

Abstract
Gene therapy by integrating vectors is promising for monogenic liver diseases, especially in children where episomal vectors remain transient. However, reaching the therapeutic threshold with genome-integrating vectors is challenging. Therefore, we developed a method to expand hepatocytes bearing therapeutic transgenes. The common fever medicine acetaminophen becomes hepatotoxic via cytochrome p450 metabolism. Lentiviral vectors with transgenes linked in cis to a Cypor shRNA were administered to neonatal mice. Hepatocytes lacking the essential cofactor of Cyp enzymes, NADPH-cytochrome p450 reductase (Cypor), were selected in vivo by acetaminophen administration, replacing up to 50% of the hepatic mass. Acetaminophen treatment of the mice resulted in over 30-fold expansion of transgene-bearing hepatocytes and achieved therapeutic thresholds in hemophilia B and phenylketonuria. We conclude that therapeutically modified hepatocytes can be selected safely and efficiently in preclinical models with a transient regimen of moderately hepatotoxic acetaminophen.
INTRODUCTION
Gene therapy is a promising approach to many previously incurable genetic disorders. Recombinant adeno-associated virus (rAAV) is currently the most commonly used vector for in vivo delivery (1). However, current rAAV gene therapies have some shortcomings. First, large viral doses may be required to transduce a curative threshold of cells. This is associated with a risk of an immune response (2, 3) and high cost. Second, because of their episomal nature, rAAVs are ideal for delivery to postmitotic cells because their expression is quickly lost in dividing cells (4), including the developing hepatocytes of children. Problems with loss of episomal gene expression can be overcome by using integrating vectors, including lentiviral or integrating rAAV vectors targeting specific gene loci by homologous recombination (5–7). However, the high efficiency of genetic modification required to reach therapeutic thresholds limits broad application of integrating vectors and precise gene editing technologies. Furthermore, it is difficult to produce lentiviral vectors in quantities sufficient for liver gene therapy in human patients. To overcome the efficiency problems, we developed a pharmacological hepatocyte selection regimen applicable to human patients. Here, we demonstrate that the widely used fever medicine acetaminophen (APAP) can be used to select therapeutically modified hepatocytes and attain therapeutic thresholds in preclinical models.
APAP is commonly used to treat mild pain and fever (8). Although most of APAP is metabolized by pathways independent of cytochrome p450 (Cyp) enzymes, a fraction is metabolized by Cyp1A2, Cyp2E1, and Cyp3A4 in zone 3 hepatocytes (9) to the intermediate N-acetyl-p-benzoquinone imine (NAPQI), which is hepatotoxic (Fig. 1A) (9). Cypor is a cofactor required by all Cyp enzymes for electron transfer from NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) (10). Because Cypor is an obligate cofactor for all Cyp enzymes, we hypothesized that loss of Cypor would abrogate Cyp-mediated metabolism, preventing the conversion of APAP to hepatotoxic NAPQI. Therefore, hepatocytes lacking Cypor would be protected from APAP-induced toxicity and not undergo necrosis (Fig. 1B). Here, we show that APAP treatment can be used to clonally expand hepatocytes with disrupted expression of Cypor. By linking therapeutic transgenes in cis with a CRISPR-Cas9 guide RNA (gRNA) or a short hairpin RNA (shRNA) targeting Cypor in an integrating vector, hepatocytes with integration can be selected with APAP. This strategy allows rare populations of hepatocytes harboring a disease-curing transgene to be expanded to therapeutic levels.
Fig. 1. Principles of APAP metabolism and APAP selection.
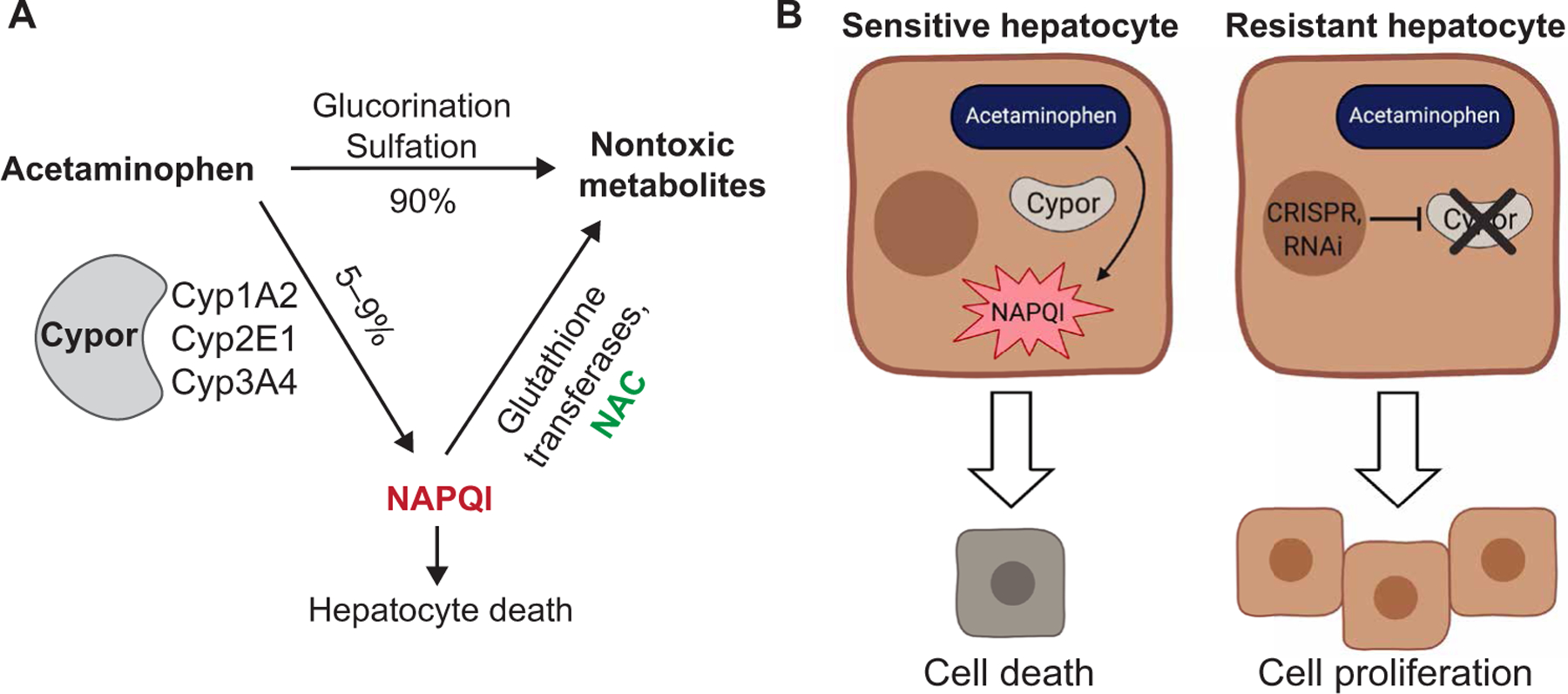
(A) Pathways of APAP metabolism. Cypor is required for the metabolism of APAP to the hepatotoxic metabolite NAPQI. N-acetylcysteine (NAC) detoxifies NAPQI. (B) Mechanism of APAP selection. Disruption of Cypor renders hepatocytes resistant to the hepatotoxic effects of APAP, resulting in a selective advantage with APAP administration. RNAi, RNA interference.
