

Abstract
We report final analysis outcomes from the phase 3 HELIOS study (NCT01611090). Patients with relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma without deletion 17p (n = 578) were randomized 1:1 to 420 mg daily ibrutinib or placebo plus ≤6 cycles of bendamustine plus rituximab (BR), followed by ibrutinib or placebo alone. Median follow-up was 63.7 months. Median investigator-assessed progression-free survival was longer with ibrutinib plus BR (65.1 months) than placebo plus BR (14.3 months; hazard ratio [HR] 0.229 [95% confidence interval (CI) 0.183–0.286]; p < .0001). Despite crossover of 63.3% of patients from the placebo plus BR arm to ibrutinib treatment upon disease progression, ibrutinib plus BR versus placebo plus BR demonstrated an overall survival benefit (HR 0.611 [95% CI 0.455–0.822]; p = 0010; median not reached in either arm). Long-term follow-up data confirm the survival benefit of ibrutinib plus BR over BR alone. Safety profiles were consistent with those known for ibrutinib and BR.
Keywords: Ibrutinib, HELIOS phase 3 trial, 5-year follow-up, overall survival, relapsed chronic lymphocytic leukemia
Introduction
Ibrutinib, administered orally once daily, is approved to treat adults with various B-cell malignancies in the United States, European Union, and other countries [1–3]. This first-in-class covalent inhibitor of Bruton’s tyrosine kinase (BTK) has changed the treatment landscape for patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) [4–7] and is one of the preferred treatments for patients with or without deletion 17p/TP53 mutation, who have previously untreated or relapsed/refractory (R/R) disease [6].
Ibrutinib was evaluated as a single-agent treatment in patients with R/R CLL/SLL in an open-label randomized, multicenter phase 3 trial (RESONATE™) [8–10]. In the final analysis of the study (median follow-up, 65.3 months in the ibrutinib arm), patients who received ibrutinib (n = 195) versus ofatumumab (n = 196), including those with high-risk genomic features, had a superior progression-free survival (PFS; hazard ratio [HR], 0.148; 95% confidence interval [CI] 0.113–0.196; p < .0001) [10]. Additionally, although 68% of patients in the ofatumumab arm crossed over to the ibrutinib arm, an overall survival (OS) benefit with ibrutinib was seen (HR 0.639; 95% CI 0.418–0.975; censored for the crossover).
Before the availability of BTK inhibitors like ibrutinib, which target the B-cell receptor signaling pathway, a chemoimmunotherapy regimen of bendamustine (an alkylating agent) plus rituximab (an anti-CD20 antibody) was commonly used for patients with R/R CLL [11,12]. In a phase 2 study of ≤6 cycles of bendamustine plus rituximab (BR) in patients with R/R CLL, overall response rate (ORR) was 59% (complete response [CR], 9%), median PFS was 15.2 months, and median OS was 33.9 months [13]. The HELIOS study was designed to determine whether ibrutinib therapy provided additional benefit when combined with BR as a chemoimmunotherapy backbone. In a prior phase 1 b multicenter study evaluating the safety and efficacy of continuous ibrutinib plus ≤6 cycles of BR in 30 patients with R/R CLL, the ORR was high (93.3%), with 70.3% of patients remaining progression free at the 36-month landmark [14].
Here, we report outcomes of the final analysis of HELIOS, a phase 3, randomized, double-blind, placebo-controlled study of ibrutinib plus ≤6 cycles of BR in 578 patients with R/R CLL/SLL (median follow-up, 63.7 months); findings from interim [15] and 3-year analyses [16] were previously reported.
Patients and methods
The HELIOS study (NCT01611090) was conducted at 133 sites in 21 countries. The protocol was approved by an independent ethics committee/institutional review board at each site [15] and performed according to principles of the Declaration of Helsinki and guidelines for Good Clinical Practice. Patients provided informed consent before participation.
Patient eligibility
As previously described [15,16], eligible patients were aged ≥18 years with active CLL/SLL disease meeting the International Workshop on Chronic Lymphocytic Leukemia 2008 criteria [17] for treatment. Included patients also had R/R disease following ≥1 prior lines of therapy (including 3% of patients in each arm who received BR [15]); an Eastern Cooperative Oncology Group performance status of 0–1; measurable lymph node disease (>1.5 cm) by computed tomography (CT) scan; and adequate liver and kidney function. Patients with deletion 17p (≥20% of blood/bone marrow cells examined by fluorescent in situ hybridization) were excluded due to the known poor response to BR by patients with this deletion. TP53 mutational testing was not performed or included in the study’s exclusion criteria.
Study design
Patients were randomly assigned (1:1) to treatment with ibrutinib (420 mg daily) plus BR (≤6 cycles of bendamustine [70 mg/m2 intravenously on Days 2–3 of Cycle 1 and Days 1–2 of Cycles 2–6] and rituximab [375 mg/m2 on Day 1 of Cycle 1 and 500 mg/m2 on Day 1 of Cycles 2–6]) or placebo plus BR (Figure 1). Patients then continued ibrutinib or placebo treatment until unacceptable toxicity or confirmed disease progression.
Figure 1.
