

Abstract
Intrauterine growth restriction (IUGR) is a common human pregnancy complication. IUGR offspring carry significant postnatal risk for early-onset metabolic syndrome, which is associated with persistent reduction in IGF-1 protein expression. We have previously shown that preadolescent IUGR male mice have decreased hepatic IGF-1 mRNA and circulating IGF-1 protein at postnatal day 21, the age when growth hormone (GH) normally upregulates hepatic IGF-1 expression. Here we studied nucleosome occupancy and CpG methylation at a putative growth hormone-responsive element in intron 2 (in2GHRE) of the hepatic IGF-1 gene in normal, sham-operated, and IUGR mice. Nucleosome occupancy and CpG methylation were determined in embryonic stem cells (ESCs) and in liver at postnatal days 14, 21, and 42. For CpG methylation, additional time points out to 2 yr were analyzed. We confirmed the putative mouse in2GHRE was GH-responsive, and in normal mice, a single nucleosome was displaced from the hepatic in2GHRE by postnatal day 21, which exposed two STAT5b DNA binding sites. Nucleosome displacement correlated with developmentally programmed CpG demethylation. Finally, IUGR significantly altered the nucleosome-depleted region (NDR) at the in2GHRE of IGF-1 on postnatal day 21, with either complete absence of the NDR or with a shifted NDR exposing only one of two STAT5b DNA binding sites. An NDR shift was also seen in offspring of sham-operated mothers. We conclude that prenatal insult such as IUGR or anesthesia/surgery could perturb the proper formation of a well-positioned NDR at the mouse hepatic IGF-1 in2GHRE necessary for transitioning to an open chromatin state.
Keywords: nucleosome, DNA methylation, intrauterine growth restriction
epidemiologic studies have linked intrauterine growth restriction (IUGR) to early-onset adult diseases such as metabolic syndrome, which includes type II diabetes mellitus, hypertension, and obesity (1, 5). Although the exact causes of IUGR-induced adult-onset morbidities are still being elucidated, a reduced circulating IGF-1 protein level is an identified risk factor for metabolic syndrome development in adults (22–24, 34, 38). IGF-1 is crucial in prenatal and postnatal life because it governs multiple developmental and metabolic processes including carbohydrate, protein, and lipid homeostasis (29). Circulating IGF-1 protein increases in adolescence due to a growth hormone-mediated surge in hepatic IGF-1 gene expression (36). Growth hormone (GH)- and STAT5b-gene knockout studies show that GH exerts its effects on IGF-1 upregulation starting around 21 days of postnatal life in mice (40, 51). GH mediates its effect on hepatic IGF-1 by binding to the GH receptor and activating the tyrosine protein kinase JAK2, which leads to phosphorylation of STAT5b (20, 46). Phospho-STAT5b then binds to multiple sites on the IGF-1 gene to upregulate expression of IGF-1 mRNA (11, 12, 41, 45). In cell culture transfection reporter assays, the growth hormone-responsive element in intron 2 (in2GHRE) of the rat hepatic IGF-1 gene, acting as an enhancer, was shown to be the most active (11). DNase1 hypersensitivity analysis has additionally shown that chromatin around in2GHRE opens following acute GH treatment of hypophysectomized rats (7). An open chromatin state in activated enhancer elements is often associated with nucleosome displacement to allow an enhanceosome complex access to DNA (35, 50).
In our laboratory's mouse model of IUGR, we have recently demonstrated that IUGR mice remain growth-restricted at postnatal day 21. This is accompanied by a persistent reduction in hepatic IGF-1 mRNA, circulating IGF-1 protein, and a perturbed IGF-1 chromatin structure across the entire gene in IUGR males (16). In this model, IUGR mice are produced from an exogenous infusion of a thromboxane A2 (TXA2) analog (U-46619) in pregnant mouse dams in the last week of gestation to mimic pregnancy-induced hypertension, which is the most common cause of human IUGR in developed countries (32). Pregnant dams with U-46619 infusion develop maternal hypertension with mean blood pressures that are 20 mmHg higher than sham-operated dams. This model produces symmetrically growth-restricted pups that have 15% lower birth weight and brain weight. IUGR pups have similar levels of dehydro-TXB2 (a TXA2 metabolite), TNF-α (induced by TXA2), and corticosterone as sham pups at birth, indicating that IUGR likely originated from maternal hypertension and subsequent uteroplacental insufficiency rather than from direct exposure of the drug itself or stress. Our findings along with other animal model and human epidemiological studies affirm the notion that the prenatal environment influences the epigenome (19, 21, 49). In light of this premise, we sought to determine whether a prenatal insult such as IUGR could also disrupt the epigenetic maturation of a DNA regulatory element of growth and metabolism that is not used until adolescence, particularly in IUGR males who have a reduction in IGF-1 mRNA and protein levels at birth and at preadolescence compared with sham males. As a model to address this problem, we used the hepatic IGF-1 in2GHRE previously characterized in rats and conserved in mice and humans but not fully characterized in the latter two species. We initially evaluated the putative mouse in2GHRE GH responsiveness in vitro. We then determined if in2GHRE underwent chromatin remodeling involving changes in nucleosome occupancy and CpG methylation at the in2GHRE in vivo. We lastly evaluated the effects of a prenatal insult of IUGR on the chromatin remodeling state of the mouse in2GHRE at postnatal day 21 when IGF-1 gene expression is known to be blunted and growth remained restricted (16).
MATERIALS AND METHODS
Animals.
All procedures were approved by the University of Utah Animal Care Committee and carried out in accordance with NIH Guide for the Care and Use of Laboratory Animals. C57Bl/6J mice (stock number 664) and C.129-Stat5btm1Hwd/J (stock number 7819) were purchased from The Jackson Laboratory (Bar Harbor, ME). Unmanipulated C57Bl/6J mice were used to characterize the normal epigenetic maturation of in2GHRE. Sham and IUGR C57Bl/6J offspring were produced according to Fung et al. (15). Briefly, timed matings were set up. At gestation day 12.5 (term gestation ∼20 days), pregnant females were anesthetized with ketamine (40 μg/g) and xylazine (8 μg/g), and a 1 cm incision was made at the right hip. Micro-osmotic pumps containing either vehicle (0.5% ethanol = sham group) or 4,000 ng/μl TXA2 analog (U-46619 dissolved in 0.5% ethanol = IUGR group; Cayman Chemical, Ann Arbor, MI) were implanted and infused continuously over the last week of gestation at 0.5 μl/h. Mice were given ad libitum access to food and water for the remainder of the pregnancy. Pups delivered spontaneously at term. Unmanipulated C57Bl/6J dams, set up at the same time as experimental dams, cross-fostered all sham and IUGR pups from birth to weaning (day 21) to minimize any potential lactation and care differences after surgery. IUGR pups defined as <1.266 g, which is <10th percentile of sham birth weights, were cross-fostered; ∼1/3 of the IUGR pups had birth weights < 1.266 g. Sham pups weighing >1.266 g were cross-fostered. Sham and IUGR livers were harvested at day 21 for comparison with livers from normal unmanipulated day 21 C57Bl/6J mice.
