

SUMMARY
The actomyosin cortex regulates the localization and function of proteins at the plasma membrane. Here, we study how membrane binding, cortical movements, and diffusion determine membrane protein distribution. In Caenorhabditis elegans zygotes, actomyosin flows transport PAR polarity proteins to establish the anterior-posterior axis. Oligomerization of a key scaffold protein, PAR-3, is required for polarization. PAR-3 oligomers are a heterogeneous population of many different sizes, and it remains unclear how oligomer size affects PAR-3 segregation. To address this question, we engineered PAR-3 to defined sizes. We report that PAR-3 trimers are necessary and sufficient for PAR-3 function during polarization and later embryo development. Quantitative analysis of PAR-3 diffusion shows that a threshold size of three subunits allows PAR-3 clusters to stably bind the membrane, where they are corralled and transported by the actomyosin cortex. Our study provides a quantitative model for size-dependent protein transportation of peripheral membrane proteins by cortical flow.
Graphical abstract
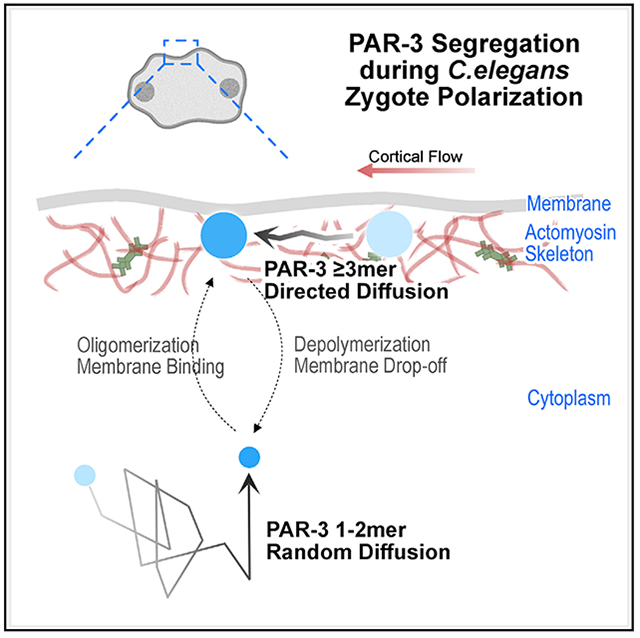
In brief
The actomyosin cytoskeleton is a major regulator of cellular organization. Chang and Dickinson develop protein-engineering and particle-tracking tools to study how clustered membrane-bound proteins are transported by actomyosin contractions in vivo. Data-driven modeling reveals how membrane binding, diffusion, and collisions with F-actin contribute to protein movement.
INTRODUCTION
Subcellular components must be properly localized for normal cellular function. Cells have evolved mechanisms to position objects as large as organelles and as small as single mRNA and protein molecules. The actomyosin cytoskeleton is actively involved in many of these mechanisms. For instance, myosin motor proteins transport large cargos, such as vesicles, by “walking” on actin filaments (Mehta, 2001), and chromosome congression in starfish oocytes is accomplished by chromosome trapping within a contracting filamentous actin meshwork (Lénárt et al., 2005). Bulk cytoplasmic and cortical flows, triggered by non-muscle myosin dependent cytoskeletal contraction, are responsible for the movement of subcellular components in a wide range of organisms, from chloroplast circulation in Elodea leaf cells (Allen and Allen, 1978) to PAR polarity protein segregation in Caenorhabditis elegans embryos (Lang and Munro, 2017). However, it has remained unclear how cortical flows can overcome the random motion caused by Brownian diffusion in order to transport macromolecular complexes over large distances.
The C. elegans zygote has been widely used to study protein segregation by cortical actomyosin flows due to its relatively large size, optical transparency, and the genetic tools that are available. During the first cell cycle of C. elegans embryonic development, distinct groups of PAR proteins are segregated to anterior and posterior poles of the cell cortex, leading to asymmetric cell division, which is essential for subsequent development. The anterior PAR complex (aPAR) consists of the oligomeric scaffold protein PAR-3, along with atypical protein kinase C (aPKC/PKC-3) and its cofactor PAR-6. These proteins are distributed uniformly throughout the entire embryo cortex before polarization starts (Cuenca et al., 2003; Lang and Munro, 2017). Just after fertilization, a gradient in actomyosin contractility along the anterior-posterior axis is introduced, leading to anterior directed cortical flows (Mayer et al., 2010; Munro et al., 2004). aPAR complexes are carried toward the anterior pole by this cortical flow and become enriched at the anterior cell cortex. Once localized, aPKC phosphorylates substrates essential for polarized cell behavior (Hong, 2018; Lang and Munro, 2017).
Multiple studies using different methods have shown that cortical flow is responsible for aPAR segregation in C. elegans zygotes. First, eliminating cortical flow with myosin light chain (MLC-4) RNAi results in PAR polarity defect (Munro et al., 2004; Shelton et al., 1999). Second, modeling approaches have demonstrated that advective flow can be sufficient to explain PAR protein partitioning in silico (Goehring et al., 2011a; Gross et al., 2019). Finally, direct induction of cytoplasmic flow by localized laser-induced heating was found to be sufficient to displace the aPAR cortical domain in vivo (Mittasch et al., 2018).
However, these studies have not addressed how cortical flows physically transport PAR proteins. The cell cortex consists of actin and myosin filaments as well as surrounding protein and water molecules, but models generally treat the entire cortex as a film of active fluid (Goehring et al., 2011a; Gross et al., 2019; Mayer et al., 2010). Therefore, it remains unclear whether cortical PAR proteins are pushed along the membrane by physical association with actin filaments or, instead, transported through viscous forces generated by cytoplasmic flow.
Clustering of aPAR, which occurs due to PAR-3 oligomerization, is critical for aPAR localization across a range of organisms and cell types (Benton and Johnston, 2003; Dickinson et al., 2017; Mizuno et al., 2003; Rodriguez et al., 2017; Sailer et al., 2015; Wang et al., 2017b). Using CRISPR-induced targeted mutagenesis and live imaging of C. elegans embryos, we previously showed that larger PAR-3 clusters move in a more directed manner due to cortical flow, while cells expressing a monomeric PAR-3 mutant are unable to effectively segregate aPAR (Dickinson et al., 2017). These results hinted at a model in which a large size enables PAR-3 clusters to be transported by cortical flow for proper polarity establishment. However, since PAR-3 oligomers have a wide range of sizes in vivo (Dickinson et al., 2017; Lang and Munro, 2017; Wang et al., 2017b), it remains unclear how oligomer size contributes to aPAR segregation.
Here, we address this question by performing a quantitative analysis of oligomerization-dependent PAR-3 segregation by actomyosin cortical flow. By engineering PAR-3 variants that form oligomers of defined sizes, we reveal that PAR-3 clusters physically collide with actomyosin cortex and segregate to the anterior cortical domain as a result of viscous friction and collisions with actin rather than a direct, stable association with actomyosin. Avidity-driven stable binding to the plasma membrane also plays a major role in successful transport of larger PAR-3 oligomers. These results provide fundamental insights into the mechanisms of polarization and of protein transport by cortical flows.
RESULTS
Engineered PAR-3 trimers are sufficient for aPAR segregation and normal development
Wild-type PAR-3 exists in vivo as a heterogeneous population of oligomers of different sizes, ranging from monomers to >15-mers (Dickinson et al., 2017). In previous work, we disrupted the PAR-3 oligomerization domain by introducing charge-reversal mutations at three positions in the endogenous par-3 gene using CRISPR. The resulting PAR-3(RRKEEE) monomeric mutant protein (here referred to as PAR-3*) does not localize stably to the cell membrane or segregate to the anterior domain (Dickinson et al., 2017; see Figures 1C and S1E). In the same study, we showed that the brightest 25% of PAR-3 clusters moved in tandem with the actomyosin cortex, while the dimmest 25% moved shorter distances in an apparently diffusive manner. Therefore, large PAR-3 clusters were presumed to play an important role in polarity establishment. However, the exact relationship between PAR-3 cluster size and segregation remained unclear. To dissect the behavior of the heterogeneous PAR-3 population and to determine how PAR-3 clusters are transported by cortical flow, we engineered PAR-3 oligomers of defined sizes.
Figure 1. Engineered PAR-3 trimers are sufficient for aPAR segregation and normal development.
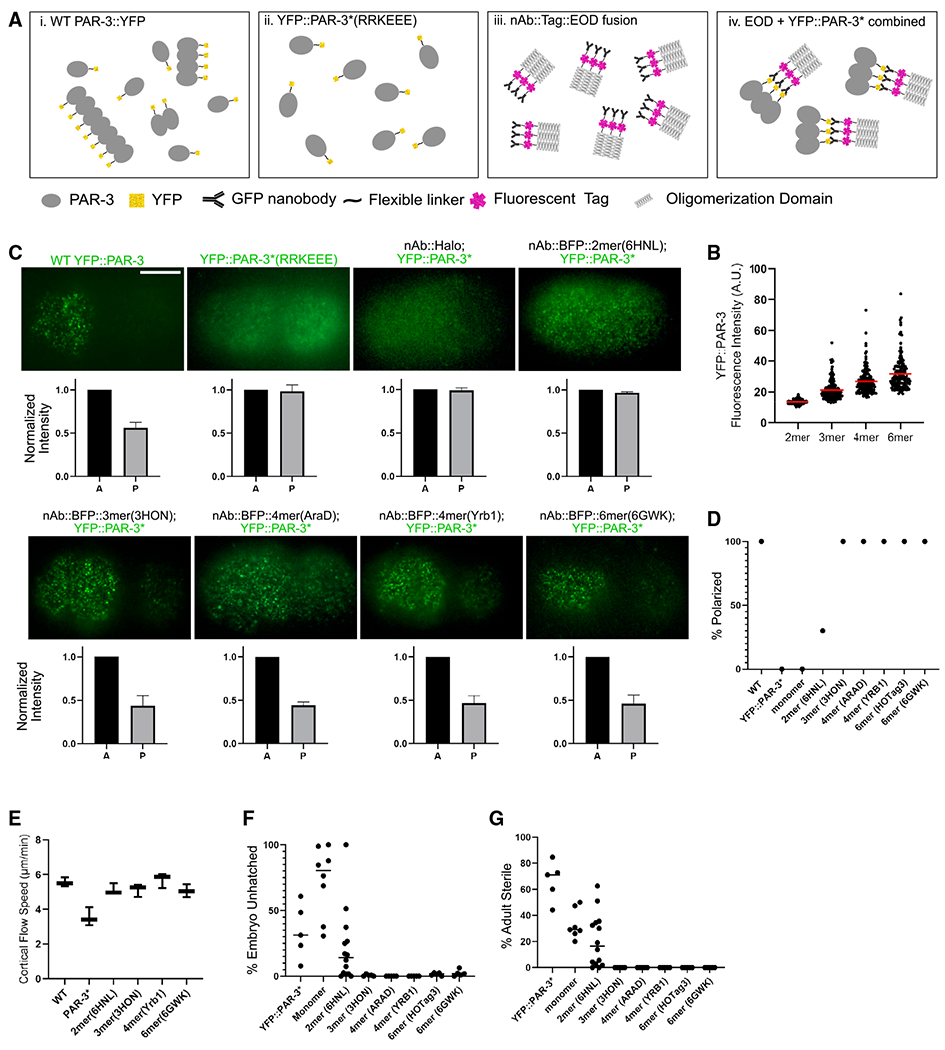
(A) Illustration of our strategy to engineer PAR-3 to specific sizes. (i) During polarity establishment, PAR-3 clusters exist as a heterogeneous population of various sizes. (ii) PAR-3* monomers in which the oligomerization domain is disrupted are shown. (iii) Transgenic nAb::EOD constructs contain the EOD (trimer is shown as an example), a fluorescent label, and a GFP-binding nanobody. (iv) PAR-3 of controlled oligomer size was generated by combining YFP::PAR-3*(RRKEEE) and EOD constructs.
(B) YFP fluorescence intensity cortical YFP::PAR-3* in EOD-expressing embryos dissected from mlc-4 RNAi-treated worms. mlc-4 RNAi was used for these measurements to prevent crowding of PAR-3 clusters into the anterior domain, which makes accurately measuring intensity more difficult. Data show only clusters that are stabilized on the cortex for more than 10 frames (0.5 s), because these are the particle tracks we used in the quantitative analysis below. We obtained equivalent results with or without the track-length filter (Figure S1F). Red bars indicate means.
(C) Upper panels: live images of cortical YFP::PAR-3* in the indicated EOD strains. Scale bar: 10 μm. Lower panels: quantification of fluorescence intensity in anterior and posterior is shown. n = 3 embryos of each strain.
(D) Percentage of embryos from each EOD strain that displays effective PAR-3 polarization. n = 10 embryos of each strain.
(E) Cortical flow speed measured from DIC videos of live embryos dissected from wild-type, YFP::PAR-3*(RRKEEE), and YFP::PAR-3*; nAB::BFP::EOD strains. n = 3 embryos from each strain. Horizontal bars indicate means.
(F) Embryo lethality of each EOD strain. “Monomer” is a control construct comprising the nanobody and fluorescent tag but no EOD. Horizontal bars indicate means.
(G) Adult sterility of each EOD strain. Monomer is a control construct comprising the nanobody and fluorescent tag but no EOD. Horizontal bars indicate means.
We first identified several protein domains that have been reported to form oligomers of defined sizes in vitro (Figures S1A and S1B; Bolten et al., 2016; Boudko et al., 2009; Büttner et al., 2012; Chik et al., 2019; Drulyte et al., 2019; Huang et al., 2014; Lee et al., 1968; Li et al., 2019; Luo et al., 2001; Parsons et al., 2002; Santiago-Frangos et al., 2019; Sun et al., 2014; Thomson et al., 2014; Veesler et al., 2010). Each oligomerization domain was tagged with a fluorescent protein and expressed in C. elegans, and the size of oligomeric protein complexes from transgenic zygotes was examined using single-cell, single-molecule pull-down (sc-SiMPull) followed by photobleaching step counting (Dickinson et al., 2017; Figure S1B). We identified dimer, trimer, tetramer, and hexamer protein domains that formed oligomers of the expected sizes in vivo.