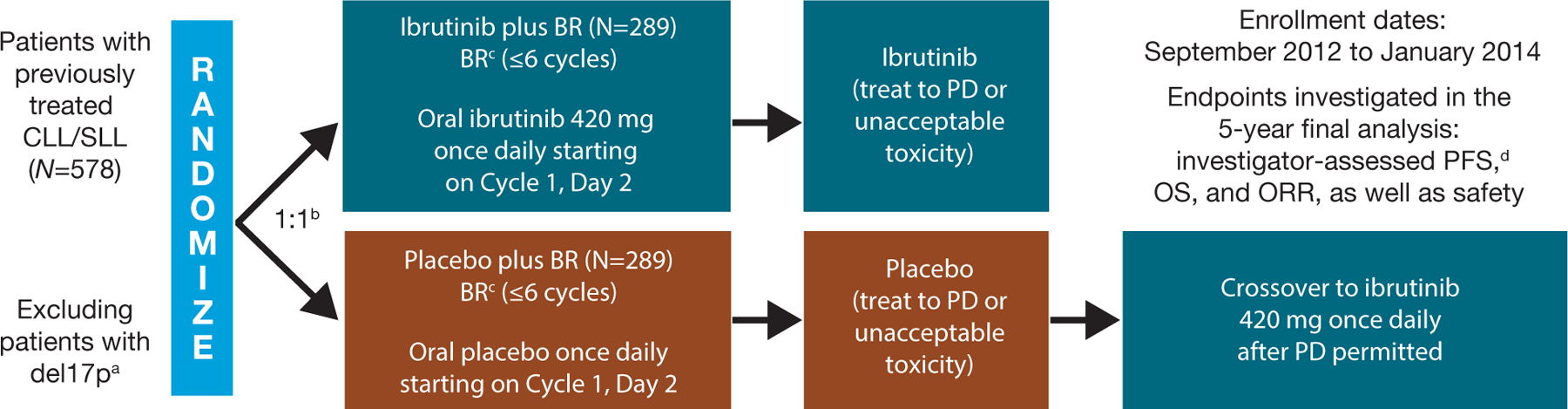
Study design and follow-up assessments. IRC: independent review committee; PD: progressive disease. aDeletion 17p in ≥20% of examined cells. bStratified by purine analog refractory status (failure to respond or relapse in ≤12 months) and prior lines of therapy (1 line versus >1 line). cSimilar dosing to Fischer et al. [13]; bendamustine: 70 mg/m2 intravenously on Days 2–3 in Cycle 1 and Days 1–2 in Cycles 2–6; rituximab: 375 mg/m2 on Day 1 of Cycle 1 and 500 mg/m2 on Day 1 of Cycles 2–6. dAccording to 2008 International Workshop on Chronic Lymphocytic Leukemia criteria (Hallek et al. [17]).
Following the prespecified interim analysis (March 2015), the Data and Safety Monitoring Board recommended unblinding the study, therefore, placebo treatment was discontinued for patients in the placebo plus BR arm. Treatment-emergent adverse event (TEAE) reporting for this arm was also discontinued at this time; these patients had continued disease evaluation and follow-up and were permitted to cross over to ibrutinib after confirmed disease progression (Figure 1). Safety data are reported for the ibrutinib plus BR arm; adverse events (AEs) occurring in patients who crossed over from the placebo plus BR arm to ibrutinib arm are excluded.
Endpoints and follow-up assessments
Endpoints investigated in this final analysis included investigator-assessed PFS, OS, ORR, and safety (Figure 1). PFS2 (time interval from randomization to either progressive disease on next-line treatment, death, or the start of subsequent antineoplastic therapy if progressive disease was not reported) was also investigated.
Minimal residual disease (MRD) testing was first performed on the bone marrow at the time of a radiologically documented CR, and subsequently on peripheral blood every 12 weeks [15,16]. Due to the long half-life of anti-CD20 monoclonal antibodies such as rituximab in peripheral blood, the first bone marrow sample was acquired to mitigate cross-reactivity [15]. Testing was done at a central laboratory by flow cytometry using an eight-color panel of antibodies in line with the EuroFlow panel [15,16,18]. A protocol amendment following the interim analysis enabled MRD analysis for all patients with partial response (PR) or better [16]. The patient proportion with undetectable MRD increased through to the 3-year follow-up but plateaued thereafter, therefore data collection was terminated shortly afterwards.
Statistical analyses
All randomized patients were included in the efficacy analysis (intent-to-treat population). Patients who received ≥1 dose of the study drug were included in the safety analysis (safety population).
In the interim analysis, overall concordance between independent review committee (IRC)-assessed and investigator-assessed progressive disease was 90% and 85% in the ibrutinib plus BR and placebo plus BR arms, respectively [15]. PFS analyses were performed using IRC assessments in the interim analysis and investigator assessments in the long-term analysis. For patients alive at the time of this analysis, OS – defined as the time interval from randomization to death, regardless of cause – was censored at the last known date they were alive.
PFS distribution was estimated using the Kaplan–Meier method and a stratified log-rank test. Cox-proportional hazards model was used to calculate HR.
Results
Patient demographics and baseline characteristics were well balanced between treatment arms and previously reported [15].
Treatment exposure and patient disposition
At the final analysis, median follow-up was 63.7 months (95% CI 62.8–64.3; range 0.1–74.5; Table 1) overall, and similar for patients in both treatment arms. Median time on treatment in the ibrutinib plus BR arm (n = 287) was 55.7 months (range 0.2–72.9). As placebo treatment was discontinued at the interim analysis, median time on treatment in the placebo plus BR arm (n = 287) was 14.3 months (range 0.2–30.6).
Table 1.
Patient disposition for the intent-to-treat population.
| Ibrutinib plus BR (n = 289) | Placebo plus BRa (n = 289) | Total (N = 578) | |
|---|---|---|---|
| Median time on study, months (95% CI) | 63.3 (62.1–64.4) | 64.0 (63.0–64.8) | 63.7 (62.8–64.3) |
| [Range] | [0.2–73.5] | [0.1–74.5] | [0.1–74.5] |
| Study treatment phase disposition, n (%) | |||
| Did not receive study drug | 2 (0.7) | 2 (0.7) | 4 (0.7) |
| Discontinued study treatment | 287 (99.3) | 287 (99.3) | 574 (99.3) |
| Primary reason for discontinuation,b n (%) | |||
| Investigator or sponsor decision (including end of follow-up on trial)c | 136 (47.1) | 84 (29.1) | 220 (38.1) |
| Progressive disease or relapse | 55 (19.0) | 148 (51.2) | 203 (35.1) |
| Adverse event | 58 (20.1) | 34 (11.8) | 92 (15.9) |
| Withdrawal of consent | 23 (8.0) | 13 (4.5) | 36 (6.2) |
| Death | 16 (5.5) | 9 (3.1) | 25 (4.3) |
| Lost to follow-up | 1 (0.3) | 1 (0.3) | 2 (0.3) |
| Crossover to ibrutinib | – | 183 (66.3) | – |
BR: bendamustine and rituximab; CI: confidence interval; TEAEs: treatment-emergent adverse events.
