

Abstract
Dysregulated microRNA (miRNA) expression in the brain can contribute to cognitive dysfunction and aberrant tau hyperphosphorylation in Alzheimer’s disease (AD). Several studies have reported a role for microRNA-23b-3p (miR-23b-3p) in various neurologic disorders; however, its involvement in cognition-related functions remains unclear. In the present study, we investigated the potential therapeutic effects and mechanisms of miR-23b-3p in AD. miRNA profiles in the cortex of amyloid precursor protein (APP)/presenilin 1 (PS1) double transgenic mice (APP/PS1 mice) demonstrated that miR-23b-3p was reduced. This decrease was verified in APPswe cells, SAMP8 mouse brains, and plasma from AD patients. Furthermore, glycogen synthase kinase-3β (GSK-3β), a major tau kinase implicated in tau pathology, was identified as a target of miR-23b-3p. Functional in vivo studies demonstrated that intracerebroventricular delivery of miR-23b-3p in APP/PS1 mice ameliorated cognitive deficits, histopathological changes, and tau phosphorylation immunoreactivity at several sites by inhibiting GSK-3β expression and activation. Similarly, the upregulation of miR-23b-3p in APPswe cells inhibited GSK-3β-mediated tau hyperphosphorylation, Aβ1-42 generation, and neuronal apoptosis, resulting in the suppression of the GSK-3β/p-tau and Bax/caspase-3 pathways. Collectively, our findings strongly support the hypothesis that miR-23b-3p plays a neuroprotective role in AD, thereby identifying miR-23b-3p as a promising therapeutic target for AD.
Keywords: MT: Oligonucleotides: Therapies and Applications, Alzheimer’s disease, cognition, glycogen synthase kinase-3β, microRNA therapy, tau protein
Graphical abstract
Jiang et al. reveal the potential therapeutic effects and mechanisms of miR-23b-3p in Alzheimer’s disease (AD), indicating that miR-23b-3p ameliorated cognitive deficits, inhibited cell apoptosis, and reduced tau hyperphosphorylation via the downregulation of GSK-3β-elicited signaling pathways against amyloid-βpeptide-induced lesions, thereby identifying miR-23b-3p as a promising therapeutic target for AD.
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disease, with clinical features characterized by memory loss, cognitive impairment, and personality change in the elderly.1 The main pathological hallmarks in the brains of AD patients are the presence of extracellular amyloid plaques composed of amyloid-β peptide (Aβ) and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein, which eventually elicit the loss of neurons.2,3 Although there is much evidence to suggest that the tau protein is a downstream target of Aβ-initiated neurodegeneration, postmortem studies have indicated that the severity of AD symptoms corresponds more to the degree of tau-related pathology than to Aβ deposition.4 While an overall kinase-phosphatase imbalance is considered to be the cause of tau hyperphosphorylation, glycogen synthase kinase-3β (GSK-3β) is generally considered a major tau kinase, modulating key phosphorylated sites in tau protein that are in close proximity to microtubule-binding domains and their amino acid residues,5, 6, 7 which are known to be vulnerable to toxic self-aggregation.8 Because hyperphosphorylated tau is highly neurotoxic through the promotion of neuronal apoptosis and neurodegeneration in AD,9 inhibiting the Aβ-induced hyperphosphorylation of tau could be an effective therapeutic strategy for the treatment of AD.
MicroRNAs (miRNAs) are short, single-stranded, non-coding RNAs (18–22 nt in length) that are involved in the posttranscriptional regulation of genes, and several are known to play crucial roles in biological processes such as apoptosis, inflammation, cell proliferation, and neuronal development.10 Recent studies on aberrantly expressed miRNAs have been encouraging, and it may soon be possible to diagnose AD pathophysiology based on an analysis of these miRNAs in human blood or AD mouse brains.11, 12, 13, 14, 15 Functional studies have demonstrated that miRNAs that correlate with AD pathology also regulate neuroinflammation, Aβ formation, tau phosphorylation, and vascular inflammation, processes that are crucial in the pathogenesis of AD.16, 17, 18, 19
miRNA-23b-3p (miR-23b-3p) belonging to the miR-23b/27b/24 cluster (9q22.32) is involved in several cellular functions, including differentiation, proliferation, development, and metabolism, and is highly expressed in the brains of humans and rodents.20, 21, 22 The overexpression of miR-23b improves learning and memory by attenuating long-term neurological deficits and inhibiting the activation of neuronal autophagy.23 miR-23b-3p, a subtype of miR-23b, possesses a biological function similar to miR-23b, modulating apoptosis in neuronal cells.24 Interestingly, the downregulation of miR-23b-3p has been reported in blood samples from AD patients.14 In our previous study, we performed a microarray-based differential gene expression analysis that revealed the low expression of miR-23b-3p in the amyloid-β protein precursor (APP) and presenilin 1 (PS1) transgenic mouse brain.25 Furthermore, miR-23b-3p was demonstrated to protect neurons against apoptosis in in vitro AD models,26,27 supporting the hypothesis that miR-23b-3p plays a beneficial role in AD pathology. However, the functional role of miR-23b-3p in AD and its underlying regulatory mechanism have not yet been elucidated.
In the present study, we investigated the function and underlying molecular mechanisms of miR-23b-3p relating to AD. miR-23b-3p was demonstrated to be downregulated during AD progression, and this was verified in cells, mice, and patients with AD. Furthermore, the upregulation of miR-23b-3p exerted neuroprotective effects by inhibiting tau hyperphosphorylation and neuronal apoptosis. Mechanistically, miR-23b-3p was revealed to protect against Aβ-induced tau hyperphosphorylation by directly targeting GSK-3β. Notably, in vivo upregulation of miR-23b-3p alleviated cognitive deficits by suppressing the GSK-3β/p-tau and Bax/caspase-3 pathways. In summary, our findings demonstrate that miR-23b-3p plays a neuroprotective role in AD by targeting GSK-3β signaling, revealing it as a potential therapeutic target in AD.
Results
miR-23b-3p expression decreases during AD progression
High-throughput sequencing analysis of the miRNA profile in APP/PS1 mouse cortex revealed aberrant expression of miR-23b-3p. As shown in Figure 1A and Table S1, the expression levels of 10 miRNAs were significantly decreased in APP/PS1 mice at different stages. Of these, miR-23b-3p was the most representative miRNA, being significantly downregulated in brain tissue from the early (3-month) and progressive (9-month) stages of AD progression. These changes were accompanied by a high value for the area under the curve (AUC: 0.917 ± 0.080) discriminating APP/PS1 mice from wild-type (WT) mice (Figure 1B; p < 0.05). To confirm these expression changes during the progression of AD, APP/PS1 mice and senescence-accelerated mouse prone 8 (SAMP8) mice were analyzed by quantitative real-time PCR. Our quantitative real-time PCR results validate the miRNA expression profile results, demonstrating that miR-23b-3p expression was decreased in the hippocampus and cortex of APP/PS1 and SAMP8 mice compared with their controls at different disease stages (Figures 1C and 1D, p < 0.01, p < 0.001 versus WT or senescence-accelerated mouse resistant 1 [SAMR1]).
Figure 1.

miR-23b-3p expression in miRNA profile, in AD cell lines, AD mouse brains, and blood samples of AD patients
(A) miR-23b-3p expression in APP/PS1 mouse cortex using miRNA sequencing analysis at different stages of the disease (n = 3). Two-way hierarchical clustering of miRNAs in the sequencing experiments was performed to differentiate the miRNAs with different expression levels at different stages of the disease. The color scale indicates the relative expression of a miRNA in a particular age group: Red, high relative expression; green, low relative expression. (B) ROC plot for differentially expressed miR-23b-3p in the cortex of APP/PS1 mice and WT mice (n = 12). (C and D) Decreased expression of miR-23b-3p in the cortex (C) and hippocampus (D) of APP/PS1 and SAMP8 mice (n = 4). (E) Decreased expression of miR-23b-3p in APPswe cells at different time points after copper treatment (n = 3). (F) Reduced level of miR-23b-3p in the plasma of AD patients compared with healthy aged volunteers (HAVs) (n = 14, HAVs; n = 22, AD patients). (G) Correlation analysis of plasma miR-23b-3p level and MMSE score using Pearson correlation (n = 14, HAVs; n = 22, AD patients). (H) ROC plot for differentially expressed miR-23b-3p in the blood of AD patients and HAVs (n = 14, HAVs; n = 22, AD patients). Comparisons between 2 groups were analyzed using an unpaired t test. Comparisons among multiple groups were analyzed using 1-way ANOVA, followed by Tukey’s post hoc testing to analyze differences between groups. Results represent means ± SEMs. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus each correspondent control.
Human neuroblastoma SH-SY5Y cells overexpressing the Swedish mutant form of human β-amyloid precursor protein (APPswe cells) treated with copper to trigger Aβ neurotoxicity are an established in vitro model (Figure S1; p < 0.001 versus control), as previously reported.28,29 miR-23b-3p expression was downregulated in APPswe cells that were treated with copper (compared with control cells that did not receive treatment with copper) at different time points (Figure 1E; p < 0.001 at 12 h, p < 0.05 at 24 h, p < 0.01 at 36 h, p < 0.001 at 48 h). Furthermore, miR-23b-3p levels were significantly reduced in plasma from AD patients (Figure 2F, p < 0.001 versus healthy age-matched volunteers [HAVs]). A statistically significant positive correlation between miR-23b-3p level and Mini-Mental Status Examination (MMSE) score in AD patients was found (Figure 1G; R2 = 0.608, p < 0.001). The diagnostic accuracy of miRNA differentially expressed in AD patients and HAVs was determined by receiver-operating characteristic (ROC) curve analysis (Figure 1H). The AUC for miR-23b-3p was 0.909 ± 0.048, indicating a good diagnostic value for AD, with 81.8% sensitivity and 92.9% specificity. These results provide evidence that miR-23b-3p is downregulated during AD progression, and that miR-23b-3p has potential diagnostic value in blood plasma from AD patients.
Figure 2.