To generate PAR-3 oligomers of defined sizes, we adopted a strategy in which PAR-3 is linked to one of these “extra oligomerization domains” (EODs) via a nanobody. We generated transgenic constructs comprising a nanobody that binds to GFP/YFP (Wang et al., 2017a), a fluorescent BFP or HaloTag, and an EOD (Figure 1Aiii). We verified that the nanobody successfully bound to GFP/YFP in C. elegans lysates in vitro (Figure S1D) and in live embryos in vivo (see below). To generate PAR-3 oligomers of defined sizes, we crossed each nanobody (nAb)::EOD transgene into a strain carrying an endogenous YFP-tagged PAR-3 monomer allele (YFP::PAR-3*, which has the same RRKEEE charge-reversal mutations identified previously; Dickinson et al., 2017; Figure 1A). In the resulting strains, YFP::PAR-3 formed clusters on the plasma membrane, which colocalized with the expressed nAb::EOD constructs (Figure S1E). We measured the YFP intensity of these induced foci on the cortex and confirmed that they exhibited the expected trend of increasing sizes (Figure 1B).
Strikingly, our trimer, tetramer, and hexamer nAb::EOD constructs fully rescued the polarity defects in the parental YFP::PAR-3* monomeric strain. One trimer nAb::EOD construct, two different tetramer constructs, and two different hexamer constructs all induced YFP::PAR-3* to form cortical clusters that segregated to the anterior (Figures 1C and 1D; Video S1). A negative control strain, expressing the YFP nAb without any EOD, closely resembled the YFP::PAR-3* mutant: PAR-3* did not bind stably to the membrane or segregate to the anterior (Figure 1C; Video S1). The engineered dimer (6HNL) PAR-3 exhibited an intermediate phenotype: PAR-3 dimers localized to the membrane but with shorter membrane binding lifetime (Video S1; see below for quantification). A minority of dimer embryos (4/14) showed some ability to polarize, but most embryos (10/14) were not polarized effectively (Figures 1D and S1G). These data indicate that a threshold size of three PAR-3 molecules per cluster is necessary and sufficient for robust polarity establishment in the C. elegans zygote.
Previous studies showed that cortical flow speed was significantly reduced in PAR-3 monomeric mutant strains, although this reduction in cortical flow was not severe enough to account for loss of polarity in these mutants (Rodriguez et al., 2017). To ensure that the differences in EOD strain polarization were not due to differences in cortical flow, we acquired differential interference contrast (DIC) videos of live EOD embryos and measured the cortical flow speed. We confirmed the slower cortical flow seen earlier in PAR-3 monomeric mutants (Rodriguez et al., 2017), but all of the EOD strains, including dimer, showed cortical flow speeds similar to wild type (Figure 1E). We conclude that the difference in polarization between dimer and larger EODs is due to factors other than cortical flow speed.
Consistent with their polarity defects, monomeric PAR-3* mutants exhibit partially penetrant embryonic lethality and adult sterility (Dickinson et al., 2017). Remarkably, these phenotypes were almost fully rescued by the trimer, tetramer, or hexamer EOD::nAb constructs and partially rescued by the dimer construct (Figures 1F and 1G). We did observe some mild phenotypes in two engineered strains: the PAR-3*; hexamer(HO-Tag3) strain had slow larval growth and maturation, and a few embryos (<10%) of the PAR-3*; tetramer(AraD) strain exhibited cytokinesis defects. These phenotypes were not present in other stains and have no obvious relationship to cell polarity in the zygote, so we presume that they are artifacts of expressing these specific EODs. We excluded these two strains from the analysis that follows. In addition, while we did not observe plate-level phenotypes in the tetramer(Yrb1) strain, we noticed that embryos dissected from this strain were more delicate; they more often died or took a longer time to polarize during fluorescence imaging. Despite these caveats of specific engineered EOD strains, the recovery of PAR-3 localization, zygote polarity, embryonic lethality, and adult sterility indicate that oligomers as small as trimers are sufficient for PAR-3 function in C. elegans.
PAR-3 trimers and larger oligomers undergo directed motion due to cortical flow, while PAR-3 dimers do not
We were intrigued by the observation that engineered trimers, but not dimers, of PAR-3 were sufficient for cell polarization. To attempt to explain why cortical flow is effective on trimers, but not on dimers, and to better define the movement of PAR-3 on the cortex, we analyzed the diffusive behavior of individual engineered PAR-3 clusters. We imaged one-cell embryos of each EOD strain during polarity establishment using total internal reflection fluorescence (TIRF) microscopy at a high frame rate (20 frames/s) to visualize the diffusion of clusters on the cortex and then computationally segmented and tracked individual particles (Figure 2A). To characterize and compare the motion of different-sized clusters, we performed a mean squared displacement (MSD) analysis.
Figure 2. PAR-3 trimers and larger oligomers undergo directed motion due to cortical flow, while PAR-3 dimers do not.
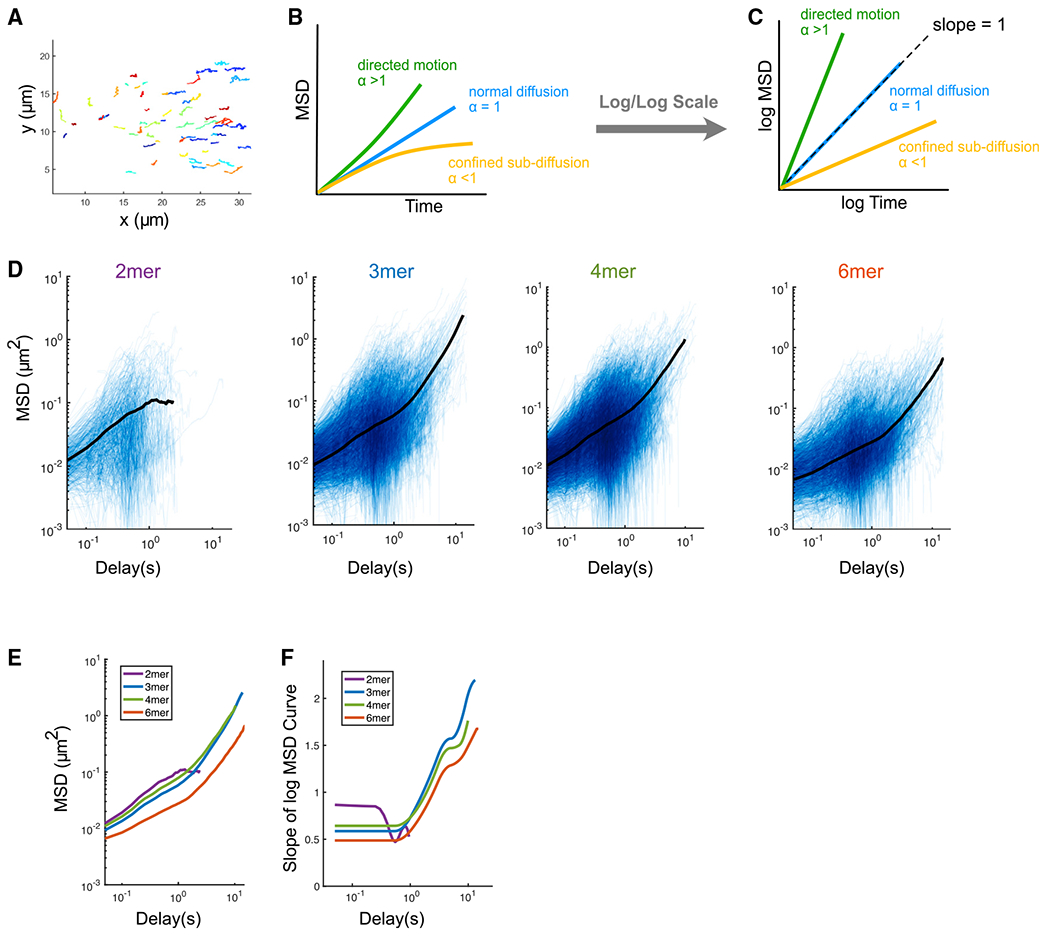
(A) Tracks of engineered PAR-3 trimers imaged at the cortex during polarity establishment. Anterior is to the left. Different colors are only used for better visualization. t = 15 s.
(B) Illustration of the interpretation of anomalous parameters.
(C) Illustration of the interpretation of the MSD curve on log/log scales.
(D) Log/log scale MSD curves for dimer, trimer, tetramer, and hexamer, each curve describing the motion of a single PAR-3 cluster. For each plot, data were acquired and pooled from three embryos. Averaged MSD curves are shown for time lags defined by at least 20 data points.
(E) Comparison of the averaged MSD curves for dimer, trimer, tetramer, and hexamer.
(F) Slopes of the averaged MSD curves for dimer, trimer, tetramer, and hexamer, estimated by fitting a smoothing spline to each curve and then taking its derivative.
The diffusion of particles on a 2D surface, such as the cell membrane, can be described as
where D is the diffusion coefficient and α is the anomalous diffusion parameter. α = 1 describes normal Brownian diffusion. An α < 1 indicates sub-diffusion, where the movement of a particle is confined, while α > 1 represents super-diffusion, which indicates directed movement of particles (Figures 2B and 2C). Therefore, the anomalous parameter α provides direct information about the type of motion a particle is experiencing. The anomalous parameter α can be easily visualized by plotting MSD versus time on a log/log scale. For particles undergoing normal Brownian diffusion, these plots are straight lines with a slope of 1. For other types of motion, the slope of the curve is equal to α.
We generated log/log MSD plots from our single-particle tracking data for PAR-3 dimer, trimer, tetramer, and hexamer EOD strains (Figure 2D). To more easily compare these plots, we overlaid the averaged MSD curves (Figure 2E) and calculated the slope of each log/log MSD curve by taking the derivative of a smoothing spline fit to the data (Figure 2F). This analysis revealed non-Brownian diffusive behaviors of PAR-3 oligomers, which are distinct between engineered dimer and larger oligomers. The log-log MSD curves for trimers, tetramers, and hexamers are not straight lines (Figures 2D and 2E), which indicates that these clusters undergo different types of motion on different timescales. On timescales shorter than 1 s, α < 1 for clusters of all sizes, indicating confined movement due to interactions with the actomyosin cortex. However, dimers had a slope closer to 1 compared with larger clusters (Figure 2F), indicating that their movement is more diffusive than that of larger clusters. On longer timescales (>1 s), larger oligomers display anomalous parameters greater than 1, which indicates a directed motion due to cortical flow (Figures 2D–2F). In contrast, dimers did not exhibit directed motion but instead dissociated from the cortex on longer timescales (Figure 2D).
The difference in diffusive behavior between dimers and larger oligomers is consistent with the polarity and embryo lethality phenotypes we observed in the dimer strain (Figure 1). We conclude that, during polarization, movement of PAR-3 dimers on the cortex is dominated by weakly confined random diffusion, while trimers, tetramers, and hexamers undergo directed movement under cortical flow at longer timescales.
PAR-3 clusters and cortical actin move in tandem but are not physically associated
To address the physical explanation for non-Brownian diffusive behavior of larger PAR-3 clusters, we examined the interaction between PAR-3 and the actomyosin cortex. Although actomyosin and PAR-3 clusters move in tandem toward the anterior pole, there is no known binding interaction between PAR-3 and F-actin. The relationship between PAR-3 and actomyosin has been visualized by observing labeled myosin II (NMY-2; Video S2; Dickinson et al., 2017), but F-actin and PAR-3 have not been visualized together in living embryos to our knowledge. Therefore, we constructed a strain carrying endogenously tagged mScarlet::PAR-3 and a transgenic GFP::utrophin reporter that binds to F-actin (Tse et al., 2012). We imaged zygotes from this strain using TIRF microscopy (Figure S2A; Video S3). In agreement with previous studies, the movement of the actin cortex and of PAR-3 were tightly coupled (Goehring et al., 2011a; Munro et al., 2004; Figures 3A and 3B; Video S3).
Figure 3. Super-resolution imaging reveals that PAR-3 moves in tandem with the actomyosin cortex despite not being physically associated with actin.
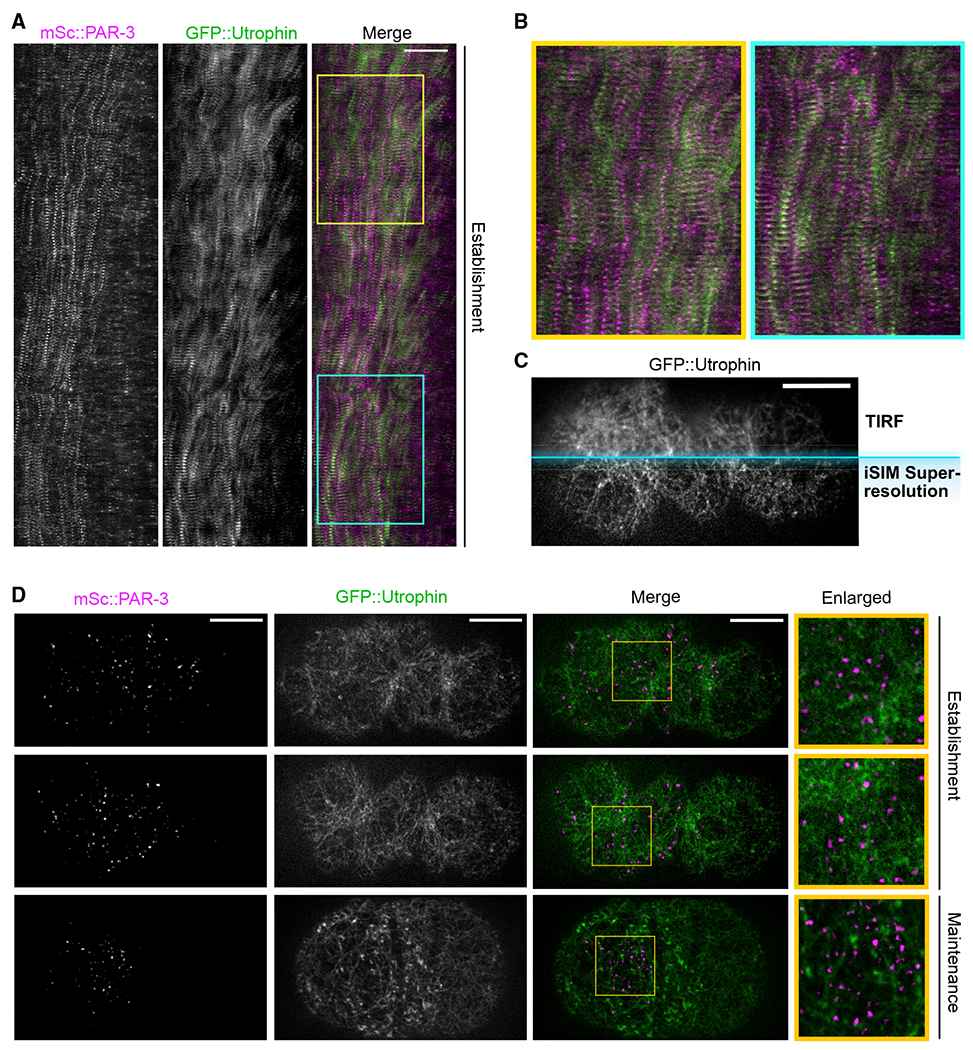
(A) Kymograph of cortical mSc::PAR-3 and utrophin::GFP during the first cell cycle. Embryos were imaged using TIRF at 3 s/frame. Anterior is to the left. Scale bar represents 10 μm. Yellow and blue boxes indicate regions enlarged in (B).
