and written informed consent was obtained from the participant and their surrogate caregivers according to the Declaration of Helsinki. A separate written consent was also obtained from the patient for the purposes of case report preparation. All authors have read and complied with the Journal's Ethical Publication Guidelines. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest
No specific funding was received for this work. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months
The authors declare that there are no additional disclosures to report.
Relevant disclosures and conflicts of interest are listed at the end of this article.
Contributor Information
Galit Kleiner, Email: gkleinerfisman@yahoo.com.
Mario Masellis, Email: mario.masellis@sunnybrook.ca.
References
- 1. Litvan I, Hauw JJ, Bartko JJ, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 1996;55(1):97–105. [DOI] [PubMed] [Google Scholar]
- 2. Ahmed Z, Josephs KA, Gonzalez J, DelleDonne A, Dickson DW. Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido‐nigro‐luysial degeneration and axonal dystrophy. Brain 2008;131(Pt 2):460–472. [DOI] [PubMed] [Google Scholar]
- 3. Quinn N, Schrag A. Huntington's disease and other choreas. J Neurol 1998;245(11):709–716. [DOI] [PubMed] [Google Scholar]
- 4. Ciubotaru CR, Esfahani F, Benedict RH, Wild LM, Baer AN. Chorea and rapidly progressive subcortical dementia in antiphospholipid syndrome. J Clin Rheumatol 2002;8(6):332–339. [DOI] [PubMed] [Google Scholar]
- 5. Wong JC, Armstrong MJ, Lang AE, Hazrati LN. Clinicopathological review of pallidonigroluysian atrophy. Mov Disord 2013;28(3):274–281. [DOI] [PubMed] [Google Scholar]
- 6. Munhoz RP, Bergeron C, Lang AE. Sporadic case of dentatorubral pallidoluysian atrophy with no CAG repeat expansion and no intranuclear inclusions. Mov Disord 2004;19:580–583. [DOI] [PubMed] [Google Scholar]
- 7. Josephs KA, Katsuse O, Beccano‐Kelly DA, et al. Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol 2006;65(4):396–405. [DOI] [PubMed] [Google Scholar]
- 8. Ahmed Z, Bigio EH, Budka H, et al. Globular glial tauopathies (GGT): Consensus recommendations. Acta Neuropathol 2013;126:537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takeuchi R, Toyoshima Y, Tada M, et al. Globular glial mixed four repeat tau and TDP‐43 Proteinopathy with motor neuron disease and frontotemporal dementia. Brain Pathol 2016;26(1):82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gelpi E, Höftberger R, Graus F, et al. Neuropathological criteria of anti‐IgLON5‐related tauopathy. Acta Neuropathol 2016;132(4):531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Im SY, Kim YE, Kim YJ. Genetics of progressive Supranuclear palsy. J Mov Disord 2015;8(3):122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]