RESULTS
Selection of Cypor knockout hepatocytes
To demonstrate that hepatocytes deficient in Cypor can be selected by APAP, Cypor was knocked out in primary hepatocytes in vivo. A plasmid expressing Streptococcus pyogenes Cas9 (SpCas9) and a U6-driven gRNA targeting Cypor (pX330-Cypor gRNA; Fig. 2A) was delivered via hydrodynamic injection (11) into wild-type adult mice. Mice were then treated biweekly with APAP by intraperitoneal injection at a dose sufficient to produce an alanine aminotransferase (ALT) elevation >800 IU/liter (250 to 450 mg/kg). Plasma ALT activity 6 hours after treatment was monitored as a measure of hepatotoxicity. ALT responses were initially highly elevated after APAP injection but diminished after repeated treatments. This effect was not seen in control mice not treated with pX330-Cypor gRNA (Fig. 2B). The decrease in ALT suggested the loss of nontransfected hepatocytes and expansion of APAP-resistant Cypor knockout hepatocytes. Liver DNA was assayed for insertion or deletion mutations (in-dels) at the Cypor gRNA cut site using the Tracking of Indels by DEcomposition (TIDE) algorithm (12). In-del analysis showed 2.6 ± 1.3% Cypor-deficient hepatocytes in unselected mice that received the Cypor gRNA plasmid without APAP treatment, and a significant (P = 0.003) expansion to 35.5 ± 2.1% Cypor-deficient hepatocytes in mice with APAP selection (Fig. 2C). This was confirmed by Cypor immunofluorescence (IF) showing large areas of clonally expanded Cypor-negative hepatocytes after selection, as compared to rare Cypor-negative hepatocytes in unselected mice (Fig. 2D). These findings demonstrate that a knockout of Cypor in hepatocytes in conjunction with APAP treatment is an efficient method to expand a small population of correctly targeted hepatocytes to a substantial portion of the liver.
Fig. 2. APAP selection of Cypor-deficient hepatocytes.
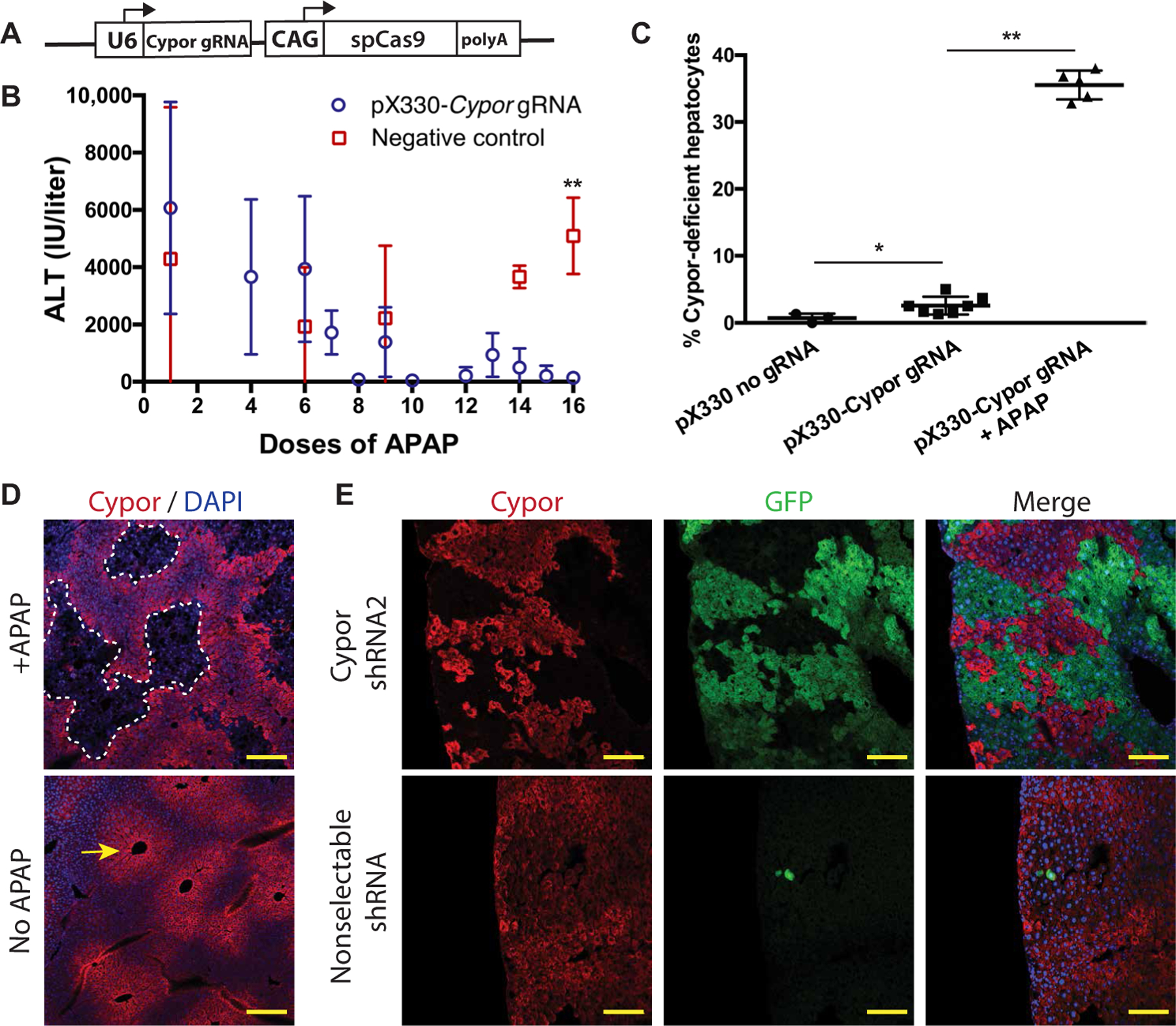
(A) Schematic of the pX330-Cypor gRNA vector. (B) ALT in APAP-treated mice 6 hours after APAP injection for mice treated with pX330-Cypor gRNA (n = 10) or negative control (n = 8). (C) Percent Cypor-deficient hepatocytes measured by in-del analysis of whole-liver homogenate gDNA in mice that received a negative control pX330 vector lacking a gRNA (n = 3), pX330-Cypor gRNA and no APAP (n = 7), or pX330-Cypor gRNA with APAP treatment (n = 5). (D) Cypor immunofluorescent staining (red) in liver sections from mice that received pX330-Cypor gRNA with or without APAP treatment. Arrow indicates zone 3 expression of Cypor. Scale bars, 200 µm. DAPI, 4′,6-diamidino-2-phenylindole. (E) Top: Cypor immunofluorescent staining and GFP expression in liver sections from mice treated with a Cypor shRNA GFP-expression transposon vector and APAP selection (n = 3). Bottom: Negative control: Mouse treated with a transposon expressing an shRNA against an unrelated gene (Hpd). Scale bars, 100 µm. Data are means ± SD. *P < 0.05 and **P < 0.01 by Mann-Whitney U test.
Selection of Cypor shRNA–expressing hepatocytes
Off-target genomic effects are a concern for clinical use of the CRISPR-Cas9 system, as is the need for codelivery of Cas9 and associated immunogenicity (13). Therefore, it may prove preferable to target Cypor via shRNA-mediated knockdown. Two shRNAs against Cypor were screened for selectability by APAP. A transposon plasmid expressing enhanced green fluorescent protein (EGFP) and a U6-driven shRNA was codelivered with Sleeping Beauty transposase via hydrodynamic injection in adult wild-type mice. Mice were treated with biweekly APAP injections to select for hepatocytes a Cypor knockdown due to shRNA expression. Mice treated with Cypor-shRNA2, but not Cypor-shRNA1 (fig. S1), showed large clonally expanded areas of EGFP-positive hepatocytes and decreased Cypor levels by IF staining comprising about 50% of the liver area (Fig. 2E and table S1). This indicates that an shRNA can provide sufficient knockdown of Cypor expression to allow the expansion of Cypor-negative hepatocytes with APAP selection. Cypor-shRNA2 was used for all future experiments using an shRNA.
