

Graphical abstract

Keywords: SARS-CoV-2, Coronavirus, COVID-19, Nsp3 macrodomain inhibitors, ADP-ribosylation
Abstract
A series of amino acid based 7H-pyrrolo[2,3–d]pyrimidines were designed and synthesized to discern the structure activity relationships against the SARS-CoV-2 nsp3 macrodomain (Mac1), an ADP-ribosylhydrolase that is critical for coronavirus replication and pathogenesis. Structure activity studies identified compound 15c as a low-micromolar inhibitor of Mac1 in two ADP-ribose binding assays. This compound also demonstrated inhibition in an enzymatic assay of Mac1 and displayed a thermal shift comparable to ADPr in the melting temperature of Mac1 supporting binding to the target protein. A structural model reproducibly predicted a binding mode where the pyrrolo pyrimidine forms a hydrogen bonding network with Asp22 and the amide backbone NH of Ile23 in the adenosine binding pocket and the carboxylate forms hydrogen bonds to the amide backbone of Phe157 and Asp156, part of the oxyanion subsite of Mac1. Compound 15c also demonstrated notable selectivity for coronavirus macrodomains when tested against a panel of ADP-ribose binding proteins. Together, this study identified several low MW, low µM Mac1 inhibitors to use as small molecule chemical probes for this potential anti-viral target and offers starting points for further optimization.
1. Introduction
The current outbreak of SARS-CoV-2 and other recent outbreaks of highly pathogenic human coronaviruses (CoVs) has exposed the challenges posed by CoVs and the lack of antivirals available to target CoVs and other viruses of pandemic potential. The development of novel antivirals targeting conserved CoV proteins that could counter emerging CoVs is greatly needed, and many different approaches are currently being pursued.1 One potential novel CoV drug target is the highly conserved macrodomain (Mac1), which is a domain within the large non-structural protein 3 (nsp3) protein. Mac1 is present in all CoVs, and contains a conserved three-layered α/β/α fold, typical of all macrodomains. All CoV Mac1 proteins tested have ADP-ribose binding (reader) and ADP-ribosylhydrolase (ARH) (eraser) activity and are members of the larger MacroD-type macrodomain family, which includes human macrodomains Mdo1 and Mdo2.2 Importantly, several studies have demonstrated that Mac1 is essential for CoV-mediated pathogenesis in multiple animal models of infection, indicating that Mac1 may be a suitable drug target.3
ADP-ribosylation is a dynamic post-translational modification catalyzed by ADP-ribosyltransferases (ARTs, a.k.a. Sirtuins, ARTCs and PARPs) where ADP-ribose is transferred from NAD+ onto target proteins or to DNA/RNA.4 ADP-ribose can be transferred as a mono-ADP-ribose (MAR), or consecutively attached units of MAR covalently attached through glycosidic bonds to preceding ADP-ribose units to form a branched or linear poly-ADP-ribose (PAR) chain. ADP-ribose can then be removed from proteins through several different eraser enzymes, such as PAR glycohydrolase (PARG), ADP-ribosylhydrolases (ARHs), and macrodomains. Although these proteins catalyze similar reactions, the families are structurally distinct.2 ADP-ribosylation is known to play important biological roles in cellular processes such as the DNA damage response, cellular signaling, cell cycle control, ER stress, immune regulation, and many others. Furthermore, both mono- and poly-ARTs can inhibit virus replication, implicating ADP-ribosylation in the host innate immune response to infection.5 Furthermore, in addition to CoVs, several families of viruses encode for macrodomains including the Coronaviridae, Togaviridae, Matonaviridae, Hepeviridae, and the Iridoviridae, indicating a significant evolutionary advantage for some viruses to directly counter ADP-ribosylation.
Multiple studies have reported that Mac1 is critical for CoV replication and pathogenesis. This has largely been accomplished using reverse genetics to mutate a highly conserved asparagine to alanine (N41A-SARS-CoV). This mutation nearly eliminates the enzymatic activity of a recombinant SARS-CoV macrodomain protein in vitro.6 This mutation had only a limited impact on CoV replication in transformed cells, but in animal models these mutant viruses proved highly attenuated, with low viral loads, increased IFN production, and the inability to cause significant disease.[6], [7], [8], [9], [10] Recombinant murine hepatitis virus strain JHM (MHV-JHM) with this same mutation (N1347A) replicated poorly in primary macrophages, but importantly this defect could be partially rescued by the PARP inhibitors or siRNA knockdown of PARP12 or PARP14.11 These data indicate that Mac1 likely counters PARP-mediated anti-viral ADP-ribosylation. Further genetic analysis found that mutations in MHV Mac1 predicted to alter ADP-ribose binding and hydrolysis resulted in severe replication defects in cell culture (D1329A) or could not be recovered (G1439V, D1329A/N1347A), indicating that for some CoVs Mac1 may be essential for replication.12 Looking beyond CoVs, mutations in Chikungunya virus, Sindbis virus, and Hepatitis E virus macrodomains also have severe phenotypic effects on virus replication and pathogenesis.[13], [14], [15], [16], [17] As viral macrodomains are clearly important virulence factors, they are potential targets for anti-viral therapeutics.3
Since the emergence of SARS-CoV-2, there has been an increased effort to identify compounds that bind to or inhibit the activity of the SARS-CoV-2 Mac1 protein. Most of these studies only involved molecular modeling of potential inhibitors, though a few have tested compound activity in biochemical assays.[18], [19], [20], [21], [22] Virdi et al. used a differential scanning fluorimetry (DSF) assay to screen ∼ 2500 compounds, and while they identified several compounds that altered the melting temperature of the protein, it’s yet unclear if any of these compounds can inhibit Mac1 activity.21 Russo et al. developed an immunofluorescence assay to measure Mac1 activity in cells. They demonstrated that poly(I:C) or IFNγ induced global MARylation in A549 cells that could be removed by expression of Mac1, but not the N1040A mutant protein that has minimal enzymatic activity.19 They utilized this assay in a small inhibitor screen but did not identify any significant hits. Despite this, the assay could be a highly useful tool for measuring compound activity in cells. Dasovich et al. developed a novel ADP-ribosylhydrolase assay that utilizes the ability of NudF to specifically cleave AMP from ADP-ribose released from ADP-ribosylated proteins in the presence of macrodomain, but not from untreated ADP-ribosylated protein.18 The generated AMP can then be measured in high-throughput fashion. The authors screened over 3000 compounds using this assay and identified 2 compounds that inhibited mono-ARH activity. One, dihydralazine, inhibited both Mdo2 and the SARS-CoV Mac1 domain with high IC50 values (∼0.5 mM), while the other compound, Dasatinib, specifically inhibited CoV Mac1 mono-ARH activity with an IC50 of ∼50 μM. While Dasatinib is toxic to cells at higher concentrations, it may be a suitable scaffold for further inhibitor development. A FRET assay developed by Sowa et al. was used to test approved drugs for their ability to inhibit several macrodomains. Suramin was found to be an inhibitor of Mac1 with an IC50 of ∼8.7 µM, and it also showed concentration dependent stabilization of the protein in DSF assay. However, suramin has multiple reported activities and therefore does not represent a high quality chemical probe for Mac1 inhibition.22
Recently, a screen was conducted in a multi-national collaboration to identify several small molecule fragments as weak inhibitors of the SARS-CoV-2 nsp3 Mac1.20 One such inhibitor, a fragment with a pyrrolo pyrimidine core (blue heterocycle, Figure 1 ), compound 1, is shown in Figure 1 below. This compound demonstrated weak potency against Mac1 in an ADP-ribose binding AlphaScreen™ (amplified luminescence proximity homogeneous assay) assay (IC50 = 180 μM). In addition, the crystal structure of this compound in the active site of the nsp3 macrodomain demonstrated a hydrogen bonding network between the pyrrolo pyrimidine and the side chain of Asp22 and the amide backbone NH of Ile23 in the adenosine binding site of the nsp3 macrodomain (Figure 1).20 In addition, one crystal structure showed the side chain carboxylate of 1 (red, Figure 1) forming hydrogen bonds with the backbone amides of Phe157 and Asp156, part of the oxyanion subsite. This pyrrolo pyrimidine core was also part of another fragment, compound 2, (IC50 = 400 μM), with the same binding mode. Taken together, these data provide a good starting point for optimization for several reasons: 1) the molecular weights of these two fragment hits are < 300, allowing for fragment growth strategies that will leave the final inhibitors within drug like parameters; 2) the pyrrolo pyrimidine core was the most common core found among the hits and it has a conserved/predictable binding mode; 3) proximity to oxyanion subsite and phosphate subsites to further block ADPr binding/recognition and improve potency; 4) derivatives with this core can be synthesized readily from commercially available starting materials.
Figure 1.

Binding mode for pyrrolo pyrimidine fragment hits generated from co-crystal structures of 1 (PDB id. 5RSG) and 2 (PDB id. 5RSE) with Mac1.
This manuscript outlines the design, synthesis and in vitro evaluation of two subseries of pyrrolo pyrimidines. The subseries derived from secondary amino acids identified two compounds 6g and 6h that improved the potency almost 10-fold over the initial fragment. The subseries derived from primary amino acids afforded compound 15c, a low micromolar derivative (IC50 = 6.1 μM) capable of causing a 4.3 °C thermal shift in the DSF assay, comparable to that of ADPr, consistent with direct binding to Mac1. Compound 15c also demonstrated the ability to inhibit enzymatic activity of Mac1. Molecular modelling allowed us to rationalize the potency observed for the derivatives as well as a rationale for the improvement in potency from compound 15c through fragment growth. Furthermore, compound 15c was selective for coronavirus macrodomains as indicated by profiling against a panel of ADP-ribose binding proteins. Taken together, these two pyrrolo pyrimidine series yielded compounds with improved potency capable of selectively inhibiting Mac1 ADP-ribose binding and ARH activity.
2. Results
2.1. Synthesis
Initial designs for inhibitors focused on attaching a carboxylic acid 1–3 atoms away from the pyrrolo pyrimidine core, similar to that of compound 1. The general synthesis of secondary cyclic amino acid derivatives is outlined in Scheme 1 below. Commercially available chloride 3 was the starting material for a nucleophilic aromatic substitution reaction with secondary amino esters 4a-b and 4f-h. This reaction was conducted with K2CO3 in DMF or with DIEA in isopropanol to afford the desired esters 5a-b and 5f-h in good yield. Ester hydrolysis in either acidic or basic conditions led to the production of carboxylic acids 6a-b and 6f-h. Alternatively, secondary amino acids 7a-c reacted with chloride 3 to afford the desired acids 6c-e directly.
Scheme 1.

Synthesis of secondary amino acid derivatives.