miR-23b-3p inhibits cell apoptosis
(A) Quantitative real-time-PCR analysis of miR-23b-3p levels after transfection of miR-23b-3p mimics (n = 4). (B) MTT assay demonstrating the effect of miR-23b-3p on cell viability in APPswe cells induced by copper (n = 6). (C) Representative cell immunofluorescence images of APPswe cell apoptosis induced by copper. Green, annexin-V; red, PI; blue, Hoechst 33342. Blue and red arrows indicate the representative normal and apoptotic cells, respectively. Bar, 100 μm. (D) Representative flow cytometry images demonstrating the effect of miR-23b-3p on cell apoptosis in APPswe cells induced by copper. (E) Quantification of the percentage of cell apoptosis induced by copper in the presence of miR-23b-3p mimics (n = 3). Comparisons between 2 groups were analyzed using an unpaired t test. Comparisons among multiple groups were analyzed by 1-way ANOVA, followed by Tukey’s post-hoc test to analyze differences between groups. Results represent means ± SEMs. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus NCM; #p < 0.05, ##p < 0.01, ###p < 0.001 versus NCM plus copper.
miR-23b-3p inhibits apoptosis in the in vitro AD model
To study the functional role of miR-23b-3p in AD, miR-23b-3p mimics, miR-23b-3p mutation (MUT), and negative control mimics (NCM) were transiently transfected into APPswe cells with or without copper treatment, and cell viability and apoptosis were evaluated. Our findings reveal that miR-23b-3p levels were significantly higher in APPswe cells after transfection with miR-23b-3p mimics (Figure 2A; p < 0.001 versus NCM). Results from the MTT assay reveal that miR-23b-3p mimics increased the viability of APPswe cells treated with copper (Figure 2B; p < 0.05 versus NCM plus copper). Next, cell immunofluorescence and flow cytometry were used to further analyze apoptosis in APPswe cells. The results reveal that the total rate of apoptosis (composed of both early and late apoptosis) in APPswe cells treated with copper was decreased in transfected cells expressing miR-23b-3p mimics (Figures 2C–2E; p < 0.05, p < 0.01, and p < 0.001 versus NCM plus copper). In contrast, miR-23b-3p MUT transfection did not increase cell viability or decrease the apoptosis of APPswe cells (with or without copper treatment) compared with cells transfected with NCM. miR-23b-3p mimics also increased cell viability in the MTT assay, suggesting that miR-23b-3p slightly improved the health of neuronal cells in the physiological state. However, the result did not reach statistical significance. miR-23b-3p mimics also inhibited apoptosis in normally cultured APPswe cells without copper treatment, as demonstrated by a reduction in propidium iodide (PI)-stained cells in the immunofluorescence assay (Figure 2C) and a reduction in apoptotic ratios in flow cytometry (Figures 2D and 2E; p < 0.05, p < 0.01 versus NCM). Thus, miR-23b-3p upregulation may protect against AD deficits by inhibiting apoptosis.
miR-23b-3p regulates GSK-3β expression by directly binding to the 3′-untranslated regions of its mRNA
To identify the underlying mechanism by which miR-23b-3p exerts neuroprotection during AD progression, we used bioinformatics analyses that incorporated online databases (TargetScan, miRDB, and Tarbase) and a high-throughput sequencing analysis comparing APP/PS1 mice with WT mice. To narrow down the predicted miR-23b-3p targets in the three databases (Figure 3A; Table S2), 14 predicted mRNA targets were excluded because they are frequently upregulated in a wide variety of tumors, and no report indicates their biological activity in neuronal function. Of the 4 remaining mRNA targets, only GSK-3β mRNA was found by quantitative real-time PCR as having a negative correlation with miR-23b-3p in APP/PS1 mice during disease progression (Figure 3B, R2 = 0.390, p < 0.05; Figures S2A–S2C). A potential target site for miR-23b-3p was identified in the 3′-untranslated regions (3′ UTR) of the GSK-3β gene, with a proper miR-support vector regression (SVR) score of −0.5377 for miR-23b-3p (Table S2). This binding site (positions 1,001–1,008) is highly conserved in mammals (Figure 3C).
Figure 3.

miR-23b-3p targets the GSK-3β gene by binding to the 3′ UTR
(A) Venn diagram of predicted targets of miR-23b-3p from 3 databases: TargetScan, miRDB, and Tarbase. A total of 18 common targets among these 3 databases was found. (B) Correlation analysis between miR-23b-3p and GSK-3β mRNA levels in APP/PS1 mouse brain at different stages of the disease. (C) A potential binding site for miR-23b-3p in the 3′ UTR of GSK-3β mRNA was computationally identified. This binding site is conserved in a number of species. (D) Construction of recombinant Luc-GSK3B-MUT and Luc-GSK3B-WT. (E) Changes in relative luciferase activity in each group after transfection. Compared with the negative control group (NCM), miR-23b-3p mimics (miR-23b-3p) exerted a significant inhibition of reporter luciferase activity in HEK293 cells when using a construct containing WT GSK3B 3′ UTR. No inhibition was observed when using a construct containing mutant GSK3B 3′ UTR. (F) GSK3β mRNA expression was quantified after transfection of APPswe cells with miR-23b-3p mimics and miR-23b-3p inhibitor. (G) Representative western blot images of GSK-3β protein expression after transfection of APPswe cells with miR-23b-3p mimics (miR-23b-3p), inhibitor (miR-23b-3p-I), NCMs, and negative control inhibitor (NCI). (H) Quantification of GSK-3β protein levels using western blotting. Correlation analysis between miR-23b-3p and GSK-3β levels was evaluated using a Pearson rank correlation coefficient. Comparisons between 2 groups were analyzed by unpaired t test. Comparisons among multiple groups were analyzed by 1-way ANOVA, followed by Tukey’s post hoc test to analyze the differences between groups. The results represent means ± SEMs. n = 3. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus NCM; &p < 0.05, &&p < 0.01 versus NCI.
In a dual-luciferase reporter assay in HEK293 cells, luciferase activity was significantly reduced in cells that were co-transfected with miR-23b-3p mimics plus pGL3-GSK-3B-3′ UTR-WT (compared with cells that were co-transfected with miR-23b-3p mimics plus pGL3-GSK-3B-3′ UTR-MUT) (Figures 3D and 3E; p < 0.001 versus NCM), suggesting that miR-23b-3p binds directly to GSK-3β through the predicted targeting sequence. The other three predicted mRNAs, carbonic anhydrase 2 (CA2), heparan sulfate 6-O-sulfotransferase 2 (HS6ST2), and myristoylated alanine-rich protein kinase C substrate-like protein 1 (MARCKSL1), demonstrated reduced luciferase activities when the luciferase reporter contained either the putative 3′ UTRs or the mutant 3′ UTR (Figures S2D–S2F). Therefore, the remaining three predicted mRNAs did not appear to be specific mRNA targets of miR-23b-3p.
Next, quantitative real-time PCR and Western blot analyses were used to identify the effects of miR-23b-3p on GSK-3β gene and protein expression. As shown in Figures 3F–3H, the expression of GSK-3β was significantly decreased at the mRNA and protein levels in miR-23b-3p mimics-transfected cells (p < 0.01 and 0.05 versus NCM), whereas its expression at both levels was increased in cells transfected with miR-23b-3p inhibitor (p < 0.01 and 0.05 versus negative control inhibitor [NCI]). In the miR-23b-3p MUT-transfected group, GSK-3β expression was not significantly changed at either the mRNA or protein level compared with the NCM group. Taken together, these results provide evidence that miR-23b-3p directly binds GSK-3β mRNA to inhibit its translation during the AD pathological process.
miR-23b-3p exerts neuroprotection by suppressing tau phosphorylation and apoptotic pathway dependent on GSK-3β expression
Next, we evaluated GSK-3β expression and its inhibitory phosphorylation level at Ser 9, GSK-3β activity, tau phosphorylation level, apoptotic markers Bax protein expression and caspase-3 activity, and Aβ1-42 level after transfection with miR-23b-3p mimics or inhibitor into APPswe cells treated with copper. miR-23b-3p mimics inhibited the expression of GSK-3β and increased the level of phosphorylated GSK-3β at Ser9, leading to the increased ratio of GSK-3β-Ser9/GSK-3β and decreased activity of GSK-3β (Figures 4A–4C, all p < 0.05 versus NCM). In addition, the reduced level of tau phosphorylation at Ser396 and Ser404, downregulated expression of Bax, suppressed activity of caspase-3, and decreased production of Aβ1-42 were observed in the miR-23b-3p mimics transfected cells (Figures 4A, 4B, 4D, and 4E, all p < 0.05 versus NCM). Conversely, an opposite trend was observed in the alteration of these proteins when cells were transfected with miR-23b-3p inhibitor (all p < 0.05 versus NCI). Together, these results suggest that miR-23b-3p may protect neuronal cells from apoptosis by inhibiting the GSK-3β/p-tau and Bax/caspase-3 signaling pathways.
Figure 4.

miR-23b-3p induces neuroprotection by inhibiting tau phosphorylation and cell apoptosis in a GSK-3β-dependent manner
(A and B) Western blot images of GSK-3β, p-GSK-3β (Ser9), p-Tau-Ser396, p-Tau-Ser404, and Bax (A), and quantification of these proteins after transfection with miR-23b-3p mimics/inhibitor (B) (n = 3). (C) GSK-3β activity assay results demonstrating the effects of miR-23b-3p mimics/inhibitor on GSK-3β activity in APPswe cells (n = 3). (D) ELISA results demonstrating the effects of miR-23b-3p mimics/inhibitor on the release of active caspase-3 in APPswe cells (n = 3). (E) ELISA results demonstrating the effects of miR-23b-3p mimics/inhibitor on Aβ1-42 production in APPswe cells (n = 3). (F) Representative flow cytometry images demonstrating the effects of miR-23b-3p and GSK-3β overexpression on APPswe cell apoptosis induced by copper. (G) Quantification of APPswe cell apoptosis induced by copper in the presence of miR-23b-3p and GSK-3β overexpression (n = 6). (H and I) Western blot images of GSK-3β, p-GSK-3β, p-Tau-Ser396, p-Tau-Ser404, and Bax (H), and bar graph demonstrating quantification of these proteins after miR-23b-3p and GSK-3β overexpression (I) (n = 3). (J) GSK-3β assay results demonstrating the effects of miR-23b-3p and GSK-3β overexpression on GSK-3β beta activity in APPswe cells (n = 3). (K) ELISA results demonstrating the effects of miR-23b-3p and GSK-3β overexpression on the release of active caspase-3 in APPswe cells (n = 3). (L) ELISA results demonstrating the effects of miR-23b-3p and GSK-3β overexpression on Aβ1-42 production in APPswe cells (n = 3). Comparisons among multiple groups were analyzed by 1-way ANOVA, followed by Tukey’s post hoc test to analyze differences between groups. The results represent means ± SEMs. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus NCM; &p < 0.05 versus NCI; #p < 0.05, ##p < 0.01, ###p < 0.001 versus miR-23b-3p.
To further investigate miR-23b-3p regulation of GSK-3β, GSK-3β was overexpressed in copper-treated APPswe cells co-transfected with or without miR-23b-3p mimics. The overexpression of GSK-3β resulted in an increased total apoptotic rate (composed of early and late apoptosis) in copper-treated APPswe cells (Figures 4F and 4G; all p < 0.001 versus NCM). Moreover, the increase in GSK-3β expression was accompanied by an increase in GSK-3β activity, upregulation of tau phosphorylation at Ser396 and Ser404, overproduction of Aβ1-42, and activation of apoptotic markers, including Bax expression and caspase-3 activity (Figures 4H–4L; p < 0.05, p < 0.01 versus NCM). As expected, miR-23b-3p mimics inhibited the apoptotic rates in copper-treated APPswe cells (Figures 4F and 4G; all p < 0.01 versus NCM). Moreover, miR-23b-3p mimics increased the inhibitory phosphorylation of GSK-3β at Ser9 and inhibited the activity of GSK-3β (Figures 4H–4J; p < 0.05, p < 0.01 versus NCM), and these changes were accompanied by a reduction in tau phosphorylation, a decrease in Aβ1-42 level, and a suppression of intrinsic apoptotic pathways (Figures 4H–4L; p < 0.05, p < 0.01, p < 0.001 versus NCM). Furthermore, when GSK-3β was overexpressed in APPswe cells transfected with miR-23b-3p mimics, GSK-3β overexpression significantly reversed the inhibitory effect of miR-23b-3p on cell apoptosis, tau phosphorylation, and Aβ1-42 production, as demonstrated by the increased responses of the apoptotic rate at different stages, GSK-3β expression and activity, tau phosphorylation levels at Ser396 and Ser404, quantity of Aβ1-42, Bax expression, and caspase-3 activity (Figures 4F–4L; p < 0.05, p < 0.01, p < 0.001 versus miR-23b-3p). Together, these results provide evidence that the miR-23b-3p/GSK-3β axis plays an important role in suppressing tau phosphorylation and the intrinsic apoptosis pathway in AD.
