

ABSTRACT
Humoral immunity is a major component of the adaptive immune response against viruses and other pathogens with pathogen-specific antibody acting as the first line of defense against infection. Virus-specific antibody levels are maintained by continual secretion of antibody by plasma cells residing in the bone marrow. This raises the important question of how the virus-specific plasma cell population is stably maintained and whether memory B cells are required to replenish plasma cells, balancing their loss arising from their intrinsic death rate. In this study, we examined the longevity of virus-specific antibody responses in the serum of mice following acute viral infection with three different viruses: lymphocytic choriomeningitis virus (LCMV), influenza virus, and vesicular stomatitis virus (VSV). To investigate the contribution of memory B cells to the maintenance of virus-specific antibody levels, we employed human CD20 transgenic mice, which allow for the efficient depletion of B cells with rituximab, a human CD20-specific monoclonal antibody. Mice that had resolved an acute infection with LCMV, influenza virus, or VSV were treated with rituximab starting at 2 months after infection, and the treatment was continued for up to a year postinfection. This treatment regimen with rituximab resulted in efficient depletion of B cells (>95%), with virus-specific memory B cells being undetectable. There was an early transient drop in the antibody levels after rituximab treatment followed by a plateauing of the curve with virus-specific antibody levels remaining relatively stable (half-life of 372 days) for up to a year after infection in the absence of memory B cells. The number of virus-specific plasma cells in the bone marrow were consistent with the changes seen in serum antibody levels. Overall, our data show that virus-specific plasma cells in the bone marrow are intrinsically long-lived and can maintain serum antibody titers for extended periods of time without requiring significant replenishment from memory B cells. These results provide insight into plasma cell longevity and have implications for B cell depletion regimens in cancer and autoimmune patients in the context of vaccination in general and especially for COVID-19 vaccines.
IMPORTANCE Following vaccination or primary virus infection, virus-specific antibodies provide the first line of defense against reinfection. Plasma cells residing in the bone marrow constitutively secrete antibodies, are long-lived, and can thus maintain serum antibody levels over extended periods of time in the absence of antigen. Our data, in the murine model system, show that virus-specific plasma cells are intrinsically long-lived but that some reseeding by memory B cells might occur. Our findings demonstrate that, due to the longevity of plasma cells, virus-specific antibody levels remain relatively stable in the absence of memory B cells and have implications for vaccination.
KEYWORDS: antibodies, B cell responses, plasma cells, immune memory, viral immunity
INTRODUCTION
Acute viral infections and vaccinations induce long-term humoral immunity through the generation of memory B cells (MBCs) and plasma cells that contribute to humoral immunity through complementary mechanisms (1). Plasma cells preferentially reside in the bone marrow (BM), which is a specialized niche for the long-term survival of these cells through provision of various signals (2–5). Importantly, plasma cells constitutively secrete large amounts of antibodies without requiring antigenic stimulation through the B cell receptor (BCR) (2–4), which allows for the maintenance of serum antibody levels for extended periods in the absence of antigen. BM plasma cells thus represent long-lived effector cells contributing to immediate protection or limitation of pathogen spread. In contrast, MBCs represent a reservoir of antigen-specific B cells capable of rapidly responding to antigen by proliferating and differentiating into antibody-secreting plasma cells, thus rapidly increasing pathogen-specific antibody titers and also giving rise to more MBCs. Some of these MBCs can also enter germinal center reactions and undergo additional somatic hypermutation, thus qualitatively improving the humoral response for the encountered pathogen. Importantly, the MBC population is also long-lived and can be maintained for extended periods of time (6, 7), with human pathogen-specific MBCs being detectable for several decades after vaccination and infection (8–10).
The long-term maintenance of virus-specific antibodies for decades or even life raises the important question of how the virus-specific plasma cell population is stably maintained. While plasma cells are intrinsically long-lived, reseeding of the plasma cell population by MBCs might still be required to maintain a stable pool of plasma cells over extended periods of time (4). However, to which degree and how MBCs can replenish the plasma cell pool in vivo is unclear. MBCs can differentiate into plasma cells through BCR-dependent as well as BCR-independent signals. Antigen persistence or reinfection can result in antigen-driven proliferation and differentiation of MBCs into plasma cells and thus contribute to the replenishment of the plasma cell pool in an antigen-dependent manner. Of note, MBCs can also be stimulated in vitro to differentiate into plasma cells by providing cytokine and/or Toll-like receptor stimulation without the need for BCR stimulation (11–13). In addition, other nonspecific stimuli such as bystander T cell help can stimulate MBCs and drive plasma cell differentiation (14). However, the importance of such antigen-independent differentiation in vivo remains unclear (14, 15).
We previously estimated the half-life of lymphocytic choriomeningitis virus (LCMV)-specific plasma cells after irradiation-mediated B cell depletion (4). A caveat of this study was that this was not a specific depletion of B cells and irradiation acting on other cells might change the environment and affect the longevity of plasma cells. Rituximab, a monoclonal antibody (MAb) specific for human CD20, allows for specific B cell depletion and is widely used in the clinic for treatment of B cell malignancies and autoimmunity (16). Of note, a study employing rituximab-mediated B cell depletion in vaccinated rhesus macaques showed that the plasma cell pool is intrinsically long-lived and can be sustained for extended periods of time in the absence of MBCs (17). Here, we wanted to extend our earlier irradiation-based study by determining the longevity of serum antibody responses in three different acute viral infections in human CD20 transgenic (hCD20tg) mice that allow for the selective depletion of B cells using rituximab (18–20). We performed longitudinal measurements of virus-specific antibodies in the serum and analyzed the kinetics of virus-specific antibodies using a mixed effects exponential change model to determine the half-life of the virus-specific antibody in the absence of B cells.
RESULTS
Acute viral infections with LCMV, VSV, and influenza virus induce virus-specific IgG antibodies that are maintained long-term.