Construction of CpG-free reporter vector.
The pCpGL-CMV/EF1 vector, a gift from Dr. Michael Rehli, was modified to delete the CMV enhancer (28). This modified vector, designated as clone 256, was completely void of CpG sites and contained a CpG-free luciferase gene driven by the human elongation factor 1α (hu EF1a) core promoter.
Cloning, mutagenesis, and methylation of the mouse in2GHRE.
A 240 bp genomic fragment was cloned from C57Bl/6J mice by PCR and inserted into the Pst1 site of clone 256. The primer sequences used to amplify the in2GHRE were forward (F): atactgcagAGGGTGGCTCACCTCATACTC and reverse (R): atactgcagGTAGCCAAATGGCACCCCTG. Bases in lower case indicate Pst1 linker sites used to insert into clone 256. In2GHRE was inserted as a single copy in sense and antisense orientations into the Pst1 site of clone 256. Both orientations were tested for enhancer function. In2GHRE in the antisense orientation, designated clone 258, produced slightly higher activity in GH response and was chosen for subsequent experiments. The two STAT5b sites within in2GHRE of clone 258 were mutated using Finnzymes Phusion Site-Directed Mutagenesis Kit (catalog #F541; Thermo Fisher Scientific, Waltham, MA). The first STAT5b site was mutated from CTTTCCTGGAA to acgCCTGGAA. The second site was mutated from CTTCTTAGAA to CTTCTTAtgc. To evaluate the effect of CpG methylation on in2GHRE activity, all constructs were methylated in vitro using MSss1 methyltransferase (NEB, Ipswich, MA) according to the manufacturer's protocol.
Internal transfection control vector for calculating reporter gene activity.
The human EF1 alpha minimal promoter from pCpFL-CMV/EF1 was cloned into the commercial vector pGL4.70 (Promega, Madison, WI), which carries the Renilla reporter gene. This construct, designated as clone 294, was then used as an internal transfection control to calculate relative light units (RLU). The light units from each in2GHRE-luciferase construct was divided by the light units from the cotransfected Renilla-based construct, clone 294.
Establishing GH-responsive cells: cell culture, transfection, and luciferase assay conditions.
in2GHRE was evaluated for GH responsiveness in CWSV1 rat liver cells, a gift from Dr. Harriet Isom (48). CWSV1 cells were initially screened by PCR to confirm they were free of mycoplasma contamination. The primers used were F-GGTGAATACGTTCTCGGGTCTTGTACACAC and R-TNCTTTTCACCTTTCCCTCACGGTAC for the 16S-23S internal transcribed spacer region and primers F-ATGGGTGCVAACATGCAACGTCAAGC and R-GCTCAHACTTCCATTTCHCCAAA and R-CGTTTTGWGCTTTACCACCCATTGGTTGTTG for the rpoB gene (42). All PCR screening results for mycoplasma were negative. in2GHRE constructs were cotransfected with a mouse GH receptor cDNA expressed in pcDNA3 (a gift from Peter Rotwein) and mouse STAT5b cDNA expressed in pcDNA (a gift from Dr. Lothar Hennighausen). Cells were transfected with Fugene HD transfection reagent (Promega) according to the manufacturer. Cell culture growth medium was Hyclone DMEM/F12 + glutamine + HEPES supplemented with 1× SITE+3 (Sigma-Aldrich, St. Louis, MO), 10 nM glucagon (Sigma), and 1 μM dexamethasone. Cells were seeded at 5 × 105 cells/well in six-well tissue culture plates. After 24 h, recombinant GH was added at a final concentration of 20 μg/ml, and cells were harvested after another 24 h. Cell lysates were prepared and assayed in triplicate for luciferase activity following the Promega Dual Luciferase protocol. Each in2GHRE construct was evaluated in three separate transfections.
Nucleosome position determination by nucleosome occupancy and methylome sequencing assay (= GpC methyltransferase foot-printing).
Nuclei were isolated from fresh tissue to avoid damage caused by snap freezing (31). Fresh livers were excised, weighed, and mashed through a 40 μm pore size Falcon cell strainer with a plunger from a 3 ml disposal syringe into a plastic petri dish filled with 4 ml of ice-cold buffer A (500 mM sucrose, 5 mM MgCl2, Tris pH 7.4) per 0.5 g of liver. The suspension was centrifuged at 600 g for 10 min at 4°C. The supernatant was removed, and the pellet resuspended using a 10 ml pipet in 14 ml of ice-cold buffer A and centrifuged again at 600 g at 4°C. The supernatant was removed, and the pellet resuspended using a 5 ml pipet in 6 ml of buffer B (2 M sucrose, 1 mM MgCl2, 10 mM Tris pH 7.4). We transferred 1 ml aliquots to 1.5 ml microfuge tubes and centrifuged them at 16,000 g for 30 min at 4°C. The supernatant was discarded. Tubes were kept in an inverted position and swabbed out with cotton-tipped applicators to get rid of excess fluid and debris. Nuclei were then collected along the side of each tube. Nuclei were washed by resuspending in 50 μl GpC buffer plus sucrose (50 mM NaCl, 10 mM DTT, 50 mM Tris pH 8.5, 1 M sucrose) and centrifuged at 16,000 g for 10 min. Washed nuclei were resuspended in GpC buffer plus sucrose and counted by diluting in GpC buffer plus sucrose and mixing 1:1 with trypan blue solution (0.9% trypan blue in PBS). Nuclei concentration was adjusted to have between 5 × 105 and 1 × 106 nuclei in 74.5 μl GpC buffer in 1 M sucrose.
