

Abstract
Objective:
Black women suffer a higher mortality from endometrial cancer (EC) than White women. Potential biological causes for this disparity include a higher prevalence of obesity and more lethal histologic/molecular subtypes. We hypothesize that another biological factor driving this racial disparity could be the EC microbiome.
Methods:
Banked tumor specimens of postmenopausal, Black and White women undergoing hysterectomy for early stage endometrioid EC were identified. The microbiota of the tumors were characterized by bacterial 16S rRNA sequencing. The microbial component of endometrioid ECs in The Cancer Genome Atlas (TCGA) database were assessed for comparison.
Results:
95 early stage ECs were evaluated: 23 Black (24%) and 72 White (76%). Microbial diversity was increased (p<0.001), and Firmicutes, Cyanobacteria and OD1 phyla abundance was higher in tumors from Black versus White women (p<0.001). Genus level abundance of Dietzia and Geobacillus were found to be lower in tumors of obese Black versus obese White women (p<0.001). Analysis of early stage ECs in TCGA found that microbial diversity was higher in ECs from Black versus White women (p<0.05). When comparing ECs from obese Black versus obese White women, 5 bacteria distributions were distinct, with higher abundance of Lactobacillus acidophilus in ECs from Black women being the most striking difference. Similarly in TCGA, Dietzia and Geobacillus were more common in ECs from White women compared to Black.
Conclusion:
Increased microbial diversity and the distinct microbial profiles between ECs of obese Black versus obese White women suggests that intra-tumoral bacteria may contribute to EC disparities and pathogenesis.
INTRODUCTION
Endometrial cancer (EC) is the most common cancer of the female reproductive tract and the fourth most common cancer among women in the United States. In 2021, approximately 66,570 new cases of EC will be diagnosed, and approximately 12,940 women will succumb to this disease[1]. Obesity, diabetes and insulin resistance are factors associated with increased mortality for this disease[2]. Obese women with EC have a 6.25-fold increased risk of death compared to their non-obese counterparts[2].
Black women suffer an overall 55% higher mortality from EC than White women[3]. It has been suggested that this differential is due to disparate access to equitable care and delayed treatment, a higher likelihood of cancer with more adverse prognostic characteristics, and other yet to be discovered risk factors. Potential biologic causes for this disparity in Black women with EC include the following: higher risk of more lethal tumor histologies (serous tumors) and genomic subtypes (TP53 mutations or copy number high [CNH]), and higher rates of obesity and diabetes. In our multi-institutional analysis of 1,400 EC patients treated over 5 years (2005–2010), 75% of Black women were obese (BMI≥30) compared to 58% of White women (p<0.0001). Black women with EC had a higher median BMI than White women with EC (p<0.001) and were twice as likely to have diabetes (p<0.001)[4]. It is unknown how obesity and diabetes impact differences in the underlying biology of EC between Black and White women, and possibly contribute to disparate outcomes by race. However, we hypothesize that another biological factor driving this racial disparity could be the EC microbiome.
The microbiome is thought to play a complex role in human health and disease, including obesity and cancer[5–7]. Microbe-driven cancers have been described such as Helicobacter pylori in gastric cancer and human papillomavirus in cervical, anal, and head and neck cancers[8–10]. Microbial organisms shape the tumor microenvironment by modulating many of the hallmarks of cancer such as resisting cell death, avoiding immune destruction, activating invasion and metastasis[6, 11]. Less is known about interactions between the molecular alterations found in cancers with neighboring microbial communities. Correlations have been described between molecular subtypes and distinct bacterial species in colon cancer[12], suggesting that an inter-relationship may exist between cancer genomics and the microbiome, although the cause and effect of these entities is unclear.
In humans and mice, Bacteroidetes and Firmicutes bacteria predominate in the gut (>90%), and obesity is associated with a dramatic change in their relative ratio: obesity generally leads to a decrease in Bacteroidetes and increase in Firmicutes species[13], although this result has not been consistently observed across all studies. This change in the gut microbial pattern corresponds with a potential greater ability to harvest dietary energy, which would be conducive to cancer development[13]. Furthermore, increased systemic inflammation, considered a hallmark of the obese state, is consistently associated with reduced health-promoting gut bacterial diversity and richness, or dysbiosis[13]. Bacteria associated with gut dysbiosis – such as pathogenic E. Coli – directly promote the development of colorectal cancer in mouse models[14]. Additionally, gut dysbiosis associated with obesity can also lead to increased estrogen deconjugation and other effects that promote carcinogenesis[5]. It is, therefore, reasonable to hypothesize that perturbations of the microbiome likely play a role in the pathogenesis of obesity-driven cancers, such as EC.
The uterine microbiota has been more difficult to study due to the lower abundance of bacteria as compared to the gut and vagina; however, Firmicutes, Bacteriodetes, Proteobacteria and Actinobacteria are the most abundant phyla in the uterus across studies[15]. Specific uterine microbiota have been linked to endometriosis, rates of in vitro fertilization success and poor reproductive outcomes[15]. In addition, microbiota profiling of swabs from the genital tract (vagina, cervix, fallopian tubes, and ovaries) in women undergoing hysterectomy for benign disease, endometrial hyperplasia and EC demonstrate a structural microbiota shift in the endometrial hyperplasia and EC cases, distinguishable from the benign cases[16]. In particular, Atopobium vaginae, a Porphyromonas sp. and a high vaginal pH in the genital tract associated with EC, although the uterine microbiome was not specifically profiled in this study[16]. Importantly, racial differences in the vaginal microbiome variability have been demonstrated, possibly contributing to increased susceptibility of Black women to bacterial vaginosis and sexually transmitted infections[17]. Given this, it is feasible that the interconnection between the microbiota and race may account, in part, for the EC disparities seen in Black women.
There is a significant gap in our knowledge characterizing the microbiota of the malignant uterus in Black and White women and its impact on the pathogenesis of EC. Therefore, the primary objective of our study was to evaluate the microbiota of early stage ECs and assess for variations by race as well as obesity status. We hypothesize that the EC microbiome may differ in tumors between Black and White women and contribute to racial disparities.
METHODS
Patient Characteristics:
The School of Medicine Institutional Review Board at the University of North Carolina Chapel Hill (UNC-CH) approved this retrospective chart review. For our UNC-CH cohort of patients, we identified and reviewed tumor specimens from EC patients who underwent hysterectomy (prior to receipt of either radiation or chemotherapy) from 2012–2019 and had tissue banked via the Tissue Procurement Core Facility at the Lineberger Comprehensive Cancer Center. Sample size and power was calculated using the PS software. In an initial pilot study, we observed differences in the EC microbiota profiles in 21 subjects according to race. From these initial preliminary findings, we determined that we would have >80% power to identify discriminating genus level taxa between the different groups with a sample size of 100 patients. The inclusion criteria for women with EC included obese and non-obese, postmenopausal, Black and White women with stage I endometrioid EC undergoing hysterectomy at UNC-CH. We restricted the cohort to endometrioid histology because it is the most common histology, comprising 80% of ECs. We identified 95 women in the EC group (23 Black, 72 White). We also included 16 women who underwent hysterectomy for benign indications for comparison (2 Black, 13 White, 1 Other race). Women were excluded if they were taking antibiotics or had taken antibiotics during the 3 months prior to their hysterectomy. We also analyzed data from 126 EC patients with available tumor microbial data in the publicly available Cancer Genome Atlas dataset (TCGA) with similar inclusion criteria (11 Black, 115 White).