Following the prespecified interim analysis, placebo treatment was discontinued on 10 March 2015, as was the reporting of TEAEs for the placebo arm; these patients had continued disease evaluation and follow-up and were permitted to cross over to ibrutinib after confirmed disease progression.
Includes patients who did not receive study medication.
Includes patients who rolled over to the phase 3b access study (CAN3001) or commercial ibrutinib.
At the final analysis, 183 (63.3%) patients with confirmed disease progression crossed over from the placebo arm to single-agent ibrutinib treatment. The most common reasons for treatment discontinuation in the ibrutinib plus BR arm were investigator/sponsor decision (136 of 289 patients [47.1%]; mainly consisting of patients reaching study end and rolling over to an open-label access study, where ibrutinib was continued), AEs (58 patients [20.1%]; Table 1), and progressive disease/relapse (55 patients [19.0%]). In the placebo plus BR arm, most common reasons for treatment discontinuation included progressive disease/relapse (148 of 289 patients [51.2%]) and investigator/sponsor decision (84 patients [29.1%]; mainly following unblinding at the interim analysis).
Progression-free survival and overall survival
Median PFS for the ibrutinib plus BR arm at final analysis was 65.1 months (n = 289); substantially longer than for the placebo plus BR arm (median 14.3 months; n = 289; HR 0.229 [95% CI 0.183–0.286]; p < .0001; Figure 2). The 60-month PFS rate was 52.7% in the ibrutinib plus BR arm and 8.2% in the placebo plus BR arm.
Figure 2.
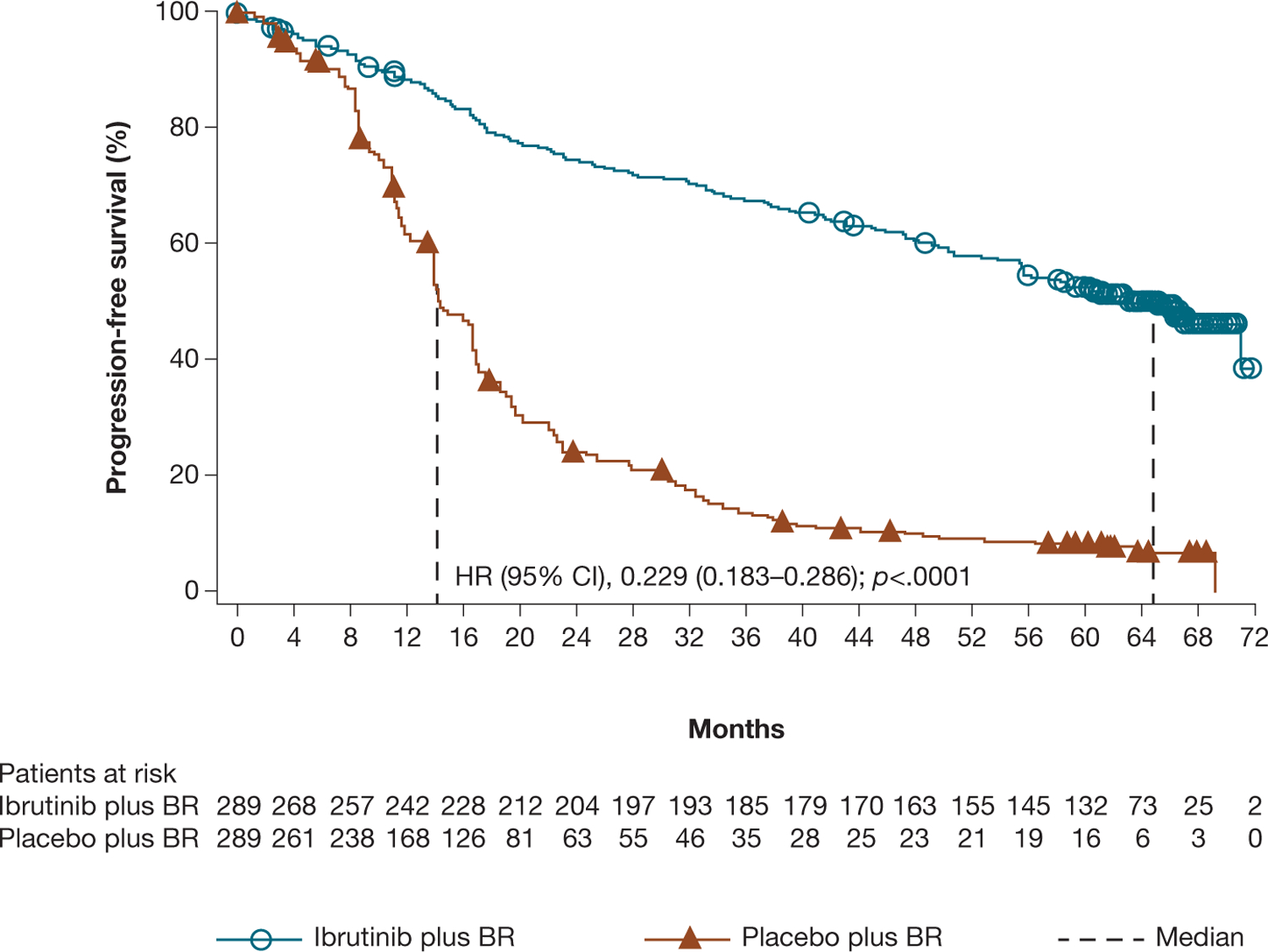
Investigator-assessed PFS for ibrutinib plus BR versus placebo plus BR.
At the final 5-year analysis, the OS advantage for patients in the ibrutinib plus BR arm versus placebo plus BR arm was maintained despite crossover of 183 patients (63.3%) from the placebo plus BR arm to ibrutinib treatment (HR 0.611 [95% CI 0.455–0.822]; p = .0010; Figure 3). Median OS was not reached in either group; the 60-month OS rate was 75.7% for ibrutinib plus BR versus 61.2% for placebo plus BR.