(B) Enlarged sections of kymograph (boxes) in (A).
(C) Comparison between TIRF imaging and high-resolution iSIM imaging. A mSc::PAR-3; GFP::UTRO zygote at polarization stage was imaged. Only the GFP::utrophin channel is shown. Scale bar represents 10 μm.
(D) Maximum projected z stacks of images of mSc::PAR-3; GFP::utrophin zygote, imaged using super-resolution iSIM imaging centered at the cortical region. Anterior is to the left. Scale bars represent 10 μm. Yellow box, region enlarged in right column.
To quantify the correlation between PAR-3 and actin movements, we selected two time points during polarity establishment: one when the cortex was actively contracting toward the anterior pole and one when the cortex was temporarily oscillating and moving toward the future dorsal side. We quantified the movements using particle image velocimetry (PIV) and compared the vector fields for PAR-3 and F-actin using Pearson’s correlation (Figure S2B and S2C). As expected, PAR-3 movement and F-actin movement were tightly correlated (Pearson’s correlation R = 0.70 for Δt1; R = 0.59 for Δt2), indicating that PAR-3 and the actomyosin cortex move in tandem. Nevertheless, PAR-3 and actin did not colocalize or overlap with each other in TIRF images (Pearson’s correlation R = 0.067 ± 0.010; Figures 3A and 3B).
Although we did not observe an obvious association between PAR-3 and actin in TIRF images, it remained possible that contacts between actin and PAR-3 might have been overlooked due to the density of the F-actin network and the diffraction-limited resolution of TIRF. Therefore, to better visualize the interaction between PAR-3 and the actin network, we performed super-resolution imaging of the cortex using instant structured illumination (iSIM) (Figure 3C). We found that the PAR-3 clusters appeared as point sources in super-resolution images, suggesting that even the largest clusters are smaller than the ~170-nm resolution of iSIM imaging (Figure 3D). In contrast, individual actin branches and pores in the actomyosin cortex were clearly visible (Figure 3D). These observations reveal important information about the scale of sizes: PAR-3 clusters are much smaller than the gaps in the actomyosin cortex. Furthermore, we observed that PAR-3 clusters were typically found within the pores of the cortex, not associated with individual filaments (Figure 3D).
In a previous study, Sailer et al. (2015) found that, during polarity maintenance, after cortical flows have ceased, wild-type PAR-3 clusters undergo weakly confined diffusion at the cortex, especially on timescales longer than 1s. Treatment with an actin depolymerizing drug eliminated this confined behavior, suggesting confinement was due to interactions between PAR-3 and F-actin (Sailer et al., 2015). These results are consistent with our finding that PAR-3 clusters are located within pores in the actin network (Figure 3D). PAR-3 clusters that diffuse within pores of the actin cortex would be expected to exhibit confined motion (“corralling”) due to collisions with actin, especially on longer timescales.
To further explore this idea, we sought to directly test whether PAR-3 oligomers are confined within the actomyosin meshwork of the cortex during polarity establishment. We repeated the imaging and MSD analysis in our EOD strains after eliminating cortical flow by depleting myosin light chain (mlc-4 RNAi), which allowed us to observe the behavior of PAR-3 clusters in the absence of cortical flow. Importantly, mlc-4 RNAi eliminated cortical flow but did not grossly disrupt the organization of the actin cortex (Figure 4A). As expected, mlc-4 depletion eliminated directed movement of larger PAR-3 clusters during polarity establishment (Figures 4B–4D; compare with Figures 2D–2F). Instead, PAR-3 clusters exhibited sub-diffusive movement that became progressively more confined on longer timescales (Figures 4C and 4D), which is nearly identical to the behavior previously reported for wild-type PAR-3 clusters during polarity maintenance (Sailer et al., 2015). We noted that, despite exhibiting sub-diffusive behavior, PAR-3 clusters remained mobile even under mlc-4 RNAi conditions. Together with our imaging results (Figure 3), these data rule out models in which PAR-3 is directly bound to F-actin or tightly encased in a dense actin meshwork. Instead, our results suggest that PAR-3 clusters diffuse within pockets in the actin cortex and are partially confined via collisions with F-actin.
Figure 4. PAR-3 clusters are loosely confined by the transient physical interactions with the actomyosin cortex.
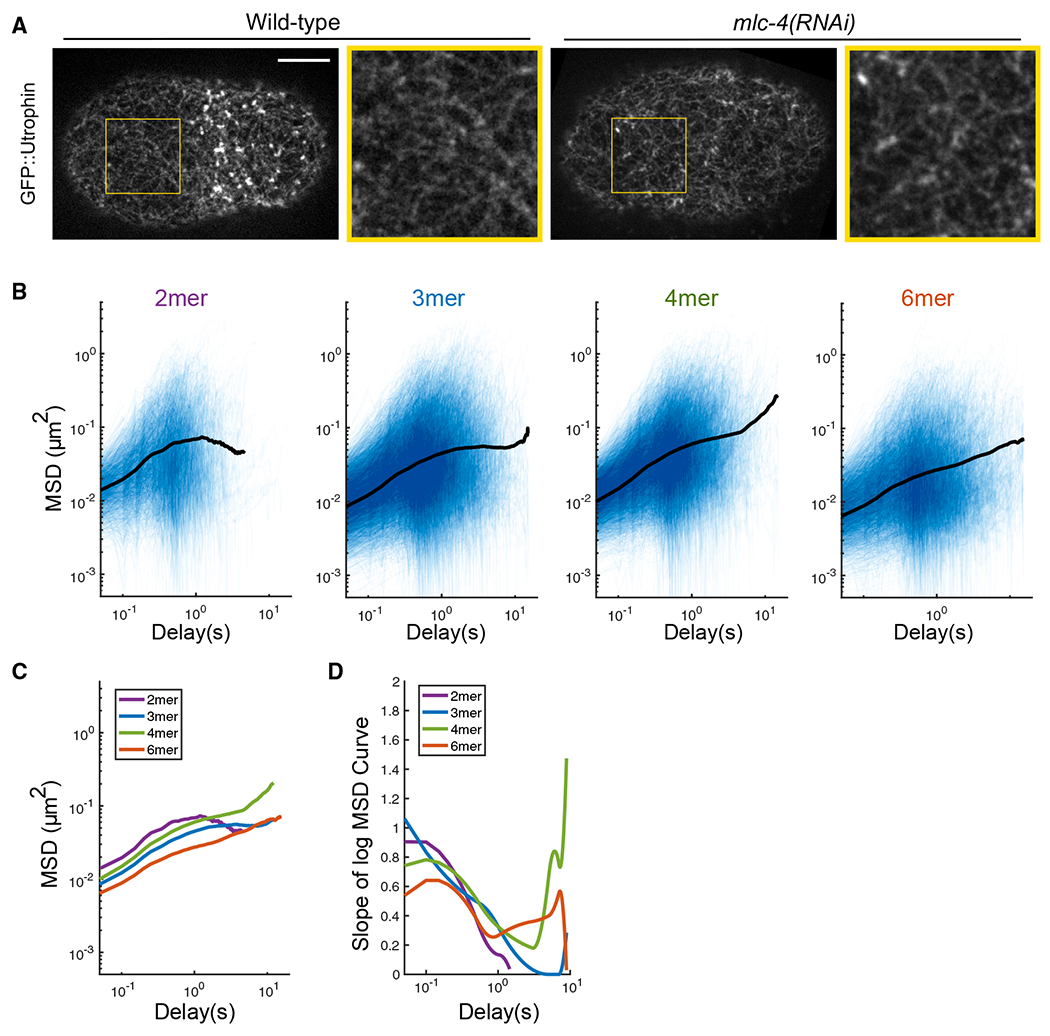
(A) Super-resolution iSIM images of GFP::utrophin zygotes at pronuclear meeting, with or without mlc-4 RNAi. The structure of the actin network is intact under mlc-4 RNAi. Yellow box, region enlarged to the right of each image. Scale bar represents 10 μm.
(B) log/log scale MSD curves for dimer, trimer, tetramer, and hexamer with mlc-4 RNAi treatment, each curve describing the motion of a single PAR-3 cluster. For each plot, data were acquired and pooled from three embryos. Averaged MSD curves are shown for time lags defined by at least 20 data points.
(C) Comparison of the averaged MSD curves for dimer, trimer, tetramer, and hexamer.
(D) Slopes of the averaged MSD curves for dimer, trimer, tetramer, and hexamer, estimated by fitting a smoothing spline to each curve and then taking its derivative.
An additional observation from these experiments was that engineered PAR-3 clusters, regardless of size, moved in a sub-diffusive fashion when mlc-4 was depleted. There was no clear difference between dimers and larger oligomers in these experiments. Therefore, the failure of the dimer construct to rescue polarity (Figure 1) cannot be explained by differences in confinement or corralling of PAR-3 dimers compared with larger oligomers. We therefore explored alternatively possible explanations for the inability of dimers to support normal polarity establishment.
Differences in diffusivity and membrane lifetime together can account for the different behaviors of dimers and larger oligomers
We next asked whether differences in diffusive behavior between dimers and larger oligomers could account for the different polarization effectiveness in EOD strains. Since all engineered PAR-3 clusters (including dimers) exhibited confined diffusion due to collisions with actin, we measured additional parameters to describe the motion of different-sized clusters. We measured diffusion coefficients by fitting the first six time steps of each averaged MSD curve, corresponding to timescales before the motion became significantly confined, to the diffusion equation (MSD = 4Dt; Figure S3A). Our measured diffusion coefficients (Figure 5A) were much smaller than those observed for another anterior PAR protein, PAR-6, in bulk fluorescence recovery after photobleaching (FRAP) experiments (Goehring et al., 2011b) but were similar to those of a slowly diffusing sub-population of PAR-6 particles that was likely associated with PAR-3 (Robin et al., 2014). We observed a trend toward slower diffusion for larger oligomers (Figure 5A).
Figure 5. Simulations reveal that the different behaviors of dimers and larger oligomers can be explained by measured diffusion and membrane binding.
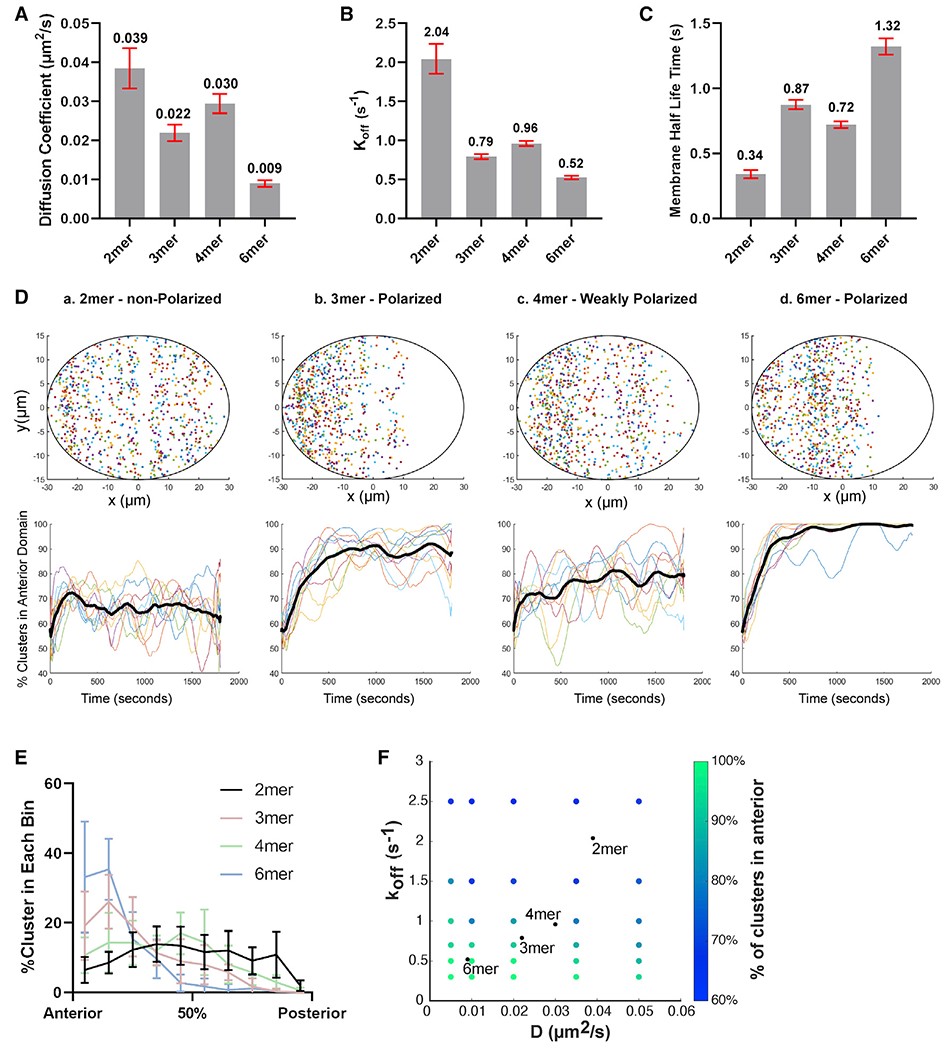
(A) The diffusion coefficient of PAR-3 in engineered EOD::PAR-3 strains (wild-type background). Red bars indicated 95% confidence interval (CI). Data points were pooled from three embryos for each strain. n = 4,237, 10,587, 12,619, and 6,724 particles for dimer, trimer, tetramer, and hexamer, respectively.