miR-23b-3p ameliorates AD-like symptoms in APP/PS1 mice
Because our previous results suggest that miR-23b-3p was involved in AD pathology, the potential therapeutic effect of miR-23b-3p on AD was evaluated in vivo (Figure 5). To accomplish this, APP/PS1 mice, which are commonly used to study pathological characteristics of AD, such as cognitive deficits, Aβ deposition, and tau pathology,30, 31, 32 were infused with adeno-associated virus (AAV) particles containing miR-23b-3p mimics or NC oligonucleotides. Remarkably, the overexpression of miR-23b-3p re-established spatial learning performance in APP/PS1 mice during the spatial learning test. Thus, the overexpression of miR-23b-3p elicited a significant time-dependent effect on escape latency within the groups (Figure 6A; F(4,220) = 290.371, p < 0.001) and a treatment-dependent effect (F(3,55) = 72.053, p < 0.001) on the escape latency. Subsequent comparisons suggest that miR-23b-3p is an effective treatment for rescuing spatial learning deficits in APP/PS1 mice (p < 0.001 versus APP/PS1 + NC). In the probe trail, miR-23b-3p-overexpressed APP/PS1 mice took more time to search the platform quadrant (Figure 6B; both p < 0.05 versus APP/PS1 + NC) and crossed the target platform area more often at 2 and 48 h posttraining (Figure 6C; both p < 0.01 versus APP/PS1 + NC). While the speed was similar for all of the mice treated with or without AAV particles containing miR-23b-3p (Figure 6D), miR-23b-3p overexpression in APP/PS1 mice elicited a less circuitous travel path that was more precise than the path followed by the NC-treated APP/PS1 controls in the probe trail, which was more irregular (Figures 6E and 6F).
Figure 5.

Scheme for the in vivo experimental procedures in APP/PS1 mice
AAVs, adeno-associated viruses; i.c.v., intracerebroventricular injection; IHC, immunohistochemistry; MWM, Morris water maze; qRT-PCR, quantitative real-time PCR; WB, western blot.
Figure 6.

miR-23b-3p ameliorates AD-like symptoms in APP/PS1 mice
(A) Comparison of latency to find the platform during 5 training days using the MWM test. (B) miR-23b-3p increased the percentage of time that the mouse spent in the target quadrant in the probe test. (C) miR-23b-3p increased the number of crossings in which the platform was previously located in the probe test. (D) Swimming speed recorded during spatial learning test. (E and F) Representative images of the trace to find the platform during the probe test at 2 and 48 h posttraining. n = 8, APP/PS1 control group; n = 17, WT group, APP/PS1 + NC group, and APP/PS1 + miR-23b-3p group. Escape latency and swimming speed in the MWM test were analyzed using ANOVA for repeated measures, and 1-way ANOVA with Tukey’s post hoc analysis was used to analyze treatment differences. Comparisons among multiple groups were analyzed by 1-way ANOVA, followed by Tukey’s post hoc test to analyze differences between groups. Results represent means ± SEMs. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus WT; &p < 0.05, &&p < 0.01, &&&p < 0.001 versus APP/PS1 mice treated with NC.
Brain morphological characteristics revealed that the overexpression of miR-23b-3p reduced neuronal degeneration, apoptosis, Aβ overproduction, and tau pathology, as assessed by histochemical staining with Nissl, TUNEL, Aβ1-42, and AT8 (phosphorylation of tau protein at Ser202/Thr205 sites) and PHF-1 (phosphorylation of tau protein at Ser396/Ser404 sites), respectively, in the hippocampus and cortex of APP/PS1 mice. Specifically, we observed a significant increase in Nissl staining (Figures 7A and 7B, p < 0.01 and 0.05 versus APP/PS1 + NC) and a decrease in the immunoreactive intensities of TUNEL, Aβ1-42, AT8, and PHF-1 in the hippocampus and cortex of APP/PS1 mice (Figures 7C–7F and 8A–8D, p < 0.05, p < 0.01, p < 0.001 versus APP/PS1 + NC). Furthermore, immunoreactivity of Tau5 (total tau protein) was almost unchanged between different treatment groups (Figures 8E and 8F). Together, these findings provide evidence that miR-23b-3p can ameliorate AD-like symptoms in APP/PS1 mice.
Figure 7.

miR-23b-3p attenuates neurodegeneration in the hippocampus and cortex of APP/PS1 mice treated with or without miR-23b-3p
(A and B) Representative images (A) and quantitative analysis of Nissl staining (B) in the cerebral cortex and hippocampus of APP/PS1 mice following miR-23b-3p or NC treatment. (C and D) Representative images (C) and quantitative analysis of TUNEL staining (D) in the cerebral cortex and hippocampus of APP/PS1 mice following miR-23b-3p or NC treatment. (E and F) Representative images (E) and quantitative analysis of Aβ1-42 immunoreactivity (F) in the cerebral cortex and hippocampus of APP/PS1 mice following miR-23b-3p or NC treatment. Comparisons among multiple groups were analyzed by 1-way ANOVA, followed by Tukey’s post hoc test to analyze differences between groups. The results represent means ± SEMs. n = 4. Bar, 50 μm. ∗∗p < 0.01, ∗∗∗p < 0.001 versus WT; &p < 0.05, &&p < 0.01 versus APP/PS1 mice treated with NC.
Figure 8.

miR-23b-3p inhibits tau phosphorylation levels in the hippocampus and cortex of APP/PS1 mice treated with or without miR-23b-3p
(A–F) Representative images and quantitative analysis of immunoreactivity of AT8 (A and B), PHF-1 (C and D), and Tau5 (E and F), in the hippocampus and cerebral cortex of APP/PS1 mice following miR-23b-3p or NC treatment. Comparisons among multiple groups were analyzed by 1-way ANOVA, followed by Tukey’s post hoc test to analyze differences between groups. The results represent means ± SEMs. n = 4. Bar, 50 μm. ∗∗p < 0.01, ∗∗∗p < 0.001 versus WT; &p < 0.05, &&p < 0.01, &&&p < 0.001 versus APP/PS1 mice treated with NC.
miR-23b-3p modulates GSK-3β-dependent tau phosphorylation and the Bax/caspase-3 apoptotic pathway during AD pathology in vivo
To elucidate the underlying molecular mechanisms by which miR-23b-3p reduces AD-like symptoms, we first examined the miR-23b-3p-related GSK-3β/tau and Bax/caspase-3 pathways in the hippocampus and cortex of APP/PS1 and SAMP8 mice at different disease stages and compared them with those of age-matched SAMR1 and WT mice. Our results reveal that the levels of GSK-3β, Tau-Ser396, Bax, and caspase-3 were increased at different disease stages (Figures S3A, S3B, S3D, S3E, S3G, S3H, S3J and S3K; p < 0.05, p < 0.01, p < 0.001 versus WT or SAMR1). These changes mirrored the decreased expression of miR-23b-3p in the hippocampus and cortex of AD mice at 3 months, 6 months, and 9 months (Figures S3C, S3F, S3I, and S3L; p < 0.05, p < 0.01, p < 0.001 versus WT or SAMR1). Moreover, these outcomes were similar to those obtained with the AD cell line.
After confirmation of miR-23b-3p upregulation and distribution in the hippocampus and cortex of AAV-miR-23b-3p-treated APP/PS1 mice (Figure S4; p < 0.01 and 0.05 versus APP/PS1 + NC), miR-23b-3p downstream signaling was evaluated. Notably, GSK-3β expression and GSK-3β activity were both reduced, and the inhibitory GSK-3β-Ser9 to GSK-3β ratio was increased in the cortex and hippocampus of AAV-miR-23b-3p-treated APP/PS1 mice (Figures 9A–9F; p < 0.05, p < 0.001 versus APP/PS1 + NC). These results suggest that the miR-23b-3p/GSK-3β axis may play a role in AD deficits. Moreover, the observed increase in tau phosphorylation at Ser 396 and Ser 404 sites in APP/PS1 mice was significantly reduced in AAV-miR-23b-3p-treated APP/PS1 mice (Figures 9A, 9B, 9D, and 9E; p < 0.05, p < 0.01 versus APP/PS1 + NC). Likewise, levels of the pro-apoptotic marker Bax were also reduced (p < 0.05, p < 0.01 versus APP/PS1 + NC). Together, these results provide evidence that miR-23b-3p can interrupt GSK-3β-dependent tau phosphorylation and apoptosis during AD pathology in vivo.
Figure 9.

miR-23b-3p regulates GSK-3β-mediated signaling pathway in the cortex and hippocampus of APP/PS1 mice treated with or without miR-23b-3p
(A and B) Western blot images of GSK-3β, p-GSK-3β, p-Tau-Ser396, p-Tau-Ser404, and Bax (A), and bar graph showing the quantification of these proteins in the cerebral cortex of APP/PS1 mice following miR-23b-3p or NC treatment (B). (C) GSK-3β activity in the cortex of APP/PS1 mice following miR-23b-3p or NC treatment. (D and E) Western blot images of GSK-3β, p-GSK-3β, p-Tau-Ser396, p-Tau-Ser404, and Bax (D), and bar graph showing the quantification of these proteins in the hippocampus of APP/PS1 mice following miR-23b-3p or NC treatment (E). (F) GSK-3β activity in the hippocampus of APP/PS1 mice following miR-23b-3p or NC treatment. Comparisons between 2 groups were analyzed by unpaired t test. The results represent means ± SEMs. n = 4. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus APP/PS1 mice treated with NC.
Discussion
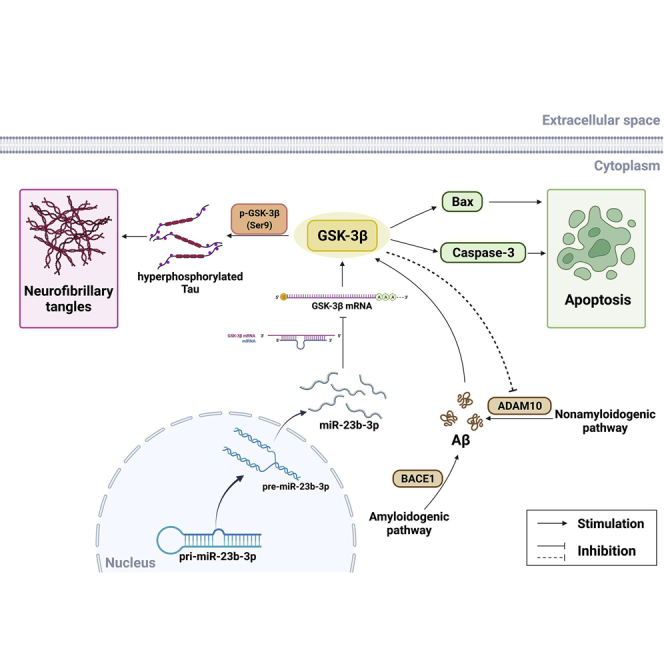
The present study reports the involvement of miR-23b-3p and its downstream signaling pathways in AD pathology, revealing miR-23b-3p as a potential therapeutic target for the treatment of AD. miRNA profiling analysis indicated that miR-23b-3p was downregulated in the APP/PS1 mouse brain, and this was subsequently confirmed in APPswe cell culture, in the SAMP8 mouse model, and in AD patients. Moreover, the upregulation of miR-23b-3p reduced neuronal apoptosis and inhibited tau hyperphosphorylation at multiple phosphorylated sites in APPswe cells subjected to Aβ toxicity by downregulating GSK-3β expression, which ultimately resulted in the suppression of the GSK-3β/tau and Bax/caspase-3 pathways in vitro. Likewise, in vivo administration of miR-23b-3p ameliorated cognitive deficits and histopathological changes and reduced tau hyperphosphorylation by downregulating GSK-3β, thereby inhibiting GSK-3β-elicited tau hyperphosphorylation and the Bax/caspase-3 apoptotic pathway (Figure 10).