We first determined the kinetics of the antibody response in three different mouse models of acute viral infections. We infected BALB/c mice with either LCMV, influenza virus, or vesicular stomatitis virus (VSV) and measured virus-specific IgG antibody titers in the serum by enzyme-linked immunosorbent assay (ELISA). Mice infected with the different viruses exhibited similar virus-specific antibody kinetics (Fig. 1A to C). As previously reported, virus-specific antibody levels increased sharply during the first 3 weeks postinfection, eventually reaching peak levels around day 35 to 50, and were afterward stably maintained long-term (4).
FIG 1.

Acute viral infections generate durable antibody responses. Mice were infected with influenza (A), LCMV (B), or VSV (C) and virus-specific antibody titers in the serum were measured by ELISA. Data (mean and SEM) from a representative experiment (n = 5 mice) are shown.
Efficient depletion of virus-specific memory B cells after treatment of human CD20 transgenic mice with rituximab.
The efficiency and specificity of rituximab, an anti-hCD20 MAb, to deplete naive and memory B cells in human CD20 transgenic (hCD20tg) mice has been extensively characterized (18–20). Importantly, hCD20 expression in hCD20tg mice has been shown to faithfully recapitulate the expression of CD20 in humans, being first expressed at the immature B cell stage, maintained at high levels in all B cell subsets, including germinal center (GC) B cells, but being absent on plasma cells (19). To study the effects of B cell depletion on virus-specific antibody titers, we initiated rituximab treatment of hCD20tg mice 2 months postinfection with LCMV (Fig. 2A), a time point at which the virus infection had been resolved and serum antibody titers were stably maintained for several weeks (Fig. 1). Of note, we previously demonstrated that the GC B cell response peaks around 2 weeks postinfection and then gradually subsides and reaches baseline levels within 4 to 6 weeks postinfection (21). Initiation of rituximab treatment at 2 months postinfection was thus unlikely to interfere significantly with virus-specific GC B cell responses. Immune mice initially received three injections of 1 mg rituximab within the first week, a regimen that was previously shown to result in efficient depletion of B cells (20), or received phosphate-buffered saline (PBS) as control. Mice were subsequently treated with weekly injections of rituximab or PBS for the remainder of the experiment to maintain B cell depletion, with B cell levels in the peripheral blood being monitored by flow cytometry. The employed treatment regimen efficiently depleted total circulating B cells (B220+ CD19+) in LCMV immune hCD20tg mice and maintained the B cell depletion to a high degree throughout the treatment period (Fig. 2B). Comparable depletion kinetics and efficiencies were also observed in influenza and VSV immune hCD20tg mice after initiation of rituximab treatment (Fig. 2C and D). The number of circulating B cells in the peripheral blood declined sharply within 1 week of treatment initiation, and maximum depletion was achieved after about a month of treatment. B cell numbers in the peripheral blood of rituximab-treated hCD20tg mice remained at almost undetectable levels for the duration of the experiments. Of note, rituximab treatment efficiently depleted circulating B cells (B220+ CD19+) in LCMV immune hCD20tg mice but did not affect B cell frequencies in LCMV immune non-hCD20 transgenic BALB/c mice that exhibited peripheral blood B cell frequencies comparable to PBS-treated controls (20 to 30% of peripheral blood mononuclear cells [PBMCs]) throughout a 100-day treatment course, underlining the specificity of rituximab for human CD20 but not mouse CD20.
FIG 2.
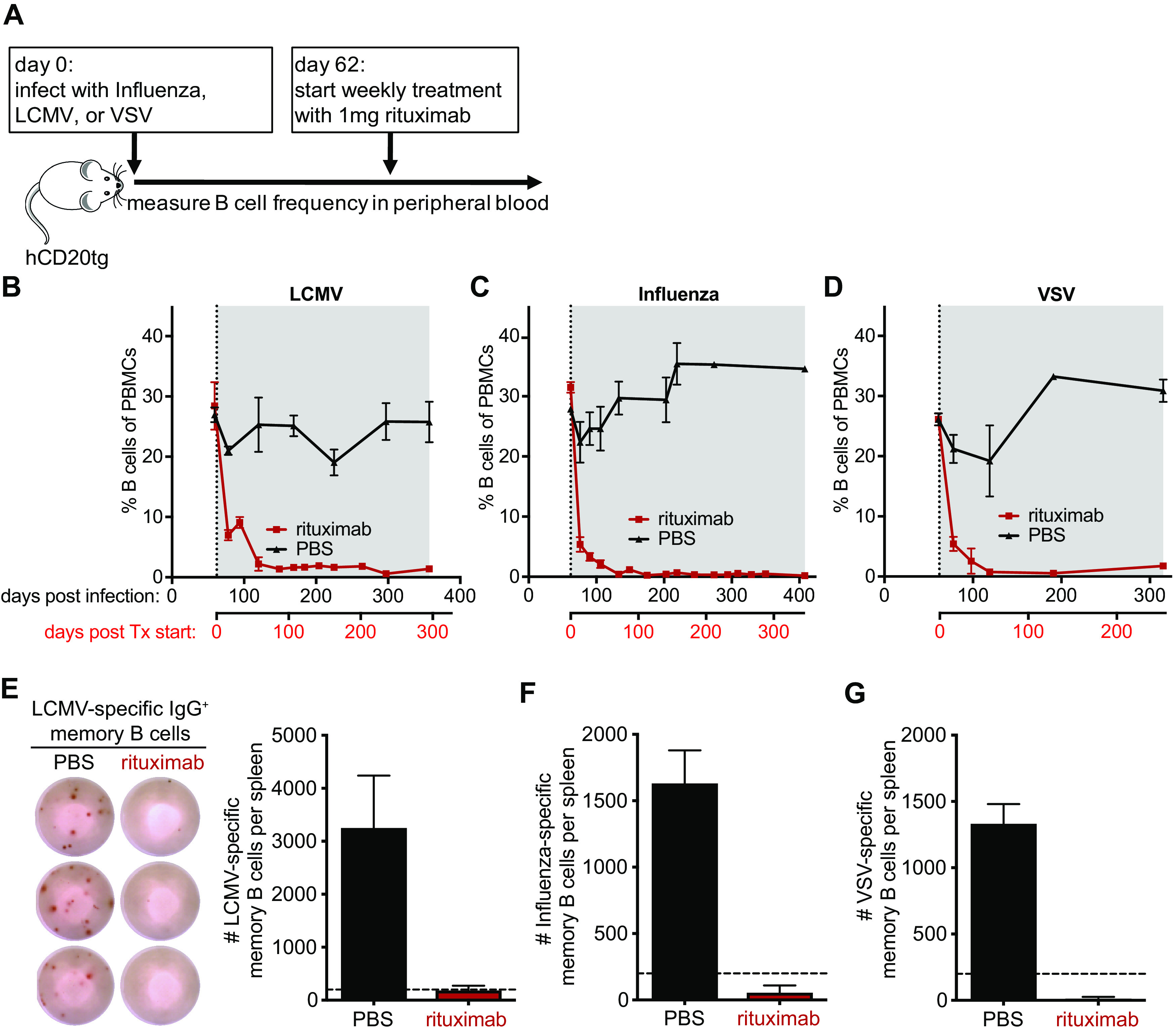