(B) The koff of PAR-3 in engineered EOD::PAR-3 strains (wild-type background). Red bars indicated 95% CI. Data points were pooled from three embryos for each strain. n = 4,237, 10,587, 12,619, and 6,724 particles for dimer, trimer, tetramer, and hexamer, respectively.
(C) The membrane binding half-life time of PAR-3 in engineered EOD::PAR-3 strains. Red bars indicated 95% CI. Data points were pooled from three embryos for each strain. n = 4,237, 10,587, 12,619, and 6,724 particles for dimer, trimer, tetramer, and hexamer, respectively.
(D) Upper panels: final cluster positions after simulating particle movement for 15 min. Ellipse, boundary of simulated embryo. x and y units, μm. Lower panels: percentage of particles in the anterior domain over time in simulated embryos is shown. Each line describes an independent repeat of the simulation. Black line, average.
(E) The number of clusters in each 6-μm-wide bin along the AP axis in simulated EOD embryos. n = 10 for each set of conditions.
(F) Dependence of the simulation outcomes on parameter values. Each point represents the average results of 10 simulation runs. The parameter values used in (D) and (E) are indicated by black points.
Next, to quantify membrane lifetime, we calculated the particle disappearance probability per imaging frame in each EOD strain, based on the distributions of particle track lengths (see STAR Methods). Using the known imaging frame rate, we converted the particle disappearance probability to a membrane unbinding rate (Figure 5B) and membrane half-life time (Figure 5C). A caveat of these estimates is that clusters can disappear due to photobleaching in addition to membrane unbinding, resulting in an overestimation of the actual koff. To evaluate the extent to which photobleaching might affect our measurements, we examined fluorescence intensity over time in our hexamer dataset. Since hexamers can undergo multiple photobleaching events without disappearing entirely, we would expect their intensity to decrease over the length of a single particle track if appreciable photobleaching were occurring. However, we found that the fluorescence intensity of most hexamers remained constant over the length of our observation; the median hexamer was 94% as bright during the last 10 frames before it disappeared as during the first 10 frames of its observation (Figure S3B). Therefore, photobleaching makes only a minor contribution to our estimates of koff and membrane lifetime.
We observed trends of decreasing diffusivity (Figure 5A) and increasing membrane lifetime (Figures 5B and 5C) as oligomer size increased. We therefore hypothesized that the slower diffusion and reduced random motion of larger PAR-3 oligomers, together with sufficient membrane dwell time to experience cortical flow, would allow larger oligomers to be efficiently segregated toward the anterior.
To test this hypothesis, we computationally simulated the movement on the plasma membrane of PAR-3 clusters with different diffusivities and membrane binding lifetimes. Prior studies have developed complex and sophisticated models of PAR protein segregation and mutual antagonism (Goehring et al., 2011a; Sailer et al., 2015; Gross et al., 2019). However, these models either did not include PAR protein clustering (Goehring et al., 2011a; Gross et al., 2019) or did not examine the process of polarity establishment (Sailer et al., 2015). Here, we took a simpler approach in order to focus on how cortical flow would be expected to segregate clustered proteins of different sizes. Each particle in our simulations underwent a biased random walk due to the combination of Brownian diffusion and cortical flow, and at each time step, particles dissociated from the membrane with a probability calculated from koff. We incorporated two additional assumptions, which are based on experimental observations. First, we assumed that the number of PAR-3 clusters at the membrane remains constant over time; that is, for each PAR-3 cluster that dissociates from the membrane, a new one appears. This assumption is justified by our observation that the amount of PAR-3 at the membrane is similar throughout polarization (Video S1). Second, we assumed that new PAR-3 clusters preferentially bind the membrane in the part of the cell where PAR-3 is present. This assumption is justified by observations that posterior PAR proteins occupy the region of the plasma membrane that has been cleared of anterior PARs (Cuenca et al., 2003) and that posterior PAR proteins prevent binding of new PAR-3 clusters to the membrane (Sailer et al., 2015). We implemented this assumption in our simulations by choosing the locations of newly binding PAR-3 particles at random from the measured distribution of existing particles at each time step. We note that this second assumption introduces positive feedback to the system, because more PAR-3 particles are added to the regions of the membrane where PAR-3 particles are already enriched. To ensure that this form of feedback was not sufficient to establish polarity on its own, we ran simulations in the absence of cortical flow and confirmed that polarization did not occur (Figure S3D). Then, we ran simulations in the presence of cortical flow, using the diffusion coefficient and koff measured in each EOD strain.
We quantified the polarization of each simulated embryo in several different ways. First, we quantified the percentage of clusters in the anterior domain over time in each simulation, and we observed an efficient polarization within the first 10 min using parameters measured for trimers and hexamers. In contrast, using parameters measured for dimers, the percentage of clusters in the anterior domain remained constant for the duration of the simulations (Figure 5D, lower panels). Second, we quantified the final cluster distribution along the anterior-posterior (AP) axis by counting the clusters in each 6-μm-wide bin along the AP axis (Figure 5E). Consistent with previous results, simulated PAR-3 trimers and hexamers were localized in the anterior domain, but simulated dimers remained evenly distributed. Using the parameters measured for tetramers, we observed polarization that was significant but weaker than that of simulated trimers and hexamers. This is consistent with the fact that our measurements of D and koff for tetramers fell outside the trend observed for dimers, trimers, and hexamers. Although the reason for faster diffusion and weaker membrane binding of tetramers is not clear, it is consistent with the mild phenotype observed for tetramer(YrbI) EOD embryos (see above). Overall, our simulation results indicate that the efficient segregation of larger PAR-3 oligomers can be explained by cooperativity between reduced random motion and membrane binding.
To more generally explore how diffusivity and membrane binding lifetime determine the efficiency of polarity establishment, we systematically tested the effect of different combinations of D and koff spanning the range measured from PAR-3 oligomers (Figure 5F). We found that the effectiveness of polarization, quantified by percentage of clusters in the anterior, decreases as koff and diffusion coefficient increase. Intuitively, for cortical flows to effectively segregate a membrane-bound protein, the cortical flow must dominate the randomization of particle positions that occurs due to diffusion and membrane unbinding and rebinding. Our simulations reveal that slower diffusion and more stable membrane association allows larger PAR-3 clusters to be efficiently polarized by cortical flow.
Tracking wild-type PAR-3 movement with a dual-labeling technique confirms the size threshold in an endogenous setting
Up to this point, we have characterized the diffusive behavior of engineered EOD::PAR-3, which revealed size-dependent polarization behavior but represents an artificial situation. To test whether our observations hold for endogenous PAR-3 clusters, we sought to measure the diffusive behaviors of a heterogeneous population of wild-type PAR-3 clusters. Tracking wild-type PAR-3 clusters tagged with fluorescent proteins is difficult due to the high density of particles and the inherent bias of particle-tracking software toward bright signals. To overcome these issues, we developed a method to track individual PAR-3 clusters in an unbiased way while also estimating their sizes. We labeled endogenous HaloTag::PAR-3 with a mixture of two different HaloTag ligand dyes (Grimm et al., 2015, 2016, 2017) at a defined ratio. Each HaloTag molecule binds to a single dye molecule so that, when a mixture of dyes is used for labeling, a chimeric population of labeled Halo::PAR-3 molecules results (Figure 6A). By adjusting the ratio of the two dyes, we achieved conditions in which the less-abundant dye was present at single-molecule levels on the cortex, yielding discrete labels that are ideal for tracking. At the same time, the more-abundant dye labels the majority of molecules, providing cluster size information (Figure 6A).
Figure 6. The PAR-3 size threshold in an endogenous setting.
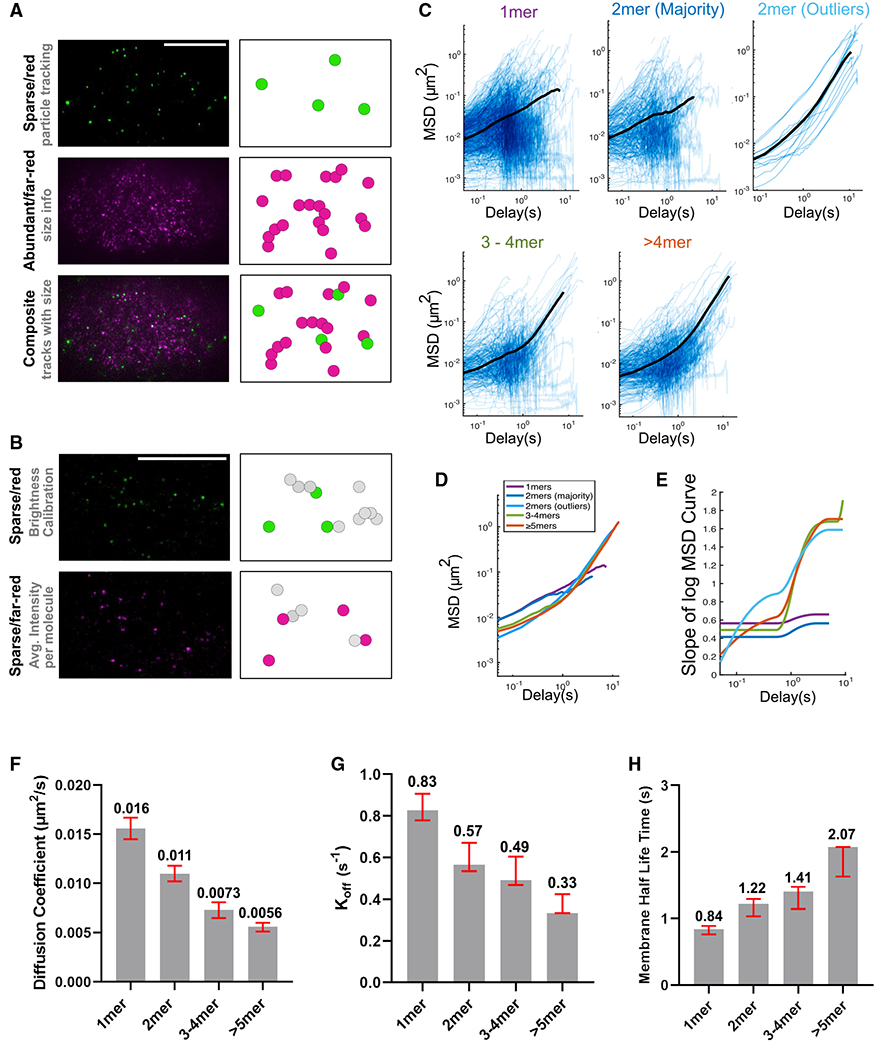
(A) TIRF images and cartoon illustrations of the dual-labeling experiment. Magenta represents far-red/abundant channel and green represents red/sparse channel. Scale bar represents 10 μm.
(B) TIRF images and cartoon illustrations of the calibration double dilution experiment, where the dyes for both channels are diluted to single-molecule levels. Scale bar represents 10 μm.
(C) Log/log scale MSD curves for each cluster group binned by estimated size, each curve describing the motion of a single PAR-3 cluster. For each plot, data were acquired and pooled from five embryos. Averaged MSD curves are shown for time lags defined by at least 20 data points except for the dimer outlier curves (top right), for which the average is over all 13 particles observed.
(D) Comparison of the averaged MSD curves for each cluster group binned by estimated size.
(E) Slopes of the averaged MSD curves for each cluster group binned by estimated size. Slopes were estimated by fitting a smoothing spline to each curve and then taking its derivative.
(F) The diffusion coefficient measured in a dual-labeling experiment. Red bars indicated 95% CI. Data points were pooled from three embryos for each strain. n = 672, 297, 244, and 322 particles for monomers, dimers, trimers to tetramers, and greater than pentamers, respectively.
(G) The koff measured in a dual-labeling experiment. Red bars indicated 95% CI. Data points were pooled from three embryos for each strain. n = 672, 297, 244, and 322 particles for monomers, dimers, trimers to tetramers, and greater than pentamers, respectively.
(H) The membrane binding lifetime measured in a dual-labeling experiment. Red bars indicated 95% CI. Data points were pooled from three embryos for each strain. n = 672, 297, 244, and 322 particles for monomers, dimers, trimers to tetramers, and greater than pentamers, respectively.
We imaged dual-labeled Halo::PAR-3 embryos on a custom-built TIRF microscope with a dual-view emission path design, allowing simultaneous imaging of both wavelengths. We first used the signals from the sparse (red) dye to perform an MSD analysis, using the same approach as for the EOD strains. Then, we measured the size of each PAR-3 cluster by fetching the fluorescence intensity of each particle in the abundant (far-red) channel (Figure 6A). To convert this measured fluorescence intensity into an estimate of molecules per cluster, we calibrated our microscope by measuring the brightness of single far-red dye molecules in embryos with both dyes diluted to single-molecule levels (Figures 6B and S4; STAR Methods). We used the red dye, which was present at single-molecule levels in both calibration and tracking experiments, as an internal standard to correct for embryo-to-embryo variations in laser power and depth of focus (Figures 6B and S4; STAR Methods).
Tracking endogenous PAR-3 clusters, while measuring their sizes in this way, confirmed the trends we found in our experiments with engineered EOD::PAR-3. First, there was a clear difference in diffusion behavior between smaller clusters (monomer and dimer) and larger oligomers (greater than or equal to trimer; Figures 6C–6E). The anomalous parameters for monomers and dimers remain below 1 at all timescales, indicating weakly confined diffusion, while anomalous parameters for larger clusters increased from <1 on short timescales to >1 on longer timescales, demonstrating that larger PAR-3 clusters display directed motions during the timescale of polarization. Second, we calculated the diffusion coefficient, koff, and membrane binding half-time for each cluster size group. In agreement with the EOD data, we observed a decrease in the diffusion coefficient, a decrease in koff, and an increase in membrane half-time with increasing cluster size (Figures 6F–6H).