Figure 10.

Potential mechanisms of miR-23b-3p inhibition of cell apoptosis and tau phosphorylation via repression of the GSK-3β signaling pathway in AD
miR-23b-3p ameliorates cognitive dysfunction in AD against tau hyperphosphorylation and neuronal apoptosis via directly targeting GSK-3β-mediated p-tau/Bax/caspase-3 pathways. In addition, miR-23b-3p reduces Aβ production, possibly via regulating the GSK-3β-associated nonamyloidogenic pathway. 3′ UTR, 3′untranslated region; Aβ, amyloid-β peptides; ADAM10, disintegrin and metalloprotease 10; BACE1, beta-site APP cleaving enzyme 1; Bax, BCL2-associated X protein; GSK-3β, glycogen synthase kinase-3β.
NFTs in the brains of AD patients are principally composed of the hyperphosphorylated microtubule-associated protein tau. These aggregates are a crucial factor in the pathogenesis of AD, causing neuronal apoptosis and contributing to the development of the disease.33 Clinical evidence reveals that abnormally phosphorylated tau develops in the early stages of AD, and cognitive dysfunction in AD patients may precede the presence of histologically identified NFTs.34 Although the precise mechanisms underlying the formation of NFTs remain elusive, the dysregulation of protein kinases and protein phosphatases is recognized as the direct cause for abnormal tau hyperphosphorylation and neuronal dysfunction in AD pathology.35
A number of phosphorylation sites on tau protein related to AD have been reported. The phospho-epitopes phosphorylated Ser202 and phosphorylated Thr205 are both pathologically relevant in AD, and their phosphorylation represents an early step from monomer to filaments.36 The region around Ser396/Ser404 is also considered a therapeutic target because of its predominant role in NFT formation, and phosphorylation at these residues is indicative of more mature hyperphosphorylation during AD progression.37,38 In addition, phosphorylation at Thr205, Ser396, and Ser404 is indicative of Alzheimer-like PHF, apoptosis occurrence, and region-specific neurodegeneration.39 In the present study, antibodies directed against AT8, PHF-1, and Tau5, which are sensitive to the phosphorylation of Thr202, Thr205, Ser396, Ser404, and total tau, were selected for immunohistochemical evaluation of the phosphorylation status of tau protein and the response of tau protein to the effect exerted by GSK-3β. As expected, hyperphosphorylation of tau at AT8 and PHF-1 was remarkably increased, and the Tau-Ser396/Tau and Tau-Ser404/Tau ratios were higher. These results are consistent with the observed enhancement in the rate of apoptosis and increased expression of apoptotic markers, including Bax and caspase-3, in cell and mouse models of AD.
More than 1,000 miRNAs have a functional role in humans, potentially regulating 30% of human genes.40 In the brains of AD patients, specific miRNAs are aberrantly expressed, suggesting that they mediate the expression of specific mRNAs that contribute to neural dysfunction.41 Although miR-15a, miR-128a, and miR-124b-3p have been identified as contributing to the phosphorylation of tau, which leads to AD pathogenesis,42, 43, 44 we focused our attention on miR-23b-3p, a member of the miR-23b family. miR-23b-3p is not only a tumor suppressor in some cancers45,46 but it is also involved in several neurologic disorders, such as intrauterine hypoxia and epilepsy.24,47 In these disorders, miR-23b-3p alleviates neuropathic pain and ameliorates the severity of seizures and abnormal electroencephalogram recordings in kainic acid-treated mice. However, the role of miR-23b-3p in AD has not yet been investigated.
In our effort to investigate the hypothetical link between miR-23b-3p and AD, miR-23b-3p expression was found to be significantly lower in AD cells and in the brains of AD mice. Furthermore, miR-23b-3p expression was found to have clinical diagnostic significance. On the one hand, miR-23b-3p was significantly underexpressed in AD plasma compared to HAVs; on the other hand, a positive correlation was detected between miR-23b-3p level and cognitive function in AD, accompanied by an excellent AUC with high sensitivity and specificity for AD. These findings suggest that miR-23b-3p could be considered a useful marker to diagnose AD. Interestingly, miR-23b-3p downregulation did not occur in an orderly time-dependent manner, as illustrated by a remarkable decrease in the precursor during the early stages of AD in APP/PS1 and SAMP8 mouse brain (at 1 or 3 months) and in APPswe cells subjected to copper (during the first 24 h). This period of time can be considered as the period in which the most relevant pathological processes of the early stage of AD were developing. These results are not in line with previous reports that cognitive deficits and pathological changes become apparent after 6 months.48 However, notwithstanding the possibility that experimental variables could account for the observed differences in the reported results, our results are in agreement with the proposal that transgenic mice and neuronal cells with mutations in APP and/or PS1 strengthen the integrated amyloid cascade hypothesis, and these genetic and molecular anomalies can be expressed differently in pre-clinical and early AD brains.49, 50, 51 More important, our results elucidating the role of miR-23b-3p in AD pathology and cognitive decline provide evidence that miR-23b-3p can exert neuroprotective effects, improve cognitive function, and prevent the onset of histopathological changes by reducing the hyperphosphorylation of tau and cell apoptosis in cell lines and in the AD mouse model. Therefore, we propose miR-23b-3p as a potential therapeutic target in AD.
The novelty of our study is represented by the discovery that miR-23b-3p is an upstream regulator of GSK-3β expression, which links our observation of miR-23b-3p downregulation during AD progression to a pathologically relevant mechanism. As an abundant miRNA in the brain, several targets of miR-23b-3p have been identified. These targets are associated with neurological disorders and include autophagy-related gene 12, leading to the activation of neuronal autophagy during the pathogenesis of traumatic brain injury;23 apoptotic protease activating factor-1, a component protein of the apoptosome allowing the recruitment and activation of caspase-9 and mediating apoptosis;24 and hairy/enhancer of split protein, acting as a basic helix-loop-helix transcriptional repressor to participate in neuronal differentiation.52
In the present study, we combined in silico prediction of miRNA targets and database analyses for annotation and visualization with integrated discovery from miRNA/RNA sequencing to predict plausible miR-23b-3p-interacting mRNA targets. After the exclusion of targets unrelated to AD, non-specific targets, including CA2, HS6ST2, and MARCKSL1, were excluded using the dual-luciferase reporter assay. Thus, the inhibitory effect of miR-23b-3p on luciferase activity could not be abolished when the miR-23b-3p binding sites on their 3′ UTRs were mutated, indicating that miR-23b-3p did not specifically bind to and decrease their expression. However, GSK-3β was identified as a novel target of miR-23b-3p using target prediction and subsequent validation in a dual-luciferase reporter assay. Moreover, the miR-23b-3p binding site in GSK-3β mRNA was strongly conserved between various animal species.
The above results suggest that miR-23b-3p may be a key target for identifying drugs for the treatment of AD that regulate the phosphorylation of tau protein. Importantly, we observed a dramatic increase in GSK-3β protein expression in the AD mouse brain and a significant negative correlation between GSK-3β and miR-23b-3p in the cortex and hippocampus, the early affected brain regions in AD. It is notable that GSK-3β regulates cell apoptosis by phosphorylating Bax and activating caspase-3 expression.53,54 It has been reported that a search for potential therapeutic candidates for AD should include interventions that attenuate tau hyperphosphorylation via the upregulation of GSK-3β phosphorylation at the inhibitory Ser9 site.55,56 However, no studies have been reported concerning the mechanistic control exerted by miR-23b-3p on GSK-3β in AD. To address this, the mechanistic control exerted by miR-23b-3p on GSK-3β in AD was elucidated.
Among the multiple kinases associated with tau pathology in AD, activation of the GSK-3β/tau hyperphosphorylation axis is crucial in controlling cognitive impairment caused by excessive stress.57 In the present study, we clearly demonstrate strong activation of GSK-3β-associated tau hyperphosphorylation at multiple sites in the cortex and hippocampus of APP/PS1 and SAMP8 mouse brain, with the upregulation of the apoptotic markers Bax and caspase-3 occurring after GSK-3β activation. The link between miR-23b-3p and GSK-3β, tau phosphorylation, and neuronal apoptosis was strongly demonstrated by the miRNA sequencing of APP/PS1 mouse brain at different stages of AD, and by upregulation and downregulation of miR-23b-3p in the in vitro AD model. Importantly, activation of the GSK-3β/tau/Bax/caspase-3 axis was inhibited by transfecting miR-23b-3p into the AD cell model. Conversely, GSK-3β upregulation inhibited the neuroprotective effects of miR-23b-3p and abolished the inhibitory effects of miR-23b-3p on the tau/Bax/caspase-3 axis. Finally, we also investigated the in vivo overexpression of miR-23b-3p in the brain of an AD mouse model to explore the proposed miR-23b-3p signaling pathway and associated therapeutic effects. Notably, miR-23b-3p overexpression inhibited tau phosphorylation and neuronal apoptosis via GSK-3β-mediated pathways, confirming that the regulatory effect on GSK-3β exerted by miR-23b-3p was beneficial to AD in the brain.
In addition to the direct effect on tau hyperphosphorylation via GSK-3β signaling, miR-23b-3p reduced Aβ levels in the AD models. Previous studies have demonstrated that GSK-3β activation is possibly involved in Aβ formation by inducing beta-site APP cleaving enzyme 1 (BACE1)-mediated APP processing in the amyloidogenic pathway and by downregulating disintegrin and metalloprotease 10 (ADAM10) in the nonamyloidogenic pathway.58,59 By acting as a feedback factor in GSK-3β activation, Aβ promotes GSK-3β activity via disruption of the Wnt pathway,60 ultimately resulting in tau hyperphosphorylation.8 Although miR-23b-3p did not significantly affect the expression of BACE1 and PS1 in copper-treated APPswe cells (Figure S5), ADAM10 was upregulated. Thus, the observed reduction in Aβ production caused by miR-23b-3p in vitro (in APPswe cells) and in vivo (in the cortex and hippocampus of APP/PS1 mice) may be related to GSK-3β-involved nonamyloidogenic pathways, supporting the hypothesis that miR-23b-3p demonstrates neuroprotective activities through the inhibition of multiple AD neuropathologies.
The present study has several limitations. First, the patient cohort size for different AD stages needs to be increased. This limitation is being addressed in our follow-up studies. Second, the biological role of miR-23b-3p should be elucidated to validate tissue samples and cerebrospinal fluid samples from AD patients. In particular, the influence of individual and environmental factors on miR-23b-3p expression should be investigated. Third, more suitable in vivo models, such as the MAPT P301S PS19 transgenic strain combined with GSK-3β siRNA silencing, should be investigated, and the relationships between miR-23b-3p expression, GSK-3β signaling, and APP processing pathways should be clarified further.