For GpC methyltransferase reactions, 49.5 μl GpC buffer without sucrose, 0.75 μl of 32 mM S-adenosyl methionine, and 25 μl CviP1 methyltransferase (M.CviP1, 4 units/μl) were added to 74.5 μl of nuclei suspension and incubated at 37°C for 15 min. Note the final sucrose concentration was 0.5 M. Sucrose concentrations > 0.5 M were inhibitory to M.CviP1. The reaction was stopped by addition of 150 μl of 2× stop buffer (400 μg/ml proteinase K, 600 mM NaCl, 1% SDS, 10 mM EDTA, 20 mM Tris pH 7.4) and incubated overnight at 55°C. DNA was isolated with Zymo ZR Genomic DNA Tissue Miniprep Kit (catalog #D3050; Zymo Research, Irvine, CA) and converted for bisulfite sequencing with a Zymo EZ DNA Methylation-Gold Kit (catalog #D5005, Zymo Research). From the isolated bisulfite converted DNA, a 380 bp region containing in2GHRE and 23 GpC sites was amplified by PCR. The primer set for the antisense strand could detect 21 of the 23 sites (forward primer #4723 F: CCAATATATCTCCRTTACCCCTTTTATA and reverse primer #4759 R: TTTTTTAGTTTATTATTAGATTGAGAGATG, Fig. 1). Two of the 23 sites (sites 3 and 5) were within the sequence GpCpG; therefore, we could not determine whether cytosine methylation was due to exogenously added GpC methyltransferase or endogenous CpG methylation. Amplicons were cloned into pSC-A-amp/kan (Stratagene, La Jolla, CA), individual colonies picked, and Sanger sequenced. A minimum of three animals was tested per group.
Fig. 1.

DNA sequence of the putative mouse IGF-1 in2GHRE. The tandem STAT5b sites are marked in rectangles, the 23 GpC sites marked with squares, and 4 CpG sites by an “*”. The location where each primer annealed is indicated by a line above the sequence for forward (for) primers and below the sequence for reverse (rev) primers.
DNA methylation analysis of CpG sites around tandem STAT5b sites within in2GHRE by sodium bisulfite sequencing.
In the mouse, the STAT5b binding sites of interest are flanked by CpG sites. The first STAT5b site has a CpG 17 bp upstream and 10 bp downstream, while the second site has a CpG 22 bp upstream and 8 bp downstream. Genomic DNA, extracted from livers, was converted for bisulfite sequencing with EZ DNA Methylation-Gold Kit (catalog #D5005, Zymo Research). Bisulfite-treated DNA was amplified by PCR with primers #4723 F and #4759 R. Amplicons were cloned into pSC-A-amp/kan, individual colonies picked, and Sanger sequenced.
Statistics.
Cell culture transfection data was analyzed for statistically significance by the t-test. DNA methylation analysis was analyzed by Fisher's exact test. Statistical significance was declared at P < 0.05.
RESULTS
Mouse IGF-1 in2GHRE is GH responsive and acts through two STAT5b binding sites.
Figure 1 shows the DNA sequence and the locations of CpG, GpC, and primer annealing sites for the mouse IGF-1 in2GHRE. To confirm that the 240 bp fragment isolated from in2GHRE was GH responsive in vitro, in2GHRE was inserted in place of the CMV enhancer within the reporter plasmid pCpGL-CMV/EF1 (Fig. 2A, clone 258). The reporter plasmid, with and without in2GHRE, was cotransfected with expression vectors for the mouse GH receptor and mouse STAT5b protein into CWSV1 rat liver cells. In the absence of in2GHRE, with or without GH, no luciferase activity was detected (Fig. 2B, clone 256). When in2GHRE was added to the luciferase reporter gene (Fig. 2B, clone 258), luciferase expression was equivalent to the internal transfection control in the presence of GH. In the absence of GH, luciferase expression from clone 258 declined to 55% of the internal control, indicating in2GHRE had some GH-independent enhancer activity. A similar finding was also reported for the rat in2GHRE (46). In2GHRE contains two STAT5b binding sites. In cell culture transfection assays each site in the rat has been shown to contribute to a GH response and to act synergistically with the other (46). To determine if the mouse STAT5b sites participated in the GH response, we mutated both sites (clone 268) and found all activity was lost with or without GH (Fig. 2B, clone 268). Taken together, our results demonstrate that the putative in2GHRE of the mouse IGF-1 gene was GH responsive and that GH acted through the two STAT5b binding sites.
Fig. 2.

Mouse IGF-1 growth hormone-responsive element in intron 2 (in2GHRE) reporter constructs and their relative luciferase activities. A: a single copy of a 240 bp mouse IGF-1 in2GHRE fragment, either with wild-type sequence (WT GHRE clone 258) or with STAT5b mutated sequence (mut GHRE clone 268), was cloned upstream of human (hu) EF1a minimal promoter driving a CpG-free luciferase reporter gene (clone 256). Wild-type tandem STAT5b sites are shown as white and black rectangles and mutated sites as Xs. B: relative light units (RLU) compared with clone 294 in the presence or absence of growth hormone (GH) for the hu EF1a minimal promoter driving a CpG-free luciferase reporter (256), for the reporter construct plus the wild-type GHRE sequence (258), for the reporter construct with a mutated GHRE (268), or for the reporter construct with wild-type GHRE methylated at all 4 CpG sites (258meth).
Nucleosome occupancy across the in2GHRE in control mice is reduced by one nucleosome at 6 wk of age.
To examine nucleosome occupancy at in2GHRE, we used the nucleosome occupancy and methylome sequencing (NOMe-seq) assay. This method involves treating isolated nuclei with a DNA methyltransferase followed by sodium bisulfite sequencing. The resulting pattern of methylated and nonmethylated DNA sites reflects regions of nucleosome-depleted and nucleosome-occupied DNA, respectively. NOMe-seq has been used to interrogate nucleosome occupancy at the single molecule level within promoters and enhancers (3, 27, 47) and is similar to DNase-seq and MNase-seq in that all three methods are based on an enzyme having access to DNA that is not blocked by proteins. With the exception of caspase-activated DNase, all three methods have the potential for limited and confounding access to core histone proteins; however, each method has been shown to have greatest access to DNA void of histone proteins (2, 26). For NOME-seq we used the GpC methyltransferase M.CviP1 to methylate accessible GpC sites in the in2GHRE within DNA accessible chromatin. M.CviP1 has been shown to have restricted access to nucleosome occupied DNA and free access to nucleosome-depleted regions (NDR) (26).