Figure 3.
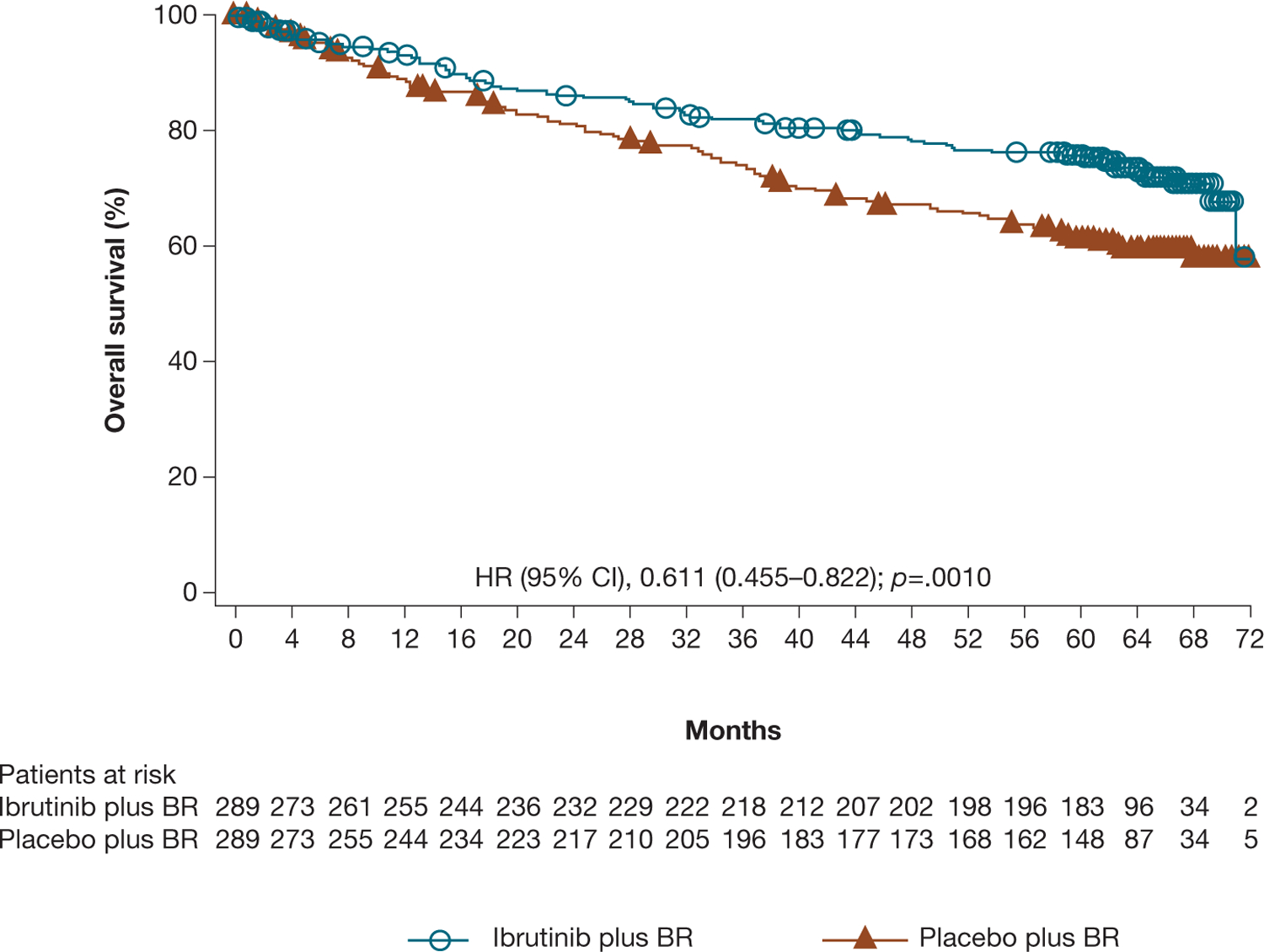
Investigator-assessed OS for ibrutinib plus BR versus placebo plus BR.
Median PFS2 was not reached in the ibrutinib plus BR arm, notably longer than in the placebo plus BR arm (63.0 months; HR 0.594 [95% CI 0.453–0.778]; p = .0001). At the time of data cutoff, 52 (18.1%) patients in the ibrutinib plus BR arm received at least one subsequent antineoplastic therapy. Of the patients in the placebo plus BR arm who did not receive ibrutinib as first subsequent therapy as part of crossover, 60 (20.9%) received other subsequent therapy. The most commonly administered subsequent therapies in the ibrutinib plus BR and placebo plus BR arms, respectively, were monoclonal antibodies (7.7% and 12.2%; mainly rituximab), nitrogen mustard analogues (7.7% and 10.8%; mainly cyclophosphamide), corticosteroids (4.2% and 10.1%), anthracyclines (1.4% and 6.6%; mainly doxorubicin), and other antineoplastic agents (6.6% and 4.2%; including venetoclax: 4.5% and 2.4%). In addition, 3 (1.0%) and 6 (2.1%) patients in the ibrutinib plus BR and placebo plus BR arms respectively, received allogenic stem cell transplant as subsequent therapy. Chemotherapy agents and anti-CD20 antibodies were commonly administered together in a variety of regimens, including R-CHOP for treatment of Richter’s transformation. Of the 13 patients in the ibrutinib plus BR arm who received subsequent venetoclax or venetoclax plus CD20 anti-body treatment, 9 patients received it as first subsequent therapy to ibrutinib; best response was CR (3 subjects), PR (3 subjects), not reported (3 subjects).
For patients who received a subsequent antineoplastic therapy, the time from start of the first subsequent therapy to progression or death was longest for patients who received ibrutinib subsequent to placebo plus BR (median not reached, 95% CI 45.57 months –not evaluable). For those who received subsequent therapy other than ibrutinib, the median time from start of the first subsequent therapy to progression or death was similar for patients previously on ibrutinib plus BR and those previously on placebo plus BR (median (95% CI) 9.43 months (3.22 – 22.08), and 9.17 months (1.68 – 15.15), respectively).