In conclusion, our in vitro and in vivo findings provide evidence that miR-23b-3p plays a protective role in AD by reducing cell apoptosis caused by the hyperphosphorylation of tau. The likely mechanism underlying miR-23b-3p-associated neuroprotection is miR-23b-3p inhibition of GSK-3β in the brain. Hence, miR-23b-3p is a potential target for new therapeutic strategies to treat AD.
Materials and methods
Animals and treatments
The transgenic mice carrying the APP gene with Swedish mutations K594 N/M595 L and PS1 with the exon-9 deletion under control of the mouse prion protein promoter (shortened to APP/PS1 mice) used in the present study are associated with early onset of AD and widely used in the study of AD.30, 31, 32,61 The SAMP8 strain is a mouse model that is frequently used in aging research because of the reduced life span and accelerated senescence of these mice.62 SAMR1 is used as a control for this mouse model because it has a similar background to that of the SAMP8 mouse strain but does not show accelerated aging. The APP/PS1 mice and age-matched WT littermates were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were divided into two parallel experiments. First, in the high-throughput next-generation sequencing experiment, APP/PS1 mice were grouped by age (1, 3, 6, and 9 months), and the same grouping was applied to the WT control mice; the cortices of 3 mice (1 male and 2 females per group) were collected and used for miRNA and mRNA sequencing analysis. Subsequently, gene expression was verified in the cortex and hippocampus of a new set of age-matched APP/PS1 and WT mice (two males and two females per group). Second, in the miR-23b-3p-infused experiment, 6-month-old APP/PS1 mice and age-matched WT mice were divided into the WT group (n = 17, 5 males and 12 females), the APP/PS1 control group (n = 8, 8 females), NC-infused APP/PS1 group (n = 17, 6 males and 11 females), and miR-23b-3p-infused APP/PS1 group (n = 17, 6 males and 11 females). SAMP8 and age-matched SAMR1 mice were provided by the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, and were divided into the following groups: 3-, 6-, and 9-month-old SAMP8 mice and age-matched SAMR1 mice controls. Each group was composed of four mice (two males and two females per group). All of the animal experiments were approved by the ethics committee of the Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences (Beijing, China), in compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Depending on the different treatments per group, brains were collected and quantitative real-time-PCR and Western blot analysis were used to evaluate the expression of genes and proteins relevant to AD during the course of the disease.
Intracerebroventricular injection and tissue preparation
Intracerebroventricular injections were performed with a stereotaxic apparatus (RWD, Nanjing, China) using the following stereotactic coordinates: AP −0.5 mm, ML 1.0 mm, and DV −3.0 mm. For miR-23b-3p upregulation experiments, AAVs (pHS-AMR vector) were used for gene delivery to the central nervous system. In brief, 6-month-old APP/PS1 mice were infused with AAV9 dissolved in phosphate-buffered solution (PBS) containing either miR-23b-3p (AAV-CAG-EGFP-miR-23b-3p) or NC oligonucleotides (AAV-CAG-EGFP-NC) (Beijing Syngentech, Beijing, China). The structural diagrams of the AAV-NC and AAV-miR-23b-3p are shown in Figure S6. APP/PS1 control mice received the same solution at equal volumes. Infusions were performed at the rate of 1.0 μL/min for a total volume of 2.0 μL.
Behavioral, histological, genetic, and molecular alterations in AAV-miR-23b-3p-injected mice were evaluated at the age of 7 months. After behavioral assessment of spatial cognition, APP/PS1 mice that were treated with or without miR-23b-3p, as well as WT mice, were randomly selected to be anesthetized using isoflurane and perfused through the heart, first with normal saline and then with 4% paraformaldehyde (PFA). For quantitative real-time-PCR and Western blot analyses, mice were sacrificed by cervical dislocation, and the cerebral cortex and hippocampus were quickly isolated and stored in liquid nitrogen for further use.
Behavioral assessment of spatial cognition
The Morris water maze (MWM) test was performed as described in our previous study.63 Briefly, during the spatial learning trial, mice were subjected to 4 training trials per day for 5 consecutive days. Both the escape latency and swimming speed were recorded. At the end of the last trial, the platform was removed for the probe trial to evaluate the spatial memory. This was performed at 2 h and 48 h posttraining. The durations that mice spent in the target quadrant and the crossings through the original location of the platform were recorded.
Histochemical analysis
The brains harvested from PFA-perfused mice were cut into 5-μm-thick sections. Neuronal degeneration, apoptosis, Aβ production, and tau pathology were detected using Nissl staining (Roche, Basel, Switzerland), TUNEL (Roche), Aβ immunohistochemistry, and tau immunohistochemistry, respectively. Routine histological protocols were used.28,63 The primary and secondary antibodies used in the histochemical analyses are listed in Table 1. The images of prefrontal cortical and hippocampal regions were acquired using a Panoramic DESK slide scanner and Caseviewer 2.3 software (3DHISTECH, Budapest, Hungary).
Table 1.
Primary and secondary antibodies used in histochemical analysis
| Primary antibody | Dilution | Source | Secondary antibody |
|---|---|---|---|
| Anti-amyloid beta1-42 rabbit pAb | 1:200 | Abcam | HRP-conjugated goat anti-rabbit IgG (H + L) (1:200, Servicebio) |
| Anti-tau (phospho S202 + T205) rabbit mAb | 1:200 | Abcam | HRP-conjugated goat anti-rabbit IgG (H + L) (1:200, Servicebio) |
| Anti-PHF-1 rabbit mAb | 1:100 | Abcam | HRP-conjugated goat anti-rabbit IgG (H + L) (1:200, Servicebio) |
| Anti-tau-5 mouse mAb | 1:200 | Millipore | HRP-conjugated goat anti-mouse IgG (H + L) (1:200, Servicebio) |
Abcam, Cambridge, MA, USA; Merck Millipore; Billerica, MA, USA; CST, Servicebio, Wuhan, China. HRP, horseradish peroxidase; IgG, immunoglobulin G; mAb, monoclonal antibody; pAb, polyclonal antibody.
Cell culture and plasmid transfection
APPswe cells were routinely cultured in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) supplemented with 2 mM l-glutamine, 10% fetal bovine serum (FBS; Gibco/Invitrogen, Grand Island, NY, USA), and 400 μM G418 (Sigma Chemical, St. Louis, MO, USA). Next, cells were divided into two groups: One group was treated with 300 μM copper, and the other was untreated (and considered to be control cells). Subsequently, the group treated with copper was divided into subgroups based on the plasmid treatment they received: miR-23b-3p mimics (chemically synthesized double strands containing a sequence consistent with the mature miR-23b-3p and a complementary sequence); miR-23b-3p inhibitor (single-chain methoxy modification); miR-23b-3p complete mutation (miR-23b-3p MUT); NCs (NCM or NCI); or NC-small interfering RNA (siRNA). These were synthesized by Genepharma (Shanghai, China) (Table 2). pCMV6-GSK-3β was purchased from Origene (Beijing, China). APPswe cells were plated into 96-well or 6-well plates, and then transfected with 100 nM miR-23b-3p mimics, inhibitor, miR-23b-3p MUT, and/or 2 μg pCMV6-GSK-3β using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. These plasmids were added at the beginning of copper-induced injury, and then cells were incubated for 24 h at 37°C. The in vitro neuroprotective effects and molecular mechanisms of miR-23b-3p action were evaluated by MTT, quantitative real-time-PCR, and Western blot analysis.
Table 2.
miRNA sequences
| miRNA name | RNA sequence |
|---|---|
| Negative control | sense: 5′-UUCUCCGAACGUG UCACGUTT-3′ antisense: 5′-ACGU GACACGUUCGGAGAATT-3′ |
| has-miR-23b-3p mimics | sense: 5′-AUCACAUUGCCAGGGAU UACCAC-3′ antisense: 5′-GGUAA UCCCUGGCAAUGUGAUUU-3′ |
| has-miR-23b-3p complete mutation | sense: 5′-AUAGGUAAGCCAGG GAUUACCAC-3′ antisense: 5′-GGUA AUCCCUGGCUUACCUAUUU-3′ |
| Inhibitor negative control | 5′-CAGUACUUUUGUGUAGUACAA-3′ |
| has-miR-23b-3p inhibitor | 5′-GUGGUAAUCCCUGGC AAUGUGAU-3′ |
Human embryonic kidney (HEK293) cells used in the construction of recombinant pGL3-GSK-3B-3′ UTR-MUT (mutant type) and pGL3-GSK-3B-3′ UTR-WT to evaluate luciferase activity were routinely cultured in DMEM supplemented with 2 mM l-glutamine and 10% FBS (Gibco/Invitrogen). Constructs containing the predicted targeting sequence (pGL3-GSK-3B-3′ UTR-WT) and the mutated targeting sequence (pGL3-GSK-3B-3′ UTR-MUT) at positions 1,001–1,008 of the GSK-3B-3′ UTR were cloned into the 3′ UTR of the reporter gene.
Cell viability assay
For the cell viability assay, APPswe cells were seeded into a 96-well plate at a density of 8,000 cells per well in 200 μL medium per well and subjected to plasmid transfection described in the section “Cell culture and plasmid transfection.” Cell survival was assessed using the MTT assay (Promega, Madison, WI, USA), according to the manufacturer’s guidelines. Absorbance was measured using a Spark 20M multimode microplate reader (Tecan Group, Mannedorf, Switzerland).
Cell apoptosis assessment
After plasmid transfection, the apoptosis of APPswe cells was measured using flow cytometry and a cellular immunofluorescence assay. In brief, cells were washed three times with ice-cold PBS and then incubated with a mixture of fluorescein isothiocyanate FITC-labeled annexin-V and PI (BD Biosciences, San Jose, CA, USA) for 30 min at 37°C in the dark. Next, cells were analyzed in a FACScan flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and the resulting data were processed using CellQuest software (BD Biosciences). The FITC-labeled annexin-V and PI-stained cells were also examined using a Spark CYTO (Tecan Group) for immunofluorescence analysis.
RNA isolation and quantitative real-time-PCR
After the treatments described above, total RNA was extracted from APPswe cells, HEK293 cells, and brain tissue (APP/PS1 mice, SAMP8 mice, and age-matched control mice) using TRIzol reagent (Invitrogen) according to the manufacturer’s guidelines. The primers used in this study are listed in Table 3. For GSK-3β mRNA expression, cDNA was synthesized from 1 μg total RNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time -PCR was performed using Fast SYBR Green Master Mix (Applied Biosystems). Relative GSK-3β gene expression was calculated using the 2−ΔΔCT method and results were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Table 3.