Efficient depletion of B cells in hCD20tg mice by rituximab. hCD20tg mice were infected with influenza virus, LCMV, or VSV, and weekly treatment with rituximab (red) or PBS (black) was initiated on day 62 postinfection. (A) Experimental design. (B to D) The frequency of B cells (B220+ CD19+ CD3−) among peripheral blood mononuclear cells (PBMCs) was monitored by flow cytometry at the indicated times after infection with LCMV (B), Influenza (C), or VSV (D). Treatment initiation and period are indicated by vertical dotted lines and gray shading, respectively. (E) Representative ELISPOT shows LCMV-specific IgG+ memory B cells in the spleen of LCMV immune mice 295 days after treatment initiation with either rituximab or PBS. Graph shows the total number of LCMV-specific IgG+ memory B cells in the spleen. Dotted line indicates the limit of detection. (F and G) Graphs show the total number of Influenza-specific (F) and VSV-specific (G) IgG+ memory B cells in the spleen at the end of the treatment period. Dotted horizontal lines indicate the limit of detection. Data (mean and SEM) from a representative experiment (n = 2 to 6 mice per group) are shown.
After determining that circulating B cells were efficiently depleted by rituximab treatment, we next wanted to investigate whether the treatment was also effective at depleting virus-specific MBCs in the spleen, the major reservoir of MBCs in mice (4). We therefore euthanized mice at the end of the treatment regimen, ranging between 185 and 346 days of continuous rituximab treatment, and quantified the number of virus-specific MBC in the spleen by enzyme-linked immunosorbent spot (ELISPOT) assay as described previously (11). Virus-specific MBCs against all three viruses were readily detected in the spleens of PBS-treated immune mice but undetectable in rituximab-treated mice (Fig. 2E to G). These results demonstrate that splenic virus-specific MBCs were efficiently depleted by rituximab.
Quantitating virus-specific serum antibody levels after depletion of memory B cells.
After confirming that rituximab treatment efficiently depleted virus-specific MBCs, we next wanted to determine its effect on virus-specific IgG antibody levels. We performed serial blood draws from immune mice treated with either rituximab or PBS and determined virus-specific IgG antibody levels in the serum by ELISA. In all immune mice, irrespective of the viral infection, we observed an early transient decline (about 3-fold) in virus-specific IgG levels within the first 3 to 4 weeks after rituximab treatment was initiated (Fig. 3A to C). The initial drop of virus-specific IgG titers in rituximab-treated animals was followed by a much slower decline over time, with antibody levels remaining relatively stable up to 1 year postinfection (Fig. 3A to C). Similar results were seen with hemagglutination inhibition (HAI) antibody to influenza virus and neutralizing antibody to VSV (Fig. 3D and E). The similar kinetics of the above assessed virus-specific antibody responses upon rituximab treatment, independent of the employed infection or assay (Fig. 3A to E), prompted us to combine all of our available data to rigorously investigate the dynamics of virus-specific antibodies following rituximab-mediated MBC depletion using a mixed effects exponential decline model. This model recapitulated the initial transient decline in virus-specific antibody with an estimated half-life of 29 days (95% confidence interval [CI], 24 to 38 days) within the first 17 days of treatment, followed by a much slower decline in virus-specific antibody in the later phase of rituximab treatment, with an estimated half-life of 372 days (95% CI, 208 to 1,751 days) after day 85 post treatment initiation (Fig. 3F). Overall, our data suggest that virus-specific serum antibody titers induced by acute viral infections can persist at high levels in the absence of virus-specific MBCs, with minimally declining titers at later time points after initiation of B cell depletion.
FIG 3.

Examining the durability of virus-specific antibody after depletion of B cells. (A to F) Virus-specific antibody responses were assessed in the serum of rituximab-treated (red) and PBS-treated (black) hCD20tg mice immune to LCMV (A), influenza (B and D), or VSV (C and E). (A to C) Virus-specific IgG was measured by ELISA. (D) Hemagglutination inhibition (HAI) titers of influenza immune mice. (E) VSV neutralization titers in VSV immune mice. Treatment initiation and period are indicated by vertical dotted line and gray shading, respectively. Horizontal dotted line indicates the limit of detection. Data (mean and SEM) from a representative experiment (n = 4 to 6 mice per group) are shown. (F) Combined estimated half-life of virus-specific antibody responses measured in panels A to E are shown for both phases of the biphasic decline.