DNA Extraction:
We characterized the EC microbiota (N=95) and benign endometrium microbiota (N=16) by 16S rRNA high throughput sequencing. Genomic DNA was extracted from tissue samples of the uterus or tumor using a modified protocol of the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Germantown, MD). Briefly, tissue samples were incubated in lysozyme (20 mg/ml) and kit buffer ATL for 30 minutes at 37°C, followed by the addition of proteinase K and incubation at 56°C overnight. Samples were further processed by bead-beating in tubes containing 0.5 mm stainless steel beads (Bullet Blender, Next Advance, Averill Park, NY) for 4 minutes. The supernatant from each sample was combined with buffer AL and 100% ethanol, and the manufacturer’s protocol was followed for the remainder of the extraction.
Library Preparation and Sequencing:
For amplicon library preparation, we amplified the V1-V3 region of the bacterial 16S rRNA using a universal reverse primer and a unique forward primer for each sample. Amplification was performed using fusion primers comprising Ion Torrent adapter 5’- CCATCTCATCCCTGCGTGTCTCCGACTCAG −3’ for the forward primer and 5’- CCTCTCTATGGGCAGTCGGTGAT −3’ for the reverse primer, and universal bacterial primer 8F 5’-AGAGTTTGATCCTGGCTCAG-3’ and 338R 5′-GCTGCCTCCCGTAGGAGT-3′. The forward primer also includes a 10bp IonXpress ™ barcode, unique to each sample. For PCR, two replicates were prepared for each sample, each containing 5x MyTaq Reaction Buffer (Bioline, Taunton, MA), 0.375 μM each of unique forward primer and universal reverse primer, MyTaq HS DNA Polymerase (Bioline, Taunton, MA), and 30 ng of DNA template. PCR was performed with an initial denaturation at 94°C for 5 minutes, followed by 35 cycles of denaturation at 94°C for 45 seconds, annealing at 55°C for 45 seconds, and extension at 72°C for 90 seconds, followed by a final 10 minute extension at 72°C.
PCR product visualization and clean-up was performed on 2% E-Gel Size Select (Life Technologies). To confirm proper band size, each cleaned PCR product was quantified using the Agilent 2100 Bioanalyzer and Quant-iT PicoGreen dsDNA Kit (Invitrogen, Waltham, MA) following the manufacturer’s protocol. Samples were pooled in equimolar ratios to make a library for sequencing on Ion Torrent NGS system. The pooled library was quantified using the Bioanalyzer and Picogreen Assays. For quality control, appropriate negative and positive controls were included in the DNA extraction, PCR, and sequencing steps.
Bacterial enzymes and pathways:
Bacterial enzymes and pathways were assessed using PICRUSt2[18].
Bioinformatics and Data Analysis:
The bacterial 16S rRNA sequences were filtered to remove low quality reads and processed through QIIME 2[19]. An average of 69,625 reads per sample was obtained after quality filtering. Sequences were assigned to operational taxonomic units (OTUs) using the Greengenes database[20]. Multivariate analyses and bacterial diversity metrics were conducted in Qiime 2 and PRIMER VII software (PRIMER-E, Plymouth Marine Laboratory). Bray Curtis similarity matrixes were used for nMDS and cluster analysis. Discriminating taxa between the groups were identified with Metastats[21], MicrobiomeAnalyst[22], and p-values were corrected for multiple hypothesis testing (p<0.05)[23].
TCGA cohort:
The microbial component of early stage (stage 1 and 2) endometrioid ECs in the TCGA database was assessed by filtering out human reads to identify phylum and genus level bacterial abundance using the methodology from similar studies in other cancers[24, 25]. Given the sparsity of the RNA sequencing data, multiple methods of assessing differential abundance were considered. In addition to analysis of composition of microbiomes (ANCOM)[26], analysis of composition of microbiomes with bias correction (ANCOM-BC)[27], which includes the prediction of structural zeros defined in ANCOM-II[28], were considered.
RESULTS
The median age for Black and White women in the EC cohort was 66 and 64 years old, respectively. The median BMI was 36 for Black women and 35 for White women. Tumor grades were similarly distributed between Black and White women with 43% of tumors of Black women being grade 1 compared to 37% of tumors of White women, and 30% of tumors of Black women were grade 3 compared to 29% of tumors of White women (Table 1). Notably, for this cohort, endometrial tumors were found for only two non-obese Black women (i.e., the majority of tumors were from obese Black women); therefore, the subsequent race comparisons were restricted to only obese Black women (N=22) versus obese White women (N=45).
Table 1:
Descriptive characteristics of endometrial cancer patients with microbiota profiling in the UNC-CH institutional database. (BMI = body mass index)
| N (%) | ||
|---|---|---|
| Black 23(24) |
White 73(76) |
|
| Median Age (years) | 66 | 64 |
| Median BMI | 36.3 | 35 |
| Tumor grade | ||
| 1 | 10 (43) | 27 (37) |
| 2 | 6 (26) | 25 (34) |
| 3 | 7 (30) | 21 (29) |
Sixteen benign hysterectomy specimens were also assessed: 2 specimens were from Black women (13%), 13 from White women (81%) and 1 from an “other race” woman (6%). The median age of women in this cohort was 46 years old, and the median BMI was 30. Microbial diversity was greater when comparing the malignant to the benign uterus (p <0.001). Furthermore, genus level abundance of Acidorovax, Bradyrhizobiu, Flavobacterium. Hyphomicrobium, Pelomonas, and Pseudomonas were increased in the ECs as compared to the benign uterus (p<0.05, Figure 1).
