

Abstract
The novel coronavirus that caused COVID-19 pandemic is SARS-CoV-2. Although various vaccines are currently being used to prevent the disease's severe consequences, there is still a need for medications for those who become infected. The SARS-CoV-2 has a variety of proteins that have been studied extensively since the virus's advent. In this review article, we looked at chemical to molecular aspects of the various structures studied that have pharmaceutical activity and attempted to find a link between drug activity and compound structure. For example, designing of the compounds which bind to the allosteric site and modify hydrogen bonds or the salt bridges can disrupt SARS-CoV2 RBD–ACE2 complex. It seems that quaternary ammonium moiety and quinolin-1-ium structure could act as a negative allosteric modulator to reduce the tendency between spike-ACE2. Pharmaceutical structures with amino heads and hydrophobic tails can block envelope protein to prevent making mature SARS-CoV-2. Also, structures based on naphthalene pharmacophores or isosteres can form a strong bond with the PLpro and form a π-π and the Mpro's active site can be occupied by octapeptide compounds or linear compounds with a similar fitting ability to octapeptide compounds. And for protein RdRp, it is critical to consider pH and pKa so that pKa regulation of compounds to comply with patients is very effective, thus, the presence of tetrazole, phenylpyrazole groups, and analogs of pyrophosphate in the designed drugs increase the likelihood of the RdRp active site inhibition. Finally, it can be deduced that designing hybrid drug molecules along with considering the aforementioned characteristics would be a suitable approach for developing medicines in order to accurate targeting and complete inhibition this virus.
Keywords: COVID-19, SARS-CoV-2, Spike-ACE2, PLpro, RdRp, 3Clpro
Abbreviations: ARTis, Respiratory tract infections; SARS-COV-2, Severe Acute Respiratory Syndrome Crona Virus 2; ORFS, Open Reading Frames; RdRp, RNA-dependent RNA polymerase; RBD, receptor-binding domain; RBM, receptor-binding motif
Graphical abstract
1. Introduction
In the twenty-first century, humans have encountered three deadly diseases which are related to the coronavirus family [1]. SARS, MERS, and COVID-19 are three deadly pandemics that cause acute respiratory tract infections (ARTIs) [2]. These highly contagious pathogenic infections have caused high mortalities so far. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is another zoonotic novel coronavirus that caused the present COVID-19 pandemic [3]. The single-stranded ribonucleic acid is the genome of the SARS-CoV-2 virus which is so identical to the SARS-CoV-1 virus that caused the 2002–2004 SARS outbreak [4]. SARS-CoV-2 is spherical and pleomorphic, measuring 80 to 125 nm in diameter, and contain single-stranded positive-sense RNA with nucleocapsids [5]. There is a polybasic cleavage site on the SARS-CoV-2 genome which is a unique feature that has made this virus highly pathogenic and susceptible to transmission. In fact, this cleavage site allows the activation of multiple proteins at the same time, effectively increasing pathogenicity, virulence, and transmission of SARS-CoV-2 [6]. The RNA genome size ranges from 26 to 32 KB, with 10 open reading frames (ORFs) that can be translated. ORF1a and ORF1b account for roughly two-thirds of the SARS-CoV-2 genome and are translated into two large replicase polypeptides called polyprotein 1a and 1ab (pp1a and pp1ab) [7]. Viral proteases process these polyproteins to produce 16 non-structural proteins (nsps) [8]. Between nsps, the RNA-dependent RNA-polymerase enzyme (RdRp) facilitates viral RNA replication via (−sense) template transcription, in which SARS-CoV-2 generates 6 sub-genomic mRNAs, which help in the translation of structural and accessory proteins located downstream of ORFs. In the ORF1ab region, NSPs has the most nonsynonymous mutations. The viral genome also encodes all of the viral structural and functional proteins required for viral replication and infection, including spike protein (S), nucleocapsid protein (N), the envelope protein (E), and membrane protein (M) [9].
The virus's basic reproduction number has been determined to be between 1.4 and 6.47 in different countries, which not only represents a larger range when compared to other corona viral infections, but also scores very close to other well-known contagious diseases such as Chickenpox, Measles, Polio, Rubella, and Mumps [10]. SARS-CoV-2 has a wavy trend of infection and mortality rates, making it critical to understand the mutations and their related epidemiology. In fact, many SARS-CoV-2 variants have arisen since the pandemic appeared in December 2019 [11]. Thus, although various vaccines have been approved and used to prevent infection, it seems that some people become still infected with different variants of this virus [12]. Accordingly; there is a need for medications for people being infected.
Different types of potential therapeutics have been evaluated and approved so far, but since new strains of the virus are constantly deriving, further medications will still be needed for accurate targeting and complete inhibition of the virus [13]. Therefore, in this review, we surveyed many pharmaceutical chemistry articles and analyzed the structure-activity relationship (SARs) of various molecules to design new anti-COVID-19 drugs to improve the treatment of COVID-19.
2. Druggable targets for SARS-CoV-2
It is obvious that understanding the life cycle of the virus is crucial to drug targets. Generally, there are two paths through which the SARS-CoV-2 can enter the host cell, including the fusion of the plasma membrane or the endosome. The spike protein of the virus uses the host cell functional receptor i.e. angiotensin-converting enzyme-2 [ACE-2] and mediates binding to the host cell membrane in both mechanisms [14,15]. Thus, the first therapeutic approach to prevent SARS-CoV-2 infection is to evaluate the effect of the pharmacological agents on the spike-ACE2 complex and to reduce their affinity to each other [4].
After forming the spike-ACE2 complex, the virus envelope is removed and the SARS-CoV-2 genome and its nucleocapsid are secreted into the cytoplasm. The pp1a and pp1b of SARS-CoV-2 hijack host cell ribosomes to help the viral translation process. The pp1a and pp1b are broken down by cysteine protease 3C (3CLPro, Mpro), and papain-like protease (PLpro) to produce viral non-structural proteins (NSPs) that perform different functions of SARS-CoV-2. In fact, M and PL proteases are vital for sustaining the basic cellular processes in replication and transcription of SARS-CoV-2, and their inhibitors could be potential druggable targets for the treatment of COVID-19. While PLpro cuts the polyprotein at three sites, Mpro is responsible for cleavage at 11 other locations that, together, produce the 16 nonstructural proteins [16].
When the SARS-CoV-2 virus begins to replicate, RdRp as one of the important NSPs forms the replicase–transcriptase complex; the negative-strand RNA is used by RdRp to synthesize a new positive RNA molecule to continue another replication and translation step and ultimately to form the newest viral particles. Therefore, it seems that inhibitory compounds of this polymerase is another druggable target for COVID-19 infection control.
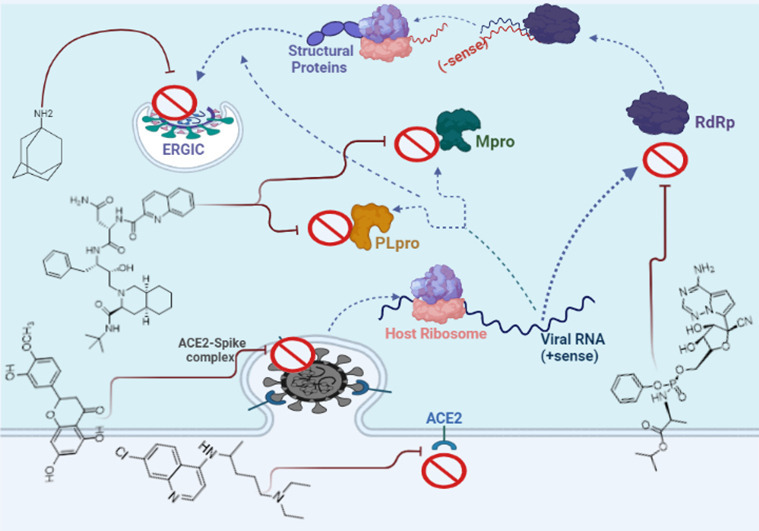
Sub-genomic RNA leads to the expression of structural proteins (SPs) including S, E, M, and N. The N and RNA stay in the cytoplasm and form a nucleoprotein complex, but S, E, and M enter the endoplasmic reticulum. Then, both complexes enter the Golgi apparatus and merge to form an adult SARS-CoV-2, ultimately, the adult virus is released from the Golgi apparatus to the extracellular region. This process is repeated many times quickly and other cells become infected. Thus, the suppressing structures of each of the S, E, M, and N proteins can also prevent further mature virus proliferation. Researchers are looking for inhibitors for these targets from three types: approved or commercially available drugs and natural compounds [[17], [18], [19]]. Fig. 1 depicts a schematic diagram of the SARS-CoV-2 life cycle in a human cell.
Fig. 1.