The next series of compounds focused on incorporating the carboxylic acid into a piperazine ring. The synthesis of this series is shown in Scheme 2 . Nucleophilic aromatic substitution of chloride 3 with (R)- and (S)-piperazine esters 8a-b afforded 9a-b in good yield. Deprotection of the Boc group from 9a-b using TFA afforded esters 10a-b as TFA salts. Basic hydrolysis of esters 9a-b led to the free carboxylates 11a-b. Deprotection of the boc group from carboxylates 11a-b afforded the TFA salts of the amino acids 12a-b in good yield.
Scheme 2.

Synthesis of piperazine based pyrrolo pyrimidines.
Synthesis of the natural and non-natural primary amino acid pyrrolo pyrimidines is outlined in Scheme 3 below. The nucleophilic aromatic substitution was conducted with either the amino esters 13a-c or in the case of glycine, with the amino acid itself to form 15d. Hydrolysis of the esters 14a-c afforded the desired amino acid pyrrolo pyrimidines 15a-c.
Scheme 3.

Synthesis of primary natural amino acid derivatives.
The beta-amino acid derivative was synthesized as outlined in Scheme 4 below. Nucleophilic aromatic substitution of amino ester 17 with chloride 3 afforded the amino ester 18 in moderate yield. Hydrolysis to the carboxylic acid was accomplished in 3 M HCl to afford the desired carboxylate 19 in 63% yield.
Scheme 4.

Synthesis of primary beta amino acid derivative 19.
2.2. Biochemical evaluation of secondary amino acid derivatives
Preliminary binding data of fragments 1 and 2 indicated that the pyrrolo pyrimidine core forms a tight hydrogen bond network with Asp,22 Ile23 and in some cases Phe156.20 Therefore, we maintained these three features while modifying the 4-position on this heterocyclic ring. Our first round of derivatization attempted to take advantage of two more hydrogen bonds that can potentially be formed by interactions with the backbone of Phe156 and Asp157 and a carboxylic acid moiety from the inhibitor (Figure 1). We hypothesized that this type of interaction would improve the potency by taking advantage of the entropic gains of constraining the carboxylic acid within a ring approximately 1–3 atoms away from the pyrrolo pyrimidine core. Based on the binding of compound 2, it is likely that rings of approximately 4–6 atoms will be tolerated within the adenosine binding site, and indeed this is what the data from the structure activity studies indicate (Table 1 ). To assess the ability for binding, we used a previously published AlphaScreen™ (AS) displacement binding assay and adapted it for SARS-CoV-2 Mac1.23 The AS assay has been used by multiple groups to determine IC50 values for inhibitors.[24], [25] This assay uses a mono-ADP-ribosylated and biotinylated peptide with a His6-tagged SARS-CoV-2 Mac1 or Mdo2 macrodomain. Importantly, the ADP-ribose is attached via an aminoxy bond, essentially eliminating the chance that the enzymatically active macrodomains would cleave it from the peptide, allowing the macrodomain protein to bind tightly to the ADP-ribosylated peptide. This binding reaction can be displaced by the addition of free ADP-ribose, validating the method as a useful way to screen for macrodomain inhibitors. The general SAR trend that was noticed from the cyclic amino esters (5a-b, d, f-h) and amino acid derivatives (6a-h) was that the ester derivatives were all less potent than their corresponding carboxylic acid derivatives as shown by comparing 5b/6b, 5f/6f, 5g/6g and 5h/6h. Often times the carboxylic acid is several-fold more potent than the ester as seen between 5g (IC50 = 127 μM) and 6g (IC50 = 21.6 μM). This trend supports our hypothesis that the carboxylic acid is important for additional binding. Another trend that was noted within this series was that carboxylic acids incorporated into 6 membered rings tended to afford compounds with greater potency. The two most potent compounds in this series, 6g and 6h were both derived from 6-membered piperidines and were several-fold more potent than any carboxylate from either a 4-membered azetidine (6a-b) or a 5-membered pyrrolidine (6c-f). In addition, 6g and 6h demonstrated selectivity for the SARS-CoV-2 Mac1 exhibiting minimal inhibition of the human macrodomain Mdo2 (>300 µM).
Table 1.
Mac1 Inhibition data for secondary amino acid derivatives. IC50s, changes in the melting temperature and corresponding SDs of three replicates are given.
| Compound | Structure | R group | Stereochem | SARS CoV2 Mac1 Alpha (µM) | Δ Tm (°C) @300 µM |
|---|---|---|---|---|---|
| 5a | 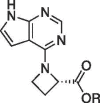 |
Me | (S) | 130 ± 5.7 | |
| 6a | H | (S) | 241 ± 5.2 | ||
| 5b | 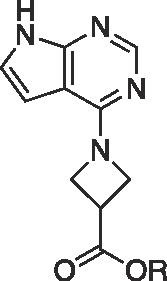 |
Me | NA | >300 | |
| 6b | H | NA | 111 ± 4.2 | ||
| 6c | 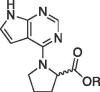 |
H | (S) | 227 ± 6.1 | |
| 5d | Me | (R) | >300 | ||
| 6d | H | (R) | 246 ± 14.3 | ||
| 6e | 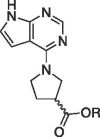 |
H | (S) | 57.8 ± 2.8 | |
| 5f | Me | (R) | 150 ± 1.6 | ||
| 6f | H | (R) | 64.7 ± 3.7 | ||
| 5g | 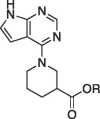 |
Et | racemic | 127 ± 12.9 | |
| 6g | H | racemic | 21.6 ± 2.1 | 1.67 ± 0.01 | |
| 10a | 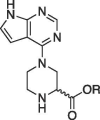 |
Me | (S) | Inactive | |
| 12a | H | (S) | 68.6 ± 2.6 | ||
| 10b | Me | (R) | Inactive | ||
| 12b | H | (R) | 266.7 ± 14.7 | ||
| 5h | 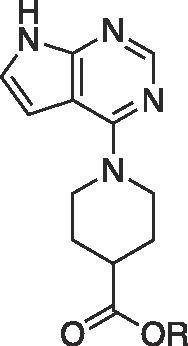 |
Et | NA | 63.8 ± 8.9 | |
| 6h | H | NA | 23.5 ± 1.5 | 1.24 ± 0.21 |
The piperazine derivatives shown in Scheme 2 were initially synthesized to provide another potential site for fragment expansion, namely the nitrogen atom not connected to the pyrrolo pyrimidine. Unfortunately, neither enantiomer of the methyl esters (10a-b) nor the carboxylic acids (12a-b) were as potent as the piperidine derivatives 6g and 6h.
2.3. Molecular modeling of secondary amino acid derivatives
Molecular modeling was performed for compounds 6g and 6h as shown in Figure 2 . The co-crystal structures with parent compound 1 and ADPr have previously been published[20], [26] and are shown (Figure 2B and D, respectively). The pyrrolo pyrimidine core for each compound, as expected, was predicted to form H-bonds with Asp22 and Ile.23 Compound 6g, which has the same number atoms between the amine and the carboxyl group, is predicted to take advantage of the same hydrogen bonds with Phe156 and Asp157 that were observed with compound 1 (Figure 2A). Compound 6h, which has an extra carbon between the amine and carboxyl, is unable to make that interaction (Figure 2C). Interestingly, it is predicted to stretch the carboxylic acid into the phosphate binding groove, forming the same hydrogen bonds previously observed with the ADPr-bound structure, including one of the conserved water molecules within that groove.
Figure 2.

Models of compounds 6g and 6h in the Mac1 active site. A) Compound 6g docked into Mac1. B) Parent fragment 1 co-crystallized structure with Mac1. C) Compound 6h docked into Mac1. D) ADPr co-crystallized with Mac1 (PDB 6WOJ). Hydrogen bonds are represented as yellow dashes, while pi-pi interactions are represented as cyan dashes.
2.4. Biochemical evaluation of primary amino acid derivatives
Primary amino acid derivatives as synthesized in Scheme 3, Scheme 4 were also analyzed in the AS assay for Mac1 binding (Table 2 ). The simplest amino acid derivative, glycinate 15d did not demonstrate any inhibitory effect on binding in the AS assay at the highest concentration tested. Some binding activity was noted with the valine ethyl ester derivative 14a (IC50 = 45 μM) and the valinate 15a (IC50 = 24 μM). Interestingly, the closely related leucine derivatives 14b/15b were completely inactive indicating that there may be a negative steric effect from the isobutyl group. The most potent derivative from the natural amino acids was the tryptophan derivative. The tryptophanate 15c was in fact the most potent compound discovered in either series (IC50 = 6.1 μM in AS) (Figure 4A). We confirmed its potency with a FRET assay measuring binding of a CFP-Mac1 to cysteine MARylated GAP-TAG fused to YFP.22 The measured IC50 (11.1 ± 3.19 µM) was in agreement with the AS assay (Fig. 4B).
Table 2.
Inhibition data for primary amino acid derivatives. IC50s, changes in the melting temperature and corresponding SDs of three replicates are listed.
| Compound | Structure | R group | SARS CoV2 Mac1 Alpha (μM) |
Δ Tm (°C) @300 µM |
|---|---|---|---|---|
| 15d | 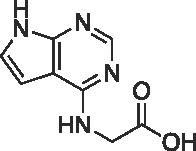 |
>300 | ||
| 14a | 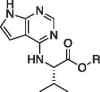 |
Et | 45 ± 0.7 | |
| 15a | H | 24 ± 0.3 | ||
| 14b | 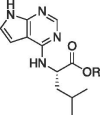 |
Et | Inactive | |
| 15b | H | >300 | ||
| 14c | 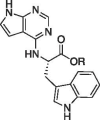 |
Me | 43.9 ± 1.2 | 0.79 ± 0.11 |
| 15c | H | 6.1 ± 0.021 | 4.32 ± 0.14 | |
| 18 | 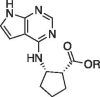 |
Me | 30 ± 0.4 | 1.51 ± 0.07 |
| 19 | H | 25.2 ± 0.2 | 1.1 ± 0.21 | |
| 20 | 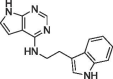 |
NA | 43 ± 5.3 |
Figure 4.

Inhibition and binding of SARS-CoV-2 Mac1-ADP-ribose binding by Compound 15c. A) Dose response curve of Mac1-ADP-ribose binding with 15c in the AS (n = 2). B) Dose response curve of Mac1-ADP-ribose binding with 15c in FRET assay (n = 3). C-D) Stabilization of SARS-CoV-2 Mac1 by Compound 15c in DSF assay (n = 2).