Figure 1:
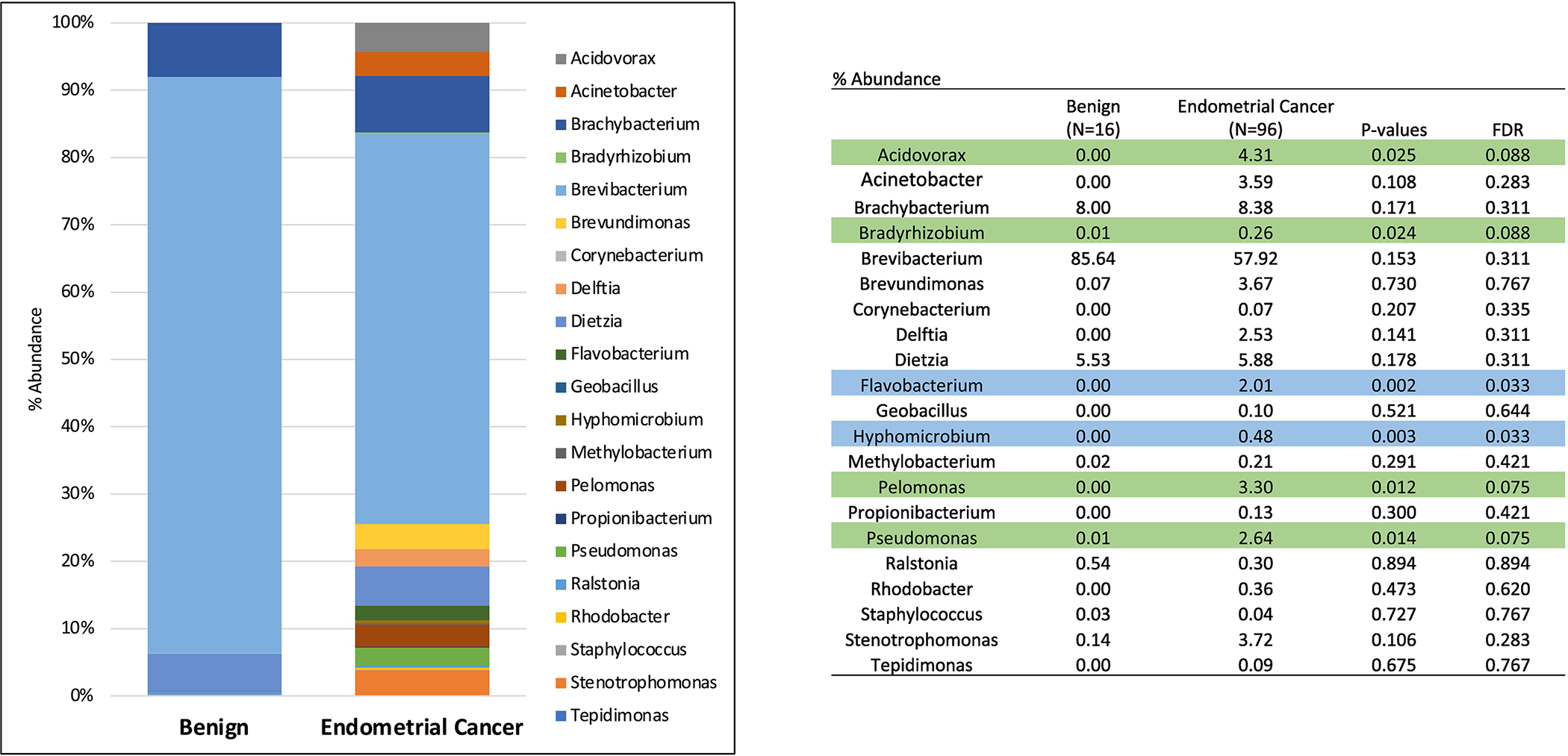
Genus-level microbiota abundance (%) between the benign uterus and endometrioid endometrial cancers. (Blue = p and FDR <0.05; Green = p<0.05 and FDR<0.10)
There were no statistically significant differences found at the phylum or genus level by obesity status when looking at the entire cohort of tumors from White and Black women combined. The tumors of White women in our cohort were then analyzed by obesity status. At the phylum-level, Firmicutes was in greater abundance in the endometrial tumors of non-obese versus obese White women whereas OD1 was in higher abundance in obese versus non-obese White women (Figure 2). Additionally, microbial diversity was increased in the ECs of obese versus non-obese White women (p<0.001).
Figure 2:
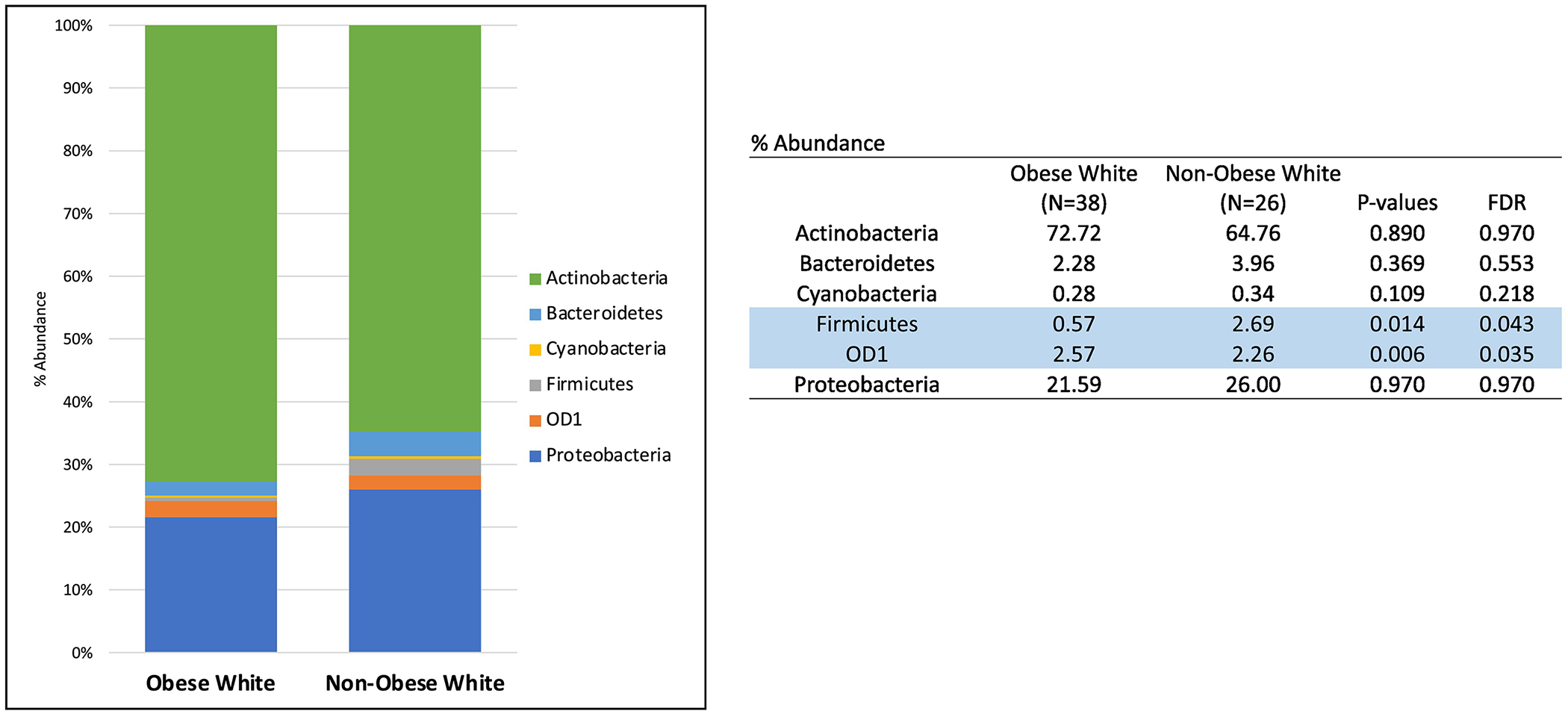
Phylum-level microbiota abundance (%) in endometrial cancer tumors of White women by obesity status. (Blue = p and FDR<0.05)
We subsequently compared the microbial profiles of the ECs from obese patients by race. Firmicutes, Cyanobacteria and OD1 phyla abundance was significantly higher in the tumors from Black versus White women (p<0.05) (Figure 3A). Additionally, microbial diversity was increased in the ECs of Black versus White women (p<0.001). The genus-level taxa displayed in Figure 3B were limited to those that contributed at least 0.1% to the total bacterial count. For clarity of visual display, we chose to display the top 26 genera. The genus-level abundance of Deitzia and Geobacillus were found to be lower in tumor of obese white versus black women. Although at a lower significance, Bradyrhizobium and Pelomonas were found to be higher in tumors of obese Black versus obese White women (p<0.05).