The life cycle of SARS-CoV-2 virus in a human cell.
3. Structure of SARS-COV-2 spike
The crystal structure of SARS-COV-2 spike receptor-binding domain [RBD] complexed with peptidase domain of ACE-2 is identified in detail [20,21]. Specifically, SARS-CoV-2 is a beta coronavirus, having similarities with SARS- CoV virus, in binding with human ACE2 receptor and spike glycoprotein for viral entry [22].
The Spike domain is cleaved by furin protease into S1 and S2; the S1 domain binds to the ACE2 receptor attachment site. RBD is the short immunologic fragment of the S1 region including residues 318 to 510, which enables it to bind to the peptidase domain of ACE2. It contains a core including a twisted five-stranded anti-parallel β sheet including β1 to β4 and β7, with three short connecting α helices containing αA to αC and an extended loop. It was reported that in SARS-CoV-2 Delta variants the overall structure of this immunologic fragment has been strictly preserved among all variants, and reoccurring surface mutations appear to be limited to several sites, consistent with its critical role in receptor binding. Thus, it is best to focus on this part of the spike to design a novel drug [23]. Indeed, the receptor-binding motif (RBM) has a high degree of structural plasticity to hold amino acid changes without distracting ACE2 binding [24]. There are few variations at the RBM that considerably alter the binding affinity of RBD to the ACE2 in SARS-CoV2 [25,26].
To stabilize the β sheet structure, there are nine cysteines in the chymotryptic fragment in the core; disulfide bonds connect cysteines 323 to 348, 366 to 419, 467 to 474 and 378 to 511 [20].In the core, between the β4–7 strands, there is a gently concave outer surface formed by a two-stranded β sheet (β 5 and β 6). This extended loop subdomain lies at one edge of the core where the RBM is, which contains the contacting residues that enable it to bind to N-terminal helix of ACE2. The RBM is particularly tyrosine-rich which present both a polar hydroxyl group and a hydrophobic aromatic ring. Multiple tyrosine residues form hydrogen-bonding interactions with the polar hydroxyl group. Thus, the networks of hydrophilic interactions which occur largely among amino acid side chains are seen in the RBM region [20]. According to the study conducted by Cong Xu et al., five tyrosine residues including (tyrosine 454,473,489,495,505) showed a vital role in the interactions of spike and ACE2 [27].
On the other hand, the crystal structure of the ACE2 ectodomain shows a claw-like N-terminal peptidase domain, with the active site at the base of a deep groove, and a C-terminal collectrin domain. This crystal structure shows residues of Ser19-Asp615 of the ACE2 N-terminal peptidase domain, one zinc ion, four N-acetyl-β-glucosaminide (NAG) glycans linked to ACE2 Asn90, Asn322, and Asn546 and RBD Asn343, as well as 80 water molecules. In the structure of the SARS-CoV-2 RBD–ACE2 complex, a chain of Asn90-linked NAG–NAG–β-D-mannose is in contact with Thr402 of the SARS-CoV-2 RBD, and this glycan–RBD interaction has been proposed to have important roles in the binding of SARS-Co- RBD by ACE2. This point was carried out in the molecular docking studies of the SARS-CoV-2 RBD–ACE2 complex with promising inhibitors [20,21,28].
On the other hand, it was recognized that a total of 18 residues of the receptor contact with 14 residues of the viral spike protein. In these complex models, a positively charged cavity at the distal end of S1 protein can bond with a highly negatively charged ridge on the top of ACE2.
It seems that the design of a novel series of ACE2 variants enhances their binding affinity for SARS-CoV-2 S trimer and their potency in blocking SARS-CoV-2 infection [29].
There are 13 hydrogen bonds and 2 salt bridges at the SARS-CoV-2 RBD–ACE2 interface, and 13 hydrogen bonds and 3 salt bridges at the SARS-CoV RBD–ACE2 interface [20,30]. Ionic bonds are formed between the ammonium ion of Lys353 and ACE2 Asp38 bearing opposite electrical charges. At position 353, the human receptor has a lysine critical for contact with Thr487 in the RBM. Particularly, a main-chain hydrogen bond between carbonyl of ACE2 Lys353 to the amide of RBD Gly488 fixes the relative positions of receptor and spike protein quite accurately. Indeed, it was reported that the presence of Thr487 appears to enhance human-to-human transmission of SARS-CoV. The methyl group of Thr487 can create hydrophobic interaction and lies in a hydrophobic pocket at the ACE2-RBD interface [20].
The salt bridges are important for SARS-CoV-2 RBD–ACE2 interaction. For example, it was reported that in the UK variant” (B.1.1.7) of SARS-CoV-2, the loss of salt bridges between Lys417 and ACE2 Asp30 and Glu484 and ACE2 Lys31 moderate in the increased receptor affinity imparted by N501Y [31].
It was recognized in the pathogenesis of the SARS-CoV-2 virus that SARS-CoV-2 RBD is located between residues Thr333-Gly526 of the S1 domain, which enables it to bind to ACE2 more strongly than SARS-CoV which may show the higher infectiousness of SARS-CoV-2 compared to SARS-CoV. Indeed, it appears that N-linked glycan at position Asn 90 of ACE2 bind to different RBDs and interferes with the infection of the cells [20].
The UK variant” (B.1.1.7) of SARS-CoV-2 is thought to be more infectious than previously circulating strains as a result of N501Y mutation that shows tyrosine 501 inserted into a cavity at the binding interface near tyrosine 41 of ACE2 using hydrophobic interactions with other aromatic side-chains of tyrosine 41. The aromatic side chain of tyrosine 501 can also mean that tyrosine is involved in possible cation-π interaction with ACE2 Lys353. This additional interaction provides a structural explanation for the increased ACE2 affinity of the N501Y mutant and likely contributes to its increased infectivity [31,32].
4. Compounds affecting spike–ACE2 complex
Based on the above, the inhibition of ACE2 could be a valid strategy for treating patients with COVID-19 [33,34]. Studies have proposed three approaches for docking some repurposed drugs to ACE2. For instance, Al-Karmalawy et al. showed that conventional ACE inhibitors (ACEIs) such as captopril, enalapril, prinivil, lisinopri, benazepril, fosinopril, and ramipril can antagonize the coupling of spike with ACE2 [35]. They showed that N-Acetyl-β-Glucosamine (NAG) specific binding site is site proximity to the hACE2-spike binding domain.
It was considered that N-linked glycan at position Asn 90 is a competitor binder and reference ligand for the ACE2 protein target, thus, researchers hypnotized that NAG specific binding site occupation with ACEIs may impact the binding mode of the spike protein [36] and investigated the binding mode of ACEIs to the NAG-specific binding site through a molecular docking strategy. Finally, Alacepril and lisinopril exhibited the best binding affinity through forming a strong hydrogen bond with Asn90, as well as significant extra interactions with other receptor-binding residues [36].
The use of ACE inhibitors for the treatment of SARS-COV-2 has been controversial. Many studies find it useful and many useless [[37], [38], [39]]. Although Tipnis et al. in human homology study of ACE in 2000 showed that ACE inhibitors could not block ACE–2 [40]. Indeed, in 2021, M. Oz et al. showed that ACE inhibitors could increase ACE2 expression in RAAS [41].
Then, Imane Abdelli et al. in their studies focused on the co-crystallized inhibitor of ACE2 i.e. β-D-mannose using Isothymol, Thymol, Limonene, P-cymene, and γ-terpine. And Isothymol binding energy was calculated at −5.78 Kcal/mol and one hydrogen interaction with THR445 amino acids of the active site was recognized, on the other hand, the β-D- Mannose-binding energy was calculated at −5. 4107 Kcal/mol; in fact, 5 interactions with GLU406, GLU406, GLN442, GLN442, LYS441 residues of the target active site were observed, furthermore, the thymol binding energy was calculated at −4, 7450 Kcal/mol, and 2 interactions with GLU406 and THR445 were considered. The study revealed that Isothymol gives the best docking scores, compared to β-D-mannose and Captropil [42]. Furthermore, Hadjer Khelfaoui et al. proposed that there are two zinc-binding sites in the ACE2 receptor. This metallic ion facilitates the viral attachment to the surface of ACE2. Thus, docking of two active sites containing Zn2+ in the ACE2 receptor and spike/ACE2 complex was chosen, then, they prepared the ACE2 receptor and spike/ACE2 complex and removed the NAG in their sequence. The results showed that Ramipril, Dalapril, and Lisinopril could bind with ACE2 receptors better than hydroxychloroquine and chloroquine [43].
Another study investigated the binding interactions of ivermectin, hydroxychloroquine, and remdesivir with human ACE-2 protein (PDB ID = 1R42) and compared it with MLN-4760 as the positive control. Docking results revealed that ivermectin had the highest binding affinity to the active site and it formed five hydrogen bonds with Arg273, Glu398, Ser511, Arg514, and Tyr515 amino acid residues present at the predicted active site. This was followed by remdesivir; it formed four hydrogen bonds with Asn394, Glu402, Glu406, and Arg514 amino acid residues present at the predicted active site. Also, MLN-4760 showed a high binding affinity by forming five H-bonds with Arg273, His345, Pro346, Thr37, and Tyr515 amino acid residues present at the predicted active site of the protein. It was notable that favipiravir achieved the lowest docking score in this study, hydroxychloroquine is bound to the human ACE-2 receptor with considerable binding force. It formed H-bonds with Arg273, Ala348, Glu375, and Arg514 amino acid residues [44]. Table 1 shows various ACE2 blockers, their chemical structures, interacting residues, and the docking software used.
Table 1.
Comparison of different blockers for ACE2, blockers' structures, the important amino acids in the reaction, and software used for docking.
| Name | Structure | Target | Interacting residues | Software package | Ref. |
|---|---|---|---|---|---|
| Chloroquine | 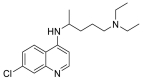 |
ACE | GLU402, GLU406 | MOE software package, MD | [45] |
| Iso thymol | 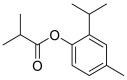 |
ACE | THR445 | MOE software package, MD simulation by iMODS | [45] |
| Trandolapril | 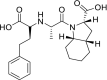 |
ACE | Asp30/H-donor | MOE, MD simulation by GROMACS | [46] |
| β-D- Mannose | 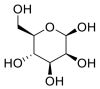 |
ACE | GLU 406, GLU 406, GLN 442, GLN 442, LYS 441 | MOE software package, MD | [45] |
| Alacepril | 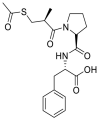 |
ACE | Asn90/H-acceptor | MOE, MD simulation by GROMACS | [46] |
| Moexipril | 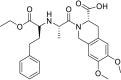 |
ACE | Asp30/H-donor | MOE, MD simulation by GROMACS | [46] |
| Fosinopril | 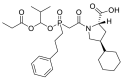 |
ACE | Gln96/H-acceptor | MOE, MD simulation by GROMACS | [46] |
| Enalapril | 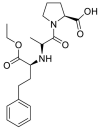 |
ACE | Asp30/H-donor | MOE, MD simulation by GROMACS | [46] |
| Enalapril | 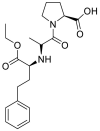 |
ACE | Asp30/H-donor | MOE, MD simulation by GROMACS | [46] |
| Thymol | 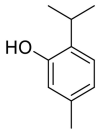 |
ACE | GLU 406,THR445 | MOE software package, MD | [45] |
| Lisinopril | 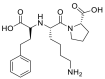 |
ACE | Asp90-H-acceptor | MOE, MD simulation by GROMACS | [46] |
| Benazepril | 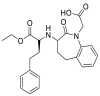 |
ACE | Lys25/H-donor | MOE, MD simulation by GROMACS | [46] |
| Zofenopril | 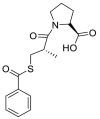 |
ACE | Asp30/H-acceptor | MOE, MD simulation by GROMACS | [46] |
| Ramipril | 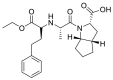 |
ACE | Lys26/H-acceptor | MOE, MD simulation by GROMACS | [46] |
| Quinapeil | 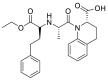 |
ACE | Pro 389/arene-H | MOE, MD simulation by GROMACS | [46] |
| Cilazapril | 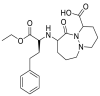 |
ACE | Pro 389/arene-H | MOE, MD simulation by GROMACS | [46] |
| Imidapril | 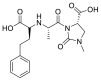 |
ACE | Asp30/H-donor | MOE, MD simulation by GROMACS | [46] |
| NAG | 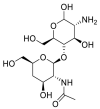 |
ACE | Asp30/H-donor | MOE, MD simulation by GROMACS | [46] |
| Perindopril | 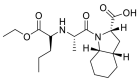 |
ACE | Asp30/H-donor Asp 30/H-acceptor |
MOE, MD simulation by GROMACS | [46] |
| Captopril | 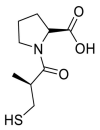 |
ACE | Asn30/H-acceptor | MOE, MD simulation by GROMACS | [46] |
Negative allosteric modulation (NMA) occurs when the binding of one substance to the complex antagonizes the affinity of the stimulus to the active site [47,48].
Regulation of spike-ACE2 attachment by binding an NMA at a site other than the ACE2 active site is a promising strategy to design novel drugs for COVID-19 treatment because when an NMA binds to spike protein fragments, a conformational change occurs and modifies the binding energy of ACE2 and spike protein complexes. Finally, the binding structure of ACE2 and spike protein fragments becomes unstable [49,50] (Fig. 2 ).
Fig. 2.