2.5. Molecular modeling of primary amino acid derivatives
Molecular modeling was performed for 15c is shown in Figure 3 along with the starting fragment 1. The pyrrolo pyrimidine core of 15c once again is predicted to form H-bonds with Asp22 and Ile23 in the Adenosine binding pocket (Figure 3A) similar to fragment 1 (Figure 3B). The carboxylate of 15c also formed a hydrogen bond network with Phe156, Asp157 and a water molecule. The indole ring of 15c was predicted to be positioned in the phosphate binding pocket, adding van der Waals interactions while essentially locking the carboxylate into position. Outlining the importance of the carboxylic acid, tryptamine derivative 20 was approximately 7-fold less potent than 15c. Beta amino acid derivatives 18 and 19 also demonstrate potency comparable to the best secondary amino acid derivatives with the methyl ester 18 (IC50 = 30 μM) almost as potent as the corresponding carboxylic acid 19 (IC50 = 25.2 μM).
Figure 3.

Model of compound 15c and fragment 1 in the active site of Mac1. Compound 15c (A) was docked into the co-crystallized structure with parent compound 1 (B). Hydrogen bonds are represented as yellow dashes, while pi-pi interactions are represented as cyan dashes. Compound 15c adopts a very similar binding pose to parent compound 1, while orienting the indole into the phosphate binding groove.
2.6. Differential scanning fluorimetry of Mac1 inhibitors
Evidence for direct Mac1 binding was obtained by using differential scanning fluorimetry (DSF) thermal shift analysis of our most potent compounds from these two series. As shown in Table 2, compounds 6h and 19 demonstrated a thermal shift of > 1 °C, while the Tm for compound 6g was ∼ 1.7 °C. The most significant thermal shift was noted with our most potent compound, 15c, and was 4.3 °C (Figure 4C-D), comparable to ADPr (Tm = 4.1 °C).
2.7. Enzymatic activity of Mac1 inhibitors
To further demonstrate the potential for this series of compounds to inhibit the enzymatic activity of Mac1, 6g and 15c were assayed using a recently developed ADPr-Glo assay.18 This assay utilizes the enzyme NudF, which is a phosphodiesterase that cleaves free ADP-ribose into AMP and diphosphate but cannot cleave ADP-ribose attached to a protein. In this assay, both compound 6g and 15c demonstrated a dose-dependent inhibition of Mac1 enzyme activity (Figure 5 ). Compound 15c demonstrated greater inhibition, consistent with its increased binding activity. These results demonstrate that both 6g and 15c can inhibit Mac1 ADP-ribosylhydrolase activity.
Figure 5.

Compounds 6g and 15c inhibit SARS-CoV-2 Mac1-ADP-ribosylhydrolase activity. A) Dose response curve for 6g and 15c against SARS-CoV-2 Mac1 in the ADPr-Glo assay (n = 2). B) Neither compound demonstrated any activity in a NudF-mediated counterscreen.
2.8. Macrodomain selectivity profiling
To determine the specificity of 15c, we utilized the FRET assay and tested 15c for its ability to inhibit MAR binding of an extended panel of MAR binding proteins from viruses and humans, including human macrodomains, the CHIKV macrodomain, and 3 separate coronavirus macrodomains. Compound 15c was remarkably selective for coronavirus macrodomains, specifically SARS-CoV-2, SARS-CoV and MERS-CoV (Figure 6 ).
Figure 6.

Inhibition profile for 15c against a panel of ADP-ribose binding proteins. Data shown are means and standard deviations of four replicates.
3. Conclusion
In conclusion, we present the design, synthesis, binding evaluation and inhibition of Mac1 by a series of primary and secondary amino acid derivatives of 7H-pyrrolo[2,3–d]pyrimidines in this manuscript. Structure activity studies were conducted against the SARS-CoV-2 macrodomain (Mac1), a potential anti-viral drug target. A molecular modelling study defined a predictive binding mode for this series outlining the importance of the pyrrolo pyrimidine core and a carboxylic acid 3–4 atoms from the 4 position of the core. Low micromolar derivatives 6g and 6h were discovered from the secondary amino acid series that also demonstrated selectivity over human macrodomains. Compound 15c from the primary amino acid series demonstrated low micromolar binding potency against Mac1 and a thermal shift comparable to ADPr. In addition, compounds 6g and 15c demonstrated the ability to inhibit the enzymatic activity of Mac1. Compound 15c also showed a clear selectivity profile towards coronavirus macrodomains, especially towards SARS-CoV-2 Mac1, which may be important when developing the compound further to study its effects on the virus infection. Taken together, many of these derivatives represent some of the most potent small molecule inhibitors of the SARS-CoV-2 Mac1 in the literature, many of which have MW < 300 leaving room for derivatization and optimization while maintaining drug like parameters.
4. Materials and methods
4.1. Plasmids
SARS-CoV-2 Mac1 (residues 205–379 of nsp3) was cloned into the pET30a + expression vector with an N-terminal His tag and a TEV cleavage site (Synbio).26 The pETM-CN Mdo2 Mac1 (residues 7-243) expression vector with an N-terminal His-TEV-V5 tag and the pGEX4T-PARP10-CD (residues 818-1025) expression vector with an N-terminal GST tag were previously described. All plasmids were confirmed by restriction digest, PCR, and direct sequencing.
4.2. Protein expression and purification
The proteins used were prepared as previously described.[22], [26], [27]
4.3. Differential scanning fluorimetry (DSF)
Thermal shift assay with DSF involved use of LightCycler® 480 Instrument (Roche Diagnostics). In total, a 15 μL mixture containing 8X SYPRO Orange (Invitrogen), and 10 μM macrodomain protein in buffer containing 20 mM HEPES, NaOH, pH 7.5 and various concentrations of ADP-ribose or hit compounds were mixed on ice in 384-well PCR plate (Roche). Fluorescent signals were measured from 25 to 85 °C in 0.2 °C/30/Sec steps (excitation, 470–505 nm; detection, 540–700 nm). The main measurements were carried out in triplicate. Data evaluation and Tm determination involved use of the Roche LightCycler® 480 Protein Melting Analysis software, and data fitting calculations involved the use of single site binding curve analysis on GraphPad Prism. The thermal shift ΔTm was calculated by subtracting the Tm values of the DMSO from the Tm values of compounds.
4.4. AlphaScreen™ assay
The AlphaScreen™ reactions were carried out in 384-well plates (Alphaplate, PerkinElmer, Waltham, MA) in a total volume of 40 μL in buffer containing 25 mM HEPES (pH 7.4), 100 mM NaCl, 0.5 mM TCEP, 0.1% BSA, and 0.05% CHAPS. All reagents were prepared as 4X stocks and 10 μL volume of each reagent was added to a final volume of 40 μL. All compounds were transferred acoustically using ECHO 555 (Beckman Inc) and preincubated after mixing with purified His-tagged macrodomain protein (250 nM) for 30 min at RT, followed by addition of a 10 amino acid biotinylated and ADP-ribosylated peptide [ARTK(Bio)QTARK(Aoa-RADP)S] (Cambridge peptides) (625 nM). After 1 h incubation at RT, streptavidin-coated donor beads (7.5 μg/ml) and nickel chelate acceptor beads (7.5 μg/mL); (PerkinElmer AlphaScreen™ Histidine Detection Kit) were added under low light conditions, and plates were shaken at 400 rpm for 60 min at RT protected from light. Plates were kept covered and protected from light at all steps and read on BioTek plate reader using an AlphaScreen™ 680 excitation/570 emission filter set. For counter screening of the compounds, 25 nM biotinylated and hexahistidine-tagged linker peptide (BIO-6His) (PerkinElmer) was added to the compounds, followed by addition of beads as described above.
4.5. ADPr Glo Assay
AThe enzyme assay was performed primarily based on a previously reported high-throughput assay.18 Briefly, the compounds of interest were diluted in 2% (v./v.) DMSO and preincubated with SARS-CoV-2 Mac1 (2 nM) and NudF ADP-ribose pyrophosphatase (125 nM) at ambient temperature for 30 minutes. Following the preincubation, 20 µM mono-ADP-ribosylated substrate was added into the reaction (30 minutes at room temperature), where the free ADP-ribose was generated by the ADP-ribosylhydrolase activity of Mac1 and converted to AMP by NudF (28, https://pubmed.ncbi.nlm.nih.gov/26669448/). The commercial kit AMP-Glo (Promega) was applied to convert AMP to detectable luminescence. An AMP standard curve was made for each experiment to convert the relative luminescence unit (RLU) into AMP concentration and used for IC50 calculation. For counter-screen, the compound were preincubated with NudF (125 nM) only at room temperature for 30 minutes and reacted with free ADP-ribose (2 µM), followed by AMP measurement as described above. Percent inhibition was calculated and non-linear regression analysis was performed in GraphPad Prism.
4.6. FRET assay
A FRET method was utilized for the profiling of 15c a panel of human and viral macrodomains to determine their specificity.[22], [27], [29] The assay is based on the site-specific introduction of cysteine-linked mono-ADP ribose to the C-terminal Gαi peptide (GAP) by Pertussis toxin subunit1 (PtxS1) fused to YFP. To generate the FRET signal ADP-ribosyl binders were fused to CFP. Samples were prepared in the assay buffer (for most binders; 10 mM Bis-Tris propane pH 7.0, 3 % (w/v) PEG 20,000, 0.01 % (v/v) Triton X-100 and 0.5 mM TCEP), (for TARG1; 10 mM Bis-Tris propane pH 7.0, 150 mM NaCl, 0.01 % (v/v) Triton X-100 and 0.5 mM TCEP), (for PARG and PARP15 MD1; 10 mM Bis-Tris propane pH 7.0, 25 mM NaCl, 0.01 % (v/v) Triton X-100 and 0.5 mM TCEP) in a 384-well black polypropylene flat-bottom plates (Greiner, Bio-one) with 10 µL reaction volume per well. The reactions consisted of 1 µM CFP-fused binders and 5 µM MARylated YFP-GAP. Reactions were excited at 410 nm (20 nm bandwidth), while the emission signal was measured at 477 nm (10 nm bandwidth) and 527 nm (10 nm bandwidth). Afterwards, blank was deducted from the individual values and the radiometric FRET (rFRET) was calculated by dividing the fluorescence intensities at 527 nm by 477 nm. Compound was dispensed with Echo acoustic liquid dispenser (Labcyte, Sunnyvate, CA). Dispensing of larger volumes of the solutions was carried out by using Microfluidic Liquid Handler (MANTIS®, Formulatrix, Beford, MA, USA). Measurements were taken with Tecan Infinite M1000 pro plate reader.
4.7. Data analysis
Percent inhibition was calculated by measuring the background subtracted assay signal with inhibitor and dividing by assay signal without inhibitor. This value was then subtracted from 100% to determine the final percent inhibition for each data point. IC50 values were determined from Four parametric Nonlinear Regression analysis (log inhibitor vs response --Variable slope [Equation: Y = Bottom + (Top-Bottom)/(1 + 10^((LogIC50-X)*HillSlope))] of the inhibition data in GraphPad Prism (GraphPad Software, San Diego, CA). At least 3 replicates were performed for each experiment and the means and standard deviations of each experiment were reported.