Figure 3:
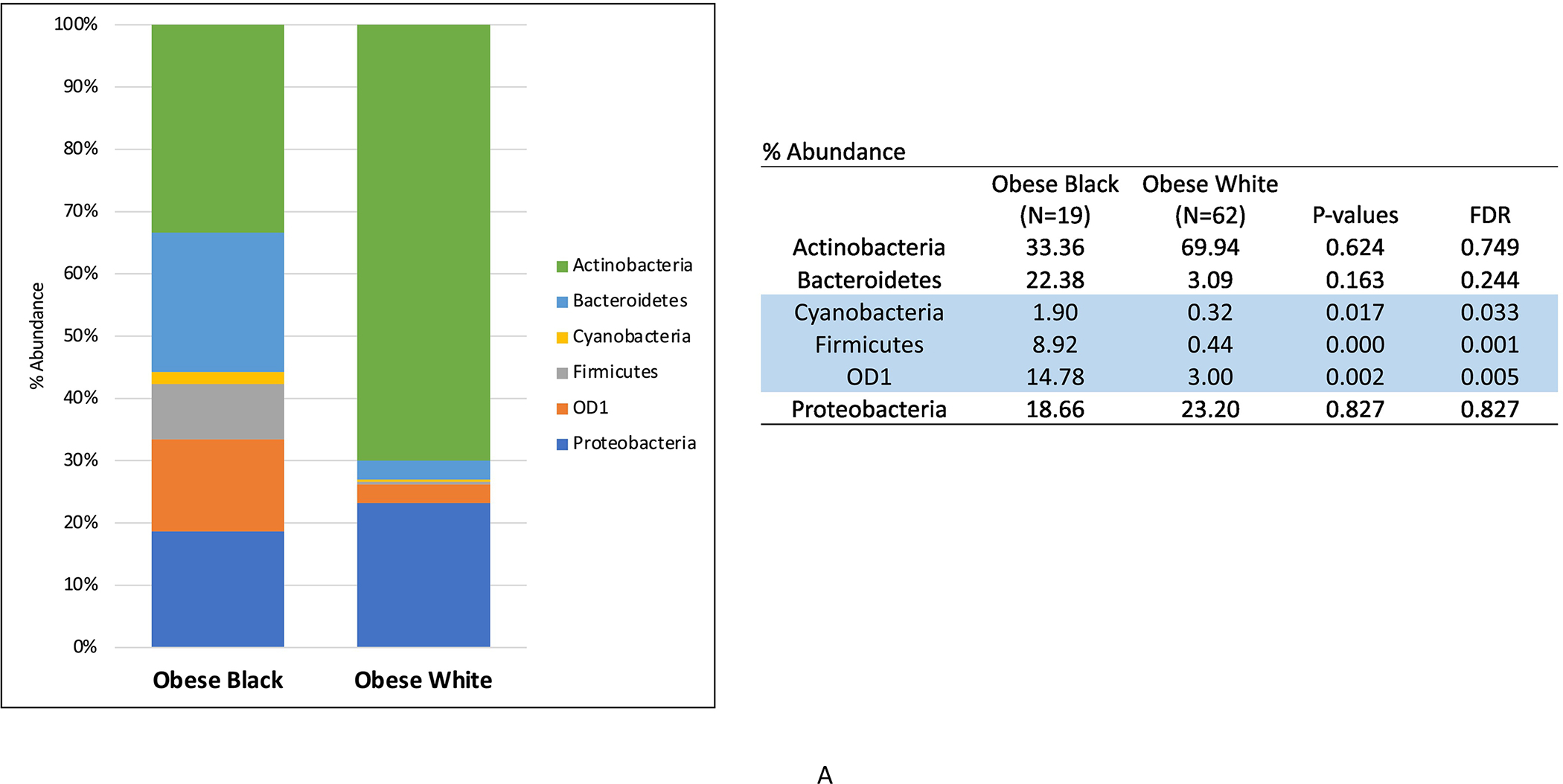
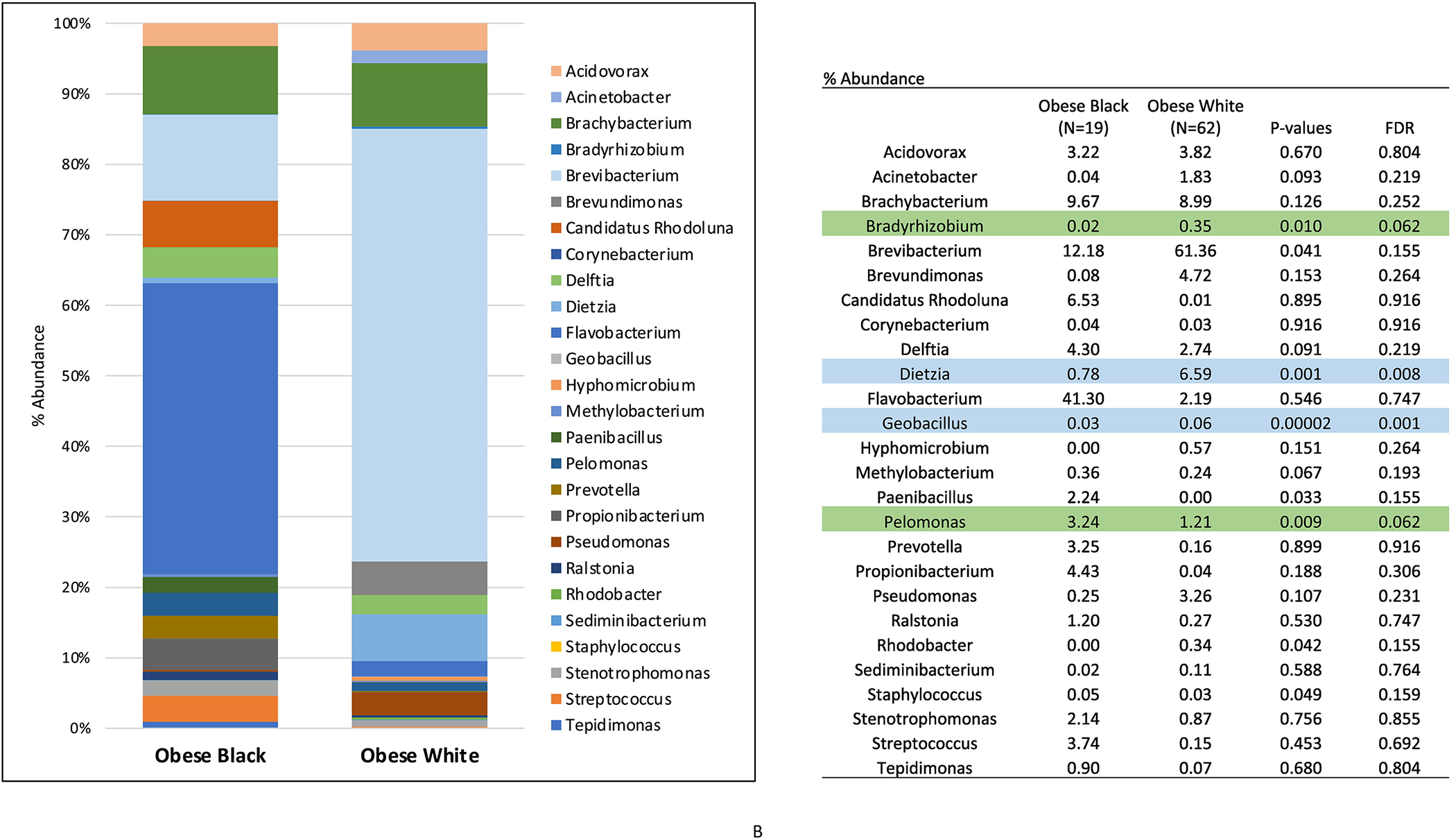
Phylum- (A) and genus-level (B) microbiota distribution (%) in endometrial tumors of obese endometrial cancer patients by race. (Blue = p and FDR<0.05; Green = p<0.05 and FDR<0.10)
We used PICRUSt2 to assess inferred functional potential from our 16S rRNA gene amplicon profiles between endometrial tumors from obese Black versus obese White women (Fig 4). The enzymes, Xanthomonalisin and peptide-aspartate beta-dioxygenase, were both significantly increased in ECs from obese Black compared to obese White women (p<0.05). In addition, several metabolic pathways were also differentially expressed according to race, including a high abundance of genes in endometrial tumors from Black women related to vitamin E synthesis, mevalonate pathway II, vitamin B6 degradation, coenzyme B biosynthesis, and 4-coumarate degradation (p<0.05).
Figure 4:
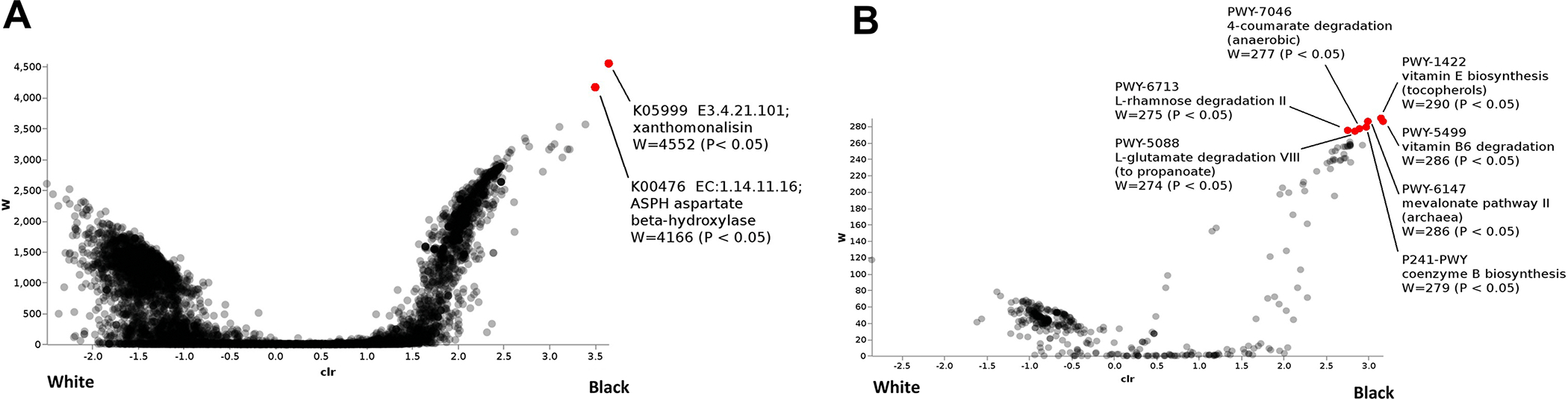
Bacterial enzymes (A) and pathways (B) that distinguished endometrial cancers from Black versus White women. (Red = p<0.05)
To confirm our microbiome findings in an independent dataset, we assessed the microbial component of early stage endometrioid ECs in the TCGA database, using the RNA sequencing for microbiome metatranscriptomics, early stage (stage 1 and 2) ECs from 11 Black and 115 White women from the TCGA cohort were included in our analysis. Similar to the UNC-CH data, increased microbial diversity was also found when comparing the ECs of Black