Selectable lentiviral vectors for hemophilia B
Standard rAAV vectors have only transient benefit in neonatal animals because they are rapidly lost by liver growth (4, 14). To determine whether our selection system could be used to treat a genetic liver disease in neonates, human factor 9 (hF9), the deficient gene in hemophilia B, was chosen. A total of 8.5 × 106 TU (transducing units) of a lentiviral vector expressing hF9 from a hepatocyte-specific mouse transthyretin (mTTR) promoter (15) and the Cypor gRNA from the U6 promoter (Fig. 3A) were delivered via facial vein injection to neonatal Cas9 transgenic mice. To select for hepatocytes that had lentiviral integrations, biweekly APAP intraperitoneal injections were started at age 8 weeks (n = 4), with half (n = 4) kept as unselected controls. After 27 APAP doses, hF9 concentrations in APAP-selected animals increased to 14,116 ± 2241 ng/ml (P = 0.029) (Fig. 3B), equivalent to a 4- to 57-fold increase, whereas unselected animals showed no significant change (fig. S2A). The percentage of Cypor-deficient hepatocytes was significantly higher at 27.2 ± 9.1% in selected mice than in unselected controls at 2.88 ± 2.82% (P = 0.029) (Fig. 3C). This finding was further corroborated by IF against Cypor and hF9, revealing clonally expanded regions of Cypor-negative, hF9-positive hepatocytes in the selection group that were not observed in unselected mice (Fig. 3D). In addition to this Cas9-dependent system, an hF9 lentiviral vector using the Cypor shRNA (Fig. 3E) was given to wild-type neonates (6.6 × 106 to 8.7 × 106 TU per pup). APAP selection was started in half of the mice at age 8 weeks, and hF9 concentrations were monitored over time. After 45 doses, the hF9 levels increased by 35- to 88-fold in APAP-selected mice (fig. S2B), with absolute hF9 levels ranging from 3790 to 14,880 ng/ml (Fig. 3F). In unselected mice, hF9 levels remained stable with no significant change from baseline (Fig. 3F). Cypor immunostaining confirmed the clonal expansion of Cypor-negative, hF9-positive regions in APAP-selected mice to about 15% of the liver area (Fig. 3G and table S1). Hence, we show that a lentiviral expression of the Cypor shRNA aids APAP selection and expansion of hepatocytes expressing hF9.
Fig. 3. Selection of hF9-expressing lentiviral vectors.
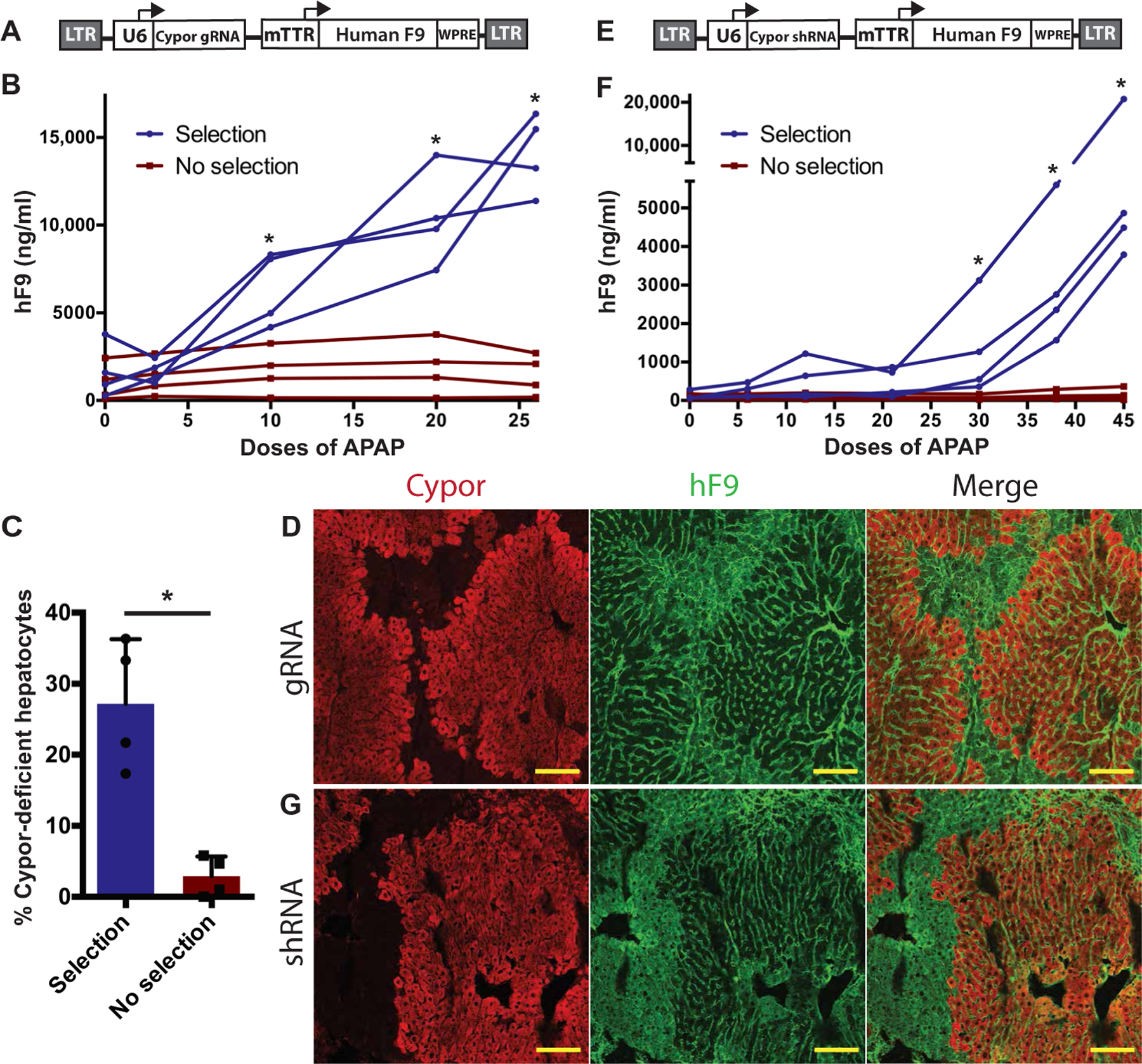
(A) Schematic of the lentiviral vector containing a U6-driven Cypor gRNA and hF9 driven by the liver-specific mTTR promoter (LV-hF9-Cypor gRNA). LTR, long terminal repeat; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element. (B) hF9 concentrations in transgenic Cas9 mice treated with LV-hF9-Cypor gRNA over the course of APAP selection. (C) Quantification of Cypor-deficient hepatocytes by in-del analysis in APAP-selected (n = 4) and unselected (n = 4) Cas9 mice treated with LV-hF9-Cypor gRNA. (D) Cypor and hF9 liver immunohistochemistry in Cas9 mice treated with LV-hF9-Cypor gRNA followed by APAP selection. Scale bars, 100 µm. (E) Schematic of the lentiviral vector expressing Cypor shRNA and hF9 (LV-hF9-Cypor shRNA). (F) hF9 concentrations in wild-type mice (n = 4) treated with LV-hF9-Cypor shRNA during APAP selection compared to unselected mice (n = 4). (G) Cypor and hF9 liver immunohistochemistry in wild-type mice treated with LV-hF9-Cypor shRNA followed by APAP selection. Scale bars, 100 µm. Data are means ± SD. *P < 0.05 by Mann-Whitney U test.
Selectable lentiviral vectors for phenylketonuria
Having shown that APAP selection can achieve therapeutic hF9 levels, we used the same strategy to treat a mouse model of phenylketonuria (PKU). PKU is an inborn error of metabolism caused by a lack of phenylalanine hydroxylase (Pah) resulting in high concentrations of circulating phenylalanine (Phe) and neurotoxicity (16). Patients with PKU are treated with a Phe-restricted diet. Although treated patients ideally have Phe concentrations between 120 and 360 µM, concentrations below 600 µM are considered acceptable (17). In the Pahenu2/enu2 PKU mouse model, Phe concentrations in untreated animals are commonly more than 1500 µM. Unlike hemophilia B, which can be corrected by transgene expression from a small percentage of hepatocytes, PKU requires at least of 10% of hepatocytes to be corrected to have an effect on disease phenotype (18, 19).