We first examined purified liver nuclei from 6 wk old male C57Bl/6 mice. This time point was chosen because disruption of the GH/IGF-1 axis at 6 wk in mutant mice results in significant growth difference compared with wild-type mice (13, 40, 51), indicating that GH-mediated regulation of IGF-1 is active at this age. After M.CviP1 treatment, a 380 bp region containing the in2GHRE was analyzed by sodium bisulfite sequencing. At 6 wk, GpC methyltransferase access was restricted to an NDR between GpC sites 4 and 16, which covers a region of 151 bp (Fig. 3A and data not shown). The average %GpC methylation at each GpC site between sites 4 and 6–16 was significantly higher at 64 ± 14 compared with the protected sites 1, 2, and 17–23, which had an average %GpC of 10 ± 4 (P < 0.05). GpC sites 3 and 5 could not be scored because they were within a GpCpG site, and we, therefore, could not determine whether the cytosine was methylated by the GpC methyltransferase or by endogenous CpG methylation. Our NOME-seq data showing an NDR at the in2GHRE at 6 wk is consistent with reported DNase hypersensitivity at in2GHRE in 7 wk old hypophysectomized rats as well as with the growth rate difference at 6 wk of age in GH knockout and STAT5b knockout mice (either of which disrupts the GH/IGF-1 axis) compared with wild type (7, 40, 51). Since we could not evaluate GpC site 3 we had to consider the distance between sites 2 and 17, a distance of 202 bp, as the maximum possible size of the NDR at 6 wk of age. Since the amount of DNA within a nucleosome core particle is conserved at 146–147 bp (10), only one nucleosome could have been displaced from an NDR of 202 bp. The two STAT5b sites within the NDR were positioned much closer to the 5′-flanking nucleosome than to the 3′-flanking nucleosome. Within the limits of resolution based on GpC site placement, the 5′-STAT5b site was at most 39 bp away from the 5′-flanking nucleosome, while the 3′-STAT5b site was at least 76 bp upstream of the 3′-flanking nucleosome.
Fig. 3.

Nucleosome position analysis by nucleosome occupancy and methylome sequencing (NOMe-seq) across the IGF-1 in2GHRE from nuclei and purified genomic DNA of 6 wk old mouse livers. The %GpC methylation results following M.CviP1 treatment and indicated presence or absence of nucleosomes on in2GHRE for nuclei of 6 wk old mice (n = 4) showing a nucleosome-depleted region of at least 151 bp (A) compared with purified liver genomic DNA of 6 wk old mice (n = 3) (B) showing all sites (380 bp) are accessible. Each GpC site is represented by a ball and stick. Results for GpC sites 3 and 5 were in the context of GCG and therefore excluded due to interpretation inability. Relative locations of the 2 STAT5b binding sites are represented by white and black rectangles.
To confirm that our GpC methylation results accurately reflected nucleosome-depleted and nucleosome-occupied regions, we analyzed in2GHRE in the context of purified genomic DNA which had no nucleosome protection. We also analyzed nuclei isolated from embryonic stem cells (ESCs) because IGF-1 expression is not dependent on GH prenatally, and, therefore, we would expect in2GHRE to be nucleosome occupied (18). When purified genomic DNA isolated from 6 wk old mouse liver was treated with M.CviP1, all GpC sites were accessible (Fig. 3B). The average %GpC methylation at each GpC site across the 21 sites was 83 ± 12. By comparison, for MCviP1-treated nuclei from three preparations of ESCs, the average %GpC methylation at each GpC site across the 21 sites was only 3 ± 5. Near complete access to all GpC sites within the in2GHRE on purified genomic DNA and minimal access at the in2GHRE in ESCs were consistent with our prediction. These results support the earlier findings that M.CviP1 methyltransferase mapping can differentiate between GpC sites within nucleosome-depleted and nucleosome-occupied DNA in vivo.
Mouse hepatic in2GHRE transitions to an open state by postnatal day 21.
Compared with wild-type mice, GH and STAT5b knockout mice begin to show decreased growth as early as postnatal day 21 (40, 51). To determine if GH-mediated upregulation of IGF-1 could be inhibited by disrupting the activation of IGF-1 GHREs, we next established when in2GHRE transitioned from a nucleosome occupied state to a nucleosome-depleted state. At postnatal day 14, M.CviP1 access to in2GHRE was still blocked by a nucleosome. A region defined by GpC sites 8–16 showed an average %GpC methylation of 29 ± 9 (Fig. 4A). The average %GpC methylation for the 5′-flanking sites 1–7 and 3′-flanking sites 17–23 was 9 ± 2 and 15 ± 12, respectively. The difference in %GpC methylation between flanking and internal sequences was not statistically significant. This GpC methylation pattern is consistent with the fact that growth rates are similar between GH knockout and STA5b knockout mice compared with wild-type mice at day 14 (40, 51). By postnatal day 21, a single NDR similar to that seen at 6 wk was detected (compare Figs. 3A and 4B). The average %GpC methylation at each GpC site in the NDR, which was positioned between GpC sites 4 and 17, a distance of 176 bp, was 54 ± 11, which was significantly higher than the %GpC methylation at each GpC site in the flanking regions defined by sites 1, 2, and 18–23, which was 17 ± 5 (P < 0.05). From our data, we conclude that the single nucleosome that covers the mouse hepatic in2GHRE is displaced by postnatal day 21.
Fig. 4.

Nucleosome position analysis by NoMe-seq across the IGF-1 in2GHRE from nuclei extracted from day (d)14 (A) and d21 mouse livers (B). At d21, a 176 bp region is fully accessible correlating with a single displaced nucleosome. Each GpC site is represented by a ball and stick. Results for GpC sites 3 and 5 were in the context of GCG and therefore excluded due to interpretation inability. Relative locations of the 2 STAT5b binding sites are represented by white and black rectangles.
CpG demethylation within the mouse hepatic in2GHRE is developmentally regulated and correlates with nucleosome displacement.
Repressive condensed chromatin is often associated with CpG hypermethylation, while active open chromatin is associated with CpG hypomethylation (25, 33). The tandem STAT5b-binding sites in mouse in2GHRE do not contain CpGs within the STAT5b consensus sequence but are flanked by CpG sites. The first STAT5b site has a CpG 17 bp upstream and 10 bp downstream of it, while the second site has a CpG 22 bp upstream and 8 bp downstream. Stat5b binding is inhibited by CpG methylation within flanking sequences (9, 39). To determine whether CpG methylation inhibited in2GHRE enhancer activity, all four CpG sites within the reporter construct clone 258 were methylated in vitro using M.Sss1 methyltransferase prior to transfection into CWSV1 cells (Fig. 2B). The four CpG sites flanking the two STAT5b sites were the only CpG sites within the entire construct. When the four CpG sites that flanked the two STAT5b binding sites in in2GHRE were methylated, enhancer activity was reduced by 90% and GH did not alter enhancer activity (Fig. 2B, clone 258meth). Our in vitro results suggest that if the nucleosome is displaced from in2GHRE but if the CpG sites remain methylated, the enhancer will not be fully functional.