Although the results from these dual-labeling experiments are broadly consistent with our data from EOD embryos, two observations merit further discussion. First, among PAR-3 particles estimated to be dimers, we observed two populations that displayed different diffusive behavior. The majority of particles (284 out of 297 tracks classified as dimers) underwent weakly confined diffusive motion, similar to engineered dimers. A small fraction (13 out of 297 tracks classified as dimers) underwent directed motion on longer timescales, similar to larger oligomers. We previously showed that, in C. elegans zygotes, 70%–80% of HaloTag molecules can be successfully labeled with ligand dyes (Dickinson et al., 2017; Sarikaya and Dickinson, 2021). Since unlabeled molecules do not contribute to the fluorescence intensity of a cluster, this minority of “dimer” tracks are very likely larger oligomers whose size was underestimated due to incomplete labeling.
Second, we tracked many particles that had no detectable intensity in the abundant (far-red) channel and were therefore classified as PAR-3 monomers (Figure 6C). This was surprising because we did not observe any significant membrane stabilization of PAR-3 monomers in the PAR-3(RRKEEE) mutant strain (Figure 1; Dickinson et al., 2017). Some fraction of the PAR-3 population with estimated size of 1 are likely to be dimers in which the second molecule of PAR-3 was not detected due to incomplete labeling. However, given the relatively large fraction of particles (672 out of 1,535 tracks) that appeared to be monomers, these data may reveal a previously unappreciated ability of wild-type PAR-3 monomers to interact with the membrane (see discussion).
Overall, we conclude that a size threshold of three to four monomers governs the diffusive behavior and advective transport of wild-type PAR-3 to establish the AP axis of the C. elegans embryo.
DISCUSSION
Cortical flow is a general cell behavior that remodels the membrane-bound proteome in a variety of cell types (Ananthakrishnan and Ehrlicher, 2007). Here, we have studied the biophysical basis for oligomerization-dependent transportation of peripheral membrane proteins by cortical flow. We developed methods to engineer PAR-3 size via EODs and to measure endogenous PAR-3 cluster sizes via dual labeling, and we used these tools to analyze the movement of PAR-3 clusters of different sizes. Both approaches revealed a shift in diffusive behavior as cluster size increases from two to four monomers. These experiments indicate that the motion of smaller PAR-3 clusters is dominated by diffusion, while larger PAR-3 clusters are effectively transported by advective flow. These trends are consistent with our previous observations (Dickinson et al., 2017) but add an additional degree of quantitative precision and raise new questions about the physical and molecular basis for the sharp shift in diffusive parameters at the size threshold.
The dual-labeling strategy that we developed to track endogenous PAR-3 clusters was inspired by earlier studies that achieved single-molecule labeling in vivo, either via low-level transgenic expression and partial photobleaching (Robin et al., 2014) or by microinjection (Yamashiro and Watanabe, 2017). Dual HaloTag labeling is easier to carry out than these earlier approaches and additionally allows tracking of sparse labels without sacrificing the ability to visualize the bulk cellular protein. This approach could be applied to study the dynamics of other membrane proteins, as many as 35% of which may be oligomeric (Goodsell and Olson, 2000). For example, E-cadherin forms clusters that are important for transmission of forces during tissue morphogenesis (Huebner et al., 2021). Although the dynamics of these clusters have been studied by bulk techniques, such as FRAP, a dual-labeling technique could reveal turnover rate and diffusivity, potentially providing new insights into how E-cadherin clustering controls its behavior. As another example, receptor tyrosine kinases (RTKs) are known to be modulated by homo-oligomerization (Schlessinger, 2000), but how oligomerization controls RTK activity is still not understood in detail. A dual-labeling technique, coupled with activity reporters, could provide insight into this question. Finally, we anticipate applying dual labeling to other oligomeric proteins within the PAR polarity system, including PAR-2 (Arata et al., 2016) and CHIN-1 (Sailer et al., 2015), to determine how clustering affects their dynamics on the plasma membrane.
We and others have shown that monomeric mutants of PAR-3 do not bind stably to the plasma membrane (Figure 1; Dickinson et al., 2017; Li et al., 2010; Rodriguez et al., 2017), so it was surprising that we observed a large population of apparent monomers at the membrane in our dual-labeling experiment (Figure 6). Notably, in PAR-3 monomeric mutants, the oligomerization domain was disrupted by charge-reversal (RRKEEE) mutations (Dickinson et al., 2017) or by deleting the entire N terminus (Li et al., 2010; Rodriguez et al., 2017). Thus, one possible explanation for our results is that positively charged residues in the PAR-3 N terminus, which are disrupted by the monomeric mutations, could directly interact with the negatively charged surface of the lipid bilayer. Consistent with the presence of additional membrane-binding interactions, wild-type PAR-3 particles had slower diffusivities and longer membrane lifetimes than engineered oligomers of the same sizes (compare Figures 5A–5C and 6F–6H). The isolated PAR-3 N terminus does not bind the membrane on its own (Dickinson et al., 2017) but still could contribute to membrane targeting in the context of the full-length protein. Future experiments will explore the relationship between PAR-3 structure, oligomerization, and membrane binding in greater detail.
In our previous study, the cell cycle kinase PLK-1 was shown to negatively regulate PAR-3 oligomerization by phosphorylating residues in the PAR-3 oligomerization domain (Dickinson et al., 2017). In that study, we attempted to isolate constitutive oligomerization mutants of PAR-3 by mutating the two identified PLK-1 target residues to alanine but were unable to generate stable lines due to highly penetrant lethality and sterility. Consistent with this result, we were unable to isolate EOD::PAR-3 direct fusion lines through CRISPR-mediated insertion of EODs into the PAR-3 locus, which necessitated using the nAb-linking strategy instead. In light of these observations, it was surprising that our EOD::nAb; YFP::PAR-3* strains not only are viable but are healthier than the PAR-3* monomeric mutants. We do not have a clear explanation for this observation. One possibility is that PAR-3 monomers–which are expected to be absent from both the direct EOD::PAR-3* fusion strains and the PLK-1 phosphorylation site mutants but present in the EOD::nAb; YFP::PAR-3* strains due to the difference in expression level between the EOD constructs and PAR-3–might have a critical, unknown role in development. Alternatively or in addition, phosphorylation of PAR-3 by PLK-1 might regulate crucial PAR-3 interactions other than homoligomerization. These hypotheses represent interesting areas for further investigation.
Developmentally, the purpose of PAR-3 clustering in the zygote is to transport the key polarity kinase, aPKC, to the anterior, allowing asymmetric division and cell fate specification. PAR-3 oligomerization and its binding to PAR-6/aPKC are cooperative (Dickinson et al., 2017): PAR-3 clusters containing three or more subunits bound strongly to aPKC/PAR-6 in sc-SiMPull experiments, while dimers bound more weakly and monomers associated only at background levels (Dickinson et al., 2017). This threshold for PAR-3 binding to PAR-6/aPKC is strikingly consistent with the observed size threshold for PAR-3 segregation by cortical flow, suggesting a possible connection between PAR-3 transportation and aPKC/PAR-6 binding. One hypothesis is that aPKC/PAR-6 binding could increase the volume of the aPAR complex, making it more susceptible to corralling effects and thus overcoming random Brownian diffusion. Alternatively, association with PAR-6/aPKC might facilitate membrane binding of PAR-3, either by inducing a PAR-3 conformational change (Chen et al., 2013) or through association with CDC-42 (Joberty et al., 2000) or other partners. Future work will attempt to test whether binding to PAR-6/aPKC is required for the shift in diffusive behavior that we report here.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for resources and further information should be directed to, and will be fulfilled by, the Lead Contact, Daniel J. Dickinson (daniel.dickinson@austin.utexas.edu).
Materials availability
C. elegans strains generated in this study are available from the lead contact upon request.
Data and code availability
Original microscopy data are available from the lead contact upon reasonable request.
All original code generated in this study is available via Github (see the Key Resources Table for links).
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-HaloTag pAb | Promega | Cat# G9281; RRID:AB_713650 |
| Anti-mNeonGreen nAb | Allele Biothechnology | Cat# ABP-NAB-MNGAB; RRID: AB_2629489 |
| Chemicals | ||
| JF585 | Laboratory of Luke D. Lavis | Grimm et al. (2017),Grimm et al. (2015),Grimm et al. (2016) |
| JF646 | Laboratory of Luke D. Lavis | Grimm et al. (2017),Grimm et al. (2015),Grimm et al. (2016) |
| DMSO | Sigma | D2650-5X5ML |
| Genetic fragments | ||
| GFP nano body | add gene | 89,367 |
| 6HNL | PDB | 6HNL |
| 6O2D | PDB | 6O2D |
| 8KY8 | PDB | 8KY8 |
| 3HON | PDB | 3HON |
| 3XF6 | PDB | 3XF6 |
| 6GWK | PDB | 6GWK |
| 3ZQM | PDB | 3ZQM |
| 5LFJ | PDB | 5LFJ |
| 3H5L | Huang et al. (2014) | 3H5L_2_mini |
| HOTag3 | Thomson et al. (2014) | HOTag3 |
| H protein | Sun et al. (2014) | H protein |
| Yrb-1 | Parsons et al. (2002) | Yrb-1 |
| AraD | Luo et al. (2001) | AraD |
| Experimental models: Organisms/strains | ||
| C. elegans wild-type strain | Caenorhabditis Genetics Center | N2 |
| C.elegans transgenic strain expressing GFP::UTRO Genotype: xsSi3 [GFP::utrophin + Cbr-unc-119(+)] |
Tse et al. (2012) | MG589 |
| C. elegans trains with endogenously tagged NMY-2::mKate2 Genotype: nmy-2(cp69[nmy-2::mkate2 + LoxP]) I |
Dickinson et al., (2017) | LP256 |
| C.elegans strain with endogenously tagged mNG::PAR-3 Genotype: par-3(cp54[mNeonGreen::3xFlag::par-3]) III |
Dickinson et al. (2017) | LP242 |
| C.elegans strain with endogenously tagged mSc::PAR-3 and transgenenic GFP::UTRO Genotype: par-3(djd18[EYFP-C1dPi::par-3]) III; xsSi3 [GFP::utrophin + Cbr-unc-119(+)] II |
this study | UTX136 |
| C.elegans strain withendogenously tagged NMY-2::mKate and mNG::PAR-3 Genotype: nmy-2(cp52[nmy-2::mkate2 + LoxP unc-119(+) LoxP]) I; par-3(cp54[mNeonGreen::3xFlag::par-3]) III |
this study | LP636 |
| C.elegans strain withendogenously tagged Halo::PAR-3 par-3(cp323[HaloTag-GLO PAR-3]) III | Dickinson et al. (2017) | LP621 |
| C.elegans transgenic strain expressing GFPnAb::HaloTag Genotypes: djdIs19 [Psun-1::GFPnb::HALO::3xFlag + SEC] II |
this study | UTX44 |
| C.elegans transgenic strain expressing nAb::HaloTag::6mer(HOTag3) Genotype:djdIs7 [Psun-1::GFPnb::HALO::3xFlag::HexamerD + SEC] II |
this study | UTX17 |
| C.elegans transgenic strain expressing nAb::HaloTag::3mer(3H5L_2_mini) Genotype: djdIs11 [Piffb-1::GFPnb::HALO::3xFlag::TrimerD + SEC] II |
this study | UTX21 |
| C.elegans transgenic strain expressing nAb::HaloTag::10mer(H protein) Genotype: djdIs15 [Piffb-1::GFPnb::HALO::3xFlag::DecamerD + SEC] II |
this study | UTX25 |
| C.elegans strain with endogenously tagged YFP::PAR-3 Genotype: par-3(djd18[EYFP-C1dPi::par-3]) III |
this study | UTX52 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 Genotype: par-3(djd5 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX30 |
| C.elegans transgenic strain expressing mNG::6HNL Genotype: djdIs22 [Pmex5::mNG::3XFLAG::6HNLdimer] II |
this study | UTX66 |
| C.elegans transgenic strain expressing mNG::3HON Genotype: djdIs24 [Pmex5::mNG::3XFLAG::3HON3mer] II |
this study | UTX68 |
| C.elegans transgenic strain expressing mNG::2XF6 Genotype: djdIs25 [Pmex5::mNG::3XFLAG::2XF6hexamer] II |
this study | UTX69 |
| C.elegans transgenic strain expressing mNG::6GWK Genotype: djdIs27 [Pmex5::mNG::3XFLAG::GWKhexamer] II |
this study | UTX71 |
| C.elegans transgenic strain expressing mNG::3ZQM Genotype: djdIs30 [Pmex5::mNG::3XFLAG::3ZQM10mer] II |
this study | UTX74 |
| C.elegans transgenic strain expressing mNG::5LFJ Genotype: djdIs32 [Pmex5::mNG::3XFLAG::5LFJ12mer] II |
this study | UTX76 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::HaloTag::6mer(HOTag3) Genotype: djdIs7 [Psun-1::GFPnb::HALO::3xFlag::HexamerD + SEC] II; par-3(djd6) [EYFP-C1dPi::PAR-3(RRKEEE)] III |
this study | UTX47 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::Halo Genotype: djdIs21 [Psun-1::GFPnb::HALO::3xFlag + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX50 |
| C.elegans transgenic strain expressing nAb::BFP::2mer(6HNL) Genotype: djdIs34 [Psun-1::GFPnb::mTagBFP2::6HNLdimer + SEC] II |
this study | UTX86 |
| C.elegans transgenic strain expressing nAb::BFP::3mer(3HON) Genotype: djdIs35 [Psun-1::GFPnb::mTagBFP2::3HONtrimer + SEC] II |
this study | UTX88 |
| C.elegans transgenic strain expressing nAb::BFP::4mer(AraD) Genotype:djdIs38 [Psun-1::GFPnb::mTagBFP2::AraD tetramer + SEC] II |
this study | UTX92 |
| C.elegans transgenic strain expressing nAb::BFP::4mer(Yrb1) Genotype: djdIs39 [Psun-1::GFPnb::mTagBFP2::Yrb1 tetramer + SEC] II |
this study | UTX94 |
| C.elegans transgenic strain expressing nAb::BFP::6mer(6GWK) Genotype: djdIs36 [Psun-1::GFPnb::mTagBFP2::2XF6hexamer + SEC] II |
this study | UTX89 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::BFP::2mer(6HNL) Genotype: djdIs34 [Psun-1::GFPnb::mTagBFP2::6HNLdimer + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX105 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::BFP::3mer(3HON) Genotype: djdIs34 [Psun-1::GFPnb::mTagBFP2::3HONtrimer + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX80 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::BFP::4mer(AraD) Genotype: djdIs38 [Psun-1::GFPnb::mTagBFP2::AraD tetramer + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX104 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::BFP::4mer(Yrb-1) djdIs34 [Psun-1::GFPnb::mTagBFP2::Yrb1 tetramer + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III | this study | UTX81 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::BFP::6mer(6GWK) Genotype: djdIs36 [Psun-1::GFPnb::mTagBFP2::6GWKhexamer + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX106 |
| C.elegans transgenic strain expressing LsfGFP::SNAPf Genotype: cpIs95[Pmex-5>sfGFP-GLO::3xFlag::SNAPf + SEC] II |
this study | LP566 |
| Software and Algorithms | ||
| Utrack | Jaqaman et al. (2008) | Version 2.2.0; https://github.com/DanuserLab/u-track |
| Msdanalyzer | Tarantino et al. (2014) | https://tinevez.github.io/msdanalyzer/ |
| Ezyfit | MATLAB File Exchange | Version 2.53; http://www.fast.u-psud.fr/ezyfit |
| GetClustersize | This paper | https://github.com/IvyChang1994/getClusterSize.git |
| PIVLab | Thielicke and Stamhuis (2014) | Version 2.50; http://pivlab.blogspot.com |
| PAR-3 diffusion simulation | This paper | https://github.com/IvyChang1994/Par3-Modeling.git |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All C. elegans strains were fed with OP50 and maintained on standard NGM growth medium. All strains were kept in a 20°C incubator unless noted otherwise. Embryos were examined before sex can be determined, however, most of embryos were likely to be hermaphrodites because the spontaneous occurance of male without mating is rare.