A lentivirus containing the Cypor shRNA and human PAH complementary DNA (cDNA) (Fig. 4A) was delivered via facial vein in Pahenu2/enu2 neonates (1.9 × 107 TU per pup). At 6 to 8 weeks of age, APAP selection was started and serum Phe concentration was measured every six APAP doses. At baseline, serum Phe concentrations were 2080 ± 372 µM in males, and 1904 ± 283 µM in females. After 42 to 48 doses of APAP, male mice showed an average serum Phe of 366 ± 166 µM, significantly lower (P < 0.001) than in unselected controls (Fig. 4B), with two of four mice showing Phe concentrations below the therapeutic threshold of 360 µM. APAP-selected female mice also showed a significant (P < 0.002) decrease as compared to unselected controls. However, selected female mice showed a final Phe concentration of 981 ± 70 µM, still above the therapeutic threshold. Selected mice showed a darkened coat color associated with restored melanin synthesis (Fig. 4C). Pah enzymatic activity in liver homogenate showed a significant increase in both female and male selected mice compared to unselected controls (Fig. 4D). Cypor staining showed that 40 to 55% of hepatocytes were Cypor negative (Fig. 4E and table S1), and immunohistochemistry revealed healthy hepatocytes (Fig. 4F). Cypor IF staining showed similar levels of selection in male and female mice (table S1). These results show a therapeutic correction of PKU using APAP selection of a Cypor shRNA in male mice and demonstrate that this method can be used in gene therapy for diseases that require a higher therapeutic threshold.
Fig. 4. APAP selection in a PKU mouse model.
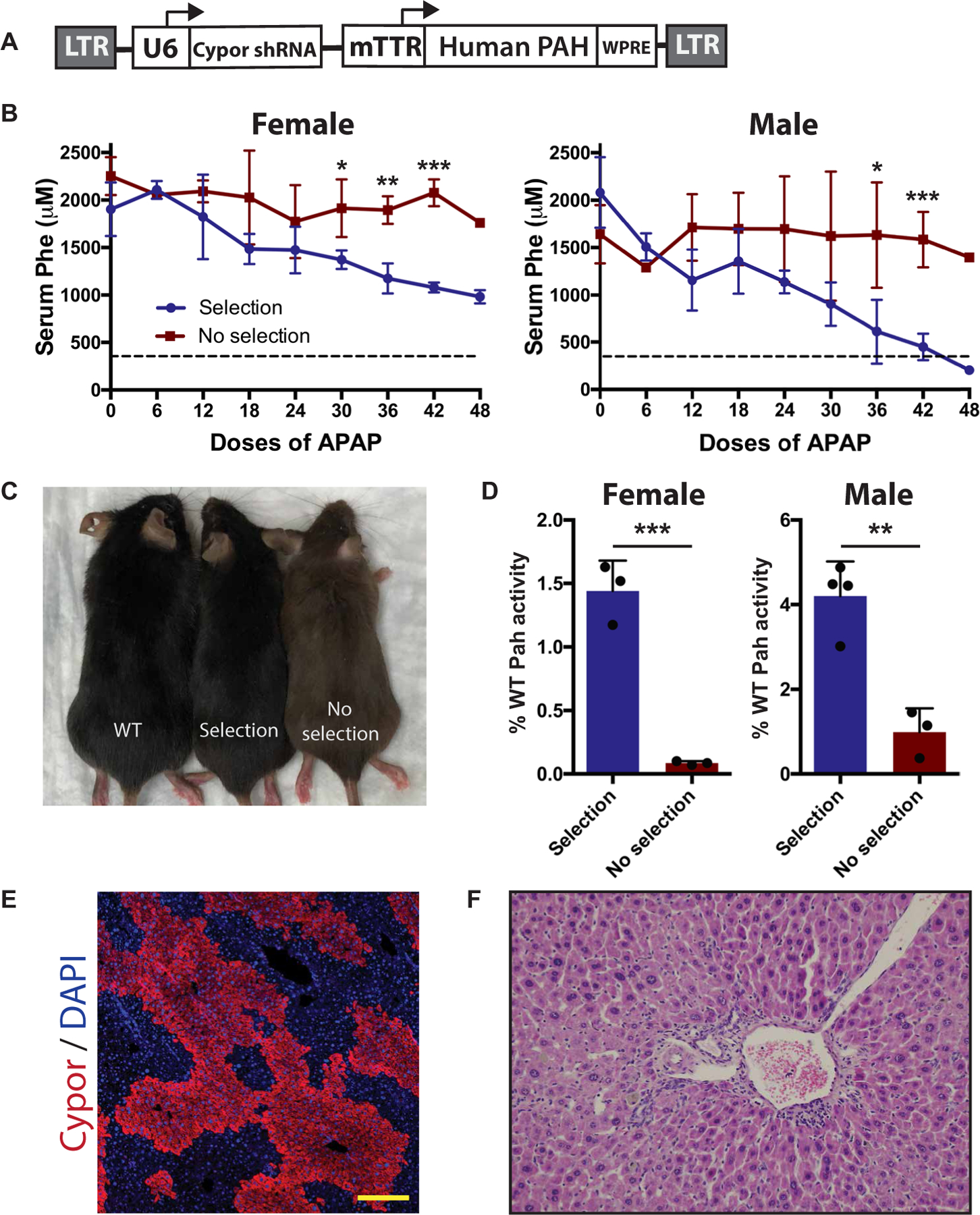
(A) Schematic of selectable lentiviral vector expressing human PAH. (B) Serum Phe concentrations in female and male Pahenu2/enu2 mice. All mice received a selectable PAH-expressing lentiviral vector as neonates, followed by either APAP selection (female n = 3, male n = 4) or no further treatment (female n = 3, male n = 3). Dashed line represents the therapeutic threshold of 360 µM. (C) Coat color comparison of wild-type (WT), APAP-selected Pahenu2/enu2, and unselected Pahenu2/enu2 mice. (D) Pah enzyme activity in liver homogenate as percentage of wild type. (E) Clonal expansion of Cypor-deficient hepatocytes in liver from a selected mouse. Scale bars, 200 µm. (F) Hematoxylin and eosin staining of liver sections in APAP-selected mice. Data are means ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001 by Student’s two-tailed t test assuming equal variance.
Partial selection
The APAP-metabolizing enzymes Cyp1A2, Cyp2E1, and Cyp3A4 are expressed in zone 3 of the hepatic lobule. Cypor is an obligate cofactor for all Cyp enzymes and functions in the metabolism of many drugs. Therefore, one potential barrier to the clinical applicability of APAP selection of Cypor-deficient hepatocytes is the theoretical loss of all Cypor-dependent zone 3 metabolic activity. To address this safety concern, we sought to demonstrate that therapeutic levels of selection can be achieved while retaining Cypor functionality in most of the zone 3 hepatocytes. Neonatal Cas9 transgenic mice were treated with a lentivirus expressing hF9 and the Cypor gRNA via facial vein injection (1.4 × 106 to 4.0 × 106 TU per pup). Upon weaning, baseline hF9 concentrations ranged from 9 to 82 ng/ml. Mice were APAP-selected with frequent monitoring of blood hF9 concentration. To achieve fully therapeutic hF9 concentrations, and to avoid hypercoagulation from superphysiologic expression, selection was halted when blood hF9 concentrations exceeded 3000 ng/ml. TIDE analysis showed 6.7 to 16.7% Cypor-deficient hepatocytes (Fig. 5A and table S2). Compared to frequencies as high as 42% in fully selected mice, this suggests that more than half of zone 3 hepatocytes retained their Cypor activity. IF staining for Cypor and Cyp2E1, a zone 3–specific gene, confirmed this result, showing that most of the Cyp2E1-expressing zone 3 hepatocytes retained Cypor expression (Fig. 5B and fig. S3). These data establish that a therapeutic threshold of gene expression can be attained with only partial loss of zone 3 Cypor-expressing hepatocytes.
Fig. 5. Safety and stability of APAP selection.