For all assays, the compounds solubilized in 100% DMSO were transferred acoustically to assay plates. All control/compound wells were backfilled with DMSO to a final concentration to 1%. Previous optimizations had shown that DMSO concentrations of up to 8% did not significantly inhibit the SARS CoV-2 Mac1 alphascreen readouts.
4.8. Modeling details
Compounds were docked into the ADPr-bound (6WOJ), 3 unique unbound conformations (7KR0, 7KR1, 6WEY) and two small molecule bound (5RSG, 5RTT) structures of Mac1.[20], [26], [30] The proteins and ligands were prepared using Schrodinger Maestro and were subsequently docked using Glide with XP precision, followed by a Prime MM-GBSA minimization, allowing flexibility for any residue within 5 Å of the ligand.[31], [32], [33], [34], [35], [36]
4.9. Chemistry general methods
All solvents were reagent grade or high performance liquid chromatography (HPLC) grade. Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. 1H NMR spectra were recorded at 400.19 MHz. All 13C spectra were recorded at 100.63 MHz. The HPLC solvent system consisted of distilled water and acetonitrile, both containing 0.1% formic acid. Analytical liquid chromatography / mass spectrometry (LC-MS) (LC/MS) were performed utilizing an Agilent Infinity 1260 DAD G7115A detector coupled with a 6120 quadrupole analyzer. Samples were run on a 5–95% gradient ACN in H2O over 6 min (both mobile phases modified w/ 0.1% TFA). All results depicted herein are (M+H)+ as ionization source is ESI. All final compounds tested were confirmed to be of ≥95% purity by the HPLC methods described above. HRMS were run in positive ion mode on a SCIEX X500B QTOF mass spectrometer coupled to an Exion LC. Each compound was resuspended in 0.1% formic acid and injected directly on to the mass spec by flow injection.
4.10. Experimental procedures
Methyl (S)-1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)azetidine-2-carboxylate (5a). 4-Chloro-7H-pyrrolo[2,3–d]-pyrimidine 3 (0.500 g, 3.26 mmol), (S)-Azetidine 2-carboxilic acid methyl ester HCl 4a (0.600 g, 3.91 mmol), isopropanol (4 mL), and DIEA (1.71 mL, 9.81 mmol) were combined in a 25 mL round bottom and left to stir under heat for 9 days. TLC showed that the reaction was complete, therefore the isopropanol was removed via rotary evaporator. EtOAc (10 mL) was added to the reaction mixture, and poured into a separatory funnel. It was washed twice with 10% aqueous sodium bicarbonate and the organic layer was dried with MgSO4.The product was purified through a column chromatography on silica gel and was characterized as the desired product. Dry yield: 0.175 g (23%). 1HNMR (CDCl3): 11.6 (br s, 1H), 8.35 (s, 1H), 7.09 (d, 1H, J = 3.6 Hz), 6.34 (d, 1H, J = 3.6 Hz), 5.11 (m, 1H), 4.54 (m, 1H), 4.38 (m, 1H), 3.79 (s, 3H), 2.82 (m, 1H), 2.50 (m, 1H); 13C NMR (CDCl3): 172.51, 156.67, 151.46, 151.35, 121.68, 102.68, 99.45, 61.86, 52.62, 49.99, 22.43; HRMS (ESI+): m/z calculated for C11H12N4O2 (M + 1) + 233.0960 found 233.1009.
Methyl 1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)azetidine-3-carboxylate (5b). 1–4-Chloro-7H-pyrrolo[2,3–d]-pyrimidine 3 (0.300 g, 1.961 mmol), methyl-azetidine-3-carboxylate-hydrochloride 4b (0.356 g, 2.35 mmol), isopropanol (3 mL) and DIEA (1.02 mL, 5.88 mmol) were combined in a 25 mL round bottom flask and stirred at 70 °C for 24 h. TLC indicated that reaction was complete. The reaction was concentrated in vacuo and the reaction mixture was diluted with ethyl acetate (10 mL) and washed with 1 M HCl (3 mL) and 10% NaHCO3 (2 × 3 mL). The organic layer was dried with MgSO4, filtered and chromatographed (EtOAc/Hexanes) to afford the desired product as a white solid with dry yield of 0.237 g, 52.1%. 1HNMR (CDCl3): 11.20 (s, 1H), 8.33 (s, 1H), 7.09 (d, 1H, J = 3.2 Hz), 6.39 (d, 1H, J = 3.2 Hz), 4.57 (d, 4H, J = 8.8 Hz), 3.78 (s, 3H), 3.67 (m, 1H); 13C NMR (DMSO‑d6): 172.93, 156.77, 150.95, 150.67, 121.95, 101.75, 98.83, 53.21, 52.02, 33.14; HRMS (ESI + ): m/z calculated for C11H12N4O2 (M + 1) + 233.0960 found 233.1019.
Methyl (R)-1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)pyrrolidine-3-carboxylate (5f). A 25.0 mL round bottom was used to combine chloride 3 (0.501 g, 3.27 mmol), (R)-methyl-pyrrolidine-3-carboxylate, HCl 4f (0.506 g, 3.92 mmol), DIEA (1.71 mL, or 9.81 mmol), and 3 mL of isopropanol. The reaction was heated for 24 h, a TLC indicated that the reaction was complete (EtOAc). The reaction was concentrated on the rotary evaporator to remove the isopropyl alcohol. EtOAc (10 mL) was added to the round bottom. The organic layer was washed with 10% NaHCO3 and the organic layer was dried with MgSO4, filtered off using vacuum filtration, chromatographed (EtOAc/hexanes). Dry yield = 0.475 g (59%). 1HNMR (CDCl3): δ 10.91 (s, 1H), 8.33 (s, 1H), 7.08 (d, 1H, J = 4.0 Hz), 6.59 (d, 1H, J = 4.0 Hz), 4.13 (m, 4H), 3.89 (s, 3H), 3.25 (m, 1H), 2.35 (m, 2H); 13C NMR (DMSO‑d6): 173.46, 154.59, 151.09, 150.96, 120.75, 102.42, 100.60, 51.85, 49.48, 46.75, 41.79, 28.08; HRMS (ESI + ): m/z calculated for C12H14N4O2 (M + 1) + 247.1117 found 247.1175.
Ethyl 1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperidine-3-carboxylate (5g). In a 25 mL round bottom flask 500 mg of the chloride 3 (3.26 mmol), 0.56 mL of racemic ethyl-piperidine-3-carboxylate HCl 4 g (3.6 mmol), and 890 mg of K2CO3 (6.53 mmol) were dissolved in 2 mL of DMF and heated to ∼ 140 °C for 1 h. A small sample of the reaction was diluted in DCM and a TLC of the reaction mixture (EtOAc) indicated that the starting material was consumed and a lower running spot was apparent. The mixture was cooled to rt for 5 min. Water (6 mL) was added to the round bottom and the mixture was stirred. A tan solid formed almost immediately and the reaction continued to stir for 10 min. The solid was filtered off and washed with dH2O (∼10–20 mL). The solid was air dried under vacuum for another 5 min and then transferred to a vial to dry overnight. Dry yield = 0.65 g (72.9%). 1HNMR (DMSO‑d6): δ 11.69 (s, 1H), 8.13 (s, 1H), 7.18 (dd, 1H, J = 3.6, 2.0 Hz), 6.55 (dd, 1H, J = 3.6, 2.0 Hz), 4.62 (dd, 1H, J = 13.6, 3.6 Hz), 4.31 (d, 1H, J = 13.6 Hz), 4.06 (q, 2H, J = 6.8 Hz), 3.34 (m, 2H), 2.54 (m, 1H), 1.97 (m, 1H), 1.76 (m, 2H), 1.52 (m, 1H), 1.15 (t, 3H, J = 6.8 Hz). 13C NMR (DMSO‑d6): 172.83, 156.22, 152.01, 150.60, 121.45, 102.19, 100.73, 60.06, 47.19, 45.81, 40.51, 26.79, 23.79, 14.04; MS (ES+, M + 1): 275.1.
Ethyl 1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperidine-4-carboxylate (5h). In a 25 mL round bottom flask 500 mg of the chloride 3 (3.26 mmol), 0.56 mL of ethyl piperidine-4-carboxylate 4 h (3.6 mmol), and 675 mg of K2CO3 (4.89 mmol) were dissolved in 2 mL of DMF and heated to ∼ 140 °C for 3 h. A small sample of the reaction was diluted in DCM and a TLC of the reaction mixture (EtOAc) indicated that the starting material was consumed and a lower running spot was apparent. The mixture was cooled to rt for 5 min. Water (6 mL) was added to the round bottom and the mixture was stirred. A tan solid formed almost immediately and the reaction continued to stir for 10 min. The solid was filtered off and washed with dH2O (∼10–20 mL). The solid was air dried under vacuum for another 5 min and then transferred to a vial to dry overnight. Dry yield = 0.657 g (72.6%). 1HNMR (CDCl3): δ 10.82 (bs, 1H), 8.34 (s, 1H), 7.11 (d, 1H, J = 3.6 Hz), 6.52 (d, 1H, J = 3.6), 4.67 (d, 2H, J = 13.0 Hz), 4.15 (q, 2H, J = 7.2 Hz), 3.30 (t, 2H, J = 13.0 Hz), 2.65 (m, 1H), 2.05 (m, 2H), 1.85 (m, 2H), 1.26 (t, 3H, J = 7.2 Hz); 13C NMR (DMSO‑d6): 173.97, 156.22, 151.94, 150.57, 121.31, 102.17, 100.77, 59.87, 44.57, 40.25, 27.63, 14.03; MS (ES+, M + 1): 275.1.
(S)-1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)azetidine-2-carboxylic acid (6a). Methyl (S)-1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)azetidine-2-carboxylate 5a (152 mg, 0.534 mmol), 3.0 M HCl (3 mL) and a stir bar were placed in a 25 mL RBF and heated to reflux for two hours. The solution was concentrated in vacuo until a brown solid could be observed in the flask. The residue was allowed to dry overnight at RT and the following morning the residue was placed in a vacuum oven at 110F for 2 h, triturated with 5 mL of Et2O, vacuum filtered and allowed to sit undisturbed in a vacuum oven at room temperature for another day. Dry yield 74 mg (54% yield). 1HNMR (DMSO‑d6): 12.84 (s, 1H), 8.35 (m, 1H), 7.48 (br s, 1H), 6.7 (br s, 1H), 5.31 (m, 1H), 4.46 (m, 2H), 2.89 (m, 1H), 2.46 (m, 1H); 13C NMR (DMSO‑d6): 171.55, 149.45, 145.36, 142.39, 125.21, 101.82, 100.41, 63.50, 51.46, 21.62; HRMS (ESI + ): m/z calculated for C10H10N4O2 (M + 1) + 219.0804 found 219.0865.