We then postulated that the in vivo CpG methylation-demethylation status at in2GHRE might correlate with closed or open chromatin. If changes in endogenous CpG methylation preceded nucleosome displacement, we could use endogenous CpG methylation to map when particular developmentally regulated epigenetic modifications are assigned to the in2GHRE nucleosome for future displacement. In mouse ESCs where our NOMe-seq data indicated the in2GHRE was completely covered by nucleosomes, in2GHRE overall %CpG methylation across the four CpG sites was 89 ± 5 (Fig. 5). We then analyzed CpG methylation in C57Bl/6J males at various time points. By embryonic day 19.5 with term gestation at ∼20 days, the overall %CpG methylation declined to 64 ± 7. At postnatal day 14, when the in2GHRE was still covered by a nucleosome, overall %CpG methylation was 62 ± 17 and remained elevated until postnatal day 16, which had an overall %CpG methylation of 60 ± 12. The overall %CpG methylation then decreased sharply to 21 ± 8 at postnatal day 17. Four days later at postnatal day 21, the in2GHRE was nucleosome free (Fig. 4B). These data indicate that the transition to an NDR occurred somewhere within the 5-day period between postnatal days 16 and 21. At day 35, which is within the mouse adolescent period, overall %CpG methylation declined to 7 ± 5 (30). At 3 mo of age, overall %CpG methylation was unchanged at 8 ± 3. To demonstrate tissue-specific CpG demethylation, we also determined whole brain in2GHRE CpG methylation at this time point and found that the overall %CpG methylation was significantly higher at 85 ± 11 (data not shown) compared with 8 ± 3 in the liver (P < 0.05). Even though we did not specifically determine nucloeosome occupancy in whole brain in2GHRE, our liver data would suggest that the hyper-CpG methylated brain in2GHRE was nucleosome occupied. Finally by 2 yr of age, which is approaching the end of a laboratory C57Bl/6 mouse life span, overall %CpG methylation at the hepatic in2GHRE was 29 ± 4, which was statistically different from the %CpG at 3 mo.
Fig. 5.

Overall %CpG methylation of the 4 CpG sites within IGF-1 in2GHRE in male mice. %CpG correlated with nucleosome occupancy at the in2GHRE as depicted in Figs. 3 and 4.
IUGR inhibits nucleosome displacement from the hepatic in2GHRE.
After characterizing the mouse hepatic in2GHRE chromatin remodeling, we next determined whether decreases in hepatic IGF-1 mRNA and circulating IGF-1 in juvenile IUGR male mice in this model could be due, in part, to disruption of in2GHRE activation. We, therefore, examined CpG methylation and nucleosome occupancy in postnatal day 21 sham-operated and IUGR male mice compared with normal male mice.
Juvenile sham and IUGR mice underwent the same programmed CpG demethylation as normal mice at postnatal day 21 (Fig. 6). As for nucleosome positioning, we analyzed six mice emanating from three separate litters and found that, in half of the IUGR mice, the nucleosome was not displaced from the in2GHRE (Fig. 7A). Concurrently, the average %GpC methylation at each of the 21 sites was 22 ± 7. In the remaining half of the IUGR mice, an NDR was created; however, it was shifted more 3′ with the NDR falling between GpC sites 6 and 19, covering a distance of 218 bp (Fig. 7B). The average %GpC methylation at sites 6–19 was 62 ± 10, while the flanking %GpC methylation at sites 1, 2, and 20–23 was 30 ± 10 (P < 0.05). In addition, an opening between GpC sites 6 and 19 would only expose the 3′-STAT5b binding site. Since GH-induced STAT5b DNA binding requires chromatin opening and the two STAT5b sites to act synergistically (37), the 3′-shift in the NDR might decrease in2GHRE activity. To our surprise, a similar 3′-shift was also seen in the offspring generated from sham-operated dams (Fig. 7C). But here, a smaller NDR was defined between GpC sites 8 and 19 with a distance of 177 bp. The average %GpC methylation at each GpC site between sites 8 and 19 was 68 ± 16, while the average %GpC methylation at the flanking sites 1–4, 6, 7, and 20–23 was 29 ± 8 (P < 0.05). Whether this smaller shift would also result in decreased in2GHRE activity is currently unknown. Collectively, our results suggest that prenatal perturbations, whether it be overt IUGR induction or sham surgery on the dams, have the potential to alter chromatin remodeling of a gene regulatory element that is not used until later in postnatal life.
Fig. 6.
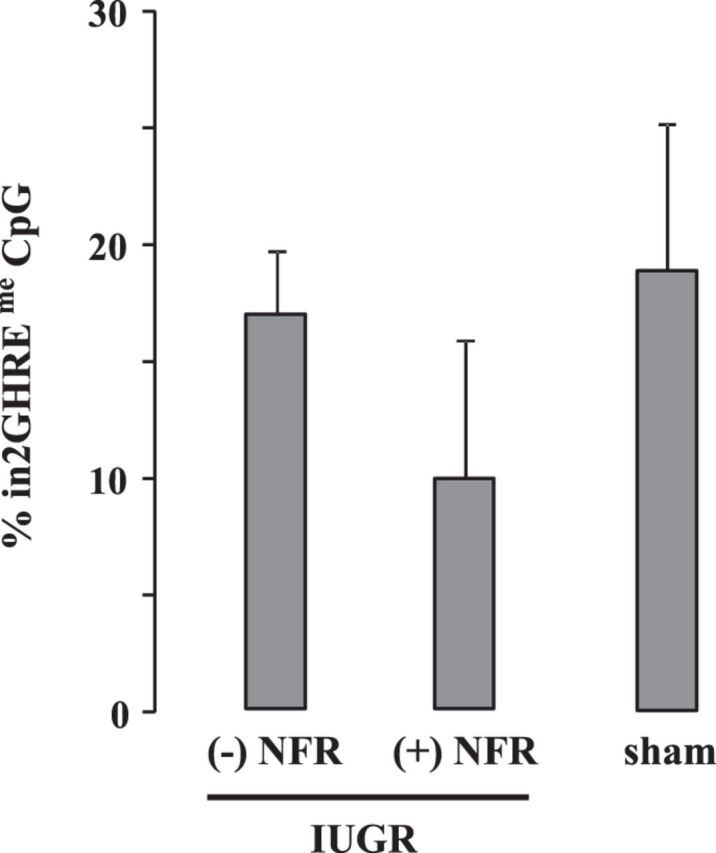
CpG demethylation at in2GHRE was unaffected by intrauterine growth restriction (IUGR) or sham surgery in d21 mice compared with control d21 mice. The IUGR group was subdivided into an absent (−) nucleosome-free region (NFR) group and a present (+) NFR group as explained in Fig. 7.
Fig. 7.

Nucleosome position analyses by NoMe-Seq across the IGF-1 in2GHRE of nuclei following M.CviP1 treatment for d21 IUGR and sham-operated mice. Either d21 IUGR mice displayed no nucleosome displacement (A), or a single nucleosome was displaced to the 3′-end of control mice compared with Fig. 4B (B). In d21 sham-operated mice, a single nucleosome was also displaced to the 3′-end compared with control mice but with a smaller nucleosome-depleted region (NDR) (C), demonstrating the effect of anesthesia/surgery alone on nucleosome displacement.