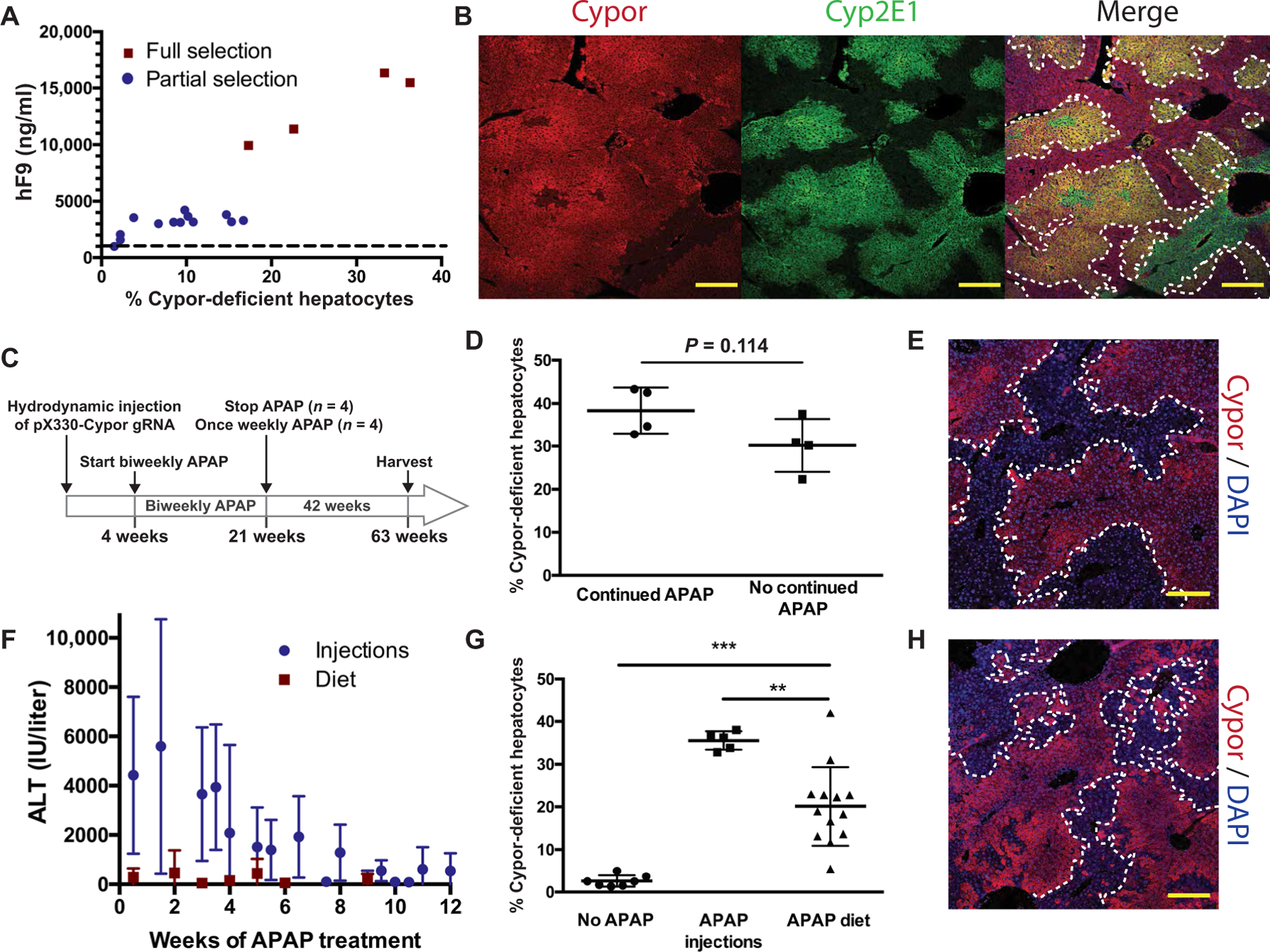
(A) Final blood hF9 concentrations and estimated percent Cypor-deficient hepatocytes in fully (n = 4) and partially (n = 13) APAP-selected mice. Dashed line represents the therapeutic hF9 threshold. (B) Representative Cypor and Cyp2E1 immunohistochemistry in a partially selected mouse (selection halted at 3181 ng/ml of blood hF9). Dotted lines surround Cypor-positive regions. Scale bars, 200 µm. (C) Experimental timeline to test long-term stability of Cypor-deficient hepatocytes. (D) Estimated percent Cypor-deficient hepatocytes in mice that received continued weekly APAP (n = 4) or no continued APAP (n = 4) over a 42-week period. (E) Representative Cypor immunohistochemistry 42 weeks after the last APAP dose. Dotted lines surround Cypor-negative regions. Scale bars, 200 µm. (F) ALT concentrations (6 hours after injection) in mice receiving pX330-Cypor gRNA and either intraperitoneal APAP (n = 19) or on an APAP-containing diet (measured 4 hours into the light cycle) (n = 10). (G) Percent Cypor-deficient hepatocytes in mice treated with pX330-Cypor gRNA followed by no further treatment (n = 7), intraperitoneal APAP injections (n = 5), or APAP diet (n = 13). (H) Representative Cypor immunohistochemistry in a mouse treated with APAP diet for 6 weeks showing areas of Cypor-negative hepatocytes. Scale bars, 200 µm. Data are means ± SD. **P < 0.01 and ***P < 0.001 by Mann-Whitney U test.
Long-term stability of Cypor-deficient hepatocytes
An additional concern for the clinical applicability of this system is the stability of Cypor-deficient hepatocytes in the absence of continuous selective pressure from APAP. To address this, mice were treated with pX330-Cypor gRNA via hydrodynamic injection and selected with biweekly APAP injections until a lack of ALT response 6 hours after injection was observed. APAP was then stopped in half of the mice (n = 4). The others (n = 4) received weekly APAP injections. Mice were harvested 42 weeks after halting APAP treatment in the first group (Fig. 5C). TIDE analysis showed no significant differences between the Cypor in-del percentages in the two groups (P = 0.114) (Fig. 5D). Cypor IF staining of livers from mice that received no continued APAP treatment showed that large areas of Cypor-deficient hepatocytes were still present (Fig. 5E). Liver function and lipid panels revealed no significant differences between the selected mice that received no continued APAP treatment and the untreated controls (fig. S4). In addition, liver histology revealed no signs of hepatocellular injury, inflammation, or necrosis (fig. S5).
Selection with modest liver damage
High doses of injected APAP were used in all experiments described above. In mice receiving intraperitoneal APAP injections, ALT responses upward of 10,000 IU/liter were observed, representing a degree of liver injury unacceptable in a clinical setting. To assess whether selection could be accomplished without extreme ALT elevation, an APAP-containing diet was formulated. Adult mice that had received hydrodynamic injections of pX330-Cypor gRNA were fed the diet for 6 to 12 weeks, with brief breaks. ALTs were monitored, and no ALTs exceeding 3000 IU/liter were observed (Fig. 5F). None of the treated animals died. Upon euthanasia, TIDE analysis showed an average of 20.7 ± 9.4% Cypor-deficient hepatocytes, a significant increase compared to mice receiving no selection (P < 0.0001) (Fig. 5G). In addition, Cypor staining in mice that received APAP diet (Fig. 5H) showed clonally expanded Cypor-deficient areas consistent with APAP selection. Hence, it is possible to select for Cypor-deficient hepatocytes gradually without periods of highly acute liver damage, suggesting that this system could be applied clinically while minimizing the risk of dangerous levels of hepatotoxicity.