1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)azetidine-3-carboxylic acid (6b). Ester 5b (0.20 g, 0.86 mmol) was suspended in 3 M HCl (3 mL) in a 25 mL round bottom flask and heated to reflux for 30 min. TLC indicated that reaction was complete. Removal of solvent and trituration with a minimal amount of cold water afforded the desired acid. Yield = 131 mg (70%). 1HNMR (DMSO‑d6): δ 12.88 (s, 1H), 8.28 (s, 1H), 7.45 (dd, 1H, J = 3.6, 2.0 Hz), 6.78 (dd, 1H, J = 3.6, 2.0 Hz), 4.4–4.8 (m, 4H), 3.72 (m, 1H); 13C NMR (DMSO‑d6): 172.85, 149.63, 146.43, 142.55, 124.83, 101.87, 100.35, 55.33, 32.97; HRMS (ESI + ): m/z calculated for C10H10N4O2 (M + 1) + 219.0804 found 219.0885.
(7H-pyrrolo[2,3–d]pyrimidin-4-yl)-l-proline (6c). A 25.0 mL round bottom was used to combine the chloride 3 (0.499 g, 3.26 mmol), l-proline (0.451 g, 3.91 mmol), DIEA (1.70 mL, 9.78 mmol), and 3.0 mL of isopropanol. The reaction was heated to ∼ 70 °C for 48 h, TLC indicated that the reaction was complete (EtOAc). The reaction was concentrated on the rotary evaporator to remove the isopropyl alcohol. The product was partitioned between 5 mL of EtOAc and 3 mL 10% NaHCO3. The bicarbonate layer was acidified to a pH of 3 with 3 M HCl, then extracted again with boiling EtOAc. Product began to crystallize in the aqueous layer, so vacuum filtration was then used to retrieve the final product. After drying, it was characterized by dry yield = 0.154 g, 20.3%. 1HNMR (DMSO‑d6): δ 12.50 (bs, 1H), 11.61 (s, 1H), 8.04 (s, 1H), 7.13 (dd, 1H, J = 3.6, 2.0 Hz), 6.56 (br s, 1H), 4.64 (m, 1H), 3.91 (m, 2H), 2.01–2.24 (m, 4H); 13C NMR (DMSO‑d6): 174.36, 154.42, 151.11, 150.82, 121.09, 102.51, 100.62, 59.85, 48.18, 28.77, 24.31; HRMS (ESI + ): m/z calculated for C11H12N4O2 (M + 1) + 233.0960 found 233.1004.
(7H-pyrrolo[2,3–d]pyrimidin-4-yl)-d-proline (6d). This compound was made in an identical manner to 6c using d-Proline as the starting material. Dry yield = 0.040 g, 29.2%. 1HNMR (DMSO‑d6): δ 12.50 (bs, 1H), 11.61 (s, 1H), 8.04 (s, 1H), 7.13 (dd, 1H, J = 3.6, 2.0 Hz), 6.56 (br s, 1H), 4.64 (m, 1H), 3.91 (m, 2H), 2.01–2.24 (m, 4H); 13C NMR (DMSO‑d6): 174.36, 154.42, 151.11, 150.82, 121.09, 102.51, 100.62, 59.85, 48.18, 28.77, 24.31; MS (ES+, M + 1): 233.1.
(S)-1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)pyrrolidine-3-carboxylate (6e). This reaction was conducted in a similar manner to 6c using (S) pyrrolidine-3-carboxylic acid as the starting material. Dry weight = 424 mg (56%). 1HNMR (DMSO‑d6): δ 11.57 (bs, 1H), 8.06 (s, 1H), 7.08 (dd, 1H, J = 3.6, 2.0 Hz), 6.53 (dd, 1H, J = 3.6, 2.0 Hz), 3.80 (m, 4H), 3.15 (m, 1H), 2.18 (m, 2H); 13C NMR (DMSO‑d6): 173.55, 148.46, 147.01, 142.68, 124.09, 103.92, 101.41, 51.39, 49.30, 42.72, 28.38; HRMS (ESI + ): m/z calculated for C11H12N4O2 (M + 1) + 233.0960 found 233.1024.
(R)-1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)pyrrolidine-3-carboxylic acid (6f). A 25.0 mL round bottom was used to combine the ester 5f (0.464 g, 1.88 mmol) and 5 mL of 3 M HCl. The reaction was heated to reflux for 2 h. It was then cooled and a white precipitate crashed out of solution and was filtered using vacuum filtration. Dry weight = 0.322 g, 69.4%. 1HNMR (DMSO‑d6): δ 11.6 (br s, 1H), 8.06 (s, 1H), 7.08 (dd, 1H, J = 3.6, 2.0 Hz), 6.53 (dd, 1H, J = 3.6, 2.0 Hz), 3.80 (m, 4H), 3.15 (m, 1H), 2.18 (m, 2H); 13CNMR (DMSO‑d6): 173.55, 148.46, 147.01, 142.68, 124.09, 103.92, 101.41, 51.39, 49.30, 42.72, 28.38; HRMS (ESI+): m/z calculated for C11H12N4O2 (M + 1) + 233.0960 found 233.1030.
1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperidine-3-carboxylic acid (6g). In a 25 mL round bottom flask, the ethyl ester 5 g (230 mg, 0.84 mmol) was dissolved in a mixture of methanol (2 mL) and 5% NaOH (2 mL). This mixture was heated to ∼ 70 °C for 1 h. The reaction was cooled to rt then the methanol was removed via rotary evaporation leaving the product in ∼ 2 mL of aqueous base. To this mixture was added 1 mL of water and 5 mL of EtOAc. The bilayer was separated using a separatory funnel and the bottom aqueous layer was acidified to pH 3 using 3 M HCl. The acidic aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organics were dried and concentrated to afford a white crystalline solid that was triturated with Et2O and collected by vacuum filtration. Dry yield = 0.11 g (52%) 1HNMR(DMSO‑d6): δ 12.39 (s, 1H), 11.69 (s, 1H), 8.13 (s, 1H), 7.17 (dd, 1H, J = 3.6, 2.0 Hz), 6.54 (dd, 1H, J = 3.6, 2.0 Hz), 4.66 (dd, 1H, J = 13.2, 4.0 Hz), 4.40 (d, 1H, J = 13.2 Hz), 3.22 (m, 2H), 2.46 (m, 1H), 1.97 (m, 1H), 1.71 (m, 2H), 1.49 (m, 1H); 13C NMR (DMSO‑d6): 174.58, 156.31, 151.99, 150.63, 121.43, 102.24, 100.73, 47.51, 45.82, 40.68, 26.95, 24.05; HRMS (ESI + ): m/z calculated for C12H14N4O2 (M + 1) + 247.1117 found 247.1181.
1-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperidine-4-carboxylic acid (6h). In a 25 mL round bottom flask, the ethyl ester 5h (320 mg, 1.17 mmol) was dissolved in a mixture of methanol (2 mL) and 5% NaOH (2 mL). This mixture was heated to ∼ 70 °C for 1 h. The reaction was cooled to rt then the methanol was removed via rotary evaporation leaving the product in ∼ 2 mL of aqueous base. To this mixture was added 1 mL of water and 5 mL of EtOAc. The bilayer was separated using a separatory funnel and the bottom aqueous layer was acidified to pH 3 using 3 M HCl. Crystals precipitated out of solution and the mixture was cooled in an ice bath for 5 min then filtered and washed with 1 mL of cold water. The solid was dried overnight to afford 0.19 g, 65%. 1HNMR (DMSO‑d6): δ 12.25 (s, 1H), 11.67 (s, 1H), 8.11 (s, 1H), 7.15 (dd, 1H, J = 3.6, 2.4 Hz), 6.56 (d, 1H, J = 3.6, 2.4 Hz), 4.52 (d, 2H, J = 13.2 Hz), 3.20 (dt, 2H, J = 12.4, 2.4 Hz), 2.56 (m, 1H), 1.91 (dd, 2H, J = 13.2, 3.6 Hz), 1.52 (m, 2H); 13CNMR (DMSO‑d6): 175.72, 156.25, 151.93, 150.58, 121.24, 102.14, 100.81, 44.70, 40.32, 27.74; HRMS (ESI + ): m/z calculated for C12H14N4O2 (M + 1) + 247.1117 found 247.1183.
1-(tert-butyl) 2-methyl (S)-4-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperazine-1,2-dicarboxylate (9a). 4-chloro-7H-pyrrolo[2,3–d]pyimidine 3 (1.0 g, 6.53 mmol), DIEA (3.42 mL, 19.61 mmol), i-PrOH (6 mL) and (S)-1-N-boc-piperazine-2-carboxylic acid methyl ester 8a (2.4 g, 9.8 mmol) were combined into a 100 mL round bottom flask and heated to reflux for 48 h. TLC (EtOAc) indicated that the reaction was complete. The i-PrOH was removed by rotary evaporation. The remaining reaction mixture was diluted in 15 mL of EtOAc, poured into a 125 mL separatory funnel and washed with 10% NaHCO3. It was dried with anhydrous MgSO4, filtered and chromatographed (70/30 EtOAc/Hexanes to 100% EtOAc) to isolate the product. Dry yield = 1.72 g (73%). 1HNMR (CDCl3): δ 11.05 (br s, 1H), 8.36 (s, 1H), 7.15 (d, 1H, J = 4.0 Hz), 6.63 (br s, 1H), 5.10 (dd, 1H, J = 51.2, 13.6 Hz), 4.91 (s, 0.5H), 4.71 (s, 0.5H), 4.61 (m, 1H), 3.98 (dd, 1H, J = 30.2, 12.4 Hz), 3.67 (s, 3H), 3.20–3.51 (m, 4H); 13C NMR (DMSO‑d6): {rotamer signals} {171.11, 170.77}, 156.28, {154.78, 154.26}, 151.91, 150.34 {121.91, 121.81}, 102.41, {100.49, 100.37}, {79.88, 79.67}, {55.18, 53.80}, 52.04, {45.54, 44.93}, 44.52, 41.14, {27.93, 27.80}; HRMS (ESI + ): m/z calculated for C17H23N5O4 (M + 1) + 362.1750 found 362.1818.