Regulation of spike-ACE2 by binding a negative allosteric modulator at the allosteric site of ACE2.
A study analyzed the interactions of quinolin-1-ium derivatives such as hydroxychloroquine and chloroquine with SARS-CoV-2 using molecular docking studies. They confirmed that hydroxychloroquine acts as an NMA. It binds to the amino acids ASP350, ASP382, ALA348, PHE40, and PHE390 on the ACE2 allosteric site rather than on the ACE2 active site [51]. It seems that quaternary ammonium moiety builds a hydrogen bond with ASP382 and a π-cation interaction with aromatic amino acids such as PHE 390 with the spike protein fragments. In addition to these interactions, chloroquine formed two hydrogen bonds with ASP 350 and ALA 348 of spike protein fragments.
Cong Xu et al. proposed a complex of spike protein trimer and the human ACE2 receptor protein, the structure was determined by electron microscopy with 3.80 Å resolution(PDB ID: 7DF4) [27]. The amino acids in the active sites included VAL-503, GLY-502, GLN-506, THR-500, PRO-499, PHE-497, LEU-455, GLY-446, ALA-475, SER-477, PHE-486, GLY-476, PHE-490, GLN-474, GLY-447, TYR-505, GLN-493, GLY-49, LEU455, PHE486, ASN501, and TYR505. Indeed, based on the interface of complex 7DF4, the key amino acid residues of ACE2 were composed of amino acid residues THR-20, ALA-386, LYS-353, GLY-352, PRO-389, PRO-499, ALA-387, SER494GLN-325, GLN-388, LYS-353, SER-19, LYS-31, GLN-24, GLY-354, GLY-326, ASP-355, SER-19, GLY-354, THR-371, GLU-375, and ALA-348. Their structural data revealed that residue tyrosine 505 plays vital roles in the engagement of SARS-CoV-2 RBD to ACE2 receptor. They demonstrated that the residue TYR 505 of SARS-CoV-2 RBD is a key amino acid required for ACE2 receptor binding. Because TYR505 could form hydrogen bonds/contacts with ALA386 (oxygen atom)/ARG393(NH2)/K353/G354 from ACE2.
Then in 2022, RanYu et al. proposed some potential spike / ACE2 dual antagonists against COVID-19 through in silico molecular docking using this PDB model [26]. The phytochemicals which have better spike protein and ACE2 inhibitory activity could block the invasion and recognition of SARS-CoV-2 at the same time [26].
They concluded that hydrogen binding interactions are necessary for connecting the phytochemicals and spike glycoprotein-ACE2. The more the hydrogen bonds, the lower the binding energy. Hydrogen bond has a strong stabilizing effect on the binding of antagonists to receptors. As mentioned above, RBM is a tyrosine rich region, the phytochemicals such as menthol or chrysophenol can interact with tyrosine residues via π-π interaction or hydrogen bond can block the invasion of the spike protein. Through the tyrosine residues, tyrosine505 showed a vital rule in connection of spike glycoprotein-ACE2. The compounds such as baicalin and Quercetin showed π-π interaction with tyrosine 505 may block ACE2 [26].
As mentioned above, a main-chain hydrogen bond between the carbonyl of ACE2 Lys to an amide of RBD Gly fixes the relative positions of receptor and spike protein quite precisely. Accordingly, the compounds which have substituents that can form a salt bridge or a hydrogen bond with LYS 562, LYS95, or His 34 are promising compounds to block ACE2 [26].
In sum, it seems phytochemicals can disrupt the hydrophilic interaction of ACE2 and the receptor-binding domain. It seems that phytochemicals modulate the binding energy of the bound structure of ACE2 and spike. This result indicates that due to the presence of phytochemicals, the bound structure of ACE2 and spike protein fragment becomes unstable [21,22].
Table 2 lists the compounds that target spike-ACE2 complex, their structures, and the important amino acids in the reaction between spike-ACE2 [26].
Table 2.
Compounds that regulate the spike-ACE2 complex, their structure, and the important amino acids in the reaction.
| Structure name | Structure | Interacting amino acids of ACE2 receptor | Interacting amino acids of the spike protein fragment | The interaction mode with spike | The interaction mode with ACE2. |
|---|---|---|---|---|---|
| Menthol | 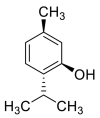 |
GLN-102 SER-77, LEU-73, TRP-69 ASN-103 | SER-494, TYR-495, TYR-453 LEU | Hydrogen bond π–π interaction Hydrophobic interaction |
Hydrophobic interaction |
| Chrysophenol | 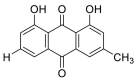 |
SER-511 HIS-505, TRY-510 | GLN-493 GLY-496, TYR-453 | Hydrogen bond π–π interaction |
Hydrogen bond π–π interaction |
| Schizandrin | 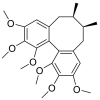 |
HIS-34, GLU-37 PHE-390 | TYR-453, TYR-495, GLY-447 TYR-449 | Hydrogen bond Hydrophobic interaction |
Hydrogen bond Hydrophobic interaction |
| Rhein | 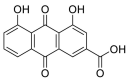 |
GLU-329, LEU-333, TRP-48 ARG-357, ASN-330 SER-331 | GLN-493, TYR-495 ARG-403 | Hydrogen bond π–π interaction |
Hydrogen bond Hydrophobic interaction |
| Scopoletin | 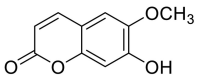 |
TRP-566, LEU-95 and LYS-562 | GLY-496 TYR-449 | Hydrogen bond | Hydrogen bond |
| Baicalin | 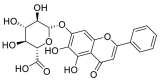 |
GLN-102 LYS-562, ALA-396 ALA-99 | GLN-493 SER-494, GLY-496, TYR-505 | Hydrogen bond π–π interaction Hydrophobic interaction |
Hydrogen bond Hydrophobic interaction |
| Shikonin | 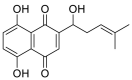 |
GLU-37, HIS-34, ARG-393, ASN-33 ALA-387 GSN-388. | TYR-453 SER-494 ARG-403 | Hydrogen bond Hydrophobic interaction |
Hydrogen bond Hydrophobic interaction |
| Oleanolic acid | 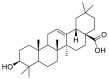 |
LYS-562, LEU-73 ALA-99 | TYR-453 GLY-496, ARG-403 SER-494 | Hydrogen bond Hydrophobic interaction |
Hydrogen bond Hydrophobic interaction |
| Tryptanthrin | 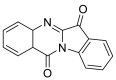 |
LYS-94 GLY-211, GLN-98, GLU-208, ASN-210 LEU-85 | GLN-493 SER-494, TYR453 TYR-449 | Hydrogen bond π–π interaction |
Hydrogen bond Hydrophobic interaction |
| Cimifugin | 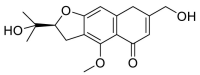 |
GLN-101, ILE-88, LEU-85 LYS-94 | GLY-496 | Hydrogen bond | Hydrogen bond |
| Berberine | 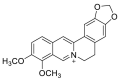 |
HIS-378 HIS-401, PHE-390 PHE-40 | GLY-496 | Hydrogen bond | π–π interaction |
| Coumarin | 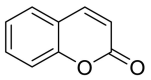 |
ASN-210 | GLN-493 TYR-453 | Hydrogen bond | Hydrogen bond |
| Perilla aldehyde | 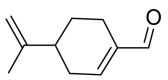 |
TRP-566 | TYR-453 SER-494 | Hydrogen bond | Hydrogen bond |
| Quercetin | 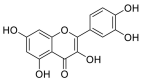 |
GLU-564, GLU-208, ASN-394 GLU-398, TRP-566 | SER-494 TYR-449 TYR-505 | Hydrogen bond π–π interaction |
Hydrogen bond π–π interaction |
| Chloroquine | 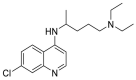 |
GLY 405, HIS 401, THR 347 | THR 467, PRO 468, CYS 469 | Hydrogen bond π–cation interaction |
Hydrogen bond |
| Hydroxy chloroquine | 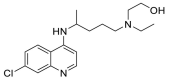 |
ASP 67, ALA 71, SER 43 | GLY 471, VAL 472, CYS 469 | Hydrogen bond π–cation interaction |
Hydrogen bond |
5. Structure of envelope protein
This protein of SARS-CoV-2 is made up of 75 amino acids that are divided into two domains: a C-terminal domain and an N-terminal transmembrane domain (TMD) [52]. It is noteworthy that C-terminal has no stable complex, thus, its structures have not been well defined. N-terminus channel entrance and lumen are its targeted inhibitor binding sites. The narrowest part of this channel could be 2.6–4.8 Å wide, with an average of 4.4 Å [53].
The envelope is a protein with diverse tasks. It has a structural role in inducing membrane curvature for viral assembly in collaboration with the viral M protein, and it also mediates host immune responses by two distinct mechanisms: a PDZ (PSD-95/Dlg/ZO-1)-binding function via its C-terminal domain, and a pore-forming TMD associated with NLRP3 inflammasome activation [54,55]. The TMD of the envelope structurally forms a pentameric ion channel [56].
In humans, each PDZ domain contains 80–110 amino acid residues and is required for the proper regulation of human immune responses [57]. SARS-CoV-2 hijacks PDZ-domain-containing proteins to increase virulence [57]. The SARS-CoV-2 envelope protein contains a PDZ-binding motif (PBM) at its C-terminus [58]. The exact mechanism is unknown, but interactions between a human cell junction protein (PALS1) and the PBM appear to show that the envelope causes PALS1 to be relocated from the cell junction to the endoplasmic reticulum–Golgi intermediate compartment site (ERGIC). In ERGIC, the envelope is localized and viral assembly occurs. Therefore, the PBM of envelope protein is involved in SARS-CoV-2 pathogenesis by binding the host's syntenin protein resulting in overexpression of inflammatory cytokines [59]. The SARS-CoV-2 envelope channel while embedding into the ERGIC/Golgi membranes can facilitate Ca2+ transport resulting in the activation of the NLRP3 inflammasome [57].
6. Compounds affecting envelope protein
It seems that the clinical utility of amantadine appears from its action as blocker of ion channels. Amantadine showed its antiviral effectiveness against influenza A with the inhibition of the M2 ion channel. It is also used for the treatment of Parkinson's Disease due to its low-potent noncompetitive inhibition of the NMDA receptor ion channel on striatal dopaminergic neurons [60].
Although there is no sequence homology between influenza A and SARS-CoV-2, amantadine inhibits two of the four known (3a and envelope proteins) or putative (Orf7b and Orf10) SARS-CoV-2 viroporins, protein E and Orf10a. The envelope protein of SARS-CoV-2 can conduct both anions and cations. Lauren Wilson et al. demonstrated that SARS-CoV E protein does form ion channels, which are more selective for monovalent cations than monovalent anions [61]. The conductivity preference is dependent on pH and the lipid constituents of the membrane. In physiological conditions, it appears to have a slight preference for cations (Na+, K+, Ca2+) [62,63].
It seems that Amantadine can build π–alkyl interactions with another aromatic amino acid [64]. Indeed, it seems that it can enter the ion channels and block them. The binding of amantadine to a synthetic TMD of envelope protein from SARS-CoV was significant enough (Kd ~ 7 mM) to completely block the ion conductance [53]. The simplest blocking mechanism for amantadine is that the cationic headgroup of amantadine is dragged into the channel by the electric field, and then occupied the channel with the hydrophobic group i.e., the adamantane group following and plugging the entry for the succeeding ions. A positively charged head group followed by a bulky hydrophobic scaffold might be the common structural feature of SARS-CoV-2 envelope protein inhibitors.
Singh Tomar et al. proposed that a 3,5-dimethyl derivative of amantadine (memantine) which is the inhibitor of NMDA receptor can inhibit the SARS-CoV-2 ion channel. They specified that gliclazide which is the potassium currents blocker can inhibit the SARS-CoV-2 ion channel [65].
On the other hand, Amiloride derivatives (in particular hexamethylene amiloride) were found to be efficient inhibitors of envelope proteins of SARS-CoV-2 [66]. Hexamethylene amiloride (HMA) like amantadine showed a positively charged headgroup and the bulky hydrophobic scaffold [67]. An investigation has demonstrated that the HMA inhibited in vitro conductance of synthetic MHV envelope and HCoV-229E envelope, which show close homology to SARS-CoV-2 envelope. They showed that a protonated form of HMA is bound to the channel [68].
To identify potential high-affinity blockers of the SARS-CoV-2 envelope channel, a screening study of approximately 6000 compounds was carried out by Chernyshev A as deposited in the ZINC database and their Vina score were compared. The study has revealed two siderophore iron chelators blocking the virus channel [53].
Liu Wenzhong in his article demonstrated that Cys44 of envelope protein is the heme iron-linked site. Indeed, they showed that heme-linked sites of E protein may be relevant to the high infectivity. It can be seen that the first two Cys40, Cys43 on this domain are connected to the carbon at the left end of the alpha position and the beta position, respectively, and the last Cys44 is related to the iron of the heme [69]. Desferrioxamine and nocardamine are siderophore iron chelators that can chelate Ferric iron at a 1:1 stoichiometry. Their chemical structures are presented in Fig. 3 .
Fig. 3.