DISCUSSION
APAP has several properties that make it ideal as an in vivo hepatocyte selection drug: It is cheap and orally available; it has been used in millions of adult and pediatric patients; its mechanism of hepatoxicity is well understood; an antidote is available; toxicity is limited to only zone 3 of the hepatic lobule; and no long-term sequelae of even severe poisoning have been reported in humans (9). Here, we show that the delivery of a therapeutic gene in cis with knockout or knockdown of Cypor is an efficient method for the selection and expansion of a small population of correctly targeted hepatocytes using moderately toxic doses of APAP. This strategy allowed us to overcome some of the current major limitations of rAAV and lentivirus liver-directed gene therapy. By permanently modifying a small starting population of hepatocytes and allowing their expansion, only one viral administration at a much lower vector dose is required and problems with the low efficiency of initial transduction are eliminated. Lower vector doses will reduce immune responses and considerably reduce the cost of vector production. Although we focused on lentiviral vectors in the work reported here, APAP-mediated selection of hepatocytes can be applied to any integrating vector, including transposons or rAAV vectors targeted at safe-harbor loci (5, 7). The only requirement is that the therapeutic transgene and the Cypor knockdown cassette are linked in cis.
Selection of gene-targeted hepatocytes was found to be successful in animals treated as neonates, allowing the treatment of inherited genetic disorders from birth. In contrast, rAAV remains largely episomal and is quickly lost during rapid cell division as shown in developing mouse and nonhuman primate livers (4, 14). Adult animals that undergo partial hepatectomy (20) also lose more than 90% of transgene expression with just one to two cell divisions. Hence, standard rAAV is inadequate for long-term treatment of genetic liver diseases in pediatric patients. Our method enables the selection of permanently integrated therapeutic transgenes that will persist for the life of the cell. Normal liver homeostasis and injury responses are largely driven by mature hepatocytes (21), and therefore, the effect of gene therapy is expected to be lifelong.
Severe APAP overdoses can cause liver failure and death (8), and this type of acute liver injury is therefore a concern for the clinical applicability of APAP selection in patients. However, we show here that selection was also successful using an only moderately hepatotoxic APAP diet and not associated with morbidity or mortality. A clinical protocol would likely involve a gradual dose escalation with careful monitoring of the degree of liver injury and rapid intervention with the APAP antidote N-acetylcysteine (9) if transaminase concentrations exceeded a defined threshold.
An additional concern associated with our protocol is the partial deficiency of hepatic Cypor itself. Several considerations indicate that this is not likely to cause clinical problems. First, human patients with germline mutations in CYPOR have the developmental disorder Antley-Bixler syndrome (22, 23); liver disease has not been reported as part of this condition. Second, liver-specific Cypor knockout mice show normal development and reproduction, although they show impaired drug metabolism and hepatic lipid accumulation (24, 25). Third, selection of Cypor-deficient hepatocytes with APAP is strictly limited to zone 3 hepatocytes because the Cyp enzymes required for APAP metabolism are expressed only in this zone. Therefore, zone 1 and 2 hepatocytes retain normal Cypor activity. Last, we demonstrated here that partial selection of only a fraction of zone 3 hepatocytes is possible (Fig. 5B). Human F9 concentrations in the fully therapeutic range were achieved while retaining intact Cyp metabolism in >50% of zone 3 hepatocytes.
Long-term stability is another important, positive feature of this system. Cypor-deficient hepatocytes persisted for the entire duration of our long-term follow-up, up to 42 weeks, without requiring selective pressure. This indicates that Cypor deficiency does not cause hepatocyte injury or cell loss. In addition, liver function tests and blood lipid tests were normal in animals that had undergone APAP selection and allowed to recover. Although additional investigations into the effects of partial Cypor deficiency in larger animals are necessary, together these findings are promising for the safe utilization of this selection strategy in human gene therapy.
In addition to demonstrating proof of principle for the secreted protein F9, we were also successful in treating PKU in male mice. The serum concentration of toxic Phe was reduced from ~2000 µM to as low as 204 µM, below the therapeutic target of 360 µM. Although the lowest Phe concentrations in two of four male mice treated with APAP selection were ~500 µM, this is within the range considered acceptable for patients with PKU treated with dietary therapy. Despite evidence of extensive selection, we were not able to achieve a therapeutic correction in female PKU mice. This sexual dimorphism in treating PKU has previously been described (26). Although the threshold for therapeutic correction in male mice was higher than expected at around 40 to 50% of hepatocytes, this is likely due to the dominant-negative effect of mutant Pah subunits in the Pahenu2/enu2 mouse model (27, 28). Pah is a homotetrameric enzyme, and thus, the wild-type protein generated by gene therapy needs to “crowd out” the endogenous mutant Pah protein, if present, to generate sufficient amounts of nonmutant homotetramers to provide enzyme activity. Much higher doses of rAAV than are typical for liver-directed gene transfer were needed to achieve correction of hyperphenylalaninemia in the Pahenu2/enu2 model, particularly in female mice (26). Hence, our results are those expected for this model. Fortunately, only a minority of human patients with PKU bear mutations that are thought to act by a dominant-negative mechanism. Hence, a standard gene addition therapy is expected to work well for most patients. In a recently reported PKU null mouse (28) obtained by a CRISPR-induced deletion of exon 1, substantially lower doses of rAAV corrected blood Phe concentrations when compared to the Pahenu2/enu2 model.
Here, two distinct approaches to rendering hepatocytes Cypor deficient were used. The CRISPR-Cas9 gene knockout approach has the advantage of creating a complete enzyme deficiency and entirely preventing APAP metabolism by Cyp enzymes. However, this method requires coadministration of Cas9, i.e., use of a second agent, be it a Cas9 rAAV or nanoparticle. This need for two drugs complicates regulatory approval. In addition, Cas9 is immunogenic and can cause off-target genome cutting (29). shRNA-mediated knockdown of Cypor avoids these issues. However, it is inherently less efficient in reducing enzyme activity because gene expression is only knocked down, not knocked out. Here, we tested two separate candidate shRNAs, and only one reduced Cypor sufficiently to allow APAP selection. A very strong RNA polymerase III promoter (U6) was used to express Cypor shRNA in our study. In prior work, we were able to achieve in vivo hepatocyte selection after integrating a promoterless Generide rAAV vector into the albumin locus (7). Drug resistance was achieved by expressing a microRNA-embedded shRNA from this strong hepatocyte promoter. It remains to be determined whether the level of Cypor shRNA expression that can be achieved by cellular promoters targeted with rAAV Generide vectors (7) is high enough to permit APAP selection.
Together, the preclinical murine data presented here are sufficiently promising to warrant an examination of our approach in nonhuman primates. Liver-directed lentiviral gene therapy has yet to be applied in human patients, even in severe disorders that require liver transplantation. One of the main reasons is the difficulty in producing clinical-scale vector doses at reasonable cost. The in vivo selection methodology described here could potentially overcome this barrier and allow gene therapy to replace orthotopic liver transplantation in liver disorders not amenable to standard rAAV therapy. Examples of such conditions include disorders that require intervention in early childhood such as Crigler-Najjar syndrome, urea cycle disorders, genetic cholestasis diseases, and severe organic acidemias. Liver gene therapy with lentiviral vectors could also enable the treatment of hemophilia in pediatric patients (15) without concerns about transgene loss by normal liver growth. Risk/benefit considerations will be important in deciding whether transient APAP toxicity and potentially permanent changes in Cyp-mediated drug metabolism are justifiable vis-à-vis the surgical risks and lifelong immune suppression inherent to orthotopic transplantation.
MATERIALS AND METHODS
Study design
This study involved comparison between cohorts of mice treated with gene therapy vectors followed by APAP selection for genomic vector integration or no further treatment. Differences between groups and success of selection were assessed quantitatively by indel analysis, blood hF9 concentration, or blood Phe concentration. Laboratory personnel performing assays [hF9 enzyme-linked immunosorbent assay (ELISA), Phe assay, and PAH activity assays] were blinded to the treatment groups. All experiments had an n of at least 3 animals per group.