PCR primer sequences
| Primer name | Primer sequence |
|---|---|
| GSK3B-F | 5′-GGCAGCATGAAAGTTAGCAGA-3′ |
| GSK3B-R | 5′-GGCGACCAGTTCTCCTGAATC-3′ |
| GAPDH-F | 5′-CGGAGTCAACGGATTTGGTCGTAT-3′ |
| GAPDH-R | 5′-AGCCTTCTCCATGGTGGTGAAGAC-3′ |
| miR-23b-3p-RT | 5′-GTCGTATCCAGTGCAGGGTCCGAG GTATTCGCACTGGATACGACGTGGTA-3′ |
| miR-23b-3p-F | 5′-CGATCACATTGCCAGGGAT-3′ |
| miR-23b-3p-R | 5′-AGTGCAGGGTCCGAGGTATT-3′ |
| miR-17-5p-RT | 5′-GTCGTATCCAGTGCAGGGTCCGAGGT ATTCGCACTGGATACGACCTACCT-3′ |
| miR-17-5p-F | 5′-GCGCAAAGTGCTTACAGTGC-3′ |
| miR-17-5p-R | 5′-AGTGCAGGGTCCGAGGTATT-3′ |
| miR-106a-5p-RT | 5′-GTCGTATCCAGTGCAGGGTCCGAGGT ATTCGCACTGGATACGACCTACCT-3′ |
| miR-106a-5p-F | 5′-CGCGAAAAGTGCTTACAGTGC-3′ |
| miR-106a-5p-R | 5′-AGTGCAGGGTCCGAGGTATT-3′ |
| U6-RT | 5′-GTCGTATCCAGTGCAGGGTCCGAGG TATTCGCACTGGATACGACAAAATA-3′ |
| U6-F | 5′-CAAATTCGTGAAGCGTTCCA-3′ |
| U6-R | 5′-AGTGCAGGGTCCGAGGTATT-3′ |
| Gsk3b-F | 5′-CCAGGAGCAGGACATTTCACC-3′ |
| Gsk3b-R | 5′-CCTGACATCACACGCCAAAG-3′ |
| Actb-F | 5′-GAGATTACTGCTCTGGCTCCTA-3′ |
| Actb-R | 5′-GGACTCATCGTACTCCTGCTTG-3′ |
F, forward primer; R, reverse primer; RT, reverse transcription primer.
miR-23b-3p levels were evaluated using the miRNA First Strand cDNA Synthesis Kit and miRNA Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China) following the manufacturer’s guidelines. Briefly, reverse transcription was performed in a 20-μL reaction mix containing 1 ng total RNA and 0.2 μM miRNA stem-loop primer mix. The reaction mixtures were incubated at 25°C for 5 min, 50°C for 15 min, and 85°C for 5 min. PCR was performed in a 20-μL reaction volume containing 0.4 μL miRNA primer, 2 μL cDNA product, and 10 μL SYBR qPCR Master Mix. The cycling conditions were as follows: 5 min at 95°C, 40 cycles of 10 s at 95°C, and 30 s at 60°C. U6 small RNA was used as an endogenous control. The 2−ΔΔCT method was used for the quantitation of gene expression.
Study population, human sample collection, miRNA isolation, and quantification of circulating miR-23b-3p in the plasma
A total of 22 patients admitted to the Xuanwu Hospital of Capital Medical University between September 2015 and October 2019 were selected and enrolled in the present study. All of the patients were diagnosed with AD based on the criteria of the National Institute of Neurological and Communicative Disorders and Stroke and the AD and Related Disorders Association (NINCDS-ADRDA) for potential AD.64 Signed informed consent was obtained from each participant. Age- and gender-matched healthy control subjects were included. The design of the present study was approved by the ethics committee of Xuanwu Hospital of Capital Medical University (Beijing, China). Apolipoprotein E (APOE) genotyping of blood genomic DNA was performed through real-time-PCR using dedicated TaqMan probes (Xiamen Memorigen, Xiamen, China). The characteristics of AD patients and healthy control subjects are summarized in Table 4.
Table 4.
Clinical information of AD patients compared with HAVs
| Group | Cases (n) | Age, y | Gender | MMSE score | APOE genetype, % |
|---|---|---|---|---|---|
| HAVs | 14 | 61.07 ± 9.24 | 7 M/7 F | – | 14.29 |
| AD | 22 | 77.32 ± 8.47 | 8 M/14 F | 18.83 ± 7.25 | 27.27 |
Results represent means ± SEMs. AD, Alzheimer’s disease patients; APOE, apolipoprotein E; F, female; HAVs, healthy age-matched volunteers; M, male; MMSE, Mini-Mental Status Examination..
Peripheral blood was collected from patients and HAVs after an overnight fast. Blood samples were taken and processed within 1 h after collection in EDTA-containing tubes. These samples were centrifuged at 1,000 × g for 10 min at 4°C. Total RNA was extracted from 200 μL blood plasma using the miRNeasy Serum/Plasma Kit (Qiagen, Valencia, CA, USA), which is designed for the simultaneous isolation of small and large RNAs, according to the manufacturer’s guidelines. Plasma miR-23b-3p levels were quantified using miRNA Universal SYBR quantitative PCR Master Mix as described in the section “RNA isolation and quantitative real-time-PCR.” Relative miR-23b-3p expression was normalized according to the 2−ΔΔCT method to the geometric mean of two internal housekeeping miRNAs (has-miR-106a-5p and has-miR-17-5p), as previously reported.12
ROC curve analysis
The ROC curve was used to assess the diagnostic power of the miR-23b-3p-based signature.65 Briefly, the expression level of miR-23b-3p in the brains of APP/PS1 and WT mice and in the plasma of AD patients and HAVs were used to generate ROC curves, which were calculated from the sections “RNA isolation and quantitative real-time-PCR” and “Study population, human sample collection, miRNA isolation, and quantification of circulating miR-23b-3p in the plasma.” The ROC curve was constructed using GraphPad Prism version 8.0 (GraphPad, La Jolla, CA, USA). The ROC curve was formed by plotting sensitivity (true positive rate) on the y axis against 1–specificity (false positive rate) on the x axis for the variables that included the expression level of miR-23b-3p as the independent variable and AD patients and HAVs (APP/PS1 and WT mice) as the dependent variable. AUC and 95% confidence intervals were calculated to assess the accuracy of each parameter (sensitivity and specificity) and to find an appropriate cutoff point.
Western blot analysis
Western blot analysis was used with a standard protocol. Protein samples were obtained from APPswe cell extractions (following plasmid transfection), the cortex and hippocampus of APP/PS1 and WT mice, and SAMP8 and SAMR1 mice, as described in the section “Animals and treatments.” A total of 20 μg protein was analyzed in each assay, as described in the study by Liu et al.28 Primary antibodies used for Western blot analyses are presented in Table 5, and β-actin and GAPDH served as loading controls.
Table 5.
Primary antibodies used in western blot analysis
| Primary antibody | Dilution | Source |
|---|---|---|
| Anti-phospho-tau (Ser396) rabbit mAb | 1:1,000 | Abcam |
| Anti-phospho-tau (Ser404) rabbit mAb | 1:1,000 | Abcam |
| Anti-tau rabbit mAb | 1:1,000 | Abcam |
| Anti-GSK-3β rabbit mAb | 1:1,000 | Abcam |
| Anti-phospho-GSK-3β (Ser9) rabbit mAb | 1:1,000 | CST |
| Anti-caspase-3 rabbit mAb | 1:1,000 | Abcam |
| Anti-Bax rabbit mAb | 1:1,000 | Abcam |
| Anti-β-actin rabbit mAb | 1:2,000 | CST |
| Anti-GAPDH rabbit mAb | 1:2,000 | CST |
Abcam, Cambridge, UK; CST, Cell Signaling Technology (Danvers, MA, USA).
GSK-3β activity, caspase-3 activity, and Aβ1-42 assay
The GSK-3β Kit (Shanghai Enzyme-linked Biotechnology, Shanghai, China) and the Human Active Caspase-3 (Asp175) SimpleStep ELISA Kit (Abcam) were used to determine the activities of GSK-3β and caspase-3 in APPswe cells after plasmid transfection and in the cortex and hippocampus of APP/PS1 and WT mice treated with or without AAV-miR-23b-3p (according to the manufacturer’s guidelines). The Human Aβ1-42 ELISA Kit (ImmunoWay Biotechnology, Plano, TX, USA) was used to determine the generation of Aβ1-42 in APPswe cells after plasmid transfection (according to the manufacturer’s guidelines).
Validation of miR-23b-3p target
Validation of the miR-23b-3p target was performed using a pGL3 construct. To generate a luciferase reporter vector, the 3′ UTR of the predicted mRNAs containing the miR-23b-3p target site was amplified using the following primers: forward, 5′-CCGCTCGAGGGTGGCTCTTTGTTTGCCTG-3′; reverse, 3′-CCGACGCGTGTGGCTATTTCTGCAAGCTCA-5′. The amplicon was then cloned into pGL3-CM (Applied Biosystems, Rockford, IL, USA). Mutant vectors of GSK-3β, CA2, HS6ST2, and MARCKSL1 3′ UTR were also constructed. Relative luciferase activity was measured using a dual-Luciferase reporter assay system (Promega, Madison, WI, USA) according to the manufacturer’s guidelines. Briefly, HEK293 cells were co-transfected with either WT or mutant recombinant pGL3 plasmids of the aforementioned mRNAs together with miR-23b-3p mimic or NC using Lipofectamine 2000 (Invitrogen). These cells were also transfected with Renilla luciferase expression plasmid as a reference control. After 36 h of transfection, cells were collected and passive lysis buffer was added to detect firefly luciferase luminescence generated by the activity of the tested mRNA. Stop & Glo reagent was subsequently added to both quench firefly luciferase and measure Renilla luciferase to normalize luminescence signals. The relative activity of the tested mRNAs was reported as the quotient of the firefly/Renilla luciferase signals.
High-throughput miRNA and mRNA sequencing
miRNA and mRNA sequence profiling was performed at Sangon Biotech (Shanghai, China) using a high-throughput sequencing method.66 The sequencing data from the present study were uploaded to the Gene Expression Omnibus, available under the accession number Database: GSE194137. Briefly, total RNA was isolated from the cortices of 1-, 3-, 6-, and 9-month-old APP/PS1 mice and age-matched control mice using TRIzol reagent (Invitrogen). RNA integrity was evaluated by 1% agarose gel electrophoresis and using an Agilent Bioanalyzer 2100 with a eukaryote Total RNA Pico kit (Sangon Biotech). For small RNA library construction, 1.5 μg total RNA with an RNA integrity number (RIN) above 8.0, an OD260/280 above 2.0, an OD260/230 above 2.0, and a concentration >100 ng/μL per sample were used.
The small RNA libraries were constructed as follows. In brief, T4 RNA ligase 2 (New England Biolabs, Ipswich, MA, USA) was used to connect the 3′ adapter. This ligation reaction was performed at 22°C for 1 h. Next, 1 μL RT primer was added to the 3′ ligation product. The reaction mixture was then incubated at 75°C for 5 min, 37°C for 30 min, and 25°C for 15 min. T4 RNA ligase 1 (New England Biolabs) was used to connect the 5′ adapter. This ligation reaction was performed at 22°C for 1 h. Reverse transcription and PCR were then performed to obtain the final library product. This product was analyzed by 12% polyacrylamide gel electrophoresis (PAGE) gel electrophoresis, and PCR product bands of ∼140–150 bp were recovered. A Qubit 2.0 DNA detection kit (Life Technologies, Carlsbad, CA, USA) was used to accurately quantify the recovered DNA to facilitate sequencing. The transcriptome sequencing libraries were generated using the VAHTS mRNA-seq V2 Library Prep Kit for Illumina® (Vazyme). For miRNA sequencing, libraries were sequenced on an Illumina Hiseq X Ten platform, generating single-end reads of 75–35 bp. Clean reads were obtained using Cutadapt version 1.14 to remove the 3′ adapter (TGGAATTCTCGGGTGCCAAGGAACTC). Trimmomatic version 0.36 was used to delete low-quality reads (Q < 20).