1-(tert-butyl) 2-methyl (R)-4-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperazine-1,2-dicarboxylate (9b). This compound was made in an identical manner to 9a using (R)-1-N-boc-piperazine-2-carboxylic acid methyl ester 8b. Dry yield = 1.84 g (60%). 1HNMR (CDCl3): δ 11.05 (br s, 1H), 8.36 (s, 1H), 7.15 (d, 1H, J = 4.0 Hz), 6.63 (br s, 1H), 5.10 (dd, 1H, J = 51.2, 13.6 Hz), 4.91 (s, 0.5H), 4.71 (s, 0.5H), 4.61 (m, 1H), 3.98 (dd, 1H, J = 30.2, 12.4 Hz), 3.67 (s, 3H), 3.20–3.51 (m, 4H); 13C NMR (CDCl3): {rotamer signals} {171.31, 170.94}, 157.30, {155.67, 155.17}, 152.21, 150.73, 121.37, 103.50, 101.35, 81.12, {55.92, 54.45}, 52.58, {46.97, 45.73}, {45.55, 44.92}, {41.81, 40.36}, 28.55; HRMS (ESI + ): m/z calculated for C17H23N5O4 (M + 1) + 362.1750 found 362.1804.
Methyl (S)-4-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperazine-2-carboxylate ●TFA (10a). 1-(tert-butyl) 2-methyl (S)-4-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperazine-1, 2-dicarboxylate 9a (1.642 g, 4.54 mmols), DCM (6 mL) and TFA (2.5 mL) were combined into a 100 mL round bottom flask and left stirring with no heat for 24hrs. TLC in a 100% EtOAc solvent system indicated that the reaction was complete. The DCM was removed by rotary evaporation. 5–10 mL of DCM and 10 mL of toluene were subsequently added to and evaporated via rotary evaporation from the round bottom containing the reaction mixture 5 times to extract the excess TFA. Dry yield (TFA salt) = 1.7 g (quant). 1HNMR (DMSO‑d6): δ 12.44 (br s, 1H), 8.42 (br s, 1H), 8.38 (s, 1H), 7.39 (dd, 1H, J = 4.0, 2.0 Hz), 6.75 (d, 1H, J = 4.0 Hz), 4.83 (dd, 1H, J = 15.2, 2.8 Hz), 4.49 (m, 2H), 3.78 (s, 3H), 3.73 (m, 2H), 3.45 (m, 1H), 3.23 (m, 1H); 13C NMR (DMSO‑d6): (TFA salt) 168.17, 167.14 (TFA), 158.27 (TFA, q, JC-F = 34.3 Hz), 155.58, 151.15, 149.66, 122.88, 102.67, 100.52, 54.31, 54.08, 53.21, 44.30, 42.28; HRMS (ESI + ): m/z calculated for C12H15N5O2 (M + 1) + 262.1226 found 262.1286.
Methyl (R)-4-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperazine-2-carboxylate ●TFA (10b). This compound was made in an identical manner to 10a using 9b as the starting material. Dry yield = 0.48 g (quant). 1HNMR (DMSO‑d6): δ 12.5 (br s, 1H), 8.42 (br s, 1H), 8.38 (s, 1H), 7.39 (dd, 1H, J = 4.0, 2.0 Hz), 6.75 (d, 1H, J = 4.0 Hz), 4.83 (dd, 1H, J = 15.2, 2.8 Hz), 4.49 (m, 2H), 3.78 (s, 3H), 3.73 (m, 2H), 3.45 (m, 1H), 3.23 (m, 1H); 13C NMR (DMSO‑d6): (TFA salt) 168.17, 167.14 (TFA), 158.27 (TFA, q, JC-F = 34.3 Hz), 155.58, 151.15, 149.66, 122.88, 102.67, 100.52, 54.31, 54.08, 53.21, 44.30, 42.28; HRMS (ESI + ): m/z calculated for C12H15N5O2 (M + 1) + 262.1226 found 262.1275.
(S)-1-(tert-butoxycarbonyl)-4-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperazine-2-carboxylic acid (11a). Compound 9a (0.5 g, 1.38 mmol), 1.25 M aq. NaOH (4 mL, 5 mmol) and MeOH (4 mL) were combined into a 50 mL round bottom flask and left stirring below the boiling point overnight. The MeOH was removed via rotary evaporation. The remaining reaction mixture was transferred to a 125 mL separatory funnel and suspended in 5–10 mL of EtOAc. The layers were separated and the basic layer was acidified drop-wise with 3 M HCl to 3–4 pH. The acidic layer was extracted twice with 10 mL of EtOAc. The combined organic layers were dried with solid MgSO4, vacuumed filtered and concentrated via rotary evaporation. Dry yield = 95 mg (19%). 1HNMR (CDCl3): δ 11.91 (bs, 1H), 8.24 (s, 1H), 7.15 (s, 1H), 6.62 (s, 1H), 4.83 (s, 1H), 4.09 (m, 2H), 3.59 (m, 4H), 1.27 (s, 9H). 13C NMR (DMSO‑d6): {172.00, 171.74}, {156.51, 156.43} {154.77, 154.51}, 151.87, 150.38, 121.74, 102.49, {100.58, 100.52}, {79.58, 79.40}, 55.20, 53.83, {45.55, 45.02}, {44.64, 44.34} 41.20, {27.99, 27.85}; HRMS (ESI + ): m/z calculated for C16H21N5O4 (M + 1) + 348.1594 found 348.1649.
(R)-1-(tert-butoxycarbonyl)-4-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperazine-2-carboxylic acid (11b). This compound was made in an identical manner to compound 11a using 10b as the starting material. Dry yield = 212 mg, 44.1%. 1HNMR (CDCl3): δ 11.91 (bs, 1H), 8.24 (s, 1H), 7.15 (s, 1H), 6.62 (s, 1H), 4.83 (s, 1H), 4.09 (m, 2H), 3.59 (m, 4H), 1.27 (s, 9H); 13C NMR (DMSO‑d6): {172.00, 171.74}, {156.51, 156.43} {154.77, 154.51}, 151.87, 150.38, 121.74, 102.49, {100.58, 100.52}, {79.58, 79.40}, 55.20, 53.83, {45.55, 45.02}, {44.64, 44.34} 41.20, {27.99, 27.85}; HRMS (ESI + ): m/z calculated for C16H21N5O4 (M + 1) + 348.1594 found 348.1652.
(S)-4-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperazine-2-carboxylic acid (12a). (S)-1-(tert-butoxycarbonyl)-4-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperazine-2-carboxylic acid (28 mg, 0.08 mmol), DCM (1 mL) and TFA (1 mL) were combined into a 25 mL round bottom flask and left stirring with no heat for 1 h. The DCM was removed via rotary evaporation. 5–10 mL of DCM were subsequently added to and evaporated via rotary evaporation from the round bottom containing the reaction mixture 7 times to extract the excess TFA. The product was characterized as the TFA salt. Dry yield = 33 mg (quant). 1HNMR (DMSO‑d6): δ 11.96 (bs, 1 M), 9.60 (br s, 2H), 8.26 (s, 1H), 7.32 (dd, 1H, J = 4.0, 2.8 Hz), 6.64 (dd,1H, J = 4.0, 2.8 Hz), 4.80 (dd, 1H, J = 14.0, 2.8 Hz), 4.43 (d, 1H, J = 14.0 Hz), 4.31 (dd, 1H, J = 10.0, 3.6 Hz), 3.64–3.77 (m, 2H), 3.40 (d, 1H, J = 12.8 Hz), 3.16 (t, 1H, 10.0 Hz); 13C NMR (DMSO‑d6): 168.21, 158.31 (TFA, q, JC-F = 33.4 Hz), 155.61, 151.19, 149.57, 122.90, 102.68, 100.54, 54.32, 44.31, 42.21, 42.01; HRMS (ESI + ): m/z calculated for C11H13N5O2 (M + 1) + 248.1069 found 248.1130.
(R)-4-(7H-pyrrolo[2,3–d]pyrimidin-4-yl)piperazine-2-carboxylic acid (12b). This compound was made in an identical manner to 12a using 11b as the starting material. Dry yield = 245 mg (quant). 12.16 (bs, 1 M), 9.60 (br s, 2H), 8.33 (s, 1H), 7.36 (d, 1H, J = 4.0 Hz), 6.70 (d,1H, J = 4.0 Hz), 4.82 (dd, 1H, J = 14.0, 2.8 Hz), 4.45 (d, 1H, J = 14.0 Hz), 4.33 (dd, 1H, J = 10.0, 3.6 Hz) 3.64–3.77 (m, 2H), 3.42 (d, 1H, J = 12.8 Hz), 3.19 (t, 1H, 10.0 Hz); 13C NMR (DMSO‑d6): 168.14, 158.23 (q, JC-F = 34.2 Hz), 155.52, 150.31, 148.92, 123.13, 102.68, 100.87, 54.35, 44.44, 42.34, 42.00; HRMS (ESI + ): m/z calculated for C11H13N5O2 (M + 1) + 248.1069 found 248.1130.
ethyl (7H-pyrrolo[2,3–d]pyrimidin-4-yl)-l-valinate (14a). 4-Chloro-7H-pyrrolo[2,3–d]-pyrimidine (0.500 g, 3.26 mmol), l-Valine ethyl ester hydrochloride (0.712 g, 3.92 mmol), isopropanol (4 mL), and DIEA (1.71 mL, 9.81 mmol) were combined in a 25 mL round bottom and left to stir under heat and water cooled condenser for 7 days. TLC showed that the reaction was complete, therefore the isopropanol was removed via rotary evaporator. EtOAc (10 mL) was added to the reaction mixture, and poured into a separatory funnel. It was washed twice with 10% aqueous sodium bicarbonate and the organic layer was dried with MgSO4. The product was purified through a column chromatography on silica gel and was characterized as the desired compound. Dry yield: 0.170 g (20%). 1HNMR (CDCl3): δ 10.6 (s, 1H), 8.35 (s, 1H), 7.05 (d, 1H, J = 4.0 Hz), 6.42 (d, 1H, J = 4.0 Hz), 5.91 (d, 1H, J = 4.0 Hz), 4.67 (q, 2H, J = 8.0 Hz), 2.20 (m, 1H), 0.82–1.27 (m, 9H); 13C NMR (DMSO‑d6): 172.61, 155.74, 150.93, 150.34, 120.98, 102.61, 99.10, 60.04, 58.85, 30.07, 19.20, 14.15; HRMS (ESI + ): m/z calculated for C13H18N4O2 (M + 1) + 263.1430 found 263.1489.