Quantitation of virus-specific BM plasma cells in rituximab-treated mice.
We next examined virus-specific plasma cells in the BM of rituximab-treated mice. This was done by quantifying the number of virus-specific IgG-producing plasma cells in the BM of immune mice by ELISPOT on viral antigen-coated plates in a short-term assay. Of note, only plasma cells actively secreting antibody are detected by this assay (11–13). The number of LCMV-specific IgG-producing BM plasma cells was assessed at two different time points (185 and 295 days) after treatment initiation. Rituximab-treated animals showed about a 3-fold reduction in the number of LCMV-specific BM plasma cells compared to those of the PBS-treated controls (Fig. 4A). However, it is worth noting that there was no drop in the number of LCMV-specific plasma cells between days 185 and 295 post rituximab treatment despite the continuous rituximab treatment and absence of virus-specific MBCs in these mice (Fig. 4A). These BM plasma cell data are consistent with the relatively stable antibody response during this period. More importantly, these results demonstrate the inherent longevity of BM plasma cells after depletion of B cells. These data also suggest that the observed reduction in plasma cells occurred early after treatment initiation with rituximab as reflected by the serological analyses (Fig. 3). Of note, we observed a similar reduction in virus-specific IgG-producing BM plasma cell in influenza and VSV-immune mice treated with rituximab (Fig. 4B and C). The number of virus-specific BM plasma cells in all three infection models accurately reflected the levels of virus-specific antibody in the serum.
FIG 4.

Analysis of virus-specific IgG+ plasma cells in the bone marrow of rituximab-treated mice. Virus-specific plasma cell numbers in the bone marrow of rituximab- (red) or PBS- (black) treated hCD20tg mice were quantified by ELISPOT. (A) Number of LCMV-specific IgG-producing bone marrow (BM) plasma cells per femur after 185 and 295 days of continuous treatment. (B) Number of influenza virus-specific BM plasma cells per femur after 342 days of continuous treatment. (C) Number of VSV-specific BM plasma cells per femur after 253 days of continuous treatment. Data (mean and SEM) from one experiment with n = 2 to 6 mice per group are shown. One-way ANOVA with Tukey’s multiple comparison test (A) and unpaired two-sided t test (B and C) was used for analyses with P values being indicated.
DISCUSSION
The data collected at different time points throughout the experiments of this study enabled us to estimate the half-life of virus-specific antibody in the absence of virus-specific MBCs. Our results demonstrate that virus-specific antibody levels in the serum remain relatively stable in the absence of MBCs with a half-life of 372 days, and that BM plasma cells, the major source of circulating IgG antibodies, are inherently long-lived. This confirms and extends our original observations in the Slifka et al. study (4) and is also consistent with the stable maintenance of plasma cells and antibody titers observed in B cell-depleted rhesus macaques (17). Interestingly, untreated immune mice showed no decline in virus-specific antibody levels, suggesting that under steady state conditions a low level of reseeding of the plasma cell pool by MBCs might occur and contribute to the stable maintenance of serum antibody levels over extended periods of time.
A surprising observation of our study was the biphasic decline of virus-specific antibody titers with an initial 3- to 4-fold drop of virus-specific antibody titers within the first 3 to 4 weeks after treatment initiation. Rituximab treatment of wild-type BALB/c mice (lacking human CD20 expression) had no effect on antibody levels showing that this drop was not due to altered virus-specific antibody half-life in the presence of rituximab. The initial drop in virus-specific antibody titers most likely reflects the immediate depletion of a subset of plasma cells that retained hCD20 expression and are thus a target of rituximab. Of note, expression of mCD20 on murine plasma cells has been previously reported (15), suggesting species-specific difference in the regulation of CD20 expression between mice and humans, which lack CD20 expression on plasmablasts and plasma cells. However, additional studies are required to elucidate the mechanisms driving these differences and determine how BM plasma cells susceptible to depletion by rituximab in the employed hCD20tg mouse model differ from refractory cells. In addition to human CD20 expression, refractory cells might also differ from susceptible cells in terms of their microanatomical location within the BM and the expression of inhibitory receptors equipping them with increased resistance to antibody-mediated depletion. The BM has been previously shown to be a niche with limited therapeutic IgG activity (22), but whether certain microanatomical niches within the BM differ in their susceptibility to antibody-mediated depletion has not been evaluated. An additional explanation for the initial drop of virus-specific antibodies following rituximab administration could be that the engagement of activating Fcγ receptors on granulocytes and macrophages induced an inflammatory state (23), impairing plasma cell survival either directly or indirectly through modulation of the supporting BM niche.
Although unlikely to be the sole mechanism due to the observed kinetics of the virus-specific GC B cell response (21), it is possible that depletion of some residual virus-specific GC B cells at day 60 postinfection could have also partially contributed to the initial drop of antibody titers upon rituximab treatment initiation. Future studies are needed to address these questions and thus provide a more in-depth analysis of the mechanisms responsible for the initial and transient drop in virus-specific antibody titers.
Two previous studies reported no decline in plasma cell numbers in mice immunized with the hapten 4-hydroxy-3-nitrophenyl-acetyl in the absence of MBCs (15, 24). Given the estimated plasma cell half-life of 372 days, these studies are in line with our data and underline the need for longitudinal studies of individual animals for extended periods of time to capture slight decreases in cell numbers due to the natural death of plasma cells. Furthermore, in line with our results, a decade-long study in nonhuman primates showed the persistence of virus-specific antibody responses in the absence of virus-specific memory B cells (17), further supporting the inherent longevity of virus-specific plasma cells. By detecting BrdU-labeled plasma cells, this study also provided direct evidence for the persistence of individual plasma cells for up to 10 years.