versus White women (Figure 5). When comparing early stage ECs of obese Black and White women, two species of Lactobacillus were identified as significant by ANCOM and as present only in Black women by ANCOM-BC: L. acidophilus and L. amylophylus. Both species were identified as structural zeros in White women by ANCOM-BC. Four additional species were identified by ANCOM as potentially significant by ANCOM: Phycisphaera mikurensis, Thioploca ingrica, Franciscella tularensis, and Alteromonas mediterranea (Figure 5). The most striking difference was the higher abundance of Lactobacillus acidophilus in tumors from Black women. Additionally, several other species of Lactobacillus were identified by ANCOM-BC as present in White women but structurally absent in Black women: L. crispatus, L. salivarius, L. plantarum, L. jensenii, L. fermentum, and L. curvatus. The species L. delbrueckii was identified by ANCOM-BC as present in both Black and White women, but more abundant in Black women; however, the statistical significance after multiple testing correction was relatively low (p=0.0088; q=0.7063). Consistent with the 16S analysis from the UNC-CH samples, the abundance of Dietzia oral taxon 368 (p<0.7739; q<0.9054) and Geobacillus thermodenitrificans (p<0.7268; q<0.9037) were found to be associated with tumors of White versus Black women, although at a lower significance than the five bacteria noted above.
Figure 5: Analysis of the microbial component of endometrial cancers in The Cancer Genome Atlas (TCGA) database.