Ethyl (7H-pyrrolo[2,3–d]pyrimidine-4-yl)-l-leucinate (14b). 4-Chloro-7H-pyrrolo[2,3–d]-pyrimidine 3 (0.5 g, 3.27 mmol), l-Leucine-Ethyl-Ester-Hydrochloride (0.767 g, 3.92 mmol), DIEA (1.71 mL, 9.81 mmol), and isopropanol (4 mL) were combined into a 25 mL round bottom flask and left to stir under heat, for approximately 48 h. TLC indicated that the reaction was complete. The isopropanol was removed with the rotary evaporator and 10 mL of Ethyl Acetate was added to the reaction mixture. It was poured into a separatory funnel and washed with 5 mL of 10% NaHCO3. The organic layer was dried with MgSO4 and filtered into a tared round bottom flask. The product was purified via column chromatography (EtOAc). Dry yield = 0.31 g (34%). 1HNMR (CDCl3): δ 8.27 (s, 1H), 7.02 (d, 1H, J = 4.0 Hz), 6.38 (s, 1H), 5.62 (s, 1H), 5.00 (s, 1H), 4.22 (m, 2H), 1.6–1.85 (m, 3H), 1.26 (t, 3H), 0.94 (d, 3H); 13C NMR (DMSO‑d6): 173.64, 155.57, 151.01, 150.23, 121.02, 102.56, 98.79, 60.14, 51.35, 24.38, 22.81, 21.26, 14.06; HRMS (ESI + ): m/z calculated for C14H20N4O2 (M + 1) + 277.1586 found 277.1649.
Methyl (7H-pyrrolo[2,3–d]pyrimidin-4-yl)-l-tryptophanate (14c). methyl 4-Chloro-7H-pyrrolo[2,3–d]-pyrimidine 3 (0.500 g, 3.27 mmol), l-Tryptophan Methyl Ester Hydrochloride (0.998 g, 3.92 mmol), isopropanol (4 mL) and DIEA (1.71 mL, 9.81 mmol) were combined in a 25 mL round bottom and left to stir under reflux for 5 days. TLC indicated that the reaction was complete, therefore the isopropanol was removed via rotary evaporator. 10 mL of EtOAc was added to the reaction mixture, and poured into a separatory funnel. It was washed twice with 10% sodium bicarbonate (3 mL), and the organic layer dried with MgSO4. The product was purified through a column chromatography on silica gel. Dry yield = 0.220 g (20.0%). 1HNMR (DMSO‑d6): δ 9.41 (s, 1H), 8.38 (s, 1H), 8.10 (s, 1H), 7.58 (d, 1H), 7.38 (d, 1H), 7.20 (m, 1H), 7.10 (t, 1H), 7.02 (m, 1H), 6.28 (s, 1H), 5.60 (s, 1H), 5.39 (m, 1H), 5.0 (m, 1H), 4.2 (m, 2H), 3.57 (s, 3H), 3.50 (m, 2H); 13C NMR (DMSO‑d6): 173.58, 155.37, 151.01, 150.23, 136.06, 127.04, 123.69, 121.15, 120.93, 118.40, 118.00, 111.43, 110.06, 102.58, 98.69, 54.02, 51.72, 27.27; HRMS (ESI + ): m/z calculated for C18H17N5O2 (M + 1) + 336.1382 found 336.1434.
(7H-pyrrolo[2,3–d]pyrimidin-4-yl)-l-valine (15a). Compound 14a (0.150 g, 0.50 mmol), 1.25 M NaOH (1.61 mL, 2.02 mmol) and 4 mL of MeOH were combined in a 25 mL round bottom flask and heated to reflux for 1 h. TLC indicated that the reaction was complete. The MeOH was removed using the rotary evaporator. The product was acidified with 3 M HCl and the solid was filtered using vacuum filtration. The dry yield was 0.113 g (96%). 1HNMR (DMSO‑d6): 11.49 (s, 1H), 8.05 (s, 1H), 7.29 (d, 1H, J = 4.0 Hz), 7.07 (s, 1H), 6.77 (d, 1H, J = 4.0 Hz), 4.61 (m, 1H), 2.19 (m, 1H), 1.00 (d, 3H, J = 6.4 Hz), 0.97 (d, 3H, J = 6.4 Hz); 13C NMR (DMSO‑d6): 173.95, 155.90, 150.94, 150.28, 120.87, 102.61, 99.10, 58.56, 29.88, 19.29, 18.98; MS (ES+, M + 1): 263.2.
(7H-pyrrolo[2,3–d]pyrimidine-4-yl)-l-leucinate (15b). The ethyl ester 14b (0.270 g, 0.77 mmol), 1.25 M NaOH (1.25 mL, 3.08 mmol) and 4 mL of MeOH were combined in a 25 mL round bottom flask and heat to boiling for 1 h, using a water cooled condenser. TLC indicated that the reaction was complete. The MeOH was removed using the rotary evaporator. The product was acidified with 3 M HCl and the solid was filtered using vacuum filtration. The dry yield was 0.082 g (43%). 1HNMR (DMSO‑d6): 11.5 (s, 1H), 8.05 (s, 1H), 7.47 (d, 1H), 7.06 (m, 1H), 6.64 (d, 1H), 4.85 (m, 1H), 1.76 (m, 2H), 1.62 (m, 1H), 0.92 (m, 6H); 13C NMR (DMSO‑d6): 175.14, 155.78, 151.04, 150.17, 120.91, 102.60, 98.86, 51.22, 40.05, 24.46, 22.95, 21.25; MS (ES+, M + 1): 249.1.
(7H-pyrrolo[2,3–d]pyrimidin-4-yl)-l-tryptophan (15c). Compound 14c (0.214 g, 0.64 mmol), 1.25 M NaOH (2 mL, 2.56 mmol) and 4 mL of MeOH were combined in a 25 mL round bottom flask and heated to boiling for 1 h. TLC indicated that the reaction was complete. The MeOH was removed using the rotary evaporator. The product was acidified with 3 M HCl and the solid was filtered using vacuum filtration. The dry yield was 0.113 g (55%). 1HNMR (DMSO‑d6): 11.46 (s, 1H), 10.75 (s, 1H), 8.01 (s, 1H). 7.57 (d, 1H, J = 8.0 Hz), 7.50 (s, 1H), 7.28 (d, 1H, J = 8.0 Hz), 7.16 (s, 1H), 7.01 (m, 3H), 6.58 (s, 1H), 4.82 (m, 1H), 3.22 (m, 2H); 13CNMR (DMSO‑d6): 174.52, 155.62, 151.06, 150.17, 136.05, 127.19, 123.60, 120.96, 120.83, 118.31, 118.19, 111.34, 110.60, 102.59, 98.70, 54.19, 27.23; HRMS (ESI + ): m/z calculated for C17H15N5O2 (M + 1) + 322.1226 found 322.1285.
(7H-pyrrolo[2,3–d]pyrimidin-4-yl)glycine (15d). 4-Chloro-7H-pyrrolo[2,3–d]-pyrimidine (0.500 g, 3.26 mmol, Glycine (0.294 g, 3.92 mmol), isopropanol (4 mL), and DIEA (1.71 mL, 9.81 mmol) were combined in a 25 mL round bottom and left to stir under heat for 9 days. TLC showed that the reaction was complete, therefore the isopropanol was removed via rotary evaporator. EtOAc (10 mL) was added to the reaction mixture, and poured into a separatory funnel. It was washed twice with 10% sodium bicarbonate (2 mL) and the aqueous layer was allowed to evaporate/concentrate overnight. The product eventually crashed out of the aqueous layer and was filtered off and characterized as the desired product. Dry yield = 0.263 g (42%). 1HNMR (DMSO‑d6): 11.5 (s, 1H), 8.05 (s, 1H), 7.58 (s, 1H), 7.07 (d, 1H), 6.53 (d, 1H), 4.05 (s, 2H); 13C NMR (DMSO‑d6): 172.18, 155.82, 151.18, 150.16, 120.98, 102.62, 98.48, 42.44; MS (ES+, M + 1): 193.1.
Methyl (1R, 2S)-2-((7H-pyrrolo[2,3–d]pyrimidin-4-yl)amino)cyclopentane-1-carboxylate (18). A 25.0 mL round bottom was used to combine chloride 3 (0.401 g, 2.62 mmol), methyl (1R, 2S)-2-aminocyclohexane-1-carboxylate 17 (0.451 g, 3.15 mmol), DIEA (1.37 mL, or 7.86 mmol), and 3.0 mL of isopropanol. The reaction was heated below the bp for 1.5 weeks, TLC indicated that the reaction was still incomplete (EtOAc). The reaction was concentrated on the rotary evaporator to remove the isopropyl alcohol. 10 mL of EtOAc was added to the round bottom. The organic layer was washed with 10% NaHCO3 and the organic layer was dried with MgSO4, filtered off using vacuum filtration and was allowed to air dry. Column chromatography was conducted (80/20% EtOAc/Hexanes → 100% EtOAc → 95/5 EtOAc/MeOH). Dry yield = 0.051 g, 12.7% yield. 1HNMR (CDCl3): δ 11.78 (s, 1H), 8.32 (s, 1H), 7.07 (d, 1H, J = 4.0 Hz), 6.39 (d, 1H, J = 4.0 Hz), 5.58 (s, 1H), 4.82 (m, 1H), 3.63 (s, 3H), 2.80 (m, 1H), 2.32 (m, 1H), 1.60–2.05 (m, 5H); 13C NMR (DMSO‑d6): 175.26, 155.59, 151.23, 150.13, 120.75, 102.43, 98.54, 55.00, 51.36, 49.61, 32.62, 28.77, 23.13; HRMS (ESI + ): m/z calculated for C13H16N4O2 (M + 1) + 261.1273 found 261.1339.
(1R, 2S)-2-((7H-pyrrolo[2,3–d]pyrimidin-4-yl)amino)cyclopentane-1-carboxylic acid (19). A 25.0 mL round bottom was used to combine the ester 18 (0.033 g, 0.127 mmol) and 10 mL of 3 M HCl. The reaction was heated to reflux for 3 h. Rotary evaporation was then used to remove the aqueous liquid. The solid was triturated with cold water and filtered to afford the product. Dry yield = 0.021 g, 63.6%. (DMSO‑d6): δ 12.79 (s, 1H), 9.98 (s, 1H), 8.30 (s, 1H), 7.37 (s, 1H), 7.15 (s, 1H), 4.65 (m, 1H), 3.19 (m, 1H), 2.06–1.64 (m, 6H); 13C NMR (DMSO‑d6): 175.00, 150.95, 150.01, 142.31, 124.19, 102.35, 101.67, 55.77, 48.53, 32.20, 28.04, 22.76; HRMS (ESI + ): m/z calculated for C12H14N4O2 (M + 1) + 247.1117 found 247.1180.