Overall response rate
Investigator-assessed ORR was 87.2% for ibrutinib plus BR versus 66.1% for placebo plus BR (p < .0001). Responses deepened over time: CR/CR with incomplete bone marrow recovery rate (CRi) in the ibrutinib arm increased from 21.5% (62/289) in the interim analysis to 38.1% (110/289) in the 3-year analysis and 40.8% (118/289) in the final analysis. The patient proportion with undetectable MRD in peripheral blood or bone marrow in the final analysis was 28.7% in the ibrutinib plus BR arm (similar to the 3-year analysis [26.3%] [16], as MRD testing was ceased shortly after the 3-year analysis).
Safety
TEAEs for the ibrutinib plus BR arm were consistent with previous reports [15, 16]. Most patients (90.2%) had at least 1 grade ≥3 TEAE and these occurred most frequently in the first 6 months of treatment (Table 2). Similar to the 3-year results, 69.0% of patients had serious TEAEs (any grade) and 20.2% had TEAEs leading to ibrutinib discontinuation (61.3% and 16.0%, respectively, in the 3-year analysis). During the first 6 months in the ibrutinib plus BR arm, 7.7% of patients discontinued ibrutinib due to AEs; this rate decreased over time with continued single-agent ibrutinib treatment (Table 2).
Table 2.
Summary of prevalence of TEAEs over time (and overall) occurring in patients in the ibrutinib plus BR arm in the final analysis.
| Ibrutinib treatment duration | ||||||||
|---|---|---|---|---|---|---|---|---|
| n (%) | ≥0–0.5 year (n = 287) |
>0.5–1 year (n = 246) |
>1–2 years (n = 216) |
>2–3 years (n = 188) |
>3–4 years (n = 171) |
>4–5 years (n = 157) |
>5–6 years (n = 129) |
Overall (n = 287) |
| Patients with any grade TEAEs | 271 (94.4) | 216 (87.8) | 180 (83.3) | 141 (75.0) | 121 (70.8) | 114 (72.6) | 49 (38.0) | 282 (98.3) |
| Patients with TEAEs of grade ≥3 | 212 (73.9) | 111 (45.1) | 87 (40.3) | 62 (33.0) | 37 (21.6) | 35 (22.3) | 17 (13.2) | 259 (90.2) |
| Patients with any treatment-related TEAEa | 225 (78.4) | 145 (58.9) | 122 (56.5) | 76 (40.4) | 68 (39.8) | 50 (31.8) | 21 (16.3) | 249 (86.8) |
| Patients with any TESAE | 104 (36.2) | 47 (19.1) | 53 (24.5) | 40 (21.3) | 29 (17.0) | 29 (18.5) | 13 (10.1) | 198 (69.0) |
| Patients with any TEAE leading to ibrutinib discontinuationb | 22 (7.7) | 15 (6.1) | 8 (3.7) | 3 (1.6) | 3 (1.8) | 2 (1.3) | 5 (3.9) | 58 (20.2) |
| Patients with any TEAEs with a fatal outcomec | 10 (3.5) | 4 (1.6) | 11 (5.1) | 3 (1.6) | 1 (0.6) | 1 (0.6) | 3 (2.3) | 33 (11.5) |
BR: bendamustine and rituximab; n: number of patients; TEAEs: treatment-emergent adverse events; TESAE: treatment-emergent serious adverse event.
Judged by the investigator to be very likely, probably, possibly, or definitely related to the study drug.
Patients who had TEAEs leading to discontinuation of ibrutinib were counted only at the interval when they discontinued ibrutinib.
Patients who had TEAE leading to death were counted only at the interval when they died.
The incidence of the TEAE of clinical interest, major hemorrhage (any grade), was 5.6% in the ibrutinib plus BR arm during the study duration, including 3 (1.0%) patients with grade 5 major hemorrhage (1 patient with a history of hypertension and an abdominal aortic aneurysm had an abdominal aortic aneurysm rupture, 1 patient had intraabdominal hemorrhage following a fall, and 1 patient had post-procedural hemorrhage following colonoscopy and colon adenoma excision; none received anticoagulants). Additionally, any grade TEAE incidences of infections, neutropenia, diarrhea, anemia, hypertension, and atrial fibrillation, respectively, were 78.7%, 59.9%, 40.4%, 25.8%, 16.7%, and 11.8% in the ibrutinib plus BR arm. Two (0.7%) patients in the ibrutinib plus BR arm had grade 3 Aspergillus infections (onset of events within the first and third year of treatment with ibrutinib), 1 (0.3%) patient experienced a grade 2 cryptococcal infection (onset of event 7 months after starting ibrutinib treatment), and 2 (0.7%) patients had Pneumocystis infections (1 grade 3; 1 grade 5; onset of both events within the second year of treatment with ibrutinib).
From the interim to final analysis, the prevalence of TEAEs generally decreased over time (Figure 4). Infections and infestations rates and other AEs declined over time. Ventricular tachyarrhythmia rates (3 patients; 1.0%), based on a Standardised MedDRA Queries narrow search, were unchanged from the interim analysis in the ibrutinib plus BR arm. In the final analysis, 3 (1.0%) patients had cerebrovascular accidents (2 grade 2; 1 grade 3). In the ibrutinib plus BR arm, 5 (1.7%) patients had Richter’s transformation (4 cases of large cell lymphoma and 1 case with ‘other’ histology). At the interim analysis, there were no transformations in the ibrutinib group and 3 in the placebo group. Overall, 74 (25.8%) patients in the ibrutinib plus BR arm died: 35 due to AEs, 11 of which were related to study treatment (5 cases of infection, 2 cases of second malignancy [chronic myelomonocytic leukemia and myelodysplastic syndrome], and 1 case each of the following: multi-organ failure, systemic inflammatory response syndrome, lung infiltration, aortic aneurysm rupture), 17 due to progressive disease, and 22 due to other reasons (i.e. >30 days after the last dose and not due to TEAEs/progressive disease).