Animal husbandry
Wild-type C57BL/6, 129S4, and B6J-Rosa-Cag-Cas9 (stock no. 026179) mice were obtained from the Jackson laboratory. Pahenu2/enu2 mice on a C57BL/6 background were obtained from the Harding Lab (30). All animal experiments were performed according to the guidelines for animal care at Oregon Health & Science University (OHSU). All animals were fed tap water and standard mouse chow (LabDiet PicoLab Rodent Diet 5LOD) unless otherwise stated. Animals were housed under a standard 12-hour on-and-off light cycle. APAP diet was created by dissolving APAP in 20 ml of 100% ethanol and adding 180 g of standard 5LOD diet preheated to 55°C. Diet was mixed until pellets were saturated with ethanol. Diet was then allowed to dry completely before being fed to mice. APAP diets [1.5, 1.6, 1.75, or 1.9% (w/w)] were created using this method. All mice were started on 1% (w/w) APAP diet and ramped up to a higher percentage diet. Mice were given 2 to 4 days on regular diet if weight loss exceeded 20%. Mice were euthanized with CO2. A cardiac puncture was performed immediately after death to deplete the liver of blood.
Plasmid construction and viral production
pX330-U6-Chimeric_BB-CBh-hSpCas9 was a gift from F. Zhang (Addgene plasmid no. 42230; http://n2t.net/addgene:42230; RRID: Addgene_42230). gRNAs were cloned into pX330 backbone according to the provided protocol (31). gRNAs were designed using http://crispor.tefor.net/ (32). The Cypor-targeting gRNA sequence was TCGTGGGGGTCCTGA CCTAC. ShRNA was based on designs by Sigma-Aldrich. The Cypor-targeting shRNA sequence was CCTGACCTACTGGTTCATCTTctcgagAAGATGAACCAGTAGGTCAGG (lower case indicates loop sequence). The transposon plasmid p/T-FAHIG (33) was used for proof-of-concept testing of candidate shRNAs. The shRNA driven by a U6 promoter was cloned using the InFusion Cloning Kit (Takara Bio) and standard restriction cloning. A validated shRNA against the mouse Hpd gene, which is not involved in APAP metabolism, was used as a nonselectable control. The transposon plasmids were codelivered with SB100X transposase plasmid.
The lentivirus backbone was received as a gift from L. Naldini (15). cDNA for codon-optimized human F9 and human PAH were added by using the InFusion Cloning Kit (Takara Bio). Lentivirus was produced by the OHSU viral production core. Lentiviral titers were determined by quantitative reverse transcription polymerase chain reaction (RT-PCR).
Delivery of plasmids and viral vectors
For neonatal delivery, wild-type, transgenic Cas9, or Pahenu2/enu2 mice P1 neonates were injected via the facial vein with lentivirus (1.4 × 106 to 1.9 × 107 TU per pup). For delivery in 6- to 8-week-old adults, hydrodynamic tail vein injections were performed to deliver plasmid in saline consisting of 10% (w/v) of the mouse’s body weight.
APAP preparation and delivery
APAP (13 mg/ml) (4-acetamidophenol, 98%; ACROS Organics) was dissolved in saline prewarmed to 50°C, sterilized with a 0.2 µM filter, aliquoted, and stored at room temperature for up to 1 month or until precipitate was observed. Mice were fasted overnight for about 16 hours before APAP administration. Mice were weighed and APAP was administered by intraperitoneal injection twice weekly. The dose of APAP started at 220 mg/kg in males and 250 mg/kg in females, and blood was drawn for ALT measurements 6 to 7 hours after APAP treatment. Subsequent APAP doses were increased by 5 to 10 mg/kg until at least two consecutive elevated ALT measurements (>800 IU/liter) were observed.
For most experiments, APAP doses were then kept constant. Pahenu2/enu2 mice that received LV-PAH-Cypor shRNA lentivirus showed ALT stabilization before the therapeutic threshold was reached and subsequently required continuous dose escalation. After fasting, if greater than 15% body weight was lost, APAP treatment was skipped for one dose.
ALT and blood chemistry
To measure blood ALT, 10 µl of blood was collected from the saphenous vein into 15 µl of heparin-containing saline. Samples were briefly centrifuged, and the supernatant was collected and stored at −20°C for up to 1 week. ALT activity was measured in quadruplicate with the ALT (SGPT) Color Endpoint kit (Teco Diagnostics) with modifications to the manufacturer’s protocol. Plasma was obtained by terminal cardiac puncture and submitted to IDEXX Laboratories for a comprehensive blood chemistry panel.
Immunohistochemistry
Liver tissues were sliced into about 5-mm sections and fixed in 4% paraformaldehyde (Sigma-Aldrich) at room temperature for 4 hours or at 4°C overnight. Liver slices were then passed through a sucrose gradient consisting of 10, 20, and 30% sucrose (w/v) in phosphate-buffered saline (PBS). Tissue slices were embedded in OCT, and 7 µM sections were cut using a cryostat onto Colorfrost Plus slides (Thermo Fisher Scientific). The following primary antibodies were used: rabbit anti-Cypor (Abcam, no. 180597), goat anti-human F9 (Affinity Biologicals, GAFIX-AP), rabbit anti-Cyp2E1 (Abcam, no. 28146), and goat anti-Cypor (Abcam, no. 166800, for Cypor/Cyp2E1 dual stain). Secondary antibodies were Alexa Fluor 555– or Alexa Fluor 647–conjugated. For visualization of GFP expression, slides were incubated with TrueBlack Lipofuscin Autofluorescence Quencher (Biotium) for 1 min before staining. Sections were permeabilized in 0.1% Triton X-100 in PBS at room temperature for 12 min, washed in 3× 5-min PBS, and then blocked in 10% normal goat or donkey serum with 0.3 M glycine for 30 min at room temperature. Slides were incubated with primary antibody for 1 hour at room temperature or overnight at 4°C. Slides were washed in 3× 5-min PBS, incubated in secondary antibody for 1 hour at room temperature, and then washed 3× 5 min. Coverslips were mounted with DAPI Fluoromount-G (SouthernBiotech). Imaging was performed on a Zeiss LSM 700, LSM 780, or LSM 900 confocal microscope.
hF9 ELISA
Blood samples for hF9 measurements were collected from the saphenous vein. hF9 concentrations were measured by using the Asserachrom IX:Ag ELISA kit (Stago) following the manufacturer’s protocol with modifications as previously described (7).
Pah enzymatic activity and serum Phe
Liver Pah enzyme activity and serum Phe were determined as previously described (30).
TIDE analysis of CRISPR/Cas9-induced in-dels
Genomic DNA extraction from homogenized liver tissue was performed using the MasterPure Complete DNA and RNA Purification Kit (Lucigen) following the manufacturer’s protocol. A 700–base pair region surrounding the PAM site of the Cypor gRNA was amplified (forward primer 5′-GTTTGCGGGTGTTAGCTCTTC-3′; reverse primer 5′-AGTCTACTTCAGTCGCAGCC-3′) using MyTaq Red Mix (Bioline). The amplicon was purified using the PCR cleanup and gel extraction kit (Macherey-Nagel) and Sanger-sequenced using the forward primer. In-dels were analyzed using the TIDE software (https://tide.nki.nl) (34). Percent Cypor-deficient hepatocytes was estimated assuming that hepatocytes account for 60% of total liver DNA (raw percent in-dels ÷ 0.6 = percent Cypor-deficient hepatocytes).
Image analysis and graphical illustrations
Image analysis (table S1) was conducted manually using ImageJ. Graphical illustrations in Fig. 1 were created with www.biorender.com.
Statistical analysis
All statistical analysis was performed using GraphPad Prism version 6.00 for Mac (GraphPad Software, La Jolla, California, www.graphpad.com). Experimental differences were evaluated by a Mann-Whitney U test. Because serum Phe levels and Pah activity in Pahenu2/enu2 mice are known to follow a normal distribution, a Student’s two-tailed t test assuming equal variance was used to analyze experimental difference in these mice. P values <0.05 were considered statistically significant. For all statistical analysis, *P < 0.05, **P < 0.01, and ***P < 0.001. All error bars indicate SD.