Structures of amantadine, HMA, Nocardamine, and Desferrioxamine as the best structures to inhibit envelope protein of SARS-CoV-2.
Overall, according to the Clifford Fong study, a predictive quantum mechanical TD DFT method is needed to describe the time-dependent behavior of inhibitors of the envelope ion channel of SARS-CoV-2. The key descriptors are the excitation energy of the first excited state of the inhibitor and the ground state HOMO-LUMO gap of the inhibitor that characterize the dynamic binding between the inhibitor and the protein target [70]. Different blockers of SARS-CoV-2 envelope protein ion channel, their chemical structures, and software used for docking are shown in Table 3 .
Table 3.
Comparison of different blockers for envelope protein (E) of SARS-CoV-2, blockers' structures, and software used for docking.
| Compound | Chemical structure | Target | Software | Ref. |
|---|---|---|---|---|
| Mefenamic acid | 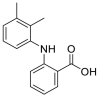 |
E protein | Docking server CB-Dock, Automated cavity-detection, AutoDock Vina, MD by GROMACS | [56] |
| Artemether |  |
E protein | Docking server CB-Dock, Automated cavity-detection, AutoDock Vina, MD by GROMACS | [56] |
| Gliclazide | 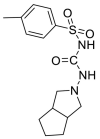 |
E protein | Experimental test | [71] |
| Memantine | 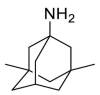 |
E protein | Experimental test | [71] |
| Wortmannin | 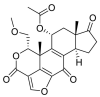 |
E protein | HDOCK web(PPD), Swiss dock web-interface, MD by Nanoscale molecular dynamics (NAMD) | [72] |
| Veliparib | 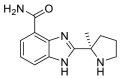 |
E protein | Schrodinger Glide software in SP mode | [73] |
| Rimantadine | 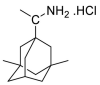 |
E protein | DOCK6 program, MD by GROMOS | [74,75] |
| Nimbolin | 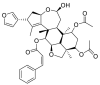 |
E protein | AutoDock Vina, MD by GROMACS | [76] |
| 7-Deacetyl-7-benzoylgedunin | 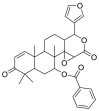 |
E protein | AutoDock Vina, MD by GROMACS | [76] |
| 24-Methylenecycloartanol | 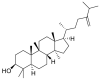 |
E protein | AutoDock Vina, MD by GROMACS | [76] |
| Plerixafor | 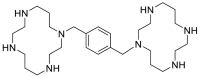 |
E protein | – | [77] |
| Mebrofenin | 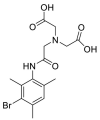 |
E protein | – | [77] |
| Kasugamycin (hydrochloride hydrate) | 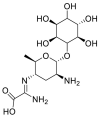 |
E protein | – | [77] |
| Saroglitazar magnesium | 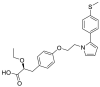 |
E protein | – | [77] |
7. Structure of PLpro
Osipiuk et al. reported the crystal structure of SARS-papain-like CoV-2's protease (PLpro) for the first time (PDB ID: 6W9C). The largest protein among all nsps, PLpro, is a multi-domain enzyme with 1945 amino acids [78]. PLpro is, in fact, one of the functional domains of nsp3. PLpro is the catalytically active domain having nine residues including Leu185, Arg166, Leu199, Glu203, Val202, Lys232, and Met206-Met 208. It seems that inhibiting the activity of PLpro will interfere with the replication cycle of the virus and reduce the infection rate because PLpro participates in the efficient cleavage of the N-terminal replicase polyproteins to produce functional proteins which play an essential role in maintaining the basic cellular process of SARS-COV-2 including viral replication [79].
PLpro can help the virus escape from the host antiviral immune response because PLpro is also implicated in cleaving proteinaceous post-translational modifications on host proteins as an evasion mechanism against host antiviral immune responses [80,81].
8. Compounds affecting on PLpro
PLpro active site contains a classic catalytic triad, composed of Cys112-His273-Asp287. The study confirmed by Delre et al. the covalent inhibitors like curcumin and afatinib, and non-covalent inhibitors like dasatinib, pexidartinib, and copanlisib (protein kinase inhibitors), amprenavir, indinavir, anagliptin, boceprevir and semagacestat (protease inhibitors), vilanterol, arformoterol, and atenolol (adrenergic receptor modulators), cilazaprilat, edoxaban and rivaroxaban (direct oral anticoagulants), ACE inhibitors, acotiamide, bentiromide, lymecycline, canagliflozin, darolutamide, lafutidine, vilazodone, and methotrexate were identified as PLpro inhibitors. It is noteworthy that the covalent inhibitor binds irreversibly to the receptor, while the non-covalent inhibitor binds reversibly to the receptor. Also, the tendency of a covalent inhibitor for binding to a target is stronger than that of a non-covalent inhibitor [82]. However, most covalent inhibitors are less attractive due to adverse drug responses, off-target side effects, toxicity, and lower potency but targeted covalent drugs have recently gained more attention. These drugs target a non-catalytic nucleophile that is unique for each target protein in contrast to the catalytic nucleophile in mechanism-based or suicide inhibitors [83,84].
For example, two thiocarbonyl-containing compounds, 6 mercaptopurine(6MP), and 6 thioguanine (6TG) were found to be slow-binding, competitive, reversible, and selective inhibitors of SARS-CoV PLpro [[85], [86], [87]]. Both 6MP and 6TG fit well into the active-site cavity of PLpro. The sulfur atom of 6MP or 6TG was juxtaposed closely with the γ-S of Cys1651 at a distance of 3.4 Å, suggesting the possible formation of a hydrogen bond. Surprisingly, in this reaction sulfur is not only a potential H-bond acceptor, but also is also a very good H-bond donor and capable of forming of H-bond [88,89].
On the other hand, the substrate-analog hexapeptidyl chloromethyl ketone (CMK) is irreversible peptidomimetic structure which have mostly five residues in length. Thiolate anion of the catalytic Cys145 residue attacks chloromethyl ketone moiety of CMK and forms a covalent bond (Fig. 4 ) [90]. The peptidic inhibitors exhibit good potency but poor pharmacokinetic profiles [84].
Fig. 4.

The reaction mechanism between the Thiolate anion of the catalytic Cys145 and the chloromethyl ketone moiety of CMK.
Indeed, the peptidic inhibitors such as Cm–FF–H, which have a highly electrophilic aldehyde group in their structure, prefer to be attacked by nucleophiles through catalytic Cys145 to form a thiohemiacetal (Fig. 5 ) [91].
Fig. 5.

The reaction mechanism between catalytic Cys145 and Cm–FF–H.
Moreover, it was suggested that inhibition of PLpro may occur through the formation of a covalent bond with the active site cysteine and α-β unsaturated carbonyl group in the inhibitor. Michael's addition reaction with the β‑carbon of the vinyl group of the vinylmethyl ester warheads from VIR251 results in the formation of a covalent thioether bond [92,93]. Importantly, the ability of unsaturated ketone to interact, via a Michael's addition reaction, with a cysteine residue has been well-documented [94]. Afatinib contains Michael acceptor group rendering it covalently reactive to a specific cysteine residue within the catalytic cleft (Fig. 6 ) [83].
Fig. 6.

The reaction mechanism between Plpro and Afatinib Michael's acceptor group.
A molecular docking study confirmed that the boronic acid moieties in peptidyl boronic acid such as Bortezomib can accept an electron pair from serine residues in Plpro to undergo the chemical reaction (Fig. 7 ). Although boron interacts with sulfur atom only weakly [84,[95], [96], [97]].
Fig. 7.

A chemical reaction between peptidyl boronic acid and serine residues in Plpro.
Also, we can see in Fig. 8 that electrophilic nitriles in nitrile containing antidiabetic gliptins can react with serine and cysteine moiety and finally can produce imidate or thioimidate [98].
Fig. 8.