Figure 4.
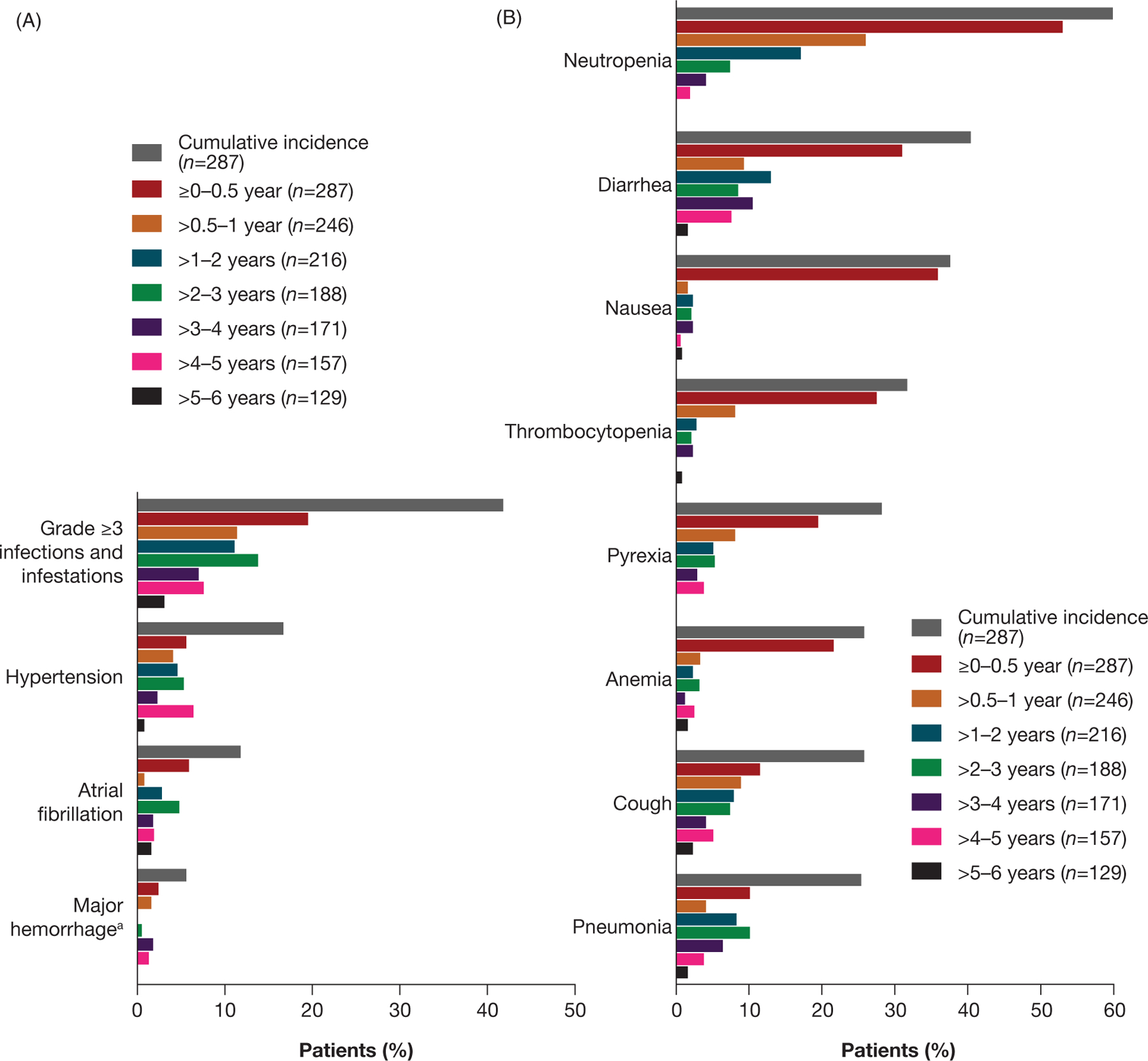
Prevalence of any grade TEAEs over time in the ibrutinib arm for (A) TEAEs of clinical interest and (B) TEAEs occurring in ≥30% of patients in the final analysis. aMajor hemorrhage TEAEs included serious/grade ≥3 hemorrhage and any grade central nervous system hemorrhage.
Discussion
In this final analysis of the HELIOS trial, median PFS for patients with R/R CLL/SLL who received a median of 2.0 prior lines of therapy (range 1–11) in the ibrutinib plus BR arm was 5.4 years. In this patient population, HELIOS is the first study showing an OS benefit with ibrutinib added to chemoimmunotherapy versus chemoimmunotherapy alone [15].
At the time of the interim analysis (median followup, 17 months), ibrutinib plus BR significantly improved PFS versus placebo plus BR for R/R CLL/SLL [15]; IRC-assessed median PFS was not reached in the ibrutinib plus BR arm versus 13.3 months in the placebo plus BR arm (HR 0.203 [95% CI 0.150–0.276]; p < .0001). This final analysis confirms the persistent benefit of ibrutinib plus chemoimmunotherapy; median investigator-assessed PFS for the ibrutinib plus BR arm was substantially longer than the placebo plus BR arm (HR 0.229 [95% CI 0.183–0.286]; p < .0001). The durability of PFS noted in our trial is consistent with previously published observations in randomized trials evaluating single-agent ibrutinib in comparable patient populations and in patients with previously untreated CLL [9,19].