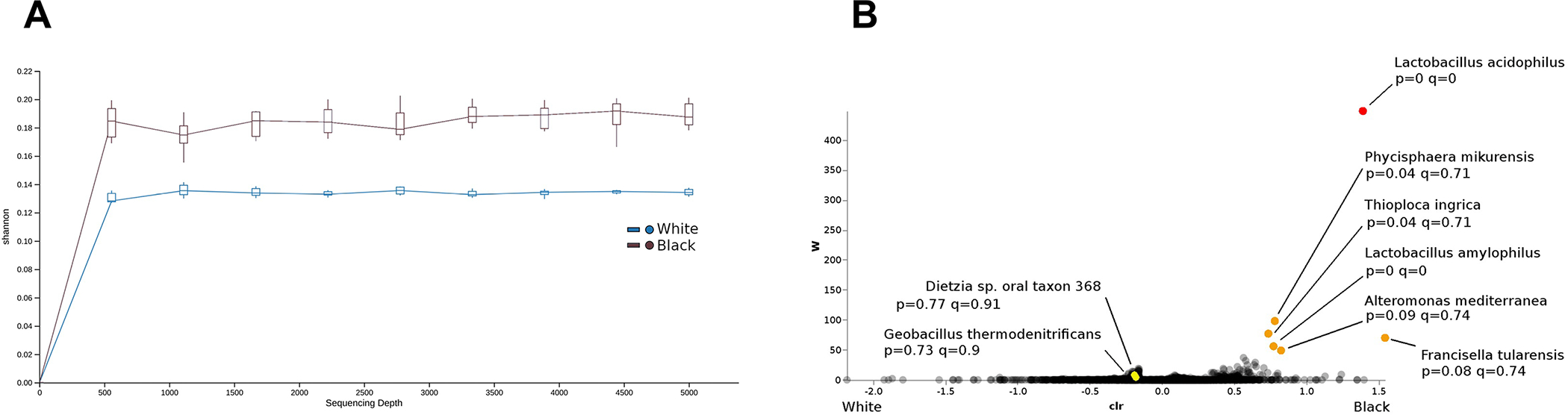
Increased microbial diversity was found when comparing the endometrial cancers of Black versus White women (A). In addition, several bacteria were found to be associated with either Black or White race (B). W scores estimated by ANCOM; additional statistical significance: p-values, q-values, and indications of structural zeros, estimated by ANCOM-BC.
DISCUSSION
The findings of this study contribute to published literature that intra-tumoral bacteria exist in the malignant uterus[15, 29]. Bacteria from the Actinobacteria, Bacteroidetes, Firmicutes, OD1 and Proteobacteria phyla were identified in both the benign and malignant uterine tissue specimens, similar to what has previously been shown in other studies assessing the microbial profile of the uterus[15]. We detected greater microbial diversity in ECs when compared to the benign uterus as well as a higher abundance of several microbes in the malignant uterus at the genus level. Significant differences based on obesity status were seen at the phylum level when the tumors of White women were examined. Additionally, increased microbial diversity was noted in the ECs in obese compared to non-obese White women.
To our knowledge, this is the first study to describe the microbial composition of ECs, with a direct assessment of differences based on race. Microbial diversity was higher in the ECs from Black versus White women as seen in both our UNC-CH dataset and TCGA. It should be noted that high microbial diversity has generally been associated with poorer reproductive tract health[29, 30]. This is opposite to what is seen in the gut where increased microbial diversity is a sign of a healthy gut microbiome, and obesity and diabetes decrease gut microbial diversity. There were distinct microbiota profiles found between ECs of obese Black versus obese White women with Firmicutes and Cyanobacteria and OD1 phyla being more abundant among the tumors from Black patients. Deitzia and Geobacillus genera were less prominent in tumors from Black women in both our UNC-CH dataset and TCGA. The potential role of the Deitzia and Geobacillus genera in cancer has largely been unexplored, although Geobacillus has been previously linked to bladder cancers[31] and its metabolites have been shown to regulate p53 function as well as apoptosis and metastasis[32].
In addition, two enzymes, Xanthomonalisin and peptide-aspartate beta-dioxygenase, and several metabolic pathways including vitamin E synthesis, mevalonate pathway II, vitamin B6 degradation, coenzyme B biosynthesis, and 4-coumarate degradation were differentially expressed according to race. The human ortholog of the bacterial enzyme peptide-aspartate beta-dioxygenase, aspartate beta-hydroxylase (ASPH), can stimulate tumor growth via its effects on angiogenesis and immunosuppression and is over-expressed in a variety cancers[33]. Furthermore, ASPH expression is upregulated via signaling through multiple pathways central to EC pathogenesis, including the insulin/insulin-like growth factor-1 and PI3K/Akt/mTOR pathways[33]. Small molecule inhibitors to ASPH are in development as cancer therapies[33]. Whether the bacterial version of ASPH would have similar effects as its human counterpart in the tumor microenvironment is unknown. However, bacterial enzymes in the human gut can impact chemotherapeutic toxicity and efficacy. For example, bacterial β-glucuronidase (GUS) can reactivate irinotecan through glucoronidation leading to dose-limiting GI toxicity that can be reversed by GUS inhibitors[34].
The mevalonate pathway is an essential metabolic pathway that begins with acetyl-CoA and utilizes HMG-CoA reductase to produce isoprenoids and cholesterol and is associated with stabilization of mutant TP53 and promotion of tumor growth[35]. This pathway can be inhibited by lipid lowering drugs such as statins via inhibition of HMG-CoA reductase, and statin use has been associated with improved survival in EC[36]. Additionally, tocotrienol, a member of the vitamin E family, acts as an inhibitor of HMG-CoA reductase[37]. Coenzyme B is an important cofactor present in archaea methanogens and is required for methane production[38]. Methanogen’s role in the gut microbiome has garnered significant interest recently via its association with several metabolic diseases, including obesity[39]. For example, methanogens are capable of interacting with bacteria that enhance production of short-chain fatty acids, which in turn provide a considerable caloric load to its host. p-Coumaric acid is widely distributed in plants, vegetables, fruits, and cereals and, along with its conjugates, has displayed immunomodulatory and anti-inflammatory properties[40, 41]. Vitamin B6, has also been shown to have anti-inflammatory properties and has the potential to modulate the host immune system and bacterial virulence[42, 43]. These pathways and their inter-relationship within the uterine microbiome are worthy of further exploration as potential targets in the prevention and treatment of EC.
Notably, there was a prominent difference in the abundance of Lactobacillus acidophilus in the tumors from Black women in the TCGA cohort. Lactobacillus species have long been associated as the key contributor in maintaining a low pH of the vaginal environment through the production of lactic acid[44]. Depletion of Lactobacillus species in the vagina has classically been associated with an opportunistic multispecies colonization resulting in bacterial vaginosis[30]. In a large study using 16S rRNA sequencing to characterize vaginal bacterial communities of a diverse ethnic population of nearly 400 asymptomatic North American women[45], five community types of vaginal microbiota were identified; four of these five community types were dominated by Lactobacillus species. Interestingly, a difference was seen in the distribution of Lactobacillus community types in those with differing racial and ethnic backgrounds. White and Asian women were more likely than Hispanic and Black women to have Lactobacillus dominant vaginal communities, and Hispanic and Black women were also more likely to have a higher average vaginal pH when compared to White and Asian women[45]. Although the uterine microbial composition differs from that of the vagina, a non-Lactobacillus-dominant (<90% Lactobacillus species) microbiota still appears to render adverse health outcomes[46]. It has been suggested that a non-Lactobacillus-dominant endometrial microbiota may be related to endometriosis and its characteristic endometrial inflammation[47]. Given these previous associations, we would have expected Lactobacillus acidophilus to be lower as opposed to higher in the ECs of Black versus White women. However, we found the opposite in that Lactobacillus acidophilus was higher in the ECs of Black women, suggesting that the inter-relationship of the Lactobacillus community on uterine health and disparities is more complex.