The chemical reaction between electrophilic nitriles in nitrile-containing and serine and cysteine moiety.
The naphthalene-based inhibitors are potent, competitive inhibitors and bind within the active site of PLpro. Because of T-shaped π–π interaction with the naphthalene group of the inhibitor as well as other van der Waals interactions [84,99,100]. The peptidic inhibitors' drawbacks such as poor pharmacokinetic profiles and in vivo instability and cell membrane impermeability unappropriate PK profiles should be kept in mind when converting these to drug molecules [84]. It seems that a combination of peptide compounds that establish covalent bonds with Plpro and naphthalene-based compounds can be considered promising Plpro inhibitors.
Disulfiram is Zn-ejector drug that can target highly conserved Zn2+-binding and/or catalytic cysteines [101]. As it is shown below in Fig. 9 .
Fig. 9.

The chemical reaction mechanism between Zn-ejector drug and catalytic cysteines.
Different blockers of SARS-CoV-2 PLpro, their chemical structures, and the software used for docking are shown in Table 4 .
Table 4.
Comparison of different blockers for PLpro of SARS-CoV-2, blockers' structures, and software used for docking.
| Compound | Chemical structure | Target | Software | Ref. |
|---|---|---|---|---|
| 6TG | 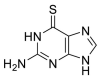 |
PLpro | – | [85] |
| 6MP | 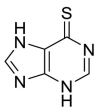 |
PLpro | – | [85] |
| CMK | 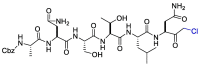 |
PLpro | – | [84] |
| Cm–FF–H | 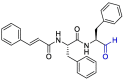 |
PLpro | – | [84] |
| VIR251 | 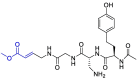 |
PLpro | OPLS force field, CovDock, Maestro (Schrödinger LLC) | [82] |
| F403_0159 | 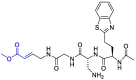 |
PLpro | OPLS force field, CovDock, Maestro (Schrödinger LLC) | [82] |
| Curcumin | 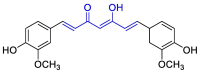 |
PLpro | OPLS force field, CovDock, Maestro (Schrödinger LLC) | [82] |
| Afatinib | 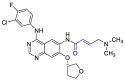 |
PLpro | OPLS force field, CovDock, Maestro (Schrödinger LLC) | [82] |
| Anagliptin | 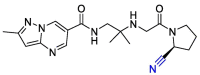 |
PLpro | OPLS force field, CovDock, Maestro (Schrödinger LLC) | [82] |
| Bortuzomib | 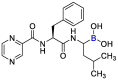 |
PLpro | – | [84] |
| Disulfiram | 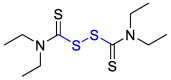 |
PLpro | – | [101] |
| Lopinavir | 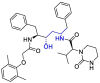 |
PLpro | – | [102] |
9. Structure of Mpro
The main protease (Mpro) of SARS-CoV-2 is a key enzyme and has a pivotal role in mediating viral replication and transcription, making it an attractive drug target for SARS-CoV-2 [103]. Mpro, a cysteine protease, mediates the maturation cleavage of polyproteins during virus replication. Understanding the atomic-level mechanism of the peptide cleavage, catalyzed by cysteine proteases, is crucial for designing structure-based potent inhibitors.
The protease-susceptible sites in a given protein or peptide usually extend to an octapeptide region in Mpro. Occasionally, the susceptible sites in some proteins may contain one subsite less or more. However, eight amino acid residues are the most common causes. Although the protein being cleaved contains much more than eight amino acid residues, usually only the segment of an octapeptide fits and extends in the active site region of Mpro. Therefore, drug design will focus on the cleavability of an octapeptide. First, the octapeptide fits well in binding to the Mpro active region leading to cleavage at its scissile bond, then, Gly143 of Mpro forms hydrogen bonds with ligands [104].
The imidazole group of histidine polarizes and activates the SH group of the cysteine and forms a CysS−/HisH+ ion (His-Cys catalytic dyad) that has a high nucleophilic property that reacts with substrates [105]. Cysteine attacks the carbonyl carbon atom of the octapeptide after which the proton from the protonated HisH+ is transferred to the nitrogen atom of the scissile peptide bond. Therefore, the π bonding is broken and the covalent adduct is generated. Then the deacetylation stage is supposed to be assisted by a water molecule activated by histidine (Fig. 10 ) [104,106,107].
Fig. 10.

The reaction mechanism between His-Cys catalytic dyad with substrates.
According to the above, it seems that the design of peptidomimetic α-keto amid compounds is a synthetic strategy to produce broad-spectrum Inhibitors of SARS-CoV-2. The α-ketoamide is an unusual reactive proelectrophile and pronucleophile moiety, displaying two possible nucleophilic reaction sites together with two electrophilic centers [108]. α-keto amid compounds match the H-bonding donor/acceptor properties of the catalytic center by offering two hydrogen-bond acceptors instead of one better than another compound such as aldehydes, α,β-unsaturated esters, and Michael acceptors [109]. In fact, the α-keto group of the compounds forms hydrogen bonds in the catalytic dyad to establish, and then another carbonyl carbon atom is attacked [110]. Finally, it seems that all potent SARS-CoV-2 Mpro inhibitors contain α-ketoamide reactive warheads (boceprevir) [111])Fig. 11 ).
Fig. 11.

The α-ketoamide reactive warheads are present in potent SARS-CoV-2 Mpro inhibitors.
The α-ketoamide moiety has been considered for its ability to adjust the conformation of lead compounds by increasing or decreasing their structural rigidity or by conferring the capacity to establish hydrogen bonds, in order to improve their potency and pharmacokinetic profile (108). Studies showed that peptidomimetic structures can fill and extend in the catalytic site of the enzyme. Computational studies elucidated that the α-ketoamide moiety with bulk substitutions prefers to adopt a planar conformation, with the nitrogen center on the same plane of the two carbonyls disposed in trans conformation [108].
In sum; it seems that the reactive warheads might be essential for SARS-CoV-2 Mpro inhibition. Fluoromethyl ketones, Aziridinyl Peptide and Aza-peptide Epoxide are electrophilic building blocks that react with nucleophilic amino acids within the active site of proteases. In An aza-peptide epoxide; the epoxide ring of the inhibitor opens during Cys attacking, leaving a hydroxyl group on the C2 atom to form hydrogen bonds with the Asn142 of the protease and the P2-Phe carbonyl O atom of the inhibitor (Fig. 12 ). The configurations of the C2 and C3 atoms are inverted from S, S to R, R [112].
Fig. 12.

Fluoromethyl ketones, Aziridinyl Peptide and Aza-peptide Epoxide that react with nucleophilic amino acids within the active site of proteases.
In some compounds, aldehyde bisulfate warhead and vinyl sulfone were replaced with keto group (Fig. 13 ), indeed, instead of polarized nitrogen, there should be aromatic groups such as styrene not to break during the deacetylation reaction [111,113].
Fig. 13.

The reaction mechanism between vinyl sulfone and thiol moiety of cystein group.
On the other hand, some articles proposed that fluorinated ketone moiety could be utilized as a warhead for targeting proteases, because it forms a thermodynamically stable hemiketal or hemithioketal after nucleophilic attack by Ser-OH or Cys-SH residues, which are present in the active sites of serine or cysteine proteases, respectively [[114], [115], [116], [117], [118]]. (Fig. 14 ).
Fig. 14.

The reaction between fluorinated ketone and Ser-OH or Cys-SH to form a thermodynamically stable hemiketal or hemithioketal after nucleophilic attack.
Linlin Zhang et al. decided to use a 5-membered ring (γ-lactam) derivative of glutamine (henceforth called Gln Lactam) in all α-ketoamides. The aldehyde can react with the thiol moiety of cysteine. This moiety enhanced the electrophilic power of the inhibitors by up to 10-fold, most probably because, the more rigid lactam leads to a reduction of the loss of entropy upon binding to the target protease [110]. The studies showed that the γ-lactam ring can fill the S1 site of M pro.
To improve the half-life of the compound in plasma, the amide bond within needs to be modified with a pyridone ring. This might prevent cellular proteases from accessing this bond and cleaving it. This compound showed favorable pharmacokinetic results. It seems administration of nebulized pyridone-containing α-ketoamides to the lungs would be possible [109]. In a research, to increase the solubility of the compound in plasma and to reduce its binding to plasma proteins, they added hydrophobic groups for example they replaced the hydrophobic cinnamoyl moiety with the somewhat less hydrophobic tert-Butyloxycarbonyl protecting (Boc) group. The studies showed that Boc group is necessary to enter the cells. Cyclohexyl moiety in these structures can fill the S2 pocket and produce broad-spectrum inhibitors. Indeed 3 fluro-phenyl can be replaced with cyclohexyl moiety and can increase lung tropism [116,119]. The synthetic strategy to produce inhibitors of SARS-CoV-2 Mpro is presented in Fig. 15 .
Fig. 15.

The synthetic strategy to produce the main protease (Mpro) inhibitors of SARS-CoV-2.
The α,β-epoxyketone tripeptide compounds inhibit Mpro via the S-alkylation of the active site cysteine and then the ring-opening reaction of epoxy ketones which produce both diastereomers of the targeted epoxy ketones (R and S) [120]. (Fig. 16 ).
Fig. 16.

Alkylation of the active site cysteine and then the ring-opening reaction of epoxy ketones.
10. Compounds affecting Mpro
The Mpro active site contains a catalytic dyad in which a cysteine residue (Cys145) acts as a nucleophile, and histidine residue (His41) acts as a base [121]. Cys145 is a chemical species that form bonds with electrophiles by donating an electron pair.
Cysteine thiol is a highly reactive moiety due to its high electron density and polarizability and can, therefore, be targeted by less reactive ligands i.e. cysteine-targeted electrophiles such as vinyl sulfones, epoxides, isothiazolones, and ketoamides.
It seems that ketoamide groups of boceprevir and telaprevir can react covalently with cysteine thiol moiety of COVID as well as Hepatitis C virus (HCV) protease which is also targeted covalently by these drugs [122].
Indeed, cyclopropanesulfonamide moiety in Paritaprevir and Simeprevir can be targeted by cysteine residue (Cys145) [123]. In a study conducted by Duc Duy Nguyen et.al, They concluded that the strongest bond is formed between the boronic acid moiety of bortezomib(lewis acid) and Cys 145(base) [124].
On the other hand, it seems that Mpro histidine has a positively charged imidazole functional group. The unprotonated imidazole is nucleophilic and can serve as a general base. Thus, compound 621 can produce cation-π interaction with His 41 [125].
Abdusalam et.al in a virtual screening study recognized that a π -alkyl interaction between His 41 and the morpholine ring of ZINC03231196 was formed. Compound ZINC33173588 exhibited a π - π T-shaped bond with a first benzene ring and His41, in fact, two π -alkyl bonds were formed between amino acids Cys145, His41, and morpholine ring [126].
It was described that SARS-CoV-2 Mpro is a target for Atazanavir. Atazanavir could inhibit viral replication in cell culture models of infection that also prevented the release of a cytokine storm-associated mediators. The appropriate binding of Atazanavir, suggested by its lower energy score, may be related to its projected ability to form hydrogens bonds with the amino acid residues Asn142, His164, and Glu166 in Mpro [16].
Lopinavir, initially developed as an anti-HIV drug, inhibits the Mpro because of the analogy of the drug with the transition state of the hydrolysis reaction. Lopinavir is a substrate of the CYP3A4 cytochrome, thus it is physically combined with ritonavir, which is a suicide cytochrome inhibitor that acts by the metabolic generation of an isocyanate intermediate that then carbamoylates a nucleophilic residue at the cytochrome [102]. (Fig. 17 ).
Fig. 17.