Unlike the interim analysis of HELIOS, the 3-year analysis showed an improved median OS with ibrutinib plus BR (HR 0.652 [95% CI 0.454–0.935]; p = .019). In this final analysis, the long median PFS with ibrutinib plus BR translated into an OS benefit (HR 0.611 [95% CI 0.455–0.822]; p = .0010), despite 63.3% of patients crossing over from the placebo plus BR arm to ibrutinib treatment. The RESONATE study also demonstrated an OS benefit with ibrutinib versus ofatumumab for R/R CLL/SLL (HR 0.639 [95% CI 0.418–0.975]) [10]. Additionally, median PFS2 was substantially longer for patients in the ibrutinib plus BR arm despite crossover, further supporting the benefit of earlier treatment with ibrutinib. Among the subgroups of patients who received subsequent therapies, those who received ibrutinib as next treatment after placebo plus BR had the longest time to next progression or death. For those who received other antineoplastic subsequent therapies, there was no meaningful difference in time to next progression or death for those who previously received ibrutinib plus BR or placebo plus BR, indicating that prior ibrutinib treatment did not impact efficacy of subsequent therapy. The small individual numbers of patients treated with specific alternative therapies do not allow a recommendation of a particular treatment for patients relapsing after ibrutinib-based therapy, although responses, including complete remissions, were observed in patients treated with venetoclax regimens after ibrutinib plus BR.
Responses to ibrutinib plus BR in the HELIOS study were durable and deepened over time. The final analysis also showed a significant increase in CR/CRi rate (40.8%) compared with the interim analysis (21%), reflecting the ongoing benefit of continuous treatment with ibrutinib. The rate of undetectable MRD in peripheral blood and bone marrow plateaued and testing was discontinued shortly after the 3-year analysis, but 29% of patients had undetectable MRD at the last analysis. The PFS curve did not show a plateau at the final analysis, with a duration of follow-up very close to the median PFS, despite deepening responses over time.
Safety findings were consistent with known safety profiles of ibrutinib and BR in patients with CLL [9,20–22], and there were no unexpected findings at the latest follow-up analysis compared with the 3-year analysis. Consistent with prior analyses [15,16], this extended 5-year follow-up analysis demonstrates the manageable safety profile of ibrutinib. From the interim results to final analysis, the prevalence of TEAEs including serious TEAEs trended lower over time with ibrutinib. However, it is notable that the number of patients on treatment also decreased, partly because of discontinuations due to AEs. Overall, the prevalence of hypertension, atrial fibrillation, and major hemorrhage events decreased over time throughout the study. Rates of infections and infestations, including pneumonia, also declined over time and the incidence of serious opportunistic infections, e.g Aspergillus, Cryptococcus, and Pneumocystis, was low.
Our study is limited as it did not evaluate whether ibrutinib plus BR is more beneficial than single-agent ibrutinib in a relapsed setting. Nonetheless, an indirect comparison of the RESONATE and HELIOS trials, after adjusting for known confounding variables, has previously been published [23]. That analysis comparing single-agent ibrutinib with ibrutinib plus BR suggested that single-agent ibrutinib was superior for PFS and OS. The analysis also suggested that an induction period with BR did not improve outcomes, however, only short-term follow-up of both trials was available at the time of the published comparison, preventing firm conclusions. Similarly, in a recently published randomized trial in 208 patients with CLL (most [181] with R/R CLL), ibrutinib plus rituximab did not improve PFS versus single-agent ibrutinib despite faster remissions and lower levels of residual disease in patients receiving the combination. A recent study (Alliance A041202) in patients with previously untreated CLL also demonstrated no additional benefit of adding rituximab to ibrutinib for PFS, in the first-line setting [19]. However, due to differences in patient populations, study design, and treatment regimens, it is difficult to make indirect cross-trial comparisons.
Lack of resistance testing is another limitation of this trial, as only a few genomic biomarker samples were available. However, CLL remains an incurable disease and eventually the disease progresses further in most patients with R/R CLL despite the therapy advances of previous years. Therefore, it is important to assess adverse biologic features and resistance mechanisms leading to treatment resistance in current and future CLL studies to establish predictive biomarkers, customize therapy, and enhance therapy outcomes.
In conclusion, with an extended median follow-up of 63.7 months, this final analysis confirms the long-term safety and efficacy of ibrutinib plus BR in patients with R/R CLL/SLL. In this patient population with a median of 2.0 prior lines of therapy, median PFS in the ibrutinib plus BR arm was 5.4 years and the OS rate at 5 years was 75.7%. Long-term safety findings for the ibrutinib plus BR arm were also consistent with the known safety profiles of ibrutinib and BR and support a positive benefit/risk profile for continuous ibrutinib treatment.
Acknowledgments
Writing assistance was provided by Sally Hassan, PhD, CMPP of Parexel and funded by Janssen Global Services, LLC. The authors would like to thank patients who participated in this trial, their families, investigators, study coordinators, study teams, and nurses.
Footnotes
Presented at the XVIII International Workshop on CLL (iwCLL) meeting, 20–23 September 2019, Edinburgh, Scotland.