In a study by Walsh et al, microbiome samples were prospectively collected in a sterile fashion from the lower (i.e. vagina and cervix) and upper (i.e. uterus and ovaries/fallopian tubes) reproductive tract of women undergoing hysterectomy for either EC or a benign uterine condition, including 75 women without EC and 66 with EC. For women without EC, postmenopausal status was associated with increased α- and β-microbial diversity in the lower reproductive tract and increased α-microbial diversity in the uterus, and BMI was associated with increased microbial α-diversity in the lower tract[29]. Relative to women without EC, those with EC had greater microbial β-diversity of the lower tract, with 17 taxa significantly associated with EC[29]. Porphyromonas somerae was the most significantly enriched species in women with EC[29]. Differences in the microbiota of the uterus between women with and without EC did not reach statistical significance (p=0.07), likely due to the small sample size of uterine samples (18 benign and 16 ECs). The majority of the uterine sampling for this study was uterine swabs as opposed to our study which profiled the microbiota of the endometrial tumors. Thus, postmenopausal state, obesity, high vaginal pH and EC all increased microbial diversity in the reproductive tract, further supporting our findings of increased microbial diversity being associated with EC and obesity in our analysis. It should be noted that impact of race on the microbiota of the reproductive tract was not assessed, as no Black EC patients were included in this study.
There are several limitations to our study. This is a retrospective study utilizing banked tumor specimens in both the UNC-CH samples and TCGA, which could raise concern for specimen contamination through processing and handling. For our UNC-CH data, we relied on provider-documented antibiotic use in the medical record, medication administration records (MAR) at the time of presentation for initial consultation and other medical encounters limited to UNC-CH but could not assess antibiotic use at the time of surgery, which could alter the relative abundance of native microbiota in patients. Antibiotic data was also not available for the TCGA cohort. We were unable to evaluate differences in the microbiota profiles of ECs of obese versus non-obese Black women in the same manner as we evaluated such differences for White women, given the lack of ECs from lean women that had been previously banked at UNC-CH. Additionally, different methodologies were used to analyze the data from both cohorts in our study, bacterial 16S rRNA for the UNC-CH cohort and RNA sequencing in the TCGA cohort). This difference in methodology may explain the differences in observed taxa between tumors of White vs. Black (i.e., Lactobacillus). The sequencing of the TCGA samples was originally intended to target human RNA and was therefore not optimized for the microbial taxa. The metatrascriptomic approach, however, does allow for estimates of taxonomic profile at the species resolution rather than the genera resolution available with 16S and will identify non-bacterial species including fungal and viral species. Lastly, both cohorts include a small sample size with proportionally fewer tumors from Black women. Therefore, we are in the process of implementing a prospective study to assess the EC microbiome that controls for the collection and processing of EC specimens in a sterile fashion. This will be done in the following steps at the time of surgery: 1. The hysterectomy specimen will be placed in a sterile container by the surgeon. 2. The specimen will be immediately transported to Surgical Pathology where it will be processed in a dedicated sterile hood to avoid contamination. 3. A Pathologists’ Assistant will be readily available when surgical specimens arrive into Surgical Pathology ensuring rapid processing of specimens limiting ischemia time and preservation of nucleic acids. 4. The sterile container with the hysterectomy specimen will be opened and the uterus will be cut open to obtain samples aseptically. 5. Airborne contamination will be monitored by placing Luria-Bertani (LB) agar plates in the hood and room during processing of specimens. After specimen processing, the plates will be swabbed and the LB swabs will be processed alongside the samples for microbiome analysis. Our quality control protocols will also include sampling controls and positive and negative controls in the DNA extraction, PCR, and sequencing steps. We also plan to expand our study beyond early stage disease to advanced stage disease, as this is what ultimately accounts for death in Black women from EC, and assess both the EC and gut microbiome in parallel as women undergo surgery and chemotherapy. This prospective study will also collect tumor estrogen and progesterone receptor status as obesity associated dysregulation of the estrogen-gut microbiome axis may impact carcinogenesis[5]. By probing the inter-relationships of race, the microbiota of the malignant uterus and the gut, and EC progression, we hope to identify targetable pathways to improve treatment response and outcomes for Black women.
Ultimately, an improved understanding of the contribution of uterine microbial dysbiosis to EC pathogenesis, as well as the alarming disparities for Black women battling this disease, may result in novel targets for treatment and prevention. However, we also want to acknowledge that a multilevel framework will ultimately be needed to integrate our microbiota findings with that of the multiple dimensions underlying racial disparities, which include biological factors, individual factors, social/physical context and fundamental (structural and institutional) causes, or our understanding of how the microbiome fits within racial disparities will be limited.
Supplementary Material
S1. Genomic data commons manifest of TCGA samples.
S2. Operational taxonomic unit table of UNC-CH samples.
The endometrial cancer microbiome differs between obese Black women and obese White women.
Microbial diversity is increased in endometrial cancer tumors of obese Black women versus obese White women.
Intra-tumoral bacteria may contribute to the disparities and pathogenesis of endometrial cancer.
TCGA Acknowledgement:
The results published here are in whole or part based upon data generated by The Cancer Genome Atlas managed by the NCI and NHGRI. Information about TCGA can be found at http://cancergenome.nih.gov.