Antibody-mediated B cell depletion offers an elegant and direct way to deplete MBCs; however, the efficiency of antibody-mediated depletion depends on intravascular access, with tissue-resident cells being only inefficiently depleted (19). While the vast majority of virus-specific MBCs are located in the spleen, virus-specific MBCs have also been found in various tissues, such as the lungs (25). As we only assessed virus-specific MBCs in the spleen, we cannot entirely exclude the possibility that a small subset of tissue-resident MBCs in other tissues remained and potentially gave rise to new plasma cells in our study. However, the continued administration of rituximab in our study was aimed at depleting newly differentiated plasma cells that retained hCD20 surface expression and reached the circulation.
Rituximab is used for the treatment of lymphomas, leukemias, and various autoimmune disorders, such as rheumatoid arthritis and pemphigus (16). The effect of rituximab on serum antibody levels has been reported to range from no significant decreases in serum antibody levels to severe pan-hypogammaglobulinemia (26–36). Importantly, antibody responses to common virus and vaccine antigens have been demonstrated to be relatively stable after rituximab treatment, suggesting that rituximab treatment preferentially affects short-lived antibody responses, such as auto-reactive antibodies (36).
Infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) induces SARS-CoV-2-specific plasma cells that are detectable in the BM of convalescent individuals for at least 11 months postinfection (37). However, little is known about the longevity of SARS-CoV-2-specific plasma cells and whether SARS-CoV-2 mRNA vaccines can induce a robust and long-lived plasma cell population in the BM. Our data suggest that preexisting antibody levels against SARS-CoV-2 should be closely monitored in cancer and autoimmune patients treated with B cell-depleting agents such as rituximab. Monitoring might be especially important in patients who are treated long-term and/or initiated treatment shortly after mRNA vaccination, which poses the risk of disrupting the SARS-CoV-2-specific plasma cell pool through abrogating novel plasma cell formation in persisting GC reactions (38). However, further research is required to determine the relative contribution of newly generated plasma cells for the maintenance of the overall plasma cell pool over time in order to guide timing-related treatment decisions in mRNA-vaccinated patients.
Overall, our study demonstrates that virus-specific plasma cells in the bone marrow are intrinsically long-lived with a half-life of about 1 year, allowing the relatively stable maintenance of antibody levels in the absence of memory B cells. These results provide insights into plasma cell longevity and have implications for antibody-based B cell depletion strategies in cancer and autoimmune patients in the context of vaccination in general and especially for COVID-19 vaccines.
MATERIALS AND METHODS
Mice.
The generation and characterization of hCD20tg mice has been described previously (18). Briefly, the hCD20 locus was incorporated into bacterial artificial chromosomes and incorporated into pronuclei of F2 embryos by the Yale Genomics Transgenic Mouse Service. Mice expressing hCD20 were further characterized and backcrossed to a BALB/c background. hCD20tg mice were maintained and bred at the Emory University School of Medicine animal facility. All experiments were performed in accordance with approved IACUC protocols.
Infections, antibody treatment, and lymphocyte isolation.
Mice were infected at 6 to 8 weeks of age. For influenza infections, anesthetized mice were administered 5 × 103 PFU of A/WSN/33 (H1N1) diluted to 30 μL in PBS intranasally. Acute LCMV infections were accomplished by injecting 2 × 105 PFU of LCMV Armstrong diluted to 500 μL in PBS intraperitoneally, and 2 × 106 PFU of VSV-Indiana diluted to 500 μL in PBS was given intravenously for VSV infections. Peripheral blood was collected at the indicated times via submandibular bleeding. Lymphocytes were isolated from peripheral blood, spleens, and bone marrow as described previously (39). Rituxan (rituximab) was obtained from the Emory University Hospital pharmacy, and treatment consisted of injecting the mice intraperitoneally with 1 mg/mouse on days one, three, and five, followed by weekly injections of 1 mg as described previously (20).
Hemagglutinin inhibition assay.
One part serum was added to three parts receptor destroying enzyme (RDE) (Accurate Chemical & Scientific) and incubated at 37°C overnight. The RDE was inactivated the following morning by incubating the samples at 56°C for 1 h. Samples were then serially diluted with PBS in 96-well v-bottom plates, and eight hemagglutination units (as determined by incubation with 0.5% turkey red blood cells [RBCs] in the absence of serum) of influenza virus were added to each well. After 30 min at room temperature, 50 μL of 0.5% turkey RBCs (Lampire Biological Laboratories) suspended in PBS-0.5% bovine serum albumin (BSA) was added to each well, and the plates were shaken manually. After an additional 30 min at room temperature, the serum titers were read as the reciprocal of the final dilution for which no hemagglutination was observed.
VSV neutralization assay.
Serum was heat inactivated at 56°C for 30 min before being diluted 1:40 in serum-free Dulbecco modified Eagle medium (DMEM). Serial dilutions were performed and mixed with 100 PFU VSV-Indiana at 37°C for 1 h. These were then placed on confluent Vero cells in 6-well plates and incubated at 37°C for 45 min. After the incubation, the cells were covered with an agar overlay supplemented with 10% DMEM and incubated at 37°C. After 2 days, the overlay was removed, and the cell monolayers were stained with 1% crystal violet. The neutralization titer was reported as the reciprocal of the dilution that inhibited 50% of the virus from forming plaques.
Antibodies and flow cytometry.
For flow cytometry analysis of B cell depletion in the blood, isolated peripheral blood mononuclear cells were stained with antibodies against CD3 (17A2), B220 (RA3-6B2), CD19 (ID3), and hCD20 (2H7). Samples were acquired using a BD FACSCalibur or BD FACSCanto II, followed by analysis using FlowJo.
ELISPOT.