Disclosure statement
GAMF had a consultant/advisory role with AbbVie and Janssen. FD had a consultant/advisory role with AbbVie and Roche, received research funding from AbbVie and Janssen, honoraria from Amgen and Janssen, and was on a speakers’ bureau for Amgen and Janssen. RSS had a consultant/advisory role with AstraZeneca and Janssen and was on a speakers’ bureau for Janssen. AJ had a consultant/advisory role with Gilead Sciences, Janssen, Roche, and Sanofi Genzyme, was on a speakers’ bureau for AbbVie, Amgen, and Novartis, and received other financial/material support from Celgene. JM received research funding from Janssen-Cilag. NLB had a consultant/advisory role with ADC Therapeutics, Acerta, and BTG Therapeutics and received research funding from Autolus, Bristol Meyers Squibb, Celgene, Forty Seven, Genentech, Immune Design, Janssen, Kite Pharma, Merck, Millennium, Pharmacyclics, and Seattle Genetics. M-SD had a consultant/advisory role with AbbVie and Janssen and received honoraria from AbbVie and Janssen. JL had a consultant/advisory role with AbbVie, AstraZeneca, Gilead Sciences, Janssen, and Roche. SR had a consultant/advisory role with AstraZeneca, BeiGene, Celgene, Celltrion, Gilead Sciences, Janssen, Kite Pharma, Roche, and Sunesis Pharmaceuticals, received research funding from Janssen and Pharmacyclics, and was on a speakers’ bureau for Janssen and Roche. MAP had a consultant/advisory role with AstraZeneca and Janssen, and was on a speakers’ bureau for AbbVie, Janssen, Novartis, and Roche. AG had a consultant/advisory role with and received honoraria from Acerta, Celgene, Gilead Sciences/Kite Pharma, and Janssen, received research funding from Acerta, AstraZeneca, Bayer, CALBG, Celgene, Genentech, Hoffman-La Roche, Janssen, Kite Pharma, MD Anderson, MorphoSys AG, Pharmacyclics, and the University of Nebraska, and is on the board and a shareholder of COTA. AM had a consultant role with and received research funding from AbbVie, Acerta, Janssen, Loxo Oncology, Pharmacyclics, Genentech, Sunesis Pharmaceuticals, and TG Therapeutics, received research funding from DTRM and Regeneron, and was on the Data and Safety Monitoring Board for Celgene and TG Therapeutics. MH had a consultant/advisory role with AbbVie, Celgene, Gilead Sciences, Janssen, Mundipharma, Pharmacyclics, and Roche, received research funding from AbbVie, Celgene, Gilead Sciences, Janssen, Mundipharma, Pharmacyclics, and Roche, and was on a speakers’ bureau for AbbVie, Celgene, Gilead Sciences, Janssen, Mundipharma, Pharmacyclics, and Roche. MS, AC, KN, SB, and AH hold J&J stock and are employed by Janssen R&D. SB also holds AbbVie stock. MT and SS are also employees of Janssen. NS holds J&J stock and is employed by Janssen-Cilag. PC had a consultant/advisory role with AbbVie, Acerta Pharma, AstraZeneca, Janssen-Cilag, and Novartis, was on a speakers’ bureau for AbbVie, F. Hoffmann-LaRoche, and Janssen-Cilag, and received other financial/material support from AbbVie, Acerta Pharma, AstraZenca, F. Hoffmann-LaRoche, Gilead Sciences, GlaxoSmithKline, Janssen-Cilag, and Novartis. AC-K, SG, AA, and OS have no conflicts of interest to disclose.
Data availability
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for study data access can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
References
- [1].IMBRUVICA® (ibrutinib) [prescribing information]. Sunnyvale, CA: Pharmacyclics LLC; Janssen Biotech, Inc.: Horsham, PA; 2019. [Google Scholar]
- [2].IMBRUVICA (ibrutinib) [summary of product characteristics]. Belgium: Janssen Pharmaceutica NV; 2019. [Google Scholar]
- [3].Gayko U, Fung M, Clow F, et al. Development of the Bruton’s tyrosine kinase inhibitor ibrutinib for B cell malignancies. Ann N Y Acad Sci 2015;1358:82–94. [DOI] [PubMed] [Google Scholar]
- [4].Byrd JC, Jones JJ, Woyach JA, et al. Entering the era of targeted therapy for chronic lymphocytic leukemia: impact on the practicing clinician. J Clin Oncol 2014; 32(27):3039–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dias AL, Jain D. Ibrutinib: a new frontier in the treatment of chronic lymphocytic leukemia by Bruton’s tyrosine kinase inhibition. Cardiovasc Hematol Agents Med Chem 2014;11(4):265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma NCCN Evidence Blocks™; Version 1.2020; [cited 2019 Sep 18]. Available from: https://www.nccn.org/professionals/physician_gls/pdf/cll_blocks.pdf. [Google Scholar]
- [7].Molica S. Ibrutinib continues to influence the therapeutic landscape of chronic lymphocytic leukemia: new data presented at ASCO 2017. BMC Med 2017; 15(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med 2014;371(3):213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Byrd JC, Hillmen P, O’Brien S, et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs ofatumumab. Blood 2019;133(19):2031–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Munir T, Brown JR, O’Brien S, et al. Final analysis from RESONATE: up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am J Hematol 2019;94(12):1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Eichhorst B, Dreyling M, Robak T, et al. Chronic lymphocytic leukemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2011;22(Suppl 6):vi50–vi54. [DOI] [PubMed] [Google Scholar]
- [12].Gordon MJ, Lewis LD, Brown JR, et al. Bendamustine hydrochloride in patients with B-cell malignancies who have comorbidities – is there an optimal dose? Expert Rev Hematol 2017;10(8):707–718. [DOI] [PubMed] [Google Scholar]
- [13].Fischer K, Cramer P, Busch R, et al. Bendamustine combined with rituximab in patients with relapsed and/or refractory chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2011;29(26):3559–3566. [DOI] [PubMed] [Google Scholar]
- [14].Brown JR, Barrientos JC, Barr PM, et al. The Bruton tyrosine kinase inhibitor ibrutinib with chemoimmunotherapy in patients with chronic lymphocytic leukemia. Blood 2015;125(19):2915–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chanan-Khan A, Cramer P, Demirkan F, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double-blind, phase 3 study. Lancet Oncol 2016;17(2): 200–211. [DOI] [PubMed] [Google Scholar]
- [16].Fraser G, Cramer P, Demirkan F, et al. Updated results from the phase 3 HELIOS study of ibrutinib, bendamustine, and rituximab in relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma. Leukemia 2019;33(4):969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008;111(12):5446–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].van Dongen JJ, Lhermitte L, Bottcher S, et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 2012;26(9):1908–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Woyach JA, Ruppert AS, Heerema NA, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med 2018; 379(26):2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125(16):2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369(1):32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Eichhorst B, Fink AM, Bahlo J, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol 2016;17(7):928–942. [DOI] [PubMed] [Google Scholar]
- [23].Hillmen P, Fraser G, Jones J, et al. Comparing single-agent ibrutinib, bendamustine plus rituximab (BR) and ibrutinib plus BR in patients with previously treated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL): an indirect comparison of the RESONATE and HELIOS trials [abstract] 57th ASH Annual Meeting; 5–8 December 2015; Orlando, FL; Abstract 2944. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for study data access can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.