The mechanism of action of Ritonavir and Lopinavir.
Different blockers of SARS-CoV-2 Mpro, their chemical structures, and software used for docking are shown in Table 5 .
Table 5.
Comparison of different blockers for Mpro of SARS-CoV-2, blockers' structures, and software used for docking.
| Compound | Chemical structure | Target | Software | Ref. |
|---|---|---|---|---|
| Telaprevir | 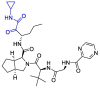 |
Mpro | MOE software package, MD by Schrödinger, Maestro software | [127] |
| Boceprevir | 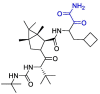 |
Mpro | MD by GROMACS and Schrödinger, AutoDock Vina | [128] |
| Beclabuvir | 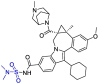 |
Mpro | Autodock vina & SMINA Pymol & Rasmol |
[1] |
| Compound 621 | 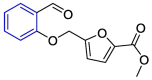 |
Mpro | AutoDock Vina, AMBER 18 simulation package | [3] |
| Paritaprevir | 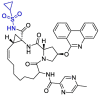 |
Mpro | AutoDock Vina, AMBER 18 simulation package | [3,12] |
| Simeprevir | 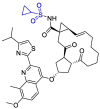 |
Mpro | AutoDock Vina, AMBER 18 simulation package | [3,11] |
| Atazanavir | 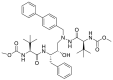 |
Mpro | Autodock tools | [16,129] |
| Efavirenz |  |
Mpro | AutoDock Vina | [130] |
| Grazoprevir |  |
Mpro | AutoDock Vina, MD by GROMACS | [131] |
| Ritonavir | 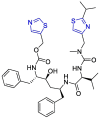 |
Mpro | mathematical pose (MathPose), mathemat ical deep learning (MathDL) | [14] |
| Bortezomib | 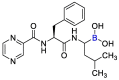 |
Mpro | mathematical pose (MathPose), mathemat ical deep learning (MathDL) | [14] |
| Carfilzomib | 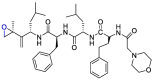 |
Mpro | Schrodinger, AMBER, Glide7 flexible docking program | [19] |
| Valrubicin | 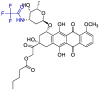 |
Mpro | Schrodinger, AMBER, Glide7 flexible docking program | [19] |
| Pitavastatin | 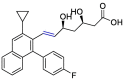 |
Mpro | PyMol, SMINA forked of AutoDock Vina | [47] |
| Perampanel | 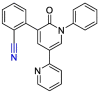 |
Mpro | PyMol, SMINA forked of AutoDock Vina | [47] |
| Curcumin | 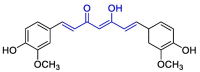 |
Mpro | Glide docking module of Maestro, OPLS3e force field, CovDock module of Schrödinger Suite, MD by GROMACS | [132] |
| ZINC03231196 | 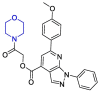 |
Mpro | Maestro software, MD by AMBER16 | [133] |
| Nirmatrelvir | 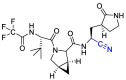 |
Mpro | [134] |
11. Structure of RdRp
The virus RdRp, which, as previously stated, plays an important role in the transcription and replication of the SARS-CoV-2, is known as nsp12 [135,136]. Other cofactors for nsp12 in polymerase activities are nsp7 and nsp8, and RdRp would not have much catalytic activity without them. As a result, the RdRp active unit is made up of an nsp7-nsp8 heterodimer, an nsp12 nucleus catalytic unit, and an extra nsp8 subunit [137].
The RdRp active site is composed of seven conserved catalytic motifs (motifs A to G). Motifs A to E is in the palm subdomain, while motifs F and G are in the finger subdomain [138]. The catalytic motif DX2-4D is found in motif A [139]. Also, the first aspartic acid 618 in this motif is invariant in most viral polymerases [140]. In motif B, there is a flexible loop that acts as a hinge to take the arrangement of configuration related to substrate binding and template RNA [140]. Residues phenylalanine753 to aspargine767 are Motif C which contains the catalytic motif SDD. Motif SDD is from residues serine759 to aspartic acid761 and is necessary for the attachment of the metal ion [139].
Aspartic acid760 and aspartic acid761 in the RdRp structures also are in charge of two magnesium ions' coordination at the catalytic center [140]. the conserved aspartic acids in catalytic motifs DX2-4D and SDD determine catalytic activity [141]. Residues from leucine544 to valine 557 are Motif F which interacts with the phosphate group of incoming NTP [140]. Also, residues from aspartic acid 499 to leucine 514 are Motif G that interacts with the RNA template strand and probably can take it to the active catalytic site [142]. It seems that the active catalytic motifs of the RdRp are highly conserved in most SARS-CoV-2 strains, therefore, RdRp is another promising drug target [140].
12. Compounds affecting on RdRp
Remdesivir (RDV, GS-5734) is a pro-drug that is a 1′-Ribose cyano substitution of adenosine nucleotide analog. Its adenine fragment is modified, and The presence of a cyano group at the anomeric carbon is unusual, and it leads to a lower activity on mammal RNA polymerases [102]. Upon entering the body, Remdesivir is hydrolyzed and phosphorylated through metabolism, Remdesivir monophosphate (RDV-MP) is its active form of Remdesivir with a negatively charged and polar monophosphate moiety, actually, this monophosphate increases the active triphosphate metabolite (RDV-TP) [143,144]. The salt bridge interaction occurred between the residues of the basic amino acids lysine545 and Arginine 555 in RdRp in motif F with RDV-MP. There are two ionic bond interactions between magnesium ions and pyrophosphate near RDV-MP that their density is not present in other structures of SARS-CoV-2 RdRp-RNA complexes. This pyrophosphate may obstruct nucleotide three phosphates (NTP) entry into the active site by occupying the nucleotide input. The 10-cyano substituent of incorporated RDV sterically clashed with the side chain of serine 861, which inhibits the chain termination reaction [145].
RMP interacts with the upstream bases from the primer chains to form base stacking and forms two hydrogen bonds with the uridine groups from the template chains [145]. Thus, it seems that modifying the structures to make phosphate more favorable for covalent incorporation at the extending strand terminus and designing some analogous pyrophosphate bound with other functionalities such as fluorine functional groups to improve its hydrogen bond acceptivity can cause the compounds to interact with RdRp more favourably. According to virtual screening studies, the presence of tetrazole or phenylpyrazole groups in the designed drugs increases the chances of inhibiting RdRp [146]. An investigation confirmed Aryl di-keto acid as a specific and reversible inhibitor of RdRp of HCV. They demonstrated that Aryl di-keto acids, like pyrophosphate mimetic inhibitors, act as product-like analogues and chelate the two divalent cations (Mg2+ ions) at the active site of HCV. Due to the chemical and biological instability and poor membrane permeability of diketo acid group, they replaced the free carboxylic acid of Aryl di-keto acids with their bioisosteres triazoles (like ribavirin) or tetrazoles. Although a similar replacement of carboxylic acid with triazole or tetrazole was successfully observed in the research of integrase as anti-HIV agents [147]. (Fig. 18 ).
Fig. 18.