DISCUSSION
In this study we have shown for the first time that the conserved hepatic in2GHRE of the IGF-1 gene is covered by a single nucleosome, which is displaced by postnatal day 21 in the mouse to create an NDR. The resultant NDR exposes two STAT5b binding sites positioned toward the 5′-side of the opening and is flanked by well-positioned nucleosomes. The closed and open chromatin states of in2GHRE correlate well with CpG hyper- and hypomethylation, respectively. CpG remethylation with aging suggests that the NDR within in2GHRE may begin to close in late adult life, alluding to a decreasing GH responsiveness at that time. In the face of a prenatal insult such as IUGR, preadolescent mouse offspring either exhibit no NDR formation within in2GHRE or form an NDR that is shifted toward the 3′-end resulting in the exposure of only one of two STAT5b-binding sites. Surprisingly, mouse dams who underwent sham surgery also produced preadolescent offspring with a 3′-shifted NDR that was smaller in size and exposed only the 3′-STAT5b site. These findings are intriguing because they illustrate that prenatal perturbations may portend longer-term consequences when the gene is epigenetically dysregulated in prenatal life.
We recognize that we have examined only one of the many GHREs within the mouse IGF-1 gene and expect that the eventual IGF-1 expression is likely modulated by the integrated activities of all GHREs within the gene. The in2GHRE that we have chosen to examine represents one of the eight GH-responsive elements identified in the rat (12). All eight sites are conserved between rats and mice but only six are conserved in humans. Current literature has not defined whether these GHREs are redundant, but some may have unique functions (41). Of the six sites conserved among rats, mice, and humans, three contain tandem STAT5b sites, while the other three contain single STAT5b-binding sites. One GHRE, which lies 71 kb upstream of exon 1 in the mouse (73 kb in the rat and 77 kb in humans), contains two STAT5b sites, and the spacing between these two sites is 223 bp (225 bp in the rat and 220 bp in humans). If both of these STAT5b sites are used, the increased spacing might require displacing more than one nucleosome. We believe that the differently sized NDRs at each IGF-1 GHRE are necessary to allow for unique enhanceosome complexes to bind. When NOMe-seq was used to interrogate nucleosome occupancy in androgen response elements (AREs), the NDRs of AREs on three different androgen-responsive target genes were found to range from 200 to 400 bp, indicating that different numbers of displaced nucleosomes were required for gene activation (3).
Given our data, we postulate that the asymmetrical positioning of the NDR closer to the 5′-flanking nucleosome at in2GHRE of the hepatic IGF-1 gene is a prerequisite for a specific enhanceosome complex to bind for activation. Studies have shown that depending on the DNA binding protein, the binding site can be centered within an NDR far from the next closest nucleosome, placed up against a nucleosome, or even within a nucleosome (8). Transcription factors in general are thought to require a properly sized NDR in order to bind to DNA. In a study involving the insulator binding protein CCCTC-binding factor (CTCF), CTCF was found to bind a 60 bp region of DNA centered in a 118 bp NDR that was flanked by 10 ordered nucleosomes on each side with linker spacing of 35 bp each (14). In some cases, both the DNA sequence and associated histones are used to define binding specificity. For p53, a 5 bp shift in the orientation of a p53-binding site relative to the dyad or center of a nucleosome makes the p53-binding site more or less accessible. The more accessible binding site is often associated with cell cycle regulatory genes, which are p53-activated to repair damaged DNA. The less accessible p53-binding site is found in proapoptotic genes, which become active following extensive DNA damage. It has been proposed that the two orientations of the p53-binding site provide a mechanism to give DNA repair a head start over cell termination. The progesterone receptor also binds its recognition site more strongly when the site is in the context of a nucleosome. One study proposes that the progesterone site recognition and stable binding are in fact enhanced by using information from both DNA and histones (4). In addition to the two STAT5b sites, our in vitro data suggest that other regulatory elements exist within the in2GHRE that regulate GH responsiveness. Methylation of the four CpG sites flanking the tandem STAT5b sites significantly inhibited reporter activity in transient transfections. However, a screen for transcription factor-binding sites using the JASPAR database did not identify any putative DNA binding sites containing the methylated CpG sites, suggesting that GH activation may involve other epigenetic regulation in addition to CpG methylation.
A number of studies have been conducted to define the epigenetic signature for specific regulatory elements. For example, the repressed and active state of an element is often marked by H3K27me and H3K4me3, respectively (6, 17). Although informative, most of these studies on histone modifications cannot be used to define the in vivo epigenetic progression throughout development because they deal with either cell culture conditions or a process that happens over too short of a time period to delineate individual steps in progression. Based on our observations with prenatal insults, we can conclude that critical epigenetic modifications are assigned in utero to the IGF-1 in2GHRE to allow for IGF-1 upregulation at adolescence. The protracted time before in2GHRE is fully open, therefore, provides an opportunity to determine when particular epigenetic modifications are assigned prenatally and early postnatally. To this end, we used CpG methylation of the in2GHRE at various time points between ES cells and 2 yr of age to denote developmental changes in the epigenetic maturation of in2GHRE. in2GHRE is hypermethylated in ES cells and remains relatively hypermethylated until postnatal day 16. Methylation then declines to ∼20% and remains there until postnatal day 28. In between this time period, the nucleosome covering the in2GHRE is removed. Methylation decreases further in early adult life to <10% and then increases again by 2 yr. Demethylation is not driving activation of the in2GHRE because although the location within intron 2 and the spacing between the STAT5b-binding sites are conserved among humans, rats, and mice, the CpG sites are not conserved between humans and rodents. However, the DNA methylation status of the in2GHRE at any one time point may reflect when certain other epigenetic modifications are being assigned. In our study, we found a sex difference in CpG methylation levels between d7 males and females where females had increased CpG methylation, but both sexes’ CpG methylation levels were similar to prenatal time points when GH is inactive (Fig. 8A). Indeed, a large number of hepatic genes, under the control of GH, have been shown to be differentially expressed based on sex (43). This is because GH expression differs between males and females. Males have higher pulsatile circulating levels, while females have lower constant levels. Evidence indicates that pulsatile expression in males may be one of the contributing factors for this differential gene expression (44). Our observation in sex difference at day 7 could suggest a delay in programmed demethylation in females. However, by day 21 and day 35, when GH is acting on in2GHRE, the methylation differences became statistically insignificant. In addition, at day 35 when GH is acting through STAT5b proteins, eliminating STAT5b in knockout mice did not affect programmed CpG demethylation (Fig. 8B). Collectively, our findings suggest epigenetic remodeling of the in2GHRE that leads to CpG demethylation around the time that GH is acting on the in2GHRE is independent of sex.