For mRNA sequencing, libraries were sequenced using the Hiseq X Ten platform, generating paired-end reads of 150 bp. Clean reads were obtained by removing adapter (forward, AGATCGGAAGAGCACACGTCTGAAC; reverse, AGATCGGAAGAGCGTCGTGTAGGGA) sequences and Poly-Ns. The low-quality reads (Q < 20) were then deleted from the raw reads. Reads counts were analyzed using the Empirical Analysis of Digital Gene Expression Data in R (edgeR) version 3.18.1, and the frequency of miRNA counts was normalized as reads per million (RPM). Expression differences between APP/PS1 mice and WT control mice were evaluated using a Student’s t test.
Bioinformatic analysis
The miR-23b-3p targets predicted by computer-aided algorithms were obtained using the following datasets: TargetScan (http://www.targetscan.org/), for searching for conserved 8-mer, 7-mer, and 6-mer sites in human, mouse, rat, chimpanzee, rhesus, cow, dog, opossum, chicken, and frog that match each miRNA seed region; miRanda (http://www.microrna.org/microrna/home.do), for estimating the degree of sequence complementarity matching between miRNA and mRNA and the free energy of the composite structures formed; the species (and number) of corresponding miRNAs were Homo sapiens (1,100), Mus musculus (717), Rattus norvegicus (387), Drosphila melanogaster (186), and Caenorhabditis elegans (233). TarBase (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8%2Findex) was used for experimental verification, with a total of 28 species, 4,296 miRNAs, 23,426 genes, and 430,392 target gene interactions. The target genes selected in these three databases were intersected, and the common target genes were subsequently screened according to binding score and correlation with AD. Finally, we obtained the target genes of miR-23b-3p in AD.
Statistical analysis
The statistical analysis was performed using SPSS software (version 18.0, SPSS, Chicago, IL, USA). The results are presented as means ± standard errors of the mean (SEMs). Escape latency and swimming speed during the acquisition trial of the MWM test were analyzed using ANOVA for repeated measures; one-way ANOVA with Tukey’s post hoc analysis was used to analyze treatment differences. The results from the probe trial, biochemical assays, molecular assays, and in vitro studies were analyzed using a one-way ANOVA, followed by Tukey’s post hoc testing to analyze differences between groups, or an unpaired t test. Differentially expressed miRNAs in APP/PS1 mice and age-matched WT mice were identified through fold change ≥ 2.0 and p ≤ 0.05. The correlation between miR-23b-3p and GSK-3β or MMSE score was evaluated using a Pearson rank correlation coefficient. Differences with a p < 0.05 were considered statistically significant.
Acknowledgments
We appreciate the guidance of Prof. Hong You and Prof. Li Min, Department of Gastroenterology, Beijing Friendship Hospital, Capital Medical University. We also are grateful for the sample preparation by Ms. Qian Wang. This research has been given ethical approval. This study was supported by the National Natural Science Foundation of China (82073709, U1803281, and 82173806), China and the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Science (2021-1-I2M-030), China.
Author contributions
R.L. and Z.L. proposed the scientific hypothesis, designed the research, and had primary responsibility for the final content. R.L. wrote the manuscript. H.J. performed the miR-23b-3p-infused in vivo experiments and analyzed the data. J.L. and S.G. analyzed the miRNA and mRNA sequencing data and verified the level of miR-23b-3p in the in vitro model and in the APP/PS1 and SAMP8 mouse brains. J.L. performed the collection of blood of AD patients and healthy aged volunteers and detected the level of miR-23b-3p in the plasma of AD patients. S.G. performed the histochemical analyses for the in vivo experiments. L.Z. performed the western blot experiment for the miR-23b-3p/GSK-3β signaling pathways in the brains of APP/PS1 and SAMP8 mice. L.Z., Z.C., and J.Z. predicted and verified the target of miR-23b-3p. L.Z. and L.W. detected the effect of miR-23b-3p in the AD cell model.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.04.008.
Contributor Information
Zhuorong Li, Email: lizhuorong@imb.pumc.edu.cn.
Rui Liu, Email: liurui@imb.pumc.edu.cn.
Supplemental information
References
- 1.Alexiou A., Kamal M.A., Ashraf G.M. Editorial: the Alzheimer's disease challenge. Front. Neurosci. 2019;13:768. doi: 10.3389/fnins.2019.00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashraf G.M., Greig N.H., Khan T.A., Hassan I., Tabrez S., Shakil S., Sheikh I.A., Zaidi S.K., Akram M., Jabir N.R., et al. Protein misfolding and aggregation in Alzheimer's disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets. 2014;13:1280–1293. doi: 10.2174/1871527313666140917095514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mamun A.A., Uddin M.S., Mathew B., Ashraf G.M. Toxic tau: structural origins of tau aggregation in Alzheimer's disease. Neural Regen. Res. 2020;15:1417–1420. doi: 10.4103/1673-5374.274329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Churcher I. Tau therapeutic strategies for the treatment of Alzheimer's disease. Curr. Top. Med. Chem. 2006;6:579–595. doi: 10.2174/156802606776743057. [DOI] [PubMed] [Google Scholar]
- 5.Jackson G.R., Wiedau-Pazos M., Sang T.K., Wagle N., Brown C.A., Massachi S., Geschwind D.H. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 6.Ochalek A., Mihalik B., Avci H.X., Chandrasekaran A., Téglási A., Bock I., Giudice M.L., Táncos Z., Molnár K., László L., et al. Neurons derived from sporadic Alzheimer's disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res. Ther. 2017;9:90. doi: 10.1186/s13195-017-0317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pérez M., Hernández F., Gómez-Ramos A., Smith M., Perry G., Avila J. Formation of aberrant phosphotau fibrillar polymers in neural cultured cells. Eur. J. Biochem. 2002;269:1484–1489. doi: 10.1046/j.1432-1033.2002.02794.x. [DOI] [PubMed] [Google Scholar]
- 8.Hernández F., Gómez de Barreda E., Fuster-Matanzo A., Lucas J.J., Avila J. GSK3: a possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010;223:322–325. doi: 10.1016/j.expneurol.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Balaraman Y., Limaye A.R., Levey A.I., Srinivasan S. Glycogen synthase kinase 3β and Alzheimer’s disease: pathophysiological and therapeutic significance. Cell Mol. Life Sci. 2006;63:1226–1235. doi: 10.1007/s00018-005-5597-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dehghani R., Rahmani F., Rezaei N. MicroRNA in Alzheimer's disease revisited: implications for major neuropathological mechanisms. Rev. Neurosci. 2018;29:161–182. doi: 10.1515/revneuro-2017-0042. [DOI] [PubMed] [Google Scholar]
- 11.Ansari A., Maffioletti E., Milanesi E., Marizzoni M., Frisoni G.B., Blin O., Richardson J.C., Bordet R., Forloni G., Gennarelli M., Bocchio-Chiavetto L. miR-146a and miR-181a are involved in the progression of mild cognitive impairment to Alzheimer's disease. Neurobiol. Aging. 2019;82:102–109. doi: 10.1016/j.neurobiolaging.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Cosín-Tomás M., Antonell A., Lladó A., Alcolea D., Fortea J., Ezquerra M., Lleó A., Martí M.J., Pallàs M., Sanchez-Valle R., et al. Plasma miR-34a-5p and miR-545-3p as early biomarkers of Alzheimer's disease: potential and limitations. Mol. Neurobiol. 2017;54:5550–5562. doi: 10.1007/s12035-016-0088-8. [DOI] [PubMed] [Google Scholar]
- 13.Gasiorowski K., Brokos B., Leszek J., Tarasov V.V., Ashraf G.M., Aliev G. Insulin resistance in alzheimer disease: p53 and MicroRNAs as important players. Curr. Top. Med. Chem. 2017;17:1429–1437. doi: 10.2174/1568026617666170103161233. [DOI] [PubMed] [Google Scholar]
- 14.Lugli G., Cohen A.M., Bennett D.A., Shah R.C., Fields C.J., Hernandez A.G., Smalheiser N.R. Plasma exosomal miRNAs in persons with and without alzheimer disease: altered expression and prospects for biomarkers. PLoS One. 2015;10:e0139233. doi: 10.1371/journal.pone.0139233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L.L., Min L., Guo Q.D., Zhang J.X., Jiang H.L., Shao S., Xing J.G., Yin L.L., Liu J.H., Liu R., Guo S.L. Profiling microRNA from brain by microarray in a transgenic mouse model of Alzheimer's disease. Biomed. Res. Int. 2017;2017:8030369. doi: 10.1155/2017/8030369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ali A., Sheikh I.A., Mirza Z., Gan S.H., Kamal M.A., Abuzenadah A.M., Damanhouri G.A., Ashraf G.M. Application of proteomic tools in modern nanotechnological approaches towards effective management of neurodegenerative disorders. Curr. Drug Metab. 2015;16:376–388. doi: 10.2174/1389200216666141208153303. [DOI] [PubMed] [Google Scholar]
- 17.Bekris L.M., Leverenz J.B. The biomarker and therapeutic potential of miRNA in Alzheimer's disease. Neurodegener. Dis. Manag. 2015;5:61–74. doi: 10.2217/nmt.14.52. [DOI] [PubMed] [Google Scholar]
- 18.Wang L., Liu J., Wang Q., Jiang H., Zeng L., Li Z., Liu R. MicroRNA-200a-3p mediates neuroprotection in Alzheimer-related deficits and attenuates amyloid-beta overproduction and tau hyperphosphorylation via coregulating BACE1 and PRKACB. Front. Pharmacol. 2019;10:806. doi: 10.3389/fphar.2019.00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao J., Yue D., Zhou Y., Jia L., Wang H., Guo M., Xu H., Chen C., Zhang J., Xu L. The role of MicroRNAs in Aβ deposition and tau phosphorylation in Alzheimer's diseases. Front. Neurol. 2017;8:342. doi: 10.3389/fneur.2017.00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barbollat-Boutrand L., Joly-Tonetti N., Dos Santos M., Metral E., Boher A., Masse I., Berthier-Vergnes O., Bertolino P., Damour O., Lamartine J. MicroRNA-23b-3p regulates human keratinocyte differentiation through repression of TGIF1 and activation of the TGF-ß-SMAD2 signalling pathway. Exp. Dermatol. 2017;26:51–57. doi: 10.1111/exd.13119. [DOI] [PubMed] [Google Scholar]
- 21.Mu W., Wang X., Zhang X., Zhu S., Sun D., Ka W., Sung L.A., Yao W. Fluid shear stress upregulates E-Tmod41 via miR-23b-3p and contributes to F-actin cytoskeleton remodeling during erythropoiesis. PLoS One. 2015;10:e0136607. doi: 10.1371/journal.pone.0136607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao S., Li T., Li J., Lu Q., Han C., Wang N., Qiu Q., Cao H., Xu X., Chen H., Zheng Z. miR-23b-3p induces the cellular metabolic memory of high glucose in diabetic retinopathy through a SIRT1-dependent signalling pathway. Diabetologia. 2016;59:644–654. doi: 10.1007/s00125-015-3832-0. [DOI] [PubMed] [Google Scholar]
- 23.Sun L., Liu A., Zhang J., Ji W., Li Y., Yang X., Wu Z., Guo J. miR-23b improves cognitive impairments in traumatic brain injury by targeting ATG12-mediated neuronal autophagy. Behav. Brain Res. 2018;340:126–136. doi: 10.1016/j.bbr.2016.09.020. [DOI] [PubMed] [Google Scholar]