ELISPOT assays were performed as previously described (12). Briefly, ELISPOT plates were coated with 1.5 mL viral lysate/plate for LCMV and VSV, 2.5 mg/well for influenza, or 62.5 μL per well of goat anti-mouse IgG+M+A (catalog number M30900; Caltag Laboratories) for total plasma cell numbers. PBS was added to these so that a total of 100 μL could be plated/well. The plates were incubated at 4°C overnight. The following morning, plates were washed with PBS/0.1% Tween and then PBS, and they were blocked for 2 h at room temperature (RT) with 10% RPMI. Lymphocytes were resuspended to 1 × 107 cells/mL, and 50 μL of this was added to the first well on each plate. Serial dilutions were performed, and the plates were incubated at 37°C for 5 h. Plates were washed with PBS followed by PBS/0.1% Tween, and 100 μL of biotinylated anti-IgG diluted 1:1,000 in PBS/0.1% Tween/1% fetal calf serum (FCS) was added to each well before incubating the plates overnight at 37°C. The following morning, plates were washed with PBS/0.1% Tween and 100 μL/well of horseradish peroxidase (HRP)-conjugated avidin-D (Vector Labs) diluted 1:1,000 in PBS/0.1% Tween/1% FCS before incubating the plates for 1 h at RT. Plates were washed with PBS/0.1% Tween followed by PBS, and 100 μL of the enzyme chromogen substrate (see below) was added to each well for ∼8 min to develop the spots. Plates were counted manually. The enzyme chromogen substrate consists of 20 mg/mL 3-amino-9-ethylcarbazole (Sigma) in dimethylformamide diluted 1:67 in 0.1 M Na-acetate buffer. This was filtered through a 0.2-μm membrane, and 100 μL H2O2/10 mL of substrate was added immediately before use.
Memory B cell assay.
Memory B cell assays were performed with minor modifications as previously described (11). Briefly, splenocytes were collected from immune mice and from one naive mouse/96-well plate to irradiate as feeder cells. A master mix was made (amount per plate is listed as follows: 60 μL of 1 mg/mL R595 lipopolysaccharide [LPS] [ALX-581-008-L002; Alexis Biochemicals], 2 mL ConA-stimulated supernatant [see below], 3 mL RPMI-10% FCS, 10 mL 1 × 107 cells/mL irradiated naive splenocytes [sex-matched] 1,200 rad irradiation). To each well, 150 μL of stimulant master mix and 50 μL of diluted splenocytes were added. Splenocytes were diluted as follows: 4 cell concentrations with 12 wells/concentration (across one row of plate). The first dilution was 2.5 × 105 splenocytes/well and then followed by three 5-fold dilutions (5 × 104, 1 × 104, 2 × 103/well). The plates were incubated at 37°C with 5% CO2 for 5 days. On the fourth day, ELISPOT plates were coated as described above. On day 5, the plates were blocked as described above. The cultured cells were transferred to 96-well round-bottom plates and centrifuged at 800 rpm for 3 min three times. Cells were resuspended in 200 μL RPMI + 1% FCS. Cells were transferred to ELISPOT plates and incubated overnight at 37°C with 5% CO2. The remainder of the procedure is identical to the ELISPOT assay described above. ELISPOT wells were scored positive for MBCs if there were greater than 3 plasma cell spots at a dilution where the day 0 ELISPOT assay revealed less than or equal to 1 plasma cell spot.
Concanavalin A (ConA) supernatant was prepared as follows: splenocytes were isolated from a naive mouse and suspended at 1.25 × 106/mL in DMEM-5% FCS medium. Cells were cultured in a T-75 flask at 20 mL volume with 2.5 μg/mL ConA (product number C5275; Sigma) and 20 ng/mL PMA (product number P1585; Sigma) for 48 h at 37°C. The supernatant was transferred to 50-mL tubes and spun at 1,500 rpm for 10 min to remove the cells. The supernatant was sterilized by filtering through a 0.2-μm syringe filter and aliquoted and frozen at −80°C.
ELISA.
ELISAs were performed as described previously (40). Briefly, Nunc MaxiSorp plates (catalog number 442404; Nunc) were coated with 2.5 μg per well of A/WSN/33, LCMV lysate, or VSV lysate diluted in PBS and kept at 4°C overnight. The following day, serum was added at different concentrations, and the plates were incubated at 37°C for 1 h. Plates were then washed three times with PBS/0.2% Tween before HRP-conjugated goat α-mouse IgG was added (product number A4416; Sigma). SigmaFast OPD tablets (product number P9187; Sigma) were used to develop the plates. The reactions were stopped with 0.1 N H2SO4, and the plates were read at 490 nm. The antibody concentrations were determined by endpoint titers.
Estimation of half-lives.
Mixed effects exponential change models were used to model the dynamics of antibody levels. Antibody (y) was log transformed to give linear models of the form ln(y) = a + b × time, which were fitted using lme4 package and R programming language. Models included population level fixed effects and random effects for individual time series. Half-lives were calculated using the formula half-life = ln(0.5)/slope.
Statistical analysis.
Statistical analyses were performed using GraphPad Prism 9.3 (GraphPad Software LLC). Values are expressed as mean and standard error of the mean (SEM). Unpaired two-sided t test was used for comparisons of two groups and ordinary one-way analysis of variance (ANOVA) with post hoc Tukey’s test for multiple comparisons. P values of less than 0.05 were considered statistically significant.
Data availability.
Code and data for modeling of antibody levels and calculation of half-lives are available on GitHub (https://github.com/HA2342/WL).
ACKNOWLEDGMENTS
This work was supported by NIH grant U19AI117891 to R. Antia and R. Ahmed. This study was supported in part by the Emory Flow Cytometry Core (EFCC), one of the Emory Integrated Core Facilities (EICF), and is subsidized by the Emory University School of Medicine. Additional support was provided by the National Center for Georgia Clinical & Translational Science Alliance of the National Institutes of Health under award number UL1TR002378.
The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
Contributor Information
Rustom Antia, Email: rantia@emory.edu.