Fig. 8.

CpG demethylation at the in2GHRE was independent of sex and phospho-STAT5b presence. CpG methylation within the liver GHRE was measured at d7, d21, and d35 in males and females (A) and in STAT5b knockout (KO) mice at 5 wk of life (B).
Using a mouse model mimicking human pregnancy-induced hypertension to elicit IUGR, which is the most common etiology of IUGR in the developed world, we have recently demonstrated a persistent decrease in IGF-1 mRNA and circulating IGF-1 in juvenile male mice with altered promoter DNA methylation and histone modification changes across the body of the IGF-1 gene (16). This current study extends our prior investigation by showing that IUGR affects the formation of the NDR within hepatic in2GHRE in the mouse IGF-1 gene as well. Thus far, we have not specifically determined whether this shifted NDR contributes to the IGF-1 decrease previously seen in this model, but the importance of proper nucleosome positioning as discussed here suggests that any aberrant shift could disrupt proper enhanceosome loading, potentially adversely affecting in2GHRE activity.
In conclusion, the primary goal of this study was to demonstrate that a protracted time course, beginning in prenatal development, is sometimes necessary to program epigenetically the eventual gene expression in postnatal life. We used the in2GHRE of the IGF-1 gene to highlight this fact. Epigenetic remodeling of the mouse hepatic in2GHRE for IGF-1 activation during the juvenile stage involves displacement of a single nucleosome to establish a discrete NDR and CpG demethylation to expose the two STAT5b-binding sites during development. These exposed STAT5b sites can then mediate the GH-induced upregulation of hepatic IGF-1 expression. The mouse in2GHRE remethylates toward the end of life, which may result in diminished GH responsiveness as the organism ages. Finally, any prenatal insult such as IUGR or anesthesia/surgery may potentially perturb the proper formation of a well-positioned NDR at the mouse hepatic IGF-1 in2GHRE, which normally transitions in2GHRE to an open chromatin state by postnatal day 21. The other GHREs within the IGF-1 gene may also be vulnerable to such epigenetic perturbations. Thus, the eventual offspring phenotype will be a culmination of the ability of different DNA-binding proteins/enhanceosomes to access DNA for gene transcription at developmentally regulated time points.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: R.A.M. and R.H.L. conception and design of research; R.A.M., C.C.Y., X.Y., J.E.W., C.W.C., A.S.B., and C.M.F. performed experiments; R.A.M., X.Y., J.E.W., C.W.C., R.H.L., and C.M.F. analyzed data; R.A.M. and C.W.C. interpreted results of experiments; R.A.M. prepared figures; R.A.M. drafted manuscript; R.A.M., C.C.Y., J.E.W., C.W.C., A.S.B., R.H.L., and C.M.F. edited and revised manuscript; R.A.M., C.C.Y., X.Y., J.E.W., C.W.C., A.S.B., R.H.L., and C.M.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Division of Neonatology, Department of Pediatrics at the University of Utah School of Medicine for financial support of this study.
REFERENCES
- 1.Agnoux AM, Antignac JP, Simard G, Poupeau G, Darmaun D, Parnet P, Alexandre-Gouabau MC. Time window-dependent effect of perinatal maternal protein restriction on insulin sensitivity and energy substrate oxidation in adult male offspring. Am J Physiol Regul Integr Comp Physiol 307: R184–R197, 2014. [DOI] [PubMed] [Google Scholar]
- 2.Allan J, Fraser RM, Owen-Hughes T, Keszenman-Pereyra D. Micrococcal nuclease does not substantially bias nucleosome mapping. J Mol Biol 417: 152–164, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreu-Vieyra C, Lai J, Berman BP, Frenkel B, Jia L, Jones PA, Coetzee GA. Dynamic nucleosome-depleted regions at androgen receptor enhancers in the absence of ligand in prostate cancer cells. Mol Cell Biol 31: 4648–4662, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballare C, Castellano G, Gaveglia L, Althammer S, Gonzalez-Vallinas J, Eyras E, Le Dily F, Zaurin R, Soronellas D, Vicent GP, Beato M. Nucleosome-driven transcription factor binding and gene regulation. Mol Cell 49: 67–79, 2013. [DOI] [PubMed] [Google Scholar]
- 5.Barker DJ. The developmental origins of adult disease. J Am Coll Nutr 23: 588S–595S, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, Hitz BC, Gupta R, Rando TA, Baker JC, Snyder MP, Cherry JM, Brunet A. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158: 673–688, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bichell DP, Kikuchi K, Rotwein P. Growth hormone rapidly activates insulin-like growth factor I gene transcription in vivo. Mol Endocrinol 6: 1899–1908, 1992. [DOI] [PubMed] [Google Scholar]
- 8.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Meth 10: 1213–1218, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Champagne FA, Weaver IC, Diorio J, Dymov S, Szyf M, Meaney MJ. Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology 147: 2909–2915, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Cherstvy AG, Teif VB. Electrostatic effect of H1-histone protein binding on nucleosome repeat length. Phys Biol 11: 044001, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Chia DJ, Varco-Merth B, Rotwein P. Dispersed chromosomal Stat5b-binding elements mediate growth hormone-activated insulin-like growth factor-I gene transcription. J Biol Chem 285: 17636–17647, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chia DJ, Young JJ, Mertens AR, Rotwein P. Distinct alterations in chromatin organization of the two IGF-I promoters precede growth hormone-induced activation of IGF-I gene transcription. Mol Endocrinol 24: 779–789, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engblom D, Kornfeld JW, Schwake L, Tronche F, Reimann A, Beug H, Hennighausen L, Moriggl R, Schutz G. Direct glucocorticoid receptor-Stat5 interaction in hepatocytes controls body size and maturation-related gene expression. Genes Dev 21: 1157–1162, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu Y, Sinha M, Peterson CL, Weng Z. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet 4: e1000138, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fung C, Brown A, Cox J, Callaway C, McKnight R, Lane R. Novel thromboxane A2 analog-induced IUGR mouse model. J Dev Origins Health Dis 2: 291–301, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Fung CM, Yang Y, Fu Q, Brown AS, Yu B, Callaway CW, Li J, Lane RH, McKnight RA. IUGR prevents IGF-1 upregulation in juvenile male mice by perturbing postnatal IGF-1 chromatin remodeling. Pediatr Res 78: 14–23, 2015. [DOI] [PubMed] [Google Scholar]