Similar replacement of carboxylic acid with triazole.
The ionizable groups of these compounds are blocked by lipophilic moieties, allowing a good membrane permeability [102]. Different blockers of SARS-CoV-2 RdRp, their chemical structures, and the software used for docking are shown in Table 6 .
Table 6.
Comparison of different blockers for RdRp of SARS-CoV-2, blockers' structures, and software used for docking.
| Compound | Chemical structure | Target | Active form | Software | Ref. |
|---|---|---|---|---|---|
| IDX-184 | 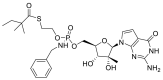 |
RdRp | 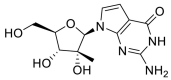 |
AutoDock Vina implemented in SCIGRESS | [2] |
| Sofosbuvir | 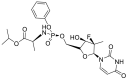 |
RdRp | 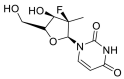 |
AutoDock Vina implemented in SCIGRESS | [2] |
| Ribavirin | 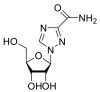 |
RdRp | – | AutoDock Vina implemented in SCIGRESS | [2] |
| Remdesivir | 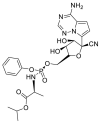 |
RdRp | 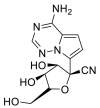 |
AutoDock Vina, RosettaCommons AutoDock Vina, RosettaCommons | [4] |
13. Discussion
Several vaccines have been developed and approved to combat SARS-CoV-2, but their effectiveness varies by individual, and after a few months, some people's immunity deteriorates, increasing their risk of COVID-19 infection. Furthermore, by arising new strains, many people all over the world become infected with this virus every day. As a result, scientists are still researching various virus proteins in order to better understand their function as well as the inhibitory effect of various synthetic or herbal compounds on these proteins [148].
In this article, we first review the latest life cycle findings of the SARS-CoV-2 and its important proteins that contribute to infection. Then, we surveyed the various structures that could have pharmaceutical activity and tried to find a structure-activity relationship (SAR).
To study the various structure against SARS-CoV-2, the molecular docking approaches have been applied to model the interaction between small molecules and various proteins of SARS-CoV-2 at the atomic level to characterize the behavior of different molecules such as repurposed drugs in the binding site of target proteins and to elucidate the ligand conformation, its docking pose, and assessment of the binding affinity [149]. As many of these studies were performed with different molecular docking software, the amount of binding energy could not be compared precisely. Also, many drugs showed an appropriate docking pose, but may not turn into their active structure in the body. Indeed, in vitro and in vivo studies are also required to know if the compounds that have been identified can inhibit COVID19 infection in a human host.
One approach to treat COVID-19 is regulating the spike-ACE2 complex by the negative allosteric modulators. [150,151]. In the beginning, SARS-CoV-2 RBD–ACE2 interface was investigated precisely. Then, studies were investigated to find some molecules that can compete with the spike to attach to the receptor. Antihypertensive drugs that block ACE receptors are the most well-known of these drugs. According to many studies, three different docking methods have been used on the receptor, but it has yet to be determined which docking method is the most appropriate. It seems all these approaches noticed N-linked glycan at position Asn 90 of ACE2 bind to different RBDs and interfere with the infection of the cell. But they ignored several key residues of ACE2 (such as lysine and histidine) that may affect the hydrogen bonding and salt bridge interaction with the spike protein of SARS-CoV-2.
Consequently, as mentioned, a main-chain hydrogen bond between the carbonyl of ACE2 Lys353 to the amide of RBD Gly488 fixes the relative positions of receptor and spike protein quite accurately. Thus, it seems the designing of the compounds which bind to the allosteric site and modify hydrogen bonds or the salt bridges in the receptor-binding domain can disrupt SARS-CoV2 RBD –ACE2 complex and is a hopeful approach to inhibit the infectiousness of SARS-COV-2.
The compounds which affected the voltage-gated ion channel of influenza and NMDA receptors seem to be effective on the channel structures of SARS-CoV-2, allowing drugs like adamantine derivative to block channels in the virus envelope. It seems that the structures with amino heads and hydrophobic tails can block envelope protein, and amine heads can react with hydrogen and metal ions to produce quaternary amines, and then, they are placed at the envelope's gate, while hydrophobic tails can block the receptor.
Also, according to the virtual screening studies in data banks, siderophores have been identified as a potential channel blocker for the envelope of SARS-CoV-2 because they have the ability to chelate the metal ions. As mentioned above, Heme-linked sites of the E protein (Cys44) may be relevant to the high infectivity [69]. Desferrioxamine and nocardamine are siderophore iron chelators that can chelate Ferric iron at a 1:1 stoichiometry to inhibit iron to bind to the heme‑iron linked site of E. Indeed, a predictive quantum mechanical TD-DFT method should be needed to describe the time-dependent behavior of these inhibitors [70].
It seems the synthesis of a naphthalene-based scaffold which can covalently bind to cysteine would be an effective strategy to antagonize the PLpro structure. Docking studies on naphthalene-based structures or their isosteres have shown that they can create a π-π interaction with the PLpro structure. Cysteine is an important amino acid in the PLpro structure because it attacks the positive charge centers in drugs, allowing cysteine to participate in nucleophilic reactions, addition, and the formation of a covalent bond between the drug and the receptor [152].
Regarding the Mpro's active site, it can be occupied by octapeptide compounds or linear compounds with a similar fitting ability to octapeptide compounds. Mpro also has a flexible structure that allows it to accommodate compounds as large as one or more amino acids. The positive charge center of the peptide structure is on the carbonyl groups. To increase the positive charge on carbon, pharmacodynamic studies have been conducted extensively. For example the carbonyl groups could be replaced with vinyl sulfone, aldehyde bisulfate, or fluorinated ketone groups. In fact, gamma-lactam rings, for example, are frequently substituted for carbonyl groups. Besides, some studies, have attempted to design drugs by adding non-polar and aromatic groups to the compounds in order to reduce the amount of drug-protein binding in the blood and the volume of distribution in the body while also increasing the drug's tropism within the lungs [153]. It is worth noting that virtual screening of potential Mpro inhibitors has also confirmed that Lewis acid structures such as bortezomib, which contains boron metal bind to M-protein with ease [154].
Various studies have shown that a key difference between RdRp and other targets is that RdRp is more polar, and many drugs must make a salt bridge with RdRp active site amino acids. Thus, the pKa of the desired compound is important to work in the active site because these compounds need to take hydrogen ions at this pH, so they can't enter the active site and form a salt bridge or hydrogen bond. Another point to consider is that the pKa of the designed drugs should be regulated to comply with patients; for example, extravasations of remdesivir into the perivascular environment due to its acidic structure will cause erythema in the vessels and skin of patients [146,155]. Therefore, in a previous research, Aryl di-keto acid was introduced as the inhibitor of RdRp of HCV and integrase enzyme of HIV. They can chelate the two divalent cations (Mg2+ ions) at the active site of RdRp. Due to their poor pharmacokinetic ability, they replaced the free carboxylic acid of Aryl di-keto acids with their bioisosteres triazoles or tetrazoles. Thus it is recommended to test the compounds introduced by Wo Hui song et al. on SARS-CoV-2 [147].
Remdesivir was the first FDA-approved drug for the treatment of SARS-CoV-2 but its effectiveness is disrupted demonstrating the need to develop a new antiviral drug. After much surveying, FDA authorized molnupiravir, paxlovid (nirmatrelvir and ritonavir), and baricitinib for emergency treatment against SARS-CoV-2 infection. Molnupiravir is an iso propyl ester prodrug of hydroxycitidine (NHC triphosphate) which target RdRp of SARS-COV-2. It actually causes an error catastrophe during viral replication [156]. The studies showed that NHC tautomers (keto-oxime, keto-hydroxylamine, and hydroxyl-oxime) through the migration of two acidic protons of the hydroxylcytosine fragment can form stable base pairs either M-G or M-A in the RdRp active center using intramolecular hydrogen bonds, explaining how the polymerase escapes proofreading and synthesizes mutated RNA; Although, the crystal structure of molnupiravir has not been reported so far, this can also be explained by intertautomer transformation of molnupiravir in solutions [156]. Furthermore, it was demonstrated that the keto-oxime tautomer of NHC interacts with Mpro through seven hydrogen bonds formed with GLY143, SER144, CYS145, HIS163, LEU141, and GLN186, two alkyl interactions with MET165, and two π-system⋯alkyl interactions with HIS41 and CYS145. The keto-hydroxylamine forms complex with Mpro due to ten hydrogen bonds with GLY143, HIS163, GLU166, LEU141, SER144, MET165, and GLN198; two alkyl interactions with MET49 and MET165; and three π-system⋯alkyl interactions with HIS41 and CYS145. Finally, the hydroxyl-oxime tautomer of molnupiravir interacts with the Mpro via four hydrogen bonds with CYS145, LEU141, and PHE140, two alkyl interactions with MET49 and MET165, and three π-system⋯alkyl interactions with HIS41 and CYS145. It seems that one of the π-system⋯alkyl interactions for all the tautomers with Mpro is formed by the π-system of the ligands [157]. (Fig. 19 ).
Fig. 19.

Molnupiravir tautomers (keto-oxime, keto-hydroxylamine, and hydroxyl-oxime) and its itramolecular hydrogen bonding.
Recently, the combination therapy of Janus kinase (JAK) inhibitor baricitinib has been granted emergency use authorization for COVID-19 hospitalized patients [158]. Emanuelle Machado Marinho et.al in their study showed that baricitinib can interact powerfully with the Mpro. The rationale for the use of these molecules for the treatment of COVID-19 is connected to the role of the JAK-STAT signaling pathway in the biosynthesis of cytokines [102] Indeed Regarding the docking pose of baricitinib and Mpro complex, the ligand has ten interactions with the amino acid residues of the enzyme, three of the conventional hydrogen bond type; one with Lys137 (2.66 Å), one with Asp197 (2.42 Å) and one with Leu287 (2.48 Å); one van der Waals interaction with Thr199; two of the carbon‑hydrogen bond type, one with Leu287 and the other with Aps289; two interactions of the π -Cation type with Arg131; a π -Anion interaction with Asp289 and interaction of the Amide- π stacked type with Thr198. They used the modern concept of hydrogen bonds [159]. Despite the conventional constant of hydrogen bond, the nature of the interaction is not constant, but its electrostatic, covalent, and dispersion contributions vary in their relative weights. The hydrogen bond has broad transition regions that merge continuously with the covalent bond, the van der Waals interaction, the ionic interaction, and also the cation–π interaction [160]. According to this theory, the hydrogen bond length of 2.2–2.5 Å is indicated as a strong covalent bond. They showed the interaction of baricitinib (and Asp197 and Leu287classified as hydrogen bonds strongly covalent because they have a bond lengths up to 2.5 Å. It was shown that pyrrolopyrimidine and ethanesulfonyl moiety of baricitinib play an important role to produce hydrogen bond interaction [159] (Fig. 20 ).
Fig. 20.

The chemical structure of baricitinib and its pyrrolopyrimidine moiety.
Water-soluble phosphate prodrug i.e. Lufotrelvir and its active form PF-00835231 were introduced as the Mpro inhibitor. This compound was replaced by its orally active analog nirmatrelvir. Which, in combination with ritonavir used by the FDA in an emergency. PF-00835231 and nirmatrelvir interact covalently, although reversibly, with the Cys-145 residue of mpro. In the case of PF-00835231, the Cysteine thiol group forms a covalent bond with the ketone group of the terminal α-hydroxyketone moiety to give a hydrogen bond-stabilized hemithioacetal, together with additional hydrogen bonds with a number of other key residues. Nirmatrelvir occupies the active site in a similar way, with Cys-145 binding covalently to its nitrile group via a Pinner-like reaction and several hydrogen bonds [102,161]. (Fig. 21 ).
Fig. 21.