All genetic modifications to the C. elegans genome were made using protocols previously published by our laboratory (Dickinson et al., 2013,2015). In brief, we used NEBuilder HiFi DNA Assembly to generate a plasmid construct that contains the homologous repair template with genome modifications and a selectable marker, flanked by 500–1500 bp of unmodified genomic homology arms. A repair template construct, a Cas9-sgRNA expressing vector, and a vector expressing extrachromosomal array fluorescent markers were co-injected into the syncytial gonads of young adult hermaphrodites. DNA repair by homologous recombination was triggered by Cas9 cleavage of the C. elegans genome, which allows the incorporation of modified repair template via homologous recombination. Knock-in animals were selected from the F2 progeny of injected animals using hygromycin selection and a phenotypic marker. After each knock-in strain was isolated, the selectable marker was excised by Cre-Lox recombination.
METHOD DETAILS
TIRF microscopy
Adult C. elegans carrying eggs were dissected in a drop of egg buffer on polylysine-coated coverslips. The embryos were gently flattened by mounting with 22.8 mm beads (Whitehouse scientific, Chester, UK) as spacers. Dual-labeling experiments (Figure 6) were carried out on a custom-built TIRF microscope (see below). All other TIRF images were acquired using a Nikon Eclipse Ti-2 microscope equipped with a 100X, 1.49 NA objective; a Photometrics Prime 95B camera; and an iLas2 circular TIRF illuminator (Roper scientific, E’ vry, France). TIRF images were magnified by a 1.5X tube lens before being collected by camera chip. The TIRF illuminator was operated in ellipse mode for acquiring images of whole embryos. mNG was excited using a 488 nm laser; YFP was excited using a 505 nm laser; mScarlet and JF585 were excited using a 561 nm laser; and JF646 was excited using a 638 nm laser.
Sc-SiMPull and photobleaching step counting
We exactly followed published protocols for sc-SiMPull (Dickinson et al., 2017; Stolpner and Dickinson, 2022). Briefly, coverslips were cleaned in a UV-ozone cleaner for 20 min to remove organic contaminants, and a PDMS microfluidic device was assembled and bonded to the coverslip to produce a narrow channel. The interior of the channel was passivated by flowing in liquid 2-[methoxy(polyethylenxy)9-12Propyl]-trimethoxysilane doped with 0.01% Biotin-PEG-Silane, washed with water, and kept desiccated until use. Just before each experiment, the device was functionalized by sequentially flowing in solutions of 0.2 mg/mL Neutravidin followed by 1 μM biotinylated antibody. The device was exhaustively washed with SiMPull buffer (10 mM Tris pH 8.0, 50 mM NaCl, 0.1% Triton X-100 and 0.1 mg/mL BSA) after each solution. Then, a single-cell embryo, dissected from an mNG-EOD expressing transgenic worm, was flowed into the center of the channel, and the device was sealed with clear tape. The embryo was crushed with the tip of a pencil to release the cellular contents, and the device was then immediately transferred to a TIRF microscope. 500 frame movies were captured at each of several stage positions to capture the full photobleaching series of each protein complex. The data were processed automatically using SiMPull analysis software (freely available at https://github.com/dickinson-lab/SiMPull-Analysis-Software; Dickinson et al., 2017) to extract the number of photobleaching steps for each particle. A detailed protocol for performing these experiments and analyzing the data is provided in Stolpner and Dickinson (2022).
Super-resolution iSIM imaging
Adult C. elegans carrying eggs were dissected in a drop of egg buffer on polylysine-coated coverslips. The embryos were gently flattened by mounting with 22.8 mm beads (Whitehouse scientific, Chester, UK) as spacers. Images were acquired using a Nikon Eclipse Ti-2 microscope equipped with a 100X, 1.49 NA objective; a Photometrics Prime BSI camera; an OptoSpin filter wheel (CAIRN Research, Kent, England), and an vt-iSIM super-resolution confocal scan head (VisiTech international, Sunderland, UK). Confocal images were magnified by a 1.5× tube lens before being collected by camera chip. During image acquisition, the focal plane was centered at the cortex, and along with 2 slices above and below, 0.25 um per slice step, 5 total slices were collected. GFP was excited using a 488 nm laser and mScarlet was excited using a 561 nm laser.
Imaging processing and display
The high-resolution iSIM images (Figures 3C and 3D) were processed through FIJI to generate Z maximum projection images. The PAR-3 channel (Figure 3D) was processed using the RF denoise command in FIJI (theta = 10) after maximum projection. Following these operations, brightness and contrast were adjusted for visibility of the signals. No other image manipulations were performed.
To generate kymographs (Figure 3A), we extracted a 15 pixel wide strip at the center of the imaged embryo image and stacked these strips on top of one another using the “montage” command in FIJI.
RNA interference
RNA interference targeting mlc-4 to eliminate cortical flow was performed by injection. We amplified 1 kb of mlc-4 from cDNA using primers that added T7 promoters to both ends of the amplified fragment. dsRNA was then synthesized using the T7 Ribomax kit (Promega) as instructed by the manufacturer. 1 mg/mL mlc-4 dsRNA was injected into young adults in either GFP:Utrophin background or EOD; PAR-3 background, and the worms were dissected 24–28 h later for embryo imaging.
Simulations of PAR-3 polarization
We developed a custom mathematical modeling script, written in MATLAB, for simulating PAR-3 cluster diffusion during polarization. The source code for our simulation and data analysis(Figure 5F) is available at: https://github.com/IvyChang1994/Par3-Modeling.git.
In brief, our simulation pipeline consists of the following steps: 1000 clusters with randomly distributed initial positions are generated. We simulated cluster positions for every time interval (0.05 s) during each 30-min simulation.
Each cluster at each time point is moved by Brownian diffusion based on the displacement calculated from diffusion coefficient measured in the EOD live embryos (Figure 5A) and a direction generated randomly.
Each cluster at each time point is moved by cortical flow. According to previous studies, the cortical flow speed decreases almost linearly from posterior pole to the anterior pole at polarization stage, with the peak flow rate to be 7.7μm/min at posterior pole (Goehring et al., 2011a; Gross et al., 2019; Mayer et al., 2010).
Each cluster at each step has a probability of dropping off the membrane. We made the assumption that the number of clusters on the membrane remains constant because of the equilibrium of membrane binding and unbinding. So each cluster that drops off the membrane will re-bind at a new location determined by the current PAR-3 distribution. To generate the PAR-3 distribution at each time point, we counted the number of PAR-3 clusters in each 0.6μm bins. This distribution was smoothed using a moving average over 3 bins, and then new PAR-3 clusters were created at locations chosen at random from this smoothed distribution.
HaloTag ligand dye feeding and dual-labeling experiments
JaneliaFluor Dyes (gifts from Luke Lavis) were dissolved in acetonitrile to 1 mM, dispensed into 2 μL aliquots in PCR tubes, dried under vacuum and stored at −20C in a desiccator upon receipt. Before use, 2 μL of DMSO was used to dissolve each single-use aliquot. 1 mL of OP50 liquid culture was spun down and resuspended in 100 μL S medium (150 mM NaCl, 1 g/L K2HPO4, 6 g/L KH2PO4, 5 μg/L cholesterol, 10 mM potassium citrate pH 6.0, 3 mM CaCl2, 3 mM MgCl2, 65 μM EDTA, 25 μM FeSO4, 10 μM MnCl2, 10 μM ZnSO4, 1 μM CuSO4) for each dye. Dissolved dye was added to the S medium-bacteria mixture. For the dual-labeling experiment, 2 μL of JF585-medium-bateria mix was added to 100 μL JF646-medium-bateria mix. For the double dilution experiment, a separate 100 μL medium-bacteria mix was made, and 2 μL of JF585-medium-bateria mix and 1 μL of JF646-medium-bateria mix was added to the 100 μL medium-bacteria mix. 30 μL of dye(s)-medium-bateria mix was dispensed into wells of a 96-well plate with round-bottom wells, and 20–30 L4-stage worms were picked into each well. Worms were grown at 20°C, with shaking at 230 rpm, for 16-24hrs before imaging. The final JaneliaFluor dye concentration used for feeding was 15uM.
Micromirror TIRF imaging
Adult C.elegans carrying eggs were dissected in a drop of egg buffer on polylysine-coated coverslips. The embryos were gently flattened by mounting with 22.8 mm beads (Whitehouse scientific, Chester, UK) as spacers. TIRF images were acquired using a custom-built TIRF microscope equipped with an Olympus APON 60X, 1.49 NA objective; a Photometrics Prime 95B camera; and a dual-view emission path design. This microscope uses a micromirror illumination path design that is optimal for imaging multiple wavelengths simultaneously (Friedman and Gelles, 2015; Friedman et al., 2006). After collection of fluorescence emission by the objective lens, an image was formed using a tube lens (Edmund Optics) with a 400 mm focal length, resulting in 133.33X magnification. The red and far-red channels were split using a T635lpxr dichroic mirror (Chroma), passed through a set of relay lenses, and collected side-by-side on the camera chip. The relay lenses introduced an additional 1.2X magnification, so that the total system magnification was 160X. JF585 and JF646 were simultaneously excited using 50mW 561 nm laser and 50mW 637 nm laser.
QUANTIFICATION AND STATISTICAL ANALYSIS
Cortical cluster intensity quantification
We used Utrack (Jaqaman et al., 2008) to detect the particle and to calculate the particle brightness in TIRF images. For particle detection, we used the point source detection algorithm with alpha = 0.01 and other parameters as default. In order to uncouple the effect on cluster size from cortical tension(Wang et al., 2017b), the movie was acquired using embryos dissected from MLC-4 RNAi treated worms (Figure 1E).
Cortical flow speed quantification
Adult C.elegans carrying eggs were dissected in a drop of egg buffer on polylysine-coated coverslips. The embryos were gently flattened by mounting with 22.8 mm beads (Whitehouse scientific, Chester, UK) as spacers. DIC movies were acquired using a Nikon Eclipse Ti-2 microscope equipped with a 100X, 1.49 NA objective; a Photometrics Prime 95B camera, and Nomarski DIC optics. DIC images were magnified by a 1.5X tube lens before being collected by camera chip. One-cell stage embryos at establishment phase were imaged at 1 s per frame. Kymographs were generated by extracting a 20 pixel wide strip at the center of the imaged embryo image and stacked these strips on top of one another using the “montage” command in FIJI. The angle of a slope on the posterior was measured and the cortical flow speed in μm/min was calculated by:
Cell polarization quantification and fluorescence measurements
To quantify the polarization ability of our EOD strains, ovals matching the shape of the anterior half and posterior half of the embryo were drawn and the mean intensity per pixel was quantified using the Measure command in FIJI. For each embryo, the anterior intensity is normalized to 1 and the ratio of posterior intensity/anterior intensity was calculated.
Embryo lethality and sterility quantification
A single young adult was picked to a new plate. After 12hrs, which allows the adult to lay eggs, the adult was removed from the plate. After another 24hrs during which the eggs were allowed to hatch, any unhatched embryos and hatched larvae were counted and embryonic lethality was calculated. The plate was maintained for additional 3 days for counting the number of fertile adults and sterile adults, which is used for calculating adult sterility.
PAR-3 particle tracking and motion analysis
We used Utrack (Jaqaman et al., 2008) to track the motion of PAR-3 particles in TIRF images. For particle detection, we used the point source detection algorithm with alpha = 0.01 for EOD data and alpha = 0.03 for dual-labeling data, with other parameters as default. For the particle tracking step, we used Brownian + directed motion mode with minimum track length = 2 and maximum GAP to close = 3 for both EOD and dual-labeling experiments.