This work is generously supported by funding from:
P30DK034987 Center for Gastrointestinal Biology and Disease
NIH/NCI – 1R21CA220269-01 Inter-relationship between microbiota diversity, obesity and race in endometrial cancer
NIH – Program in Translational Medicine T32-CA244125 to UNC/WB
Footnotes
Conflict of Interests:
The authors declare no competing financial interest for this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: A Cancer Journal for Clinicians. 2021;71:7–33. [DOI] [PubMed] [Google Scholar]
- [2].Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. New England Journal of Medicine. 2003;348:1625–38. [DOI] [PubMed] [Google Scholar]
- [3].Cote ML, Ruterbusch JJ, Olson SH, Lu K, Ali-Fehmi R. The Growing Burden of Endometrial Cancer: A Major Racial Disparity Affecting Black Women. Cancer Epidemiology Biomarkers & Prevention. 2015;24:1407–15. [DOI] [PubMed] [Google Scholar]
- [4].Ko EM, Walter Paige, Clark Leslie, Jackson Amanda, Franasiak Jason, Bolac Corey, Havrilesky Laura, Secord Angeles Alvarez, Moore Dominic T., Gehrig Paola A. Bae-Jump, Victoria L. The complex triad of obesity, diabetes and race in Type I and II endometrial cancers: Prevalence and prognostic significance. Gynecologic Oncology. 2014;133. [DOI] [PubMed] [Google Scholar]
- [5].AlHilli MM, Bae-Jump V. Diet and gut microbiome interactions in gynecologic cancer. Gynecol Oncol. 2020;159:299–308. [DOI] [PubMed] [Google Scholar]
- [6].Garrett WS. Cancer and the microbiota. Science. 2015;348:80–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lee CJ, Sears CL, Maruthur N. Gut microbiome and its role in obesity and insulin resistance. Ann N Y Acad Sci. 2020;1461:37–52. [DOI] [PubMed] [Google Scholar]
- [8].Peek RM Jr., Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. [DOI] [PubMed] [Google Scholar]
- [9].Crosbie EJ, Einstein MH, Franceschi S, Kitchener HC. Human papillomavirus and cervical cancer. The Lancet. 2013;382:889–99. [DOI] [PubMed] [Google Scholar]
- [10].Sabatini ME, Chiocca S. Human papillomavirus as a driver of head and neck cancers. Br J Cancer. 2020;122:306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12:31–46. [DOI] [PubMed] [Google Scholar]
- [12].Purcell RV, Visnovska M, Biggs PJ, Schmeier S, Frizelle FA. Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci Rep. 2017;7:11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Turnbaugh PJ, Ley Ruth E., Mahowald Michael A., Magrini Vincent, Mardis Elaine R., Gordon Jeffrey I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444. [DOI] [PubMed] [Google Scholar]
- [14].Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Baker JM, Chase DM, Herbst-Kralovetz MM. Uterine Microbiota: Residents, Tourists, or Invaders? Frontiers in Immunology. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Walther-António MR, Chen J, Multinu F, Hokenstad A, Distad TJ, Cheek EH, et al. Potential contribution of the uterine microbiome in the development of endometrial cancer. Genome Med. 2016;8:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhou X, Brown CJ, Abdo Z, Davis CC, Hansmann MA, Joyce P, et al. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. Isme j. 2007;1:121–33. [DOI] [PubMed] [Google Scholar]
- [18].Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38:685–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5:e1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chong J, Liu P, Zhou G, Xia J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat Protoc. 2020;15:799–821. [DOI] [PubMed] [Google Scholar]
- [23].Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological). 1995;57:289–300. [Google Scholar]
- [24].Robinson KM, Crabtree J, Mattick JS, Anderson KE, Dunning Hotopp JC. Distinguishing potential bacteria-tumor associations from contamination in a secondary data analysis of public cancer genome sequence data. Microbiome. 2017;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Thompson KJ, Ingle JN, Tang X, Chia N, Jeraldo PR, Walther-Antonio MR, et al. A comprehensive analysis of breast cancer microbiota and host gene expression. PLoS One. 2017;12:e0188873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mandal S, Van Treuren W, White RA, Eggesbo M, Knight R, Peddada SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis. 2015;26:27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11:3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kaul A, Mandal S, Davidov O, Peddada SD. Analysis of Microbiome Data in the Presence of Excess Zeros. Front Microbiol 2017;8:2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Walsh DM, Hokenstad AN, Chen J, Sung J, Jenkins GD, Chia N, et al. Postmenopause as a key factor in the composition of the Endometrial Cancer Microbiome (ECbiome). Sci Rep. 2019;9:19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dols JA, Smit PW, Kort R, Reid G, Schuren FH, Tempelman H, et al. Microarray-based identification of clinically relevant vaginal bacteria in relation to bacterial vaginosis. Am J Obstet Gynecol. 2011;204:305 e1–7. [DOI] [PubMed] [Google Scholar]
- [31].Liu F, Liu A, Lu X, Zhang Z, Xue Y, Xu J, et al. Dysbiosis signatures of the microbial profile in tissue from bladder cancer. Cancer Med. 2019;8:6904–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].He T, Jin M, Xu C, Ma Z, Wu F, Zhang X. The homeostasis-maintaining metabolites from bacterial stress response to bacteriophage infection suppress tumor metastasis. Oncogene. 2018;37:5766–79. [DOI] [PubMed] [Google Scholar]
- [33].Kanwal M, Smahel M, Olsen M, Smahelova J, Tachezy R. Aspartate beta-hydroxylase as a target for cancer therapy. J Exp Clin Cancer Res. 2020;39:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bhatt AP, Pellock SJ, Biernat KA, Walton WG, Wallace BD, Creekmore BC, et al. Targeted inhibition of gut bacterial beta-glucuronidase activity enhances anticancer drug efficacy. Proc Natl Acad Sci U S A. 2020;117:7374–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Parrales A, Thoenen E, Iwakuma T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018;25:460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sperling CD, Verdoodt F, Kjaer Hansen M, Dehlendorff C, Friis S, Kjaer SK. Statin use and mortality among endometrial cancer patients: a Danish nationwide cohort study. Int J Cancer. 2018;143:2668–76. [DOI] [PubMed] [Google Scholar]
- [37].Song BL, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme a reductase stimulated by delta- and gamma-tocotrienols. J Biol Chem. 2006;281:25054–61. [DOI] [PubMed] [Google Scholar]
- [38].Graham DE, White RH. Elucidation of methanogenic coenzyme biosyntheses: from spectroscopy to genomics. Nat Prod Rep. 2002;19:133–47. [DOI] [PubMed] [Google Scholar]
- [39].Chaudhary PP, Conway PL, Schlundt J. Methanogens in humans: potentially beneficial or harmful for health. Appl Microbiol Biotechnol. 2018;102:3095–104. [DOI] [PubMed] [Google Scholar]
- [40].Pei K, Ou J, Huang J, Ou S. p-Coumaric acid and its conjugates: dietary sources, pharmacokinetic properties and biological activities. J Sci Food Agric. 2016;96:2952–62. [DOI] [PubMed] [Google Scholar]
- [41].Pragasam SJ, Venkatesan V, Rasool M. Immunomodulatory and anti-inflammatory effect of p-coumaric acid, a common dietary polyphenol on experimental inflammation in rats. Inflammation. 2013;36:169–76. [DOI] [PubMed] [Google Scholar]
- [42].Bird RP. The Emerging Role of Vitamin B6 in Inflammation and Carcinogenesis. Adv Food Nutr Res. 2018;83:151–94. [DOI] [PubMed] [Google Scholar]
- [43].Miki T, Goto R, Fujimoto M, Okada N, Hardt WD. The Bactericidal Lectin RegIIIbeta Prolongs Gut Colonization and Enteropathy in the Streptomycin Mouse Model for Salmonella Diarrhea. Cell Host Microbe. 2017;21:195–207. [DOI] [PubMed] [Google Scholar]
- [44].Boskey ER, Telsch KM, Whaley KJ, Moench TR, Cone RA. Acid production by vaginal flora in vitro is consistent with the rate and extent of vaginal acidification. Infect Immun. 1999;67:5170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Moreno I, Codoner FM, Vilella F, Valbuena D, Martinez-Blanch JF, Jimenez-Almazan J, et al. Evidence that the endometrial microbiota has an effect on implantation success or failure. Am J Obstet Gynecol. 2016;215:684–703. [DOI] [PubMed] [Google Scholar]
- [47].Jiang I, Yong PJ, Allaire C, Bedaiwy MA. Intricate Connections between the Microbiota and Endometriosis. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1. Genomic data commons manifest of TCGA samples.
S2. Operational taxonomic unit table of UNC-CH samples.