Rafi Ahmed, Email: rahmed@emory.edu.
Stacey Schultz-Cherry, St. Jude Children's Research Hospital.
REFERENCES
- 1.Akkaya M, Kwak K, Pierce SK. 2020. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol 20:229–238. 10.1038/s41577-019-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Slifka MK, Matloubian M, Ahmed R. 1995. Bone marrow is a major site of long-term antibody production after acute viral infection. J Virol 69:1895–1902. 10.1128/JVI.69.3.1895-1902.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manz RA, Thiel A, Radbruch A. 1997. Lifetime of plasma cells in the bone marrow. Nature 388:133–134. 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- 4.Slifka MK, Antia R, Whitmire JK, Ahmed R. 1998. Humoral immunity due to long-lived plasma cells. Immunity 8:363–372. 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 5.Tangye SG. 2011. Staying alive: regulation of plasma cell survival. Trends Immunol 32:595–602. 10.1016/j.it.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Anderson SM, Hannum LG, Shlomchik MJ. 2006. Memory B cell survival and function in the absence of secreted antibody and immune complexes on follicular dendritic cells. J Immunol 176:4515–4519. 10.4049/jimmunol.176.8.4515. [DOI] [PubMed] [Google Scholar]
- 7.Schittek B, Rajewsky K. 1990. Maintenance of B-cell memory by long-lived cells generated from proliferating precursors. Nature 346:749–751. 10.1038/346749a0. [DOI] [PubMed] [Google Scholar]
- 8.Crotty S, Felgner P, Davies H, Glidewell J, Villarreal L, Ahmed R. 2003. Cutting edge: long-term B cell memory in humans after smallpox vaccination. J Immunol 171:4969–4973. 10.4049/jimmunol.171.10.4969. [DOI] [PubMed] [Google Scholar]
- 9.Yu X, Tsibane T, McGraw PA, House FS, Keefer CJ, Hicar MD, Tumpey TM, Pappas C, Perrone LA, Martinez O, Stevens J, Wilson IA, Aguilar PV, Altschuler EL, Basler CF, Crowe JE, Jr.. 2008. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature 455:532–536. 10.1038/nature07231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amanna IJ, Carlson NE, Slifka MK. 2007. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med 357:1903–1915. 10.1056/NEJMoa066092. [DOI] [PubMed] [Google Scholar]
- 11.Slifka MK, Ahmed R. 1996. Limiting dilution analysis of virus-specific memory B cells by an ELISPOT assay. J Immunol Methods 199:37–46. 10.1016/S0022-1759(96)00146-9. [DOI] [PubMed] [Google Scholar]
- 12.Crotty S, Aubert RD, Glidewell J, Ahmed R. 2004. Tracking human antigen-specific memory B cells: a sensitive and generalized ELISPOT system. J Immunol Methods 286:111–122. 10.1016/j.jim.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 13.Jahnmatz M, Kesa G, Netterlid E, Buisman AM, Thorstensson R, Ahlborg N. 2013. Optimization of a human IgG B-cell ELISpot assay for the analysis of vaccine-induced B-cell responses. J Immunol Methods 391:50–59. 10.1016/j.jim.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Bernasconi NL, Traggiai E, Lanzavecchia A. 2002. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 298:2199–2202. 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 15.DiLillo DJ, Hamaguchi Y, Ueda Y, Yang K, Uchida J, Haas KM, Kelsoe G, Tedder TF. 2008. Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol 180:361–371. 10.4049/jimmunol.180.1.361. [DOI] [PubMed] [Google Scholar]
- 16.Pierpont TM, Limper CB, Richards KL. 2018. Past, present, and future of rituximab-the world's first oncology monoclonal antibody therapy. Front Oncol 8:163. 10.3389/fonc.2018.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammarlund E, Thomas A, Amanna IJ, Holden LA, Slayden OD, Park B, Gao L, Slifka MK. 2017. Plasma cell survival in the absence of B cell memory. Nat Commun 8:1781. 10.1038/s41467-017-01901-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ. 2007. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol 179:3351–3361. 10.4049/jimmunol.179.5.3351. [DOI] [PubMed] [Google Scholar]
- 19.Gong Q, Ou Q, Ye S, Lee WP, Cornelius J, Diehl L, Lin WY, Hu Z, Lu Y, Chen Y, Wu Y, Meng YG, Gribling P, Lin Z, Nguyen K, Tran T, Zhang Y, Rosen H, Martin F, Chan AC. 2005. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J Immunol 174:817–826. 10.4049/jimmunol.174.2.817. [DOI] [PubMed] [Google Scholar]
- 20.Wieland A, Shashidharamurthy R, Kamphorst AO, Han JH, Aubert RD, Choudhury BP, Stowell SR, Lee J, Punkosdy GA, Shlomchik MJ, Selvaraj P, Ahmed R. 2015. Antibody effector functions mediated by Fcγ-receptors are compromised during persistent viral infection. Immunity 42:367–378. 10.1016/j.immuni.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasheed MA, Latner DR, Aubert RD, Gourley T, Spolski R, Davis CW, Langley WA, Ha SJ, Ye L, Sarkar S, Kalia V, Konieczny BT, Leonard WJ, Ahmed R. 2013. Interleukin-21 is a critical cytokine for the generation of virus-specific long-lived plasma cells. J Virol 87:7737–7746. 10.1128/JVI.00063-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lux A, Seeling M, Baerenwaldt A, Lehmann B, Schwab I, Repp R, Meidenbauer N, Mackensen A, Hartmann A, Heidkamp G, Dudziak D, Nimmerjahn F. 2014. A humanized mouse identifies the bone marrow as a niche with low therapeutic IgG activity. Cell Rep 7:236–248. 10.1016/j.celrep.2014.02.041. [DOI] [PubMed] [Google Scholar]