Supplementary Material
Acknowledgments:
We thank L. Wakefield and S. Dudley for assisting with animal husbandry. We thank C. Singleton of the OHSU viral production core for lentivirus production and titering, and B. Jenkins of the OHSU Advanced Light Microscopy Core for microscopy assistance. We also thank A. Major at Baylor University for assistance with tissue histology. We thank N. Chau for providing comments on the manuscript.
Funding:
This work was sponsored by a research agreement from LogicBio Therapeutics. A.V. was supported by an NIH Ruth L Kirschstein T32 Program in Enhanced Research Training (PERT) training grant (grant number T32 GM 71338–15).
Footnotes
Competing interests:
OHSU and M.G. have a significant financial interest in Yecuris Corporation, a company that may have a commercial interest in the results of this research and technology. M.G. is a paid consultant of Yecuris Corp., LogicBio Therapeutics, and Ambys Medicines. A patent application on the APAP selection technology described herein has been filed by OHSU (Title: Methods of Gene Therapy. Filing number: PCT/US19/29890. M.G. and A.T. are coinventors). These potential conflicts of interest have been reviewed and managed by OHSU.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary materials. Plasmids are available upon the completion of a standard academic material transfer agreement.
REFERENCES AND NOTES
- 1.Palaschak B, Herzog RW, Markusic DM, AAV-mediated gene delivery to the liver: Overview of current technologies and methods. Methods Mol. Biol 1950, 333–360 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Chirmule N, Propert KJ, Magosin SA, Qian Y, Qian R, Wilson JM, Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther 6, 1574–1583 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Shirley JL, de Jong YP, Terhorst C, Herzog RW, Immune responses to viral gene therapy vectors. Mol. Ther 28, 709–722 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cunningham SC, Dane AP, Spinoulas A, Alexander IE, Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol. Ther 16, 1081–1088 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Barzel A, Paulk NK, Shi Y, Huang Y, Chu K, Zhang F, Valdmanis PN, Spector LP, Porteus MH, Gaensler KM, Kay MA, Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature 517, 360–364 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milone MC, O’Doherty U, Clinical use of lentiviral vectors. Leukemia 32, 1529–1541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nygaard S, Barzel A, Haft A, Major A, Finegold M, Kay MA, Grompe M, A universal system to select gene-modified hepatocytes in vivo. Sci. Transl. Med 8, 342ra79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee WM, Acetaminophen (APAP) hepatotoxicity—Isn’t it time for APAP to go away? J. Hepatol 67, 1324–1331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon E, Babar A, Choudhary M, Kutner M, Pyrsopoulos N, Acetaminophen-induced hepatotoxicity: A comprehensive update. J. Clin. Transl. Hepatol 4, 131–142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyanagi T, Xia C, Kim J-JP, NADPH-cytochrome P450 oxidoreductase: Prototypic member of the diflavin reductase family. Arch. Biochem. Biophys 528, 72–89 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crespo A, Peydró A, Dasí F, Benet M, Calvete JJ, Revert F, Aliño SF, Hydrodynamic liver gene transfer mechanism involves transient sinusoidal blood stasis and massive hepatocyte endocytic vesicles. Gene Ther 12, 927–935 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Brinkman EK, Chen T, Amendola M, van Steensel B, Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D, Mou H, Li S, Li Y, Hough S, Tran K, Li J, Yin H, Anderson DG, Sontheimer EJ, Weng Z, Gao G, Xue W, Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum. Gene Ther 26, 432–442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Bell P, Lin J, Calcedo R, Tarantal AF, Wilson JM, AAV8-mediated hepatic gene transfer in infant rhesus monkeys (Macaca mulatta). Mol. Ther 19, 2012–2020 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cantore A, Ranzani M, Bartholomae CC, Volpin M, Valle PD, Sanvito F, Sergi LS, Gallina P, Benedicenti F, Bellinger D, Raymer R, Merricks E, Bellintani F, Martin S, Doglioni C, D’Angelo A, VandenDriessche T, Chuah MK, Schmidt M, Nichols T, Montini E, Naldini L, Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci. Transl. Med 7, 277ra28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harding CO, Gibson KM, Therapeutic liver repopulation for phenylketonuria. J. Inherit. Metab. Dis 33, 681–687 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Grisch-Chan HM, Schwank G, Harding CO, Thöny B, State-of-the-art 2019 on gene therapy for phenylketonuria. Hum. Gene Ther 30, 1274–1283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamman K, Clark H, Montini E, Al-Dhalimy M, Grompe M, Finegold M, Harding CO, Low therapeutic threshold for hepatocyte replacement in murine phenylketonuria. Mol. Ther 12, 337–344 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang B, Eisensmith RC, Li XHC, Finegold MJ, Shedlovsky A, Dove W, Woo SLC, Gene therapy for phenylketonuria: Phenotypic correction in a genetically deficient mouse model by adenovirus-mediated hepatic gene transfer. Gene Ther 1, 247–254 (1994). [PubMed] [Google Scholar]
- 20.Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA, Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol 75, 6969–6976 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopp JL, Grompe M, Sander M, Stem cells versus plasticity in liver and pancreas regeneration. Nat. Cell Biol 18, 238–245 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Bonamichi BDSF, Santiago SLM, Bertola DR, Kim CA, Alonso N, Mendonca BB, Bachega TASS, Gomes LG, Long-term follow-up of a female with congenital adrenal hyperplasia due to P450-oxidoreductase deficiency. Arch. Endocrinol. Metab 60, 500–504 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Unal E, Demiral M, Yildirim R, Taş FF, Ceylaner S, Özbek MN, Cytochrome P450 oxidoreductase deficiency caused by a novel mutation in the POR gene in two siblings: Case report and literature review. Hormones 20, 293–298 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Gu J, Weng Y, Zhang Q-Y, Cui H, Behr M, Wu L, Yang W, Zhang L, Ding X, Liver-specific deletion of the NADPH-cytochrome P450 reductase gene. Impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J. Biol. Chem 278, 25895–25901 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Henderson CJ, Pass GJ, Wolf CR, The hepatic cytochrome P450 reductase null mouse as a tool to identify a successful candidate entity. Toxicol. Lett 162, 111–117 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Mochizuki S, Mizukami H, Ogura T, Kure S, Ichinohe A, Kojima K, Matsubara Y, Kobayahi E, Okada T, Hoshika A, Ozawa K, Kume A, Long-term correction of hyperphenylalaninemia by AAV-mediated gene transfer leads to behavioral recovery in phenylketonuria mice. Gene Ther 11, 1081–1086 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Hamman KJ, Winn SR, Harding CO, Hepatocytes from wild-type or heterozygous donors are equally effective in achieving successful therapeutic liver repopulation in murine phenylketonuria (PKU). Mol. Genet. Metab 104, 235–240 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richards DY, Winn SR, Dudley S, Fedorov L, Rimann N, Thöny B, Harding CO, A novel Pah-exon1 deleted murine model of phenylalanine hydroxylase (PAH) deficiency. Mol. Genet. Metab 131, 306–315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD, High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol 31, 822–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richards DY, Winn SR, Dudley S, Nygaard S, Mighell TL, Grompe M, Harding CO, AAV-mediated CRISPR/Cas9 gene editing in murine phenylketonuria. Mol. Ther. Methods Clin. Dev 17, 234–245 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F, Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud J-B, Schneider-Maunoury S, Shkumatava, Teboul L, Kent J, Joly J-S, Concordet J-P, Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17, 148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wuestefeld T, Pesic M, Rudalska R, Dauch D, Longerich T, Kang T-W, Yevsa T, Heinzmann F, Hoenicke L, Hohmeyer A, Potapova A, Rittelmeier I, Jarek M, Geffers R, Scharfe M, Klawonn F, Schirmacher P, Malek NP, Ott M, Nordheim A, Vogel A, Manns MP, Zender L, A direct in vivo RNAi screen identifies MKK4 as a key regulator of liver regeneration. Cell 153, 389–401 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Brinkman EK, Kousholt AN, Harmsen T, Leemans C, Chen T, Jonkers J, van Steensel B, Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res 46, e58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.