MSD analysis and anomalous parameter visualization
Data from UTrack were converted to a suitable format using a custom MATLAB script, and then fed into the MSDanalyzer MATLAB function (Tarantino et al., 2014) to generate MSD plots. We eliminated tracks shorter than 10 frames (0.5 s of data), because we observed that UTrack sometimes erroneously identified short-lived particles in the noise of the images. To estimate slopes, we fit each averaged log-log MSD curve to a smoothing spline using the SLM toolbox for MATLAB. To avoid overfitting noisy data, the ends of each curve were trimmed prior to fitting. Fits were constrained to be increasing functions and, where appropriate, concave-up. The slope of the curve is then estimated by taking the first derivative of the fitted spline.
Particle image velocimetry quantification
To quantify the coupled motion of PAR-3 and actin filaments network (Figure S2B), we applied velocimetry (PIV) to the mSc:PAR-3 and GFP:Utrophin image channel using the PIVlab MATLAB plugin (Thielicke and Stamhuis, 2014). For mSc:PAR-3 channel: Images were pre-processed with the high-pass filter = 50, Wiener2 denoise filter = 5. The contrast was manually adjusted. Other preprocessing filters were disabled. Flow detection used the default FFT phase-space algorithm with 3 passes (window sizes 64, 32 and 16 pixels) and linear window deformation. Post-processing was done with velocity limits drawn manually to exclude outliers. Vectors that were rejected by these filters were replaced by interpolation. For Utrophin channel: Images were pre-processed with the high-pass filter = 400, Wiener2 denoise filter = 2. Other processing methods were the same as for mSc:PAR-3 channel.
Pearson’s correlation
To quantify the correlation of velocity in PAR-3 and cortex (Figure S2C), the data was organized to a three dimensional matrix, with size of (x-dimension, y-dimension, 2 (x-vector/y-vector)). Pearson’s correlation was calculated by MATLAB Command: corrcoef (Utrophin velocity matrix, PAR-3 velocity matrix).
To quantify the colocalization of PAR-3 and actin, we imported the image sequence of mSc:PAR-3 and GFP:Utrophin into the MATLAB as a numerical matrix. Data was organized to a three dimensional matrix, with size of (x-pixel position, y-pixel position, t-time resolution). Pearson’s correlation was calculated by MATLAB Command: corrcoef (Utrophin velocity matrix, PAR-3 velocity matrix).
Calculating diffusion coefficients
The PAR-3 tracks information generated by Utrack was reformatted for the ‘msdanalyzer’ MATLAB function, and the results were processed by ‘msdanalyzer’ for generating MSD curves (Tarantino et al., 2014). The built-in ‘fitMeanMSD’ function of msdanalyzer was used to fit the first 6 time steps of each MSD curve to straight line, which has a slope of 4*D.
Membrane half-life and koff quantification
To quantify the membrane half-life of EOD:PAR-3 clusters (Figure 5E), we tracked YFP:PAR-3 clusters using Utrack. The Utrack output was translated into a more readable format using a custom-written MATLAB script. The membrane half was calculated from the dwell times of individual particles at the membrane, as follows (Kinz-Thompson et al., 2016):
where the ndisappear is the number of particle disappearance events, and nnondisappear is the sum of track length of all tracks;
where τ is the measurement interval, which is 50ms for our experiments; and
The 95% confidence intervals were calculated from the β distribution using the MATLAB function betaincinv (w, y, z, ‘lower/upper’). Where w = 0.025 for defining a 95% confidence interval, y = 1 + #of disappearing events(# of tracks), z = 1 + #of non-disappearing events.
Polarity state quantification for simulated embryos
To characterize the polarization state of a simulated embryo (Figure 5D), we calculated the percentage of clusters present in the anterior domain as a function of time. The anterior domain is defined as the left side of the cleavage furrow, which is 57% percent of the embryo length (Dickinson et al., 2017). There are 36,000 time intervals in each simulated segregation, which causes sharp fluctuations. We therefore smooth the data before plotting using MATLAB function ‘smooth’, with smooth range set to 3600. The average at each time point across 10 simulations were calculated for generating the black average line.
As a second approach (Figure 5E), we extracted the x coordinates from final cluster positions. The coordinates were binned into 6μm x axis ranges and the number of clusters in each bin were counted.
Dual-labeling experiments data analysis
We calibrated JF646 intensity using control experiments in which both JF585 and JF646 were diluted to single-molecule levels. As expected, the measured fluorescence intensities of single dye molecules followed a log-normal distribution (Figure S4A and S4B) (Mutch et al., 2007). The measured single-fluorophore intensities varied from embryo to embryo, which is not surprising and likely reflects variations in mounting and eggshell thickness; importantly, however, the intensities of the red and far-red channels varied in tandem (Figure S4A). We therefore adopted the following calibration procedure, in which we used the red dye intensity as an internal standard, since it is present at single-molecule levels in both sets of experiments. First, we normalized our double-dilution datasets to the red dye intensity. Normalization was done in log space, to account for the log-normal shape of the intensity distribution, and was effective at correcting for embryo-to-embryo variations in fluorescence intensity (Figures S4A and S4C). Second, we calculated the ratio of the average far-red dye intensity to the average red dye intensity (Figure S4D). Third, the intensity of red dyes in each sparse/abundant dual-labeling experiment was measured, and the intensities were normalized to the mean red dye intensity to allow comparison between embryos (Figures S4E–S4H). The cluster size information of PAR-3 labeled by red (JF585) fluorophore was acquired by a custom-written MATLAB script. This script draws a box in the far-red channel at the location where the red signal is located, and calculates the fluorescence intensity in the far-red channel by subtracting local background from a larger box. Fourth, from the calibrated red/far-red intensity ratio, an expected mean far-red intensity per molecule was calculated. The source code for our software is available at https://github.com/IvyChang1994/getClusterSize.git. Finally, this calculated far-red intensity per molecule was used to estimate the number of far-red labeled Halo:PAR-3 molecules in each tracked cluster. This calibration procedure accounts for the differences in global fluorescence intensity between embryos, presumably caused by the differences in laser penetration depth and eggshell thickness, and allows us to estimate the number of PAR-3 molecules per cluster.
Supplementary Material
Highlights.
PAR-3 trimers are necessary and sufficient for C. elegans zygote polarization
Monomers and dimers of PAR-3 bind transiently to the plasma membrane and diffuse freely
PAR-3 trimers bind the membrane more strongly and experience advective transport
New techniques facilitate diffusion analysis of native membrane-bound oligomers
ACKNOWLEDGMENTS
We thank Luke Lavis for sharing JaneliaFluor dyes prior to publication and Charles Lang, Edwin Munro, Jeanne Stachowiak, John Wallingford, and members of the Dickinson lab for helpful discussions and comments on the manuscript. This work was supported by a Provost’s Graduate Education Fellowship from the University of Texas at Austin (Y.C.), by a research grant from the Mallinckrodt Foundation (D.J.D.), and by NIH R00 GM115964 and R01 GM138443 (D.J.D.). D.J.D. is a CPRIT Scholar supported by the Cancer Prevention and Research Institute of Texas (RR170054). Some strains were provided by the Caenorhabditis Genetics Center, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110652.
REFERENCES
- Allen NS, and Allen RD (1978). Cytoplasmic streaming in green plants. Annu. Rev. Biophys. Bio 7, 497–526. [DOI] [PubMed] [Google Scholar]
- Ananthakrishnan R, and Ehrlicher A (2007). The forces behind cell movement. Int. J. Biol. Sci 3, 303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arata Y, Hiroshima M, Pack C-G, Ramanujam R, Motegi F, Nakazato K, Shindo Y, Wiseman PW, Sawa H, Kobayashi TJ, et al. (2016). Cortical polarity of the RING protein PAR-2 is maintained by exchange rate kinetics at the cortical-cytoplasmic boundary. Cell Rep. 16, 2156–2168. [DOI] [PubMed] [Google Scholar]
- Benton R, and Johnston DSt. (2003).A conserved oligomerization domain in Drosophila bazooka/PAR-3 is important for apical localization and epithelial polarity. Curr. Biol 13, 1330–1334. [DOI] [PubMed] [Google Scholar]
- Bolten M, Delley CL, Leibundgut M, Boehringer D, Ban N, and Weber-Ban E (2016). Structural analysis of the bacterial proteasome activator bpa in complex with the 20S proteasome. Structure 24, 2138–2151. [DOI] [PubMed] [Google Scholar]
- Boudko SP, Sasaki T, Engel J, Lerch TF, Nix J, Chapman MS, and Bächinger HP (2009). Crystal structure of human collagen XVIII trimerization domain: a novel collagen trimerization fold. J. Mol. Biol 392, 787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner CR, Chechik M, Ortiz-Lombardía M, Smits C, Ebong I-O, Chechik V, Jeschke G, Dykeman E, Benini S, Robinson CV, et al. (2012). Structural basis for DNA recognition and loading into a viral packaging motor. Proc. Natl. Acad Sci 109, 811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Chen J, Shi H, Wei M, Castaneda-Castellanos DR, Bultje RS, Pei X, Kriegstein AR, Zhang M, and Shi S-H (2013). Regulation of microtubule stability and organization by mammalian Par3 in specifying neuronal polarity. Dev. Cell 24, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chik JK, Moiseeva V, Goel PK, Meinen BA, Koldewey P, An S, Mellone BG, Subramanian L, and Cho U-S (2019). Structures of CENP-C cupin domains at regional centromeres reveal unique patterns of dimerization and recruitment functions for the inner pocket. J. Biol. Chem 294, 14119–14134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenca AA, Schetter A, Aceto D, Kemphues K, and Seydoux G (2003). Polarization of the C. elegans zygote proceeds via distinct establishment and maintenance phases. Development 130, 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Pani AM, Heppert JK, Higgins CD, and Goldstein B (2015). Streamlined genome engineering with a self-excisingdrug selection cassette. Genetics 200, 1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Schwager F, Pintard L, Gotta M, and Goldstein B (2017). A single-cell biochemistry approach reveals PAR complex dynamics during cell polarization. Dev. Cell 42, 416–34.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, and Goldstein B (2013). Engineering the Caenorhabditis elegan genome using Cas9-triggered homologus recombination. Nature Methods 10, 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drulyte I, Obajdin J, Trinh CH, Kalverda AP, van der Kamp MW, Hemsworth GR, and Berry A (2019). Crystal structure of the putative cyclase IdmH from the indanomycin nonribosomal peptide synthase/polyketide synthase. Iucrj 6, 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LJ, and Gelles J (2015). Multi-wavelength single-molecule fluorescence analysis of transcription mechanisms. Methods 86, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LJ, Chung J, and Gelles J (2006). Viewing dynamic assembly of molecular complexes by multi-wavelength single-molecule fluorescence. Biophys. J 91, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring NW, Trong PK, Bois JS, Chowdhury D, Nicola EM, Hyman AA, and Grill SW (2011a). Polarization of PAR proteins by advective triggering of a pattern-forming system. Science 334, 1137–1141. [DOI] [PubMed] [Google Scholar]
- Goehring NW, Hoege C, Grill SW, and Hyman AA (2011b). PAR proteins diffuse freely across the anterior-posterior boundary in polarized C. elegans embryos. J. Cell Biol 193, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell DS, and Olson AJ (2000). Structural symmetry and protein function. Annu. Rev. Bioph Biom 29, 105–153. [DOI] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. (2015). A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 12, 244–250, 3 p following 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Choi H, Muthusamy AK, Mehl BP, Dong P, Brown TA, Lippincott-Schwartz J, Liu Z, Lionnet T, et al. (2016). Bright photoactivatable fluorophores for single-molecule imaging. Nat. Methods 13, 985–988. [DOI] [PubMed] [Google Scholar]
- Grimm JB, Muthusamy AK, Liang Y, Brown TA, Lemon WC, Patel R, Lu R, Macklin JJ, Keller PJ, Ji N, et al. (2017). A general method to finetune fluorophores for live-cell and in vivo imaging. Nat. Methods 14, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross P, Kumar KV, Goehring NW, Bois JS, Hoege C, Jülicher F, and Grill SW (2019). Guiding self-organized pattern formation in cell polarity establishment. Nat. Phys 15, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y (2018). aPKC: the kinase that phosphorylates cell polarity. F1000research 7, F1000, Faculty Rev-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P-S, Oberdorfer G, Xu C, Pei XY, Nannenga BL, Rogers JM, DiMaio F, Gonen T, Luisi B, and Baker D (2014). High thermodynamic stability of parametrically designed helical bundles. Science 346, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner RJ, Malmi-Kakkada AN, Sarikaya S, Weng S, Thirumalai D, and Wallingford JB (2021). Mechanical heterogeneity along single cell-cell junctions is driven by lateral clustering of cadherins during vertebrate axis elongation. Elife 10, e65390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, and Danuser G (2008). Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 5, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joberty G, Petersen C, Gao L, and Macara IG (2000). The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat. Cell Biol 2, 531–539. [DOI] [PubMed] [Google Scholar]