- 17.Gaydos LJ, Wang W, Strome S. Gene repression H3K27me and PRC2 transmit a memory of repression across generations and during development. Science 345: 1515–1518, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gluckman PD, Pinal CS. Regulation of fetal growth by the somatotrophic axis. J Nutr 133: 1741S–1746S, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, Rodford J, Slater-Jefferies JL, Garratt E, Crozier SR, Emerald BS, Gale CR, Inskip HM, Cooper C, Hanson MA. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes 60: 1528–1534, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herrington J, Carter-Su C. Signaling pathways activated by the growth hormone receptor. Trends Endocrinol Metab 12: 252–257, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Hogg K, Price EM, Hanna CW, Robinson WP. Prenatal and perinatal environmental influences on the human fetal and placental epigenome. Clin Pharmacol Ther 92: 716–726, 2012. [DOI] [PubMed] [Google Scholar]
- 22.Janssen JA, Stolk RP, Pols HA, Grobbee DE, Lamberts SW. Serum total IGF-I, free IGF-I, and IGFB-1 levels in an elderly population: relation to cardiovascular risk factors and disease. Arterioscler Thromb Vasc Biol 18: 277–282, 1998. [DOI] [PubMed] [Google Scholar]
- 23.Johnsen SP, Hundborg HH, Sorensen HT, Orskov H, Tjonneland A, Overvad K, Jorgensen JO. Insulin-like growth factor (IGF) I, -II, and IGF binding protein-3 and risk of ischemic stroke. J Clin Endocrinol Metab 90: 5937–5941, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Juul A, Scheike T, Davidsen M, Gyllenborg J, Jorgensen T. Low serum insulin-like growth factor I is associated with increased risk of ischemic heart disease: a population-based case-control study. Circulation 106: 939–944, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Kashiwagi K, Nimura K, Ura K, Kaneda Y. DNA methyltransferase 3b preferentially associates with condensed chromatin. Nucleic Acids Res 39: 874–888, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res 22: 2497–2506, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly TK, Miranda TB, Liang G, Berman BP, Lin JC, Tanay A, Jones PA. H2A.Z maintenance during mitosis reveals nucleosome shifting on mitotically silenced genes. Mol Cell 39: 901–911, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klug M, Rehli M. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 1: 127–130, 2006. [DOI] [PubMed] [Google Scholar]
- 29.LeRoith D, Yakar S. Mechanisms of disease: metabolic effects of growth hormone and insulin-like growth factor 1. Nat Clin Pract Endocrinol Metab 3: 302–310, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Mitchell NC, Gould GG, Smolik CM, Koek W, Daws LC. Antidepressant-like drug effects in juvenile and adolescent mice in the tail suspension test: Relationship with hippocampal serotonin and norepinephrine transporter expression and function. Front Pharmacol 4: 131, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagata T, Redman RS, Lakshman R. Isolation of intact nuclei of high purity from mouse liver. Anal Biochem 398: 178–184, 2010. [DOI] [PubMed] [Google Scholar]
- 32.Ness RB, Roberts JM. Heterogeneous causes constituting the single syndrome of preeclampsia: a hypothesis and its implications. Am J Obstet Gynecol 175: 1365–1370, 1996. [DOI] [PubMed] [Google Scholar]
- 33.Robertson KD. DNA methylation and chromatin - unraveling the tangled web. Oncogene 21: 5361–5379, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Saydah S, Ballard-Barbash R, Potischman N. Association of metabolic syndrome with insulin-like growth factors among adults in the US. Cancer Causes Control 20: 1309–1316, 2009. [DOI] [PubMed] [Google Scholar]
- 35.Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell 132: 887–898, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A, Efstratiadis A. The hormonal action of IGF1 in postnatal mouse growth. Proc Natl Acad Sci USA 105: 19378–19383, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugathan A, Waxman DJ. Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol Cell Biol 33: 3594–3610, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tong PC, Ho CS, Yeung VT, Ng MC, So WY, Ozaki R, Ko GT, Ma RC, Poon E, Chan NN, Lam CW, Chan JC. Association of testosterone, insulin-like growth factor-I, and C-reactive protein with metabolic syndrome in Chinese middle-aged men with a family history of type 2 diabetes. J Clin Endocrinol Metab 90: 6418–6423, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Tsuji-Takayama K, Suzuki M, Yamamoto M, Harashima A, Okochi A, Otani T, Inoue T, Sugimoto A, Toraya T, Takeuchi M, Yamasaki F, Nakamura S, Kibata M. The production of IL-10 by human regulatory T cells is enhanced by IL-2 through a STAT5-responsive intronic enhancer in the IL-10 locus. J Immunol 181: 3897–3905, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci USA 94: 7239–7244, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Varco-Merth B, Mirza K, Alzhanov DT, Chia DJ, Rotwein P. Biochemical characterization of diverse Stat5b-binding enhancers that mediate growth hormone-activated insulin-like growth factor-I gene transcription. PLoS One 7: e50278, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volokhov DV, Kong H, George J, Anderson C, Chizhikov VE. Biological enrichment of Mycoplasma agents by cocultivation with permissive cell cultures. Appl Environ Microbiol 74: 5383–5391, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waxman DJ, Holloway MG. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol 76: 215–228, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waxman DJ, Pampori NA, Ram PA, Agrawal AK, Shapiro BH. Interpulse interval in circulating growth hormone patterns regulates sexually dimorphic expression of hepatic cytochrome P450. Proc Natl Acad Sci USA 88: 6868–6872, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woelfle J, Billiard J, Rotwein P. Acute control of insulin-like growth factor-I gene transcription by growth hormone through Stat5b. J Biol Chem 278: 22696–22702, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Woelfle J, Chia DJ, Rotwein P. Mechanisms of growth hormone (GH) action. Identification of conserved Stat5 binding sites that mediate GH-induced insulin-like growth factor-I gene activation. J Biol Chem 278: 51261–51266, 2003. [DOI] [PubMed] [Google Scholar]
- 47.Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, Yang AS, Jones PA, Liang G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet 6: e1000917, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woodworth C, Secott T, Isom HC. Transformation of rat hepatocytes by transfection with simian virus 40 DNA to yield proliferating differentiated cells. Cancer Res 46: 4018–4026, 1986. [PubMed] [Google Scholar]
- 49.Xu XF, Hu QY, Liang LF, Wu L, Gu WZ, Tang LL, Fu LC, Du LZ. Epigenetics of hyper-responsiveness to allergen challenge following intrauterine growth retardation rat. Respir Res 15: 137, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yadon AN, Van de Mark D, Basom R, Delrow J, Whitehouse I, Tsukiyama T. Chromatin remodeling around nucleosome-free regions leads to repression of noncoding RNA transcription. Mol Cell Biol 30: 5110–5122, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, Okada S, Cataldo L, Coschigamo K, Wagner TE, Baumann G, Kopchick JJ. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc Natl Acad Sci USA 94: 13215–13220, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]