- 23.Wieland A, Ahmed R. 2019. Fc receptors in antimicrobial protection. Curr Top Microbiol Immunol 423:119–150. 10.1007/82_2019_154. [DOI] [PubMed] [Google Scholar]
- 24.Ahuja A, Anderson SM, Khalil A, Shlomchik MJ. 2008. Maintenance of the plasma cell pool is independent of memory B cells. Proc Natl Acad Sci USA 105:4802–4807. 10.1073/pnas.0800555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joo HM, He Y, Sangster MY. 2008. Broad dispersion and lung localization of virus-specific memory B cells induced by influenza pneumonia. Proc Natl Acad Sci USA 105:3485–3490. 10.1073/pnas.0800003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett CM, Rogers ZR, Kinnamon DD, Bussel JB, Mahoney DH, Abshire TC, Sawaf H, Moore TB, Loh ML, Glader BE, McCarthy MC, Mueller BU, Olson TA, Lorenzana AN, Mentzer WC, Buchanan GR, Feldman HA, Neufeld EJ. 2006. Prospective phase 1/2 study of rituximab in childhood and adolescent chronic immune thrombocytopenic purpura. Blood 107:2639–2642. 10.1182/blood-2005-08-3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Looney RJ, Anolik JH, Campbell D, Felgar RE, Young F, Arend LJ, Sloand JA, Rosenblatt J, Sanz I. 2004. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial of rituximab. Arthritis Rheum 50:2580–2589. 10.1002/art.20430. [DOI] [PubMed] [Google Scholar]
- 28.Edwards JCW, Szczepański L, Szechiński J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. 2004. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med 350:2572–2581. 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 29.Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. 2006. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum 54:723–732. 10.1002/art.21650. [DOI] [PubMed] [Google Scholar]
- 30.Keystone E, Fleischmann R, Emery P, Furst DE, van Vollenhoven R, Bathon J, Dougados M, Baldassare A, Ferraccioli G, Chubick A, Udell J, Cravets MW, Agarwal S, Cooper S, Magrini F. 2007. Safety and efficacy of additional courses of rituximab in patients with active rheumatoid arthritis: an open-label extension analysis. Arthritis Rheum 56:3896–3908. 10.1002/art.23059. [DOI] [PubMed] [Google Scholar]
- 31.Lim SH, Esler WV, Zhang Y, Zhang J, Periman PO, Burris C, Townsend M. 2008. B-cell depletion for 2 years after autologous stem cell transplant for NHL induces prolonged hypogammaglobulinemia beyond the rituximab maintenance period. Leuk Lymphoma 49:152–153. 10.1080/10428190701742506. [DOI] [PubMed] [Google Scholar]
- 32.Lim SH, Zhang Y, Wang Z, Esler WV, Beggs D, Pruitt B, Hancock P, Townsend M. 2005. Maintenance rituximab after autologous stem cell transplant for high-risk B-cell lymphoma induces prolonged and severe hypogammaglobulinemia. Bone Marrow Transplant 35:207–208. 10.1038/sj.bmt.1704742. [DOI] [PubMed] [Google Scholar]
- 33.Miles SA, McGratten M. 2005. Persistent panhypogammaglobulinemia after CHOP-rituximab for HIV-related lymphoma. J Clin Oncol 23:247–248. 10.1200/JCO.2005.05.282. [DOI] [PubMed] [Google Scholar]
- 34.Nishio M, Endo T, Fujimoto K, Fujimoto K, Sato N, Sakai T, Obara M, Kumano K, Kumano K, Minauchi K, Koike T. 2005. Persistent panhypogammaglobulinemia with selected loss of memory B cells and impaired isotype expression after rituximab therapy for post-transplant EBV-associated autoimmune hemolytic anemia. Eur J Haematol 75:527–529. 10.1111/j.1600-0609.2005.00552.x. [DOI] [PubMed] [Google Scholar]
- 35.Cambridge G, Leandro MJ, Edwards JC, Ehrenstein MR, Salden M, Bodman-Smith M, Webster AD. 2003. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum 48:2146–2154. 10.1002/art.11181. [DOI] [PubMed] [Google Scholar]
- 36.Teng YK, Wheater G, Hogan VE, Stocks P, Levarht EW, Huizinga TW, Toes RE, van Laar JM. 2012. Induction of long-term B-cell depletion in refractory rheumatoid arthritis patients preferentially affects autoreactive more than protective humoral immunity. Arthritis Res Ther 14:R57. 10.1186/ar3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner JS, Kim W, Kalaidina E, Goss CW, Rauseo AM, Schmitz AJ, Hansen L, Haile A, Klebert MK, Pusic I, O'Halloran JA, Presti RM, Ellebedy AH. 2021. SARS-CoV-2 infection induces long-lived bone marrow plasma cells in humans. Nature 595:421–425. 10.1038/s41586-021-03647-4. [DOI] [PubMed] [Google Scholar]
- 38.Turner JS, O'Halloran JA, Kalaidina E, Kim W, Schmitz AJ, Zhou JQ, Lei T, Thapa M, Chen RE, Case JB, Amanat F, Rauseo AM, Haile A, Xie X, Klebert MK, Suessen T, Middleton WD, Shi PY, Krammer F, Teefey SA, Diamond MS, Presti RM, Ellebedy AH. 2021. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature 596:109–113. 10.1038/s41586-021-03738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687. 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 40.Li ZN, Mueller SN, Ye L, Bu Z, Yang C, Ahmed R, Steinhauer DA. 2005. Chimeric influenza virus hemagglutinin proteins containing large domains of the Bacillus anthracis protective antigen: protein characterization, incorporation into infectious influenza viruses, and antigenicity. J Virol 79:10003–10012. 10.1128/JVI.79.15.10003-10012.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Code and data for modeling of antibody levels and calculation of half-lives are available on GitHub (https://github.com/HA2342/WL).