- 24.Chen Q., Xu J., Li L., Li H., Mao S., Zhang F., Zen K., Zhang C.Y., Zhang Q. MicroRNA-23a/b and microRNA-27a/b suppress Apaf-1 protein and alleviate hypoxia-induced neuronal apoptosis. Cell Death Dis. 2014;5:e1132. doi: 10.1038/cddis.2014.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu R., Li Z., Jiang H., Zeng L., Zhang J. miRNA Marker for Diagnosis and Treatment of Alzheimer’s Disease. Australia Patent. 2021;2020103707 http://pericles.ipaustralia.gov.au/ols/auspat/applicationDetails.do?applicationNo=2020103707 [Google Scholar]
- 26.2021. https://patentscope2.wipo.int/search/zh/detail.jsf?docId=WO2021088317&_cid=JP2-L14J1V-72944-1
- 27.Wang L., Zeng L., Jiang H., Li Z., Liu R. Microarray profile of long noncoding RNA and messenger RNA expression in a model of Alzheimer’s disease. Life (Basel) 2020;10:64. doi: 10.3390/life10050064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Q.S., Jiang H.L., Wang Y., Wang L.L., Zhang J.X., He C.H., Shao S., Zhang T.T., Xing J.G., Liu R. Total flavonoid extract from Dracoephalum moldavica L. attenuates beta-amyloid-induced toxicity through anti-amyloidogenesic and neurotrophic pathways. Life Sci. 2018;193:214–225. doi: 10.1016/j.lfs.2017.10.041. [DOI] [PubMed] [Google Scholar]
- 29.Liu R., Wu C.X., Zhou D., Yang F., Tian S., Zhang L., Zhang T.T., Du G.H. Pinocembrin protects against beta-amyloid-induced toxicity in neurons through inhibiting receptor for advanced glycation end products (RAGE)-independent signaling pathways and regulating mitochondrion-mediated apoptosis. BMC Med. 2012;10:105. doi: 10.1186/1741-7015-10-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crouch P.J., Hung L.W., Adlard P.A., Cortes M., Lal V., Filiz G., Perez K.A., Nurjono M., Caragounis A., Du T., et al. Increasing Cu bioavailability inhibits Aβ oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. U S A. 2009;106:381–386. doi: 10.1073/pnas.0809057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giraldo E., Lloret A., Fuchsberger T., Viña J. Aβ and tau toxicities in Alzheimer's are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. 2014;2:873–877. doi: 10.1016/j.redox.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu M., Zhou H., Liu Y., Sun J., Xie W., Zhao P., Liu J. Ultrasound-excited protoporphyrin IX-modified multifunctional nanoparticles as a strong inhibitor of tau phosphorylation and β-amyloid aggregation. ACS Appl. Mater. Interfaces. 2018;10:32965–32980. doi: 10.1021/acsami.8b08230. [DOI] [PubMed] [Google Scholar]
- 33.Hasegawa M. Structure of NFT: biochemical approach. Adv. Exp. Med. Biol. 2019;1184:23–34. doi: 10.1007/978-981-32-9358-8_2. [DOI] [PubMed] [Google Scholar]
- 34.Hogervorst E., Bandelow S., Combrinck M., Irani S.R., Smith A.D. The validity and reliability of 6 sets of clinical criteria to classify Alzheimer's disease and vascular dementia in cases confirmed post-mortem: added value of a decision tree approach. Dement. Geriatr. Cogn. Disord. 2003;16:170–180. doi: 10.1159/000071006. [DOI] [PubMed] [Google Scholar]
- 35.Martin L., Latypova X., Wilson C.M., Magnaudeix A., Perrin M.L., Terro F. Tau protein phosphatases in Alzheimer's disease: the leading role of PP2A. Ageing Res. Rev. 2013;12:39–49. doi: 10.1016/j.arr.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 36.Rankin C.A., Sun Q., Gamblin T.C. Pseudo-phosphorylation of tau at Ser202 and Thr205 affects tau filament formation. Brain Res. Mol. Brain Res. 2005;138:84–93. doi: 10.1016/j.molbrainres.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 37.Greenberg S.G., Davies P., Schein J.D., Binder L.I. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J. Biol. Chem. 1992;267:564–569. doi: 10.1016/s0021-9258(18)48531-6. [DOI] [PubMed] [Google Scholar]
- 38.Mondragon-Rodriguez S., Perry G., Luna-Munoz J., Acevedo-Aquino M.C., Williams S. Phosphorylation of tau protein at sites Ser (396-404) is one of the earliest events in Alzheimer's disease and Down syndrome. Neuropathol. Appl. Neurobiol. 2014;40:121–135. doi: 10.1111/nan.12084. [DOI] [PubMed] [Google Scholar]
- 39.Lovestone S., Reynolds C.H. The phosphorylation of tau: a critical stage in neurodevelopment and neurodegenerative processes. Neuroscience. 1997;78:309–324. doi: 10.1016/s0306-4522(96)00577-5. [DOI] [PubMed] [Google Scholar]
- 40.Pillai R.S., Bhattacharyya S.N., Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17:118–126. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Lukiw W.J. Micro-RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/wnr.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- 42.Carrettiero D.C., Hernandez I., Neveu P., Papagiannakopoulos T., Kosik K.S. The cochaperone BAG2 sweeps paired helical filament- insoluble tau from the microtubule. J. Neurosci. 2009;29:2151–2161. doi: 10.1523/jneurosci.4660-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hebert S.S., Papadopoulou A.S., Smith P., Galas M.C., Planel E., Silahtaroglu A.N., Sergeant N., Buee L., De Strooper B. Genetic ablation of dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 2010;19:3959–3969. doi: 10.1093/hmg/ddq311. [DOI] [PubMed] [Google Scholar]
- 44.Kang Q., Xiang Y., Li D., Liang J., Zhang X., Zhou F., Qiao M., Nie Y., He Y., Cheng J., Dai Y. MiR-124-3p attenuates hyperphosphorylation of tau protein-induced apoptosis via caveolin-1-PI3K/Akt/GSK3β pathway in N2a/APP695swe cells. Oncotarget. 2017;8:24314–24326. doi: 10.18632/oncotarget.15149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kou C.H., Zhou T., Han X.L., Zhuang H.J., Qian H.X. Downregulation of mir-23b in plasma is associated with poor prognosis in patients with colorectal cancer. Oncol. Lett. 2016;12:4838–4844. doi: 10.3892/ol.2016.5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang W., Li Y., Liu N., Gao Y., Li L. MiR-23b controls ALDH1A1 expression in cervical cancer stem cells. BMC Cancer. 2017;17:292. doi: 10.1186/s12885-017-3192-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhan L., Yao Y., Fu H., Li Z., Wang F., Zhang X., He W., Zheng W., Zhang Y., Zheng H. Protective role of miR-23b-3p in kainic acid-induced seizure. Neuroreport. 2016;27:764–768. doi: 10.1097/wnr.0000000000000610. [DOI] [PubMed] [Google Scholar]
- 48.D'Amelio M., Cavallucci V., Middei S., Marchetti C., Pacioni S., Ferri A., Diamantini A., De Zio D., Carrara P., Battistini L., et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat. Neurosci. 2011;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- 49.Goate A., Chartier-Harlin M.C., Mullan M., Brown J., Crawford F., Fidani L., Giuffra L., Haynes A., Irving N., James L., et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 50.Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 51.Murrell J., Farlow M., Ghetti B., Benson M.D. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- 52.Kimura H., Kawasaki H., Taira K. Mouse microRNA-23b regulates expression of Hes1 gene in P19 cells. Nucleic Acids Symp. Ser. (Oxf). 2004;48:213–214. doi: 10.1093/nass/48.1.213. [DOI] [PubMed] [Google Scholar]
- 53.Takahashi-Yanaga F. Activator or inhibitor? GSK-3 as a new drug target. Biochem. Pharmacol. 2013;86:191–199. doi: 10.1016/j.bcp.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 54.Zhang X., Jiang D., Jiang W., Zhao M., Gan J. Role of TLR4-mediated PI3K/AKT/GSK-3βSignaling pathway in apoptosis of rat hepatocytes. Biomed. Res. Int. 2015;2015:631326. doi: 10.1155/2015/631326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khan I., Tantray M.A., Alam M.S., Hamid H. Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur. J. Med. Chem. 2017;125:464–477. doi: 10.1016/j.ejmech.2016.09.058. [DOI] [PubMed] [Google Scholar]
- 56.O'Leary O., Nolan Y. Glycogen synthase kinase-3 as a therapeutic target for cognitive dysfunction in neuropsychiatric disorders. CNS Drugs. 2015;29:1–15. doi: 10.1007/s40263-014-0213-z. [DOI] [PubMed] [Google Scholar]
- 57.Jope R.S., Cheng Y., Lowell J.A., Worthen R.J., Sitbon Y.H., Beurel E. Stressed and inflamed, can GSK3 Be blamed? Trends Biochem. Sci. 2017;42:180–192. doi: 10.1016/j.tibs.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ly P.T., Wu Y., Zou H., Wang R., Zhou W., Kinoshita A., Zhang M., Yang Y., Cai F., Woodgett J., Song W. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 2013;123:224–235. doi: 10.1172/jci64516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chinchalongporn V., Shukla M., Govitrapong P. Melatonin ameliorates Aβ42-induced alteration of βAPP-processing secretases via the melatonin receptor through the Pin1/GSK3β/NF-κB pathway in SH-SY5Y cells. J. Pineal Res. 2018;64:e12470. doi: 10.1111/jpi.12470. [DOI] [PubMed] [Google Scholar]
- 60.Magdesian M.H., Carvalho M.M.V., Mendes F.A., Saraiva L.M., Juliano M.A., Juliano L., Garcia-Abreu J., Ferreira S.T. Amyloid-β binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/β-catenin signaling. J. Biol. Chem. 2008;283:9359–9368. doi: 10.1074/jbc.m707108200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ding Y., Qiao A., Wang Z., Goodwin J.S., Lee E.S., Block M.L., Allsbrook M., McDonald M.P., Fan G.H. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer's disease transgenic mouse model. J. Neurosci. 2008;28:11622–11634. doi: 10.1523/jneurosci.3153-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akiguchi I., Pallàs M., Budka H., Akiyama H., Ueno M., Han J., Yagi H., Nishikawa T., Chiba Y., Sugiyama H., et al. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: toshio Takeda's legacy and future directions. Neuropathology. 2017;37:293–305. doi: 10.1111/neup.12373. [DOI] [PubMed] [Google Scholar]
- 63.Wang L., Fang J., Jiang H., Wang Q., Xue S., Li Z., Liu R. 7-pyrrolidinethoxy-4'-methoxyisoflavone prevents amyloid β–induced injury by regulating histamine H3 receptor-mediated cAMP/CREB and AKT/GSK3β pathways. Front. Neurosci. 2019;13:334. doi: 10.3389/fnins.2019.00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haroutunian V., Davies P., Vianna C., Buxbaum J.D., Purohit D.P. Tau protein abnormalities associated with the progression of Alzheimer disease type dementia. Neurobiol. Aging. 2007;28:1–7. doi: 10.1016/j.neurobiolaging.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 65.Ha T.Y. MicroRNAs in human diseases: from autoimmune diseases to skin, psychiatric and neurodegenerative diseases. Immune Netw. 2011;11:227–244. doi: 10.4110/in.2011.11.5.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zeng L., Jiang H.L., Ashraf G.M., Li Z.R., Liu R. MicroRNA and mRNA profiling of cerebral cortex in a transgenic mouse model of Alzheimer's disease by RNA sequencing. Neural Regen. Res. 2021;16:2099–2108. doi: 10.4103/1673-5374.308104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.