N-(2-(1H-indol-3-yl)ethyl)-7H-pyrrolo[2,3–d]pyrimidin-4-amine (20). 4-chloro-7H-pyrrolo[2,3–d]pyrimidine 3 (201 mg, 1.31 mmol), tryptamine (253 mg, 1.58 mmol), isopropanol (3.5 mL), and DIEA (0.35 mL, 2.01 mmol) and a stir bar were placed into a 25 mL round bottom flask and allowed to stir at 70 °C overnight. TLC provided evidence of a complete reaction (1:7 MeOH-EtOAc). The isopropanol was removed via rotary evaporator, and to the remaining brown oil was added 2 mL of EtOAc and 2 mL of dH2O. Approximately 1 h later, a tan solid began forming between the EtOAc-diH2O interface. With a small application of heat more solid began forming. Once no more solid formed the mixture was vacuum filtered to collect the solid. The solid was then placed in a vacuum oven to dry. Dry yield 126 mg (34.7% yield). 1HNMR (DMSO‑d6): δ 11.45 (s, 1H), 10.80 (s, 1H), 8.12 (s, 1H), 7.59 (d, 1H), 7.51 (m, 1H), 7.32 (d, 1H), 7.17 (d, 1H), 7.01 (m, 3H), 6.51 (dd, 1H), 3.73 (m, 2H), 2.99 (m, 2H); 13C NMR (DMSO‑d6): 156.05, 151.51, 150.06, 136.23, 127.32, 122.57, 120.88, 120.62, 118.36, 118.20, 112.10, 111.33, 102.52, 98.54, 40.86, 25.35; HRMS (ESI + ): m/z calculated for C16H15N5 (M + 1) + 278.1327 found 247.1387.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
DVF would like to acknowledge McDaniel College Student-Faculty Summer Research Fund, the Jean Richards Fund, the Schofield fund, and the Scott and Natalie Dahne fund. DVF would also like to acknowledge Mr. Kristopher Mason and Vaccitech for the use of their mass spectrometer. DVF would also like to thank Dr. Lisa Jenkins and the NIH for use of their High Resolution Mass Spectrometer. LL would like to acknowledge the use of the facilities of the Biocenter Oulu Structural Biology core facility, a member of Biocenter Finland, Instruct-ERIC Centre Finland and FINStruct. This research was funded by National Institutes of Health (NIH) grants P20 GM113117, P30GM110761, a CTSA grant from NCATS awarded to the University of Kansas for Frontiers: University of Kansas Clinical and Translational Science Institute UL1TR002366 and University of Kansas start-up funds to A.R.F, and by Sidrid Jusélius foundation grant to L.L., and Johns Hopkins Bloomberg School of Public Health Discretionary Fund to A.K.L.L.
References
- 1.Niknam Z., Jafari A., Golchin A., et al. Potential therapeutic options for COVID-19: an update on current evidence. Eur J Med Res. 2022;27:6. doi: 10.1186/s40001-021-00626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rack J.G., Perina D., Ahel I. Macrodomains: Structure, Function, Evolution, and Catalytic Activities. Annu Rev Biochem. 2016;85:431–454. doi: 10.1146/annurev-biochem-060815-014935. [DOI] [PubMed] [Google Scholar]
- 3.Leung A.K.L., Griffin D.E., Bosch J., Fehr A.R. The Conserved Macrodomain Is a Potential Therapeutic Target for Coronaviruses and Alphaviruses. Pathogens. 2022;11 doi: 10.3390/pathogens11010094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palazzo L., Mikolcevic P., Mikoc A., Ahel I. ADP-ribosylation signalling and human disease. Open Biol. 2019;9:190041. doi: 10.1098/rsob.190041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fehr A.R., Singh S.A., Kerr C.M., Mukai S., Higashi H., Aikawa M. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes Dev. 2020;34:341–359. doi: 10.1101/gad.334425.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fehr A.R., Channappanavar R., Jankevicius G., et al. The Conserved Coronavirus Macrodomain. Promotes Virulence and Suppresses the Innate Immune Response during Severe Acute Respiratory Syndrome Coronavirus Infection. mBio. 2016;7 doi: 10.1128/mBio.01721-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eriksson K.K., Cervantes-Barragan L., Ludewig B., Thiel V. Mouse hepatitis virus liver pathology is dependent on ADP-ribose-1''-phosphatase, a viral function conserved in the alpha-like supergroup. J Virol. 2008;82:12325–12334. doi: 10.1128/JVI.02082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fehr A.R., Athmer J., Channappanavar R., Phillips J.M., Meyerholz D.K., Perlman S. The nsp3 macrodomain promotes virulence in mice with coronavirus-induced encephalitis. J Virol. 2015;89:1523–1536. doi: 10.1128/JVI.02596-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuri T., Eriksson K.K., Putics A., et al. The ADP-ribose-1''-monophosphatase domains of severe acute respiratory syndrome coronavirus and human coronavirus 229E mediate resistance to antiviral interferon responses. J Gen Virol. 2011;92:1899–1905. doi: 10.1099/vir.0.031856-0. [DOI] [PubMed] [Google Scholar]
- 10.Putics A., Filipowicz W., Hall J., Gorbalenya A.E., Ziebuhr J. ADP-ribose-1“-monophosphatase: a conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J Virol. 2005;79:12721–12731. doi: 10.1128/JVI.79.20.12721-12731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grunewald M.E., Chen Y., Kuny C., et al. The coronavirus macrodomain is required to prevent PARP-mediated inhibition of virus replication and enhancement of IFN expression. PLoS Pathog. 2019;15:e1007756. doi: 10.1371/journal.ppat.1007756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voth L.S., O'Connor J.J., Kerr C.M., et al. Unique Mutations in the Murine Hepatitis Virus Macrodomain Differentially Attenuate Virus Replication, Indicating Multiple Roles for the Macrodomain in Coronavirus Replication. J Virol. 2021;95:e0076621. doi: 10.1128/JVI.00766-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abraham R., Hauer D., McPherson R.L., et al. ADP-ribosyl-binding and hydrolase activities of the alphavirus nsP3 macrodomain are critical for initiation of virus replication. Proc Natl Acad Sci USA. 2018;115:E10457–E10466. doi: 10.1073/pnas.1812130115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abraham R., McPherson R.L., Dasovich M., et al. Both ADP-Ribosyl-Binding and Hydrolase Activities of the Alphavirus nsP3 Macrodomain Affect Neurovirulence in Mice. mBio. 2020;11 doi: 10.1128/mBio.03253-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li C., Debing Y., Jankevicius G., et al. Viral Macro Domains Reverse Protein ADP-Ribosylation. J Virol. 2016;90:8478–8486. doi: 10.1128/JVI.00705-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McPherson R.L., Abraham R., Sreekumar E., et al. ADP-ribosylhydrolase activity of Chikungunya virus macrodomain is critical for virus replication and virulence. Proc Natl Acad Sci USA. 2017;114:1666–1671. doi: 10.1073/pnas.1621485114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parvez M.K. The hepatitis E virus ORF1 'X-domain' residues form a putative macrodomain protein/Appr-1''-pase catalytic-site, critical for viral RNA replication. Gene. 2015;566:47–53. doi: 10.1016/j.gene.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dasovich M., Zhuo J., Goodman J.A., et al. High-throughput Activity Assay for Screening Inhibitors of the SARS-CoV-2 Mac1 Macrodomain. ACS Chem Biol. 2022 doi: 10.1021/acschembio.1c00721. [DOI] [PubMed] [Google Scholar]
- 19.Russo L.C., Tomasin R., Matos I.A., et al. The SARS-CoV-2 Nsp3 macrodomain reverses PARP9/DTX3L-dependent ADP-ribosylation induced by interferon signaling. J Biol Chem. 2021;297:101041. doi: 10.1016/j.jbc.2021.101041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuller M., Correy G.J., Gahbauer S., et al. Fragment binding to the Nsp3 macrodomain of SARS-CoV-2 identified through crystallographic screening and computational docking. Sci Adv. 2021;7 doi: 10.1126/sciadv.abf8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Virdi R.S., Bavisotto R.V., Hopper N.C., et al. Discovery of Drug-Like Ligands for the Mac1 Domain of SARS-CoV-2 Nsp3. SLAS Discov. 2020;25:1162–1170. doi: 10.1177/2472555220960428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sowa S.T., Galera-Prat A., Wazir S., Alanen H.I., Maksimainen M.M., Lehtio L. A molecular toolbox for ADP-ribosyl binding proteins, Cell Rep. Methods. 2021:100121. doi: 10.1016/j.crmeth.2021.100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuller M., Riedel K., Gibbs-Seymour I., et al. Discovery of a Selective Allosteric Inhibitor Targeting Macrodomain 2 of Polyadenosine-Diphosphate-Ribose Polymerase 14. ACS Chem Biol. 2017;12:2866–2874. doi: 10.1021/acschembio.7b00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du Y., Khuri F.R., Fu H. A homogenous luminescent proximity assay for 14-3-3 interactions with both phosphorylated and nonphosphorylated client peptides, Curr Chem. Genomics. 2008;2:40–47. doi: 10.2174/1875397300802010040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su L., Bryan N., Battista S., et al. Identification of HMGA2 inhibitors by AlphaScreen-based ultra-high-throughput screening assays. Sci Rep. 2020;10:18850. doi: 10.1038/s41598-020-75890-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alhammad Y.M.O., Kashipathy M.M., Roy A., et al. The SARS-CoV-2 Conserved Macrodomain Is a Mono-ADP-Ribosylhydrolase. J Virol. 2021;95 doi: 10.1128/JVI.01969-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sowa S.T., Galera-Prat A., Wazir S., Alanen H.I., Maksimainen M.M., Lehtio L. Preparation of screening assays for ADP-ribosyl readers and erasers using the GAP-tag as a binding probe. STAR Protoc. 2022;3:101147. doi: 10.1016/j.xpro.2022.101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daniels C.M., Thirawatananond P., Ong S.E., Gabelli S.B., Leung A.K. Nudix hydrolases degrade protein-conjugated ADP-ribose. Sci Rep. 2015 Dec 16 doi: 10.1038/srep18271. PMID: 26669448; PMCID: PMC4680915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glumoff T., Sowa S.T., Lehtio L. Assay technologies facilitating drug discovery for ADP-ribosyl writers, readers and erasers. BioEssays. 2022;44:e2100240. doi: 10.1002/bies.202100240. [DOI] [PubMed] [Google Scholar]
- 30.Frick D.N., Virdi R.S., Vuksanovic N., Dahal N., Silvaggi N.R. Molecular Basis for ADP-Ribose Binding to the Mac1 Domain of SARS-CoV-2 nsp3. Biochemistry. 2020;59:2608–2615. doi: 10.1021/acs.biochem.0c00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friesner R.A., Banks J.L., Murphy R.B., et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 32.Friesner R.A., Murphy R.B., Repasky M.P., et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 33.Halgren T.A., Murphy R.B., Friesner R.A., et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 34.Jacobson M.P., Friesner R.A., Xiang Z., Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 35.Jacobson M.P., Pincus D.L., Rapp C.S., et al. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 36.Sastry G.M., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]