- Kinz-Thompson CD, Bailey NA, and Gonzalez RL (2016). Chapter seven precisely and accurately inferring single-molecule rate constants. Methods Enzymol. 581, 187–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CF, and Munro E (2017). The PAR proteins: from molecular circuits to dynamic self-stabilizing cell polarity. Development 144, 3405–3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Patrick JW, and Masson M (1968). Crystalline L-ribulose 5-phosphate 4-epimerase from Escherichia coli. J. Biol. Chem 243, 4700–4705. [PubMed] [Google Scholar]
- Lénárt P, Bacher CP, Daigle N, Hand AR, Eils R, Terasaki M, and Ellenberg J (2005). A contractile nuclear actin network drives chromosome congression in oocytes. Nature 436, 812–818. [DOI] [PubMed] [Google Scholar]
- Li B, Kim H, Beers M, and Kemphues K (2010). Different domains of C. elegans PAR-3 are required at different times in development. Dev. Biol 344, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Chai Y, Song H, Weng C, Qi J, Sun Y, and Gao GF (2019). Crystal structure of african swine fever virus dUTPase reveals a potential drug target. Mbio 10, e02483–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Samuel J, Mosimann SC, Lee JE, Tanner ME, and Strynadka NCJ (2001). The structure of l -Ribulose-5-Phosphate 4-epimerase: an aldolase-like platform for epimerization. Biochemistry-Us 40, 14763–14771. [DOI] [PubMed] [Google Scholar]
- Mayer M, Depken M, Bois JS, Jülicher F., and Grill SW. (2010). Anisotropies in cortical tension reveal the physical basis of polarizing cortical flows. Nature 467, 617–621. [DOI] [PubMed] [Google Scholar]
- Mehta A (2001). Myosin learns to walk. J. Cell Sci 114, 1981–1998. [DOI] [PubMed] [Google Scholar]
- Mittasch M, Gross P, Nestler M, Fritsch AW, Iserman C, Kar M, Munder M, Voigt A, Alberti S, Grill SW, et al. (2018). Non-invasive perturbations of intracellular flow reveal physical principles of cell organization. Nat. Cell Biol 20, 344–351. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Suzuki A, Hirose T, Kitamura K, Kutsuzawa K, Futaki M, Amano Y, and Ohno S (2003). Self-association of PAR-3-mediated by the conserved N-terminal domain contributes to the development of epithelial tight junctions. J. Biol. Chem 278, 31240–31250. [DOI] [PubMed] [Google Scholar]
- Munro E, Nance J, and Priess JR (2004). Cortical flows powered by asymmetrical contraction transport PAR proteins to establish and maintain anterior-posterior polarity in the early C. elegans embryo. Dev. Cell 7, 413–424. [DOI] [PubMed] [Google Scholar]
- Mutch SA, Fujimoto BS, Kuyper CL, Kuo JS, Bajjalieh SM, and Chiu DT (2007). Deconvolving single-molecule intensity distributions for quantitative microscopy measurements. Biophys. J 92, 2926–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JF, Lim K, Tempczyk A, Krajewski W, Eisenstein E, and Herzberg O (2002). From structure to function: YrbI from Haemophilus influenzae (HI1679) is a phosphatase. Proteins Struct. Funct. Bioinform 46, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin FB, McFadden WM, Yao B, and Munro EM (2014). Single-molecule analysis of cell surface dynamics in Caenorhabditis elegans embryos. Nat. Methods 11, 677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Peglion F, Martin J, Hubatsch L, Reich J, Hirani N, Gubieda AG, Roffey J, Fernandes AR, Johnston DS, et al. (2017). aPKC cycles between functionally distinct PAR protein assemblies to drive cell polarity. Dev. Cell 42, 400–415.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailer A, Anneken A, Li Y, Lee S, and Munro E (2015). Dynamic opposition of clustered proteins stabilizes cortical polarity in the C. elegans zygote. Dev. Cell 35, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Frangos A, Fröhlich KS, Jeliazkov JR, Małecka EM, Marino G, Gray JJ, Luisi BF, Woodson SA, and Hardwick SW (2019). Caulobacter crescentus Hfq structure reveals a conserved mechanism of RNA annealing regulation. Proc. Natl. Acad Sci 116, 201814428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarikaya S, and Dickinson DJ (2021). Rapid extraction and kinetic analysis of protein complexes from single cells. Biophys. J 120, 5018–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. (2000). Cell signaling by receptor tyrosine kinases. Cell 103, 211–225. [DOI] [PubMed] [Google Scholar]
- Shelton CA, Carter JC, Ellis GC, and Bowerman B (1999). The non-muscle myosin regulatory light chain gene mlc-4 is required for cytokinesis, anterior-posterior polarity, and body morphology during Caenorhabditis elegans embryogenesis. J. Cell Biol 146, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolpner N, and Dickinson DJ (2022). Single-cell single-molecule pull-down (sc-SiMPull) for detection of protein complexes from embryonic lysates. Methods Mol. Biol 2438, 59–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Young LN, Zhang X, Boudko SP, Fokine A, Zbornik E, Roznowski AP, Molineux IJ, Rossmann MG, and Fane BA (2014). Icosahedral bacteriophage ΦX174 forms a tail for DNA transport during infection. Nature 505, 432–35. [DOI] [PubMed] [Google Scholar]
- Tarantino N, Tinevez J-Y, Crowell EF, Boisson B, Henriques R, Mhlanga M, Agou F, Israël A, and Laplantine E (2014). TNF and IL-1 exhibit distinct ubiquitin requirements for inducing NEMO–IKK supramolecular structuresNEMO–IKK recruitment to supramolecular structures. J. Cell Biol 204, 231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thielicke W, and Stamhuis EJ (2014). PIVlab – towards user-friendly, affordable and accurate digital particle image velocimetry in MATLAB. J. Open Res. Softw 2, e30. [Google Scholar]
- Thomson AR, Wood CW, Burton AJ, Bartlett GJ, Sessions RB, Brady RL, and Woolfson DN (2014). Computational design of water-soluble α-helical barrels. Science 346, 485–488. [DOI] [PubMed] [Google Scholar]
- Tse YC, Werner M, Longhini KM, Labbe J-C, Goldstein B, and Glotzer M (2012). RhoA activation during polarization and cytokinesis of the early Caenorhabditis elegans embryo is differentially dependent on NOP-1 and CYK-4. Mol. Biol. Cell 23, 4020–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veesler D, Blangy S, Lichière J, Ortiz-Lombardía M, Tavares P, Campanacci V, and Cambillau C. (2010). Crystal structure of Bacillus subtilis SPP1 phage gp23.1, a putative chaperone. Protein Sci. 19, 1812–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Tang NH, Lara-Gonzalez P, Zhao Z, Cheerambathur DK, Prevo B, Chisholm AD, Desai A, and Oegema K (2017a). A toolkit for GFP-mediated tissue-specific protein degradation in C. elegans. Development 144, 150094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S-C, Low TYF, Nishimura Y, Gole L, Yu W, and Motegi F (2017b). Cortical forces and CDC-42 control clustering of PAR proteins for Caenorhabditis elegans embryonic polarization. Nat. Cell Biol 19, 988–995. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, and Watanabe N (2017). Overview of single-molecule speckle (SiMS) microscopy and its electroporation-based version with efficient labeling and improved spatiotemporal resolution. Sensors 17, 1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original microscopy data are available from the lead contact upon reasonable request.
All original code generated in this study is available via Github (see the Key Resources Table for links).
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-HaloTag pAb | Promega | Cat# G9281; RRID:AB_713650 |
| Anti-mNeonGreen nAb | Allele Biothechnology | Cat# ABP-NAB-MNGAB; RRID: AB_2629489 |
| Chemicals | ||
| JF585 | Laboratory of Luke D. Lavis | Grimm et al. (2017),Grimm et al. (2015),Grimm et al. (2016) |
| JF646 | Laboratory of Luke D. Lavis | Grimm et al. (2017),Grimm et al. (2015),Grimm et al. (2016) |
| DMSO | Sigma | D2650-5X5ML |
| Genetic fragments | ||
| GFP nano body | add gene | 89,367 |
| 6HNL | PDB | 6HNL |
| 6O2D | PDB | 6O2D |
| 8KY8 | PDB | 8KY8 |
| 3HON | PDB | 3HON |
| 3XF6 | PDB | 3XF6 |
| 6GWK | PDB | 6GWK |
| 3ZQM | PDB | 3ZQM |
| 5LFJ | PDB | 5LFJ |
| 3H5L | Huang et al. (2014) | 3H5L_2_mini |
| HOTag3 | Thomson et al. (2014) | HOTag3 |
| H protein | Sun et al. (2014) | H protein |
| Yrb-1 | Parsons et al. (2002) | Yrb-1 |
| AraD | Luo et al. (2001) | AraD |
| Experimental models: Organisms/strains | ||
| C. elegans wild-type strain | Caenorhabditis Genetics Center | N2 |
| C.elegans transgenic strain expressing GFP::UTRO Genotype: xsSi3 [GFP::utrophin + Cbr-unc-119(+)] |
Tse et al. (2012) | MG589 |
| C. elegans trains with endogenously tagged NMY-2::mKate2 Genotype: nmy-2(cp69[nmy-2::mkate2 + LoxP]) I |
Dickinson et al., (2017) | LP256 |
| C.elegans strain with endogenously tagged mNG::PAR-3 Genotype: par-3(cp54[mNeonGreen::3xFlag::par-3]) III |
Dickinson et al. (2017) | LP242 |
| C.elegans strain with endogenously tagged mSc::PAR-3 and transgenenic GFP::UTRO Genotype: par-3(djd18[EYFP-C1dPi::par-3]) III; xsSi3 [GFP::utrophin + Cbr-unc-119(+)] II |
this study | UTX136 |
| C.elegans strain withendogenously tagged NMY-2::mKate and mNG::PAR-3 Genotype: nmy-2(cp52[nmy-2::mkate2 + LoxP unc-119(+) LoxP]) I; par-3(cp54[mNeonGreen::3xFlag::par-3]) III |
this study | LP636 |
| C.elegans strain withendogenously tagged Halo::PAR-3 par-3(cp323[HaloTag-GLO PAR-3]) III | Dickinson et al. (2017) | LP621 |
| C.elegans transgenic strain expressing GFPnAb::HaloTag Genotypes: djdIs19 [Psun-1::GFPnb::HALO::3xFlag + SEC] II |
this study | UTX44 |
| C.elegans transgenic strain expressing nAb::HaloTag::6mer(HOTag3) Genotype:djdIs7 [Psun-1::GFPnb::HALO::3xFlag::HexamerD + SEC] II |
this study | UTX17 |
| C.elegans transgenic strain expressing nAb::HaloTag::3mer(3H5L_2_mini) Genotype: djdIs11 [Piffb-1::GFPnb::HALO::3xFlag::TrimerD + SEC] II |
this study | UTX21 |
| C.elegans transgenic strain expressing nAb::HaloTag::10mer(H protein) Genotype: djdIs15 [Piffb-1::GFPnb::HALO::3xFlag::DecamerD + SEC] II |
this study | UTX25 |
| C.elegans strain with endogenously tagged YFP::PAR-3 Genotype: par-3(djd18[EYFP-C1dPi::par-3]) III |
this study | UTX52 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 Genotype: par-3(djd5 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX30 |
| C.elegans transgenic strain expressing mNG::6HNL Genotype: djdIs22 [Pmex5::mNG::3XFLAG::6HNLdimer] II |
this study | UTX66 |
| C.elegans transgenic strain expressing mNG::3HON Genotype: djdIs24 [Pmex5::mNG::3XFLAG::3HON3mer] II |
this study | UTX68 |
| C.elegans transgenic strain expressing mNG::2XF6 Genotype: djdIs25 [Pmex5::mNG::3XFLAG::2XF6hexamer] II |
this study | UTX69 |
| C.elegans transgenic strain expressing mNG::6GWK Genotype: djdIs27 [Pmex5::mNG::3XFLAG::GWKhexamer] II |
this study | UTX71 |
| C.elegans transgenic strain expressing mNG::3ZQM Genotype: djdIs30 [Pmex5::mNG::3XFLAG::3ZQM10mer] II |
this study | UTX74 |
| C.elegans transgenic strain expressing mNG::5LFJ Genotype: djdIs32 [Pmex5::mNG::3XFLAG::5LFJ12mer] II |
this study | UTX76 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::HaloTag::6mer(HOTag3) Genotype: djdIs7 [Psun-1::GFPnb::HALO::3xFlag::HexamerD + SEC] II; par-3(djd6) [EYFP-C1dPi::PAR-3(RRKEEE)] III |
this study | UTX47 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::Halo Genotype: djdIs21 [Psun-1::GFPnb::HALO::3xFlag + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX50 |
| C.elegans transgenic strain expressing nAb::BFP::2mer(6HNL) Genotype: djdIs34 [Psun-1::GFPnb::mTagBFP2::6HNLdimer + SEC] II |
this study | UTX86 |
| C.elegans transgenic strain expressing nAb::BFP::3mer(3HON) Genotype: djdIs35 [Psun-1::GFPnb::mTagBFP2::3HONtrimer + SEC] II |
this study | UTX88 |
| C.elegans transgenic strain expressing nAb::BFP::4mer(AraD) Genotype:djdIs38 [Psun-1::GFPnb::mTagBFP2::AraD tetramer + SEC] II |
this study | UTX92 |
| C.elegans transgenic strain expressing nAb::BFP::4mer(Yrb1) Genotype: djdIs39 [Psun-1::GFPnb::mTagBFP2::Yrb1 tetramer + SEC] II |
this study | UTX94 |
| C.elegans transgenic strain expressing nAb::BFP::6mer(6GWK) Genotype: djdIs36 [Psun-1::GFPnb::mTagBFP2::2XF6hexamer + SEC] II |
this study | UTX89 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::BFP::2mer(6HNL) Genotype: djdIs34 [Psun-1::GFPnb::mTagBFP2::6HNLdimer + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX105 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::BFP::3mer(3HON) Genotype: djdIs34 [Psun-1::GFPnb::mTagBFP2::3HONtrimer + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX80 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::BFP::4mer(AraD) Genotype: djdIs38 [Psun-1::GFPnb::mTagBFP2::AraD tetramer + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX104 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::BFP::4mer(Yrb-1) djdIs34 [Psun-1::GFPnb::mTagBFP2::Yrb1 tetramer + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III | this study | UTX81 |
| C. elegans strain with RRKEEE mutations and an N-terminal YFP in endogenous par-3 and trasgenic nAb::BFP::6mer(6GWK) Genotype: djdIs36 [Psun-1::GFPnb::mTagBFP2::6GWKhexamer + SEC] II; par-3(djd6 [EYFP-C1dPi::PAR-3(RRKEEE)]) III |
this study | UTX106 |
| C.elegans transgenic strain expressing LsfGFP::SNAPf Genotype: cpIs95[Pmex-5>sfGFP-GLO::3xFlag::SNAPf + SEC] II |
this study | LP566 |
| Software and Algorithms | ||
| Utrack | Jaqaman et al. (2008) | Version 2.2.0; https://github.com/DanuserLab/u-track |
| Msdanalyzer | Tarantino et al. (2014) | https://tinevez.github.io/msdanalyzer/ |
| Ezyfit | MATLAB File Exchange | Version 2.53; http://www.fast.u-psud.fr/ezyfit |
| GetClustersize | This paper | https://github.com/IvyChang1994/getClusterSize.git |
| PIVLab | Thielicke and Stamhuis (2014) | Version 2.50; http://pivlab.blogspot.com |
| PAR-3 diffusion simulation | This paper | https://github.com/IvyChang1994/Par3-Modeling.git |