Design of nirmatrelvir instead of water-soluble phosphate prodrug i.e. Lufotrelvir.
Finally, it can be concluded that one of the challenges in designing drugs is that the coronavirus has genetic mutations and drug resistance that cause the treatment protocols to change easily [162]. Therefore, it would be best to add the appropriate pharmacophores to a new molecule considering the structures and points mentioned for each protein, and then, design a new hybrid molecule to increase the drug's effectiveness against genetic mutations [163,164].
For example, hydroxychloroquine and adamantane based hybrids were synthesized by Lars Herrmann et.al, but due to the QT-prolongation problems; hydroxychloroquine hybrids couldn't be considered. Indeed they have shown the positive effects of Artemisinin on SARS-CoV-2. So the hybrid based on Artemisinin derivatives, Quinoline, or adamantane moieties was synthesized [163,165]. Other studies have shown the anti-viral activity of isothiourea, and adamantane derivatives. in 2017; the isothiourea and adamantane based hybrids were synthesized but their activity against SARS-CoV-2 has not been measured yet [166]. Further; as mentioned above the active form of remdesivir showed high polarity so to improve their pharmacokinetic profile it could be better to fuse the remdesivir active form with other pharmacophores in a single multi-functional agent.
Declaration of Competing Interest
None.
References
- 1.Reyana A., Kautish S. Corona virus-related disease pandemic: a review on machine learning approaches and treatment trials on diagnosed population for future clinical decision support. Curr. Med. Imaging. 2022;18(2):104–112. doi: 10.2174/1573405617666210414101941. [DOI] [PubMed] [Google Scholar]
- 2.Khan M., Adil S.F., Alkhathlan H.Z., Tahir M.N., Saif S., Khan M., et al. COVID-19: a global challenge with old history, epidemiology and progress so far. Molecules. 2021;26(1):39. doi: 10.3390/molecules26010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones T.C., Biele G., Mühlemann B., Veith T., Schneider J., Beheim-Schwarzbach J., et al. Estimating infectiousness throughout SARS-CoV-2 infection course. Science. 2021;373(6551):eabi5273. doi: 10.1126/science.abi5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srivastava S., Banu S., Singh P., Sowpati D.T., Mishra R.K. SARS-CoV-2 genomics: an Indian perspective on sequencing viral variants. J. Biosci. 2021;46(1):1–14. doi: 10.1007/s12038-021-00145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahmoudi S., Balmeh N., Mohammadi N., Sadeghian-Rizi T. The novel drug discovery to combat COVID-19 by repressing important virus proteins involved in pathogenesis using medicinal herbal compounds. Avicenna J. Med. Biotechnol. 2021;13(3):107. doi: 10.18502/ajmb.v13i3.6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winstone H., Lista M.J., Reid A.C., Bouton C., Pickering S., Galao R.P., et al. The polybasic cleavage site in SARS-CoV-2 spike modulates viral sensitivity to type I interferon and IFITM2. J. Virol. 2021;95(9) doi: 10.1128/JVI.02422-20. e02422-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rao P., Shukla A., Parmar P., Rawal R.M., Patel B., Saraf M., et al. Reckoning a fungal metabolite, Pyranonigrin A as a potential Main protease (Mpro) inhibitor of novel SARS-CoV-2 virus identified using docking and molecular dynamics simulation. Biophys. Chem. 2020;264 doi: 10.1016/j.bpc.2020.106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahn D.-G., Shin H.-J., Kim M.-H., Lee S., Kim H.-S., Myoung J., et al. 2020. Current Status of Epidemiology, Diagnosis, Therapeutics, and Vaccines for Novel Coronavirus Disease 2019 (COVID-19) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hebbani A.V., Pulakuntla S., Pannuru P., Aramgam S., Badri K.R., Reddy V.D. COVID-19: comprehensive review on mutations and current vaccines. Arch. Microbiol. 2022;204(1):1–17. doi: 10.1007/s00203-021-02606-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y., Gayle A.At., Wilder-Smith A., Rocklöv J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J. Travel Med. 2020;27(2) doi: 10.1093/jtm/taaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biswas S.K., Mudi S.R. Genetic variation in SARS-CoV-2 may explain variable severity of COVID-19. Med. Hypotheses. 2020;143 doi: 10.1016/j.mehy.2020.109877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ita K. Coronavirus disease (COVID-19): current status and prospects for drug and vaccine development. Arch. Med. Res. 2021;52(1):15–24. doi: 10.1016/j.arcmed.2020.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sood S., Bhatia G.K., Seth P., Kumar P., Kaur J., Gupta V., et al. Efficacy and safety of new and emerging drugs for COVID-19: favipiravir and dexamethasone. Curr. Pharmacol. Rep. 2021;7(2):49–54. doi: 10.1007/s40495-021-00253-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balmeh N., Mahmoudi S., Mohammadi N., Karabedianhajiabadi A. Predicted therapeutic targets for COVID-19 disease by inhibiting SARS-CoV-2 and its related receptors. Inform. Med. Unlocked. 2020;20 doi: 10.1016/j.imu.2020.100407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fintelman-Rodrigues N., Sacramento C., Lima C., da Silva F., Ferreira A., Mattos M. Atazanavir inhibits SARS-CoV-2 replication and pro-inflammatory cytokine production. 2020. https://www.biorxiv.org/content/101101/202004.4:v2 Preprint available from. [DOI] [PMC free article] [PubMed]
- 17.Gil C., Ginex T., Maestro I., Nozal V., Barrado-Gil L., Cuesta-Geijo M.Á., et al. COVID-19: drug targets and potential treatments. J. Med. Chem. 2020;63(21):12359–12386. doi: 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- 18.Prajapat M., Sarma P., Shekhar N., Avti P., Sinha S., Kaur H., et al. Drug targets for corona virus: a systematic review. Indian J. Pharmacol. 2020;52(1):56–65. doi: 10.4103/ijp.IJP_115_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shamsi A., Mohammad T., Anwar S., Amani S., Khan M.S., Husain F.M., et al. Potential drug targets of SARS-CoV-2: from genomics to therapeutics. Int. J. Biol. Macromol. 2021;177:1–9. doi: 10.1016/j.ijbiomac.2021.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 21.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 22.Basu A., Sarkar A., Maulik U. Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci. Rep. 2020;10(1):17699. doi: 10.1038/s41598-020-74715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J., Xiao T., Cai Y., Lavine C.L., Peng H., Zhu H., et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science. 2021;374(6573):1353–1360. doi: 10.1126/science.abl9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomson E.C., Rosen L.E., Shepherd J.G., Spreafico R., da Silva Filipe A., Wojcechowskyj J.A., et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell. 2021;184(5):1171–1187. doi: 10.1016/j.cell.2021.01.037. e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakraborty S. Evolutionary and structural analysis elucidates mutations on SARS-CoV2 spike protein with altered human ACE2 binding affinity. Biochem. Biophys. Res. Commun. 2021;534:374–380. doi: 10.1016/j.bbrc.2020.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu R., Li P. Screening of potential spike glycoprotein / ACE2 dual antagonists against COVID-19 in silico molecular docking. J. Virol. Methods. 2022;301 doi: 10.1016/j.jviromet.2021.114424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu C., Wang Y., Liu C., Zhang C., Han W., Hong X., et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. bioRxiv. 2020;7(2) doi: 10.1126/sciadv.abe5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W., Zhang C., Sui J., Kuhn J.H., Moore M.J., Luo S., et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24(8):1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao T., Lu J., Zhang J., Johnson R.I., McKay L.G.A., Storm N., et al. A trimeric human angiotensin-converting enzyme 2 as an anti-SARS-CoV-2 agent. Nat. Struct. Mol. Biol. 2021;28(2):202–209. doi: 10.1038/s41594-020-00549-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y., Zheng N., Hao P., Cao Y., Zhong Y. A molecular docking model of SARS-CoV S1 protein in complex with its receptor, human ACE2. Comput. Biol. Chem. 2005;29(3):254–257. doi: 10.1016/j.compbiolchem.2005.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu X., Mannar D., Srivastava S.S., Berezuk A.M., Demers J.P., Saville J.W., et al. Cryo-electron microscopy structures of the N501Y SARS-CoV-2 spike protein in complex with ACE2 and 2 potent neutralizing antibodies. PLoS Biol. 2021;19(4) doi: 10.1371/journal.pbio.3001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai Y., Zhang J., Xiao T., Lavine C.L., Rawson S., Peng H., et al. Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science. 2021;373(6555):642–648. doi: 10.1126/science.abi9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ragia G., Manolopoulos V.G. Inhibition of SARS-CoV-2 entry through the ACE2/TMPRSS2 pathway: a promising approach for uncovering early COVID-19 drug therapies. Eur. J. Clin. Pharmacol. 2020;76(12):1623–1630. doi: 10.1007/s00228-020-02963-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang G., Tan Z., Zhou L., Yang M., Peng L., Liu J., et al. Effects of angiotensin II receptor blockers and ACE (angiotensin-converting enzyme) inhibitors on virus infection, inflammatory status, and clinical outcomes in patients with COVID-19 and hypertension: a single-center retrospective study. Hypertension. 2020;76(1):51–58. doi: 10.1161/HYPERTENSIONAHA.120.15143. [DOI] [PubMed] [Google Scholar]
- 35.Sanchis-Gomar F., Lavie C.J., Perez-Quilis C., Henry B.M., Lippi G. Mayo Clinic Proceedings. Elsevier; 2020. Angiotensin-converting enzyme 2 and antihypertensives (angiotensin receptor blockers and angiotensin-converting enzyme inhibitors) in coronavirus disease 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Karmalawy A.A., Dahab M.A., Metwaly A.M., Elhady S.S., Elkaeed E.B., Eissa I.H., et al. Molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting the hACE2 receptor. Front. Chem. 2021;9(227) doi: 10.3389/fchem.2021.661230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khashkhusha T.R., Chan J.S.K., Harky A. ACE inhibitors and COVID-19: we don’t know yet. J. Card. Surg. 2020:1172–1173. doi: 10.1111/jocs.14582. 35(6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arcos F.S., Puche A.R., Vera T.V. Controversy regarding ACE inhibitors/ARBs in COVID-19. Revista Esp. Cardiol. (Engl. ed). 2020;73(6):516. doi: 10.1016/j.rec.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hippisley-Cox J., Young D., Coupland C., Channon K.M., San Tan P., Harrison D.A., et al. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: cohort study including 8.3 million people. Heart. 2020;106(19):1503–1511. doi: 10.1136/heartjnl-2020-317393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin-converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275(43):33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 41.Oz M., Lorke D.E., Kabbani N. A comprehensive guide to the pharmacologic regulation of angiotensin converting enzyme 2 (ACE2), the SARS-CoV-2 entry receptor. Pharmacol. Ther. 2021;221 doi: 10.1016/j.pharmthera.2020.107750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abdelli I., Hassani F., Bekkel Brikci S., Ghalem S. In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. J. Biomol. Struct. Dyn. 2021;39(9):3263–3276. doi: 10.1080/07391102.2020.1763199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khelfaoui H., Harkati D., Saleh B.A. Molecular docking, molecular dynamics simulations and reactivity, studies on approved drugs library targeting ACE2 and SARS-CoV-2 binding with ACE2. J. Biomol. Struct. Dyn. 2020;1-17 doi: 10.1080/07391102.2020.1803967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eweas A.F., Alhossary A.A., Abdel-Moneim A.S. Molecular docking reveals ivermectin and remdesivir as potential repurposed drugs against SARS-CoV-2. Front. Microbiol. 2021;11(3602) doi: 10.3389/fmicb.2020.592908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abdelli I., Hassani F., Bekkel Brikci S., Ghalem S. In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. J. Biomol. Struct. Dyn. 2021;39(9):3263–3276. doi: 10.1080/07391102.2020.1763199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Karmalawy A.A., Dahab M.A., Metwaly A.M., Elhady S.S., Elkaeed E.B., Eissa I.H., et al. Molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting the hACE2 receptor. Front. Chem. 2021;9 doi: 10.3389/fchem.2021.661230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Z., Yi F., Epplin M.P., Liu D., Summer S.L., Mizu R., et al. Negative allosteric modulation of GluN1/GluN3 NMDA receptors. Neuropharmacology. 2020;176 doi: 10.1016/j.neuropharm.2020.108117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Draper-Joyce C.J., Verma R.K., Michino M., Shonberg J., Kopinathan A., Klein Herenbrink C., et al. The action of a negative allosteric modulator at the dopamine D2 receptor is dependent upon sodium ions. Sci. Rep. 2018;8(1):1208. doi: 10.1038/s41598-018-19642-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Celı K.I., Onay-Besı Kcı A., Ayhan-Kilcigı L.G. Approach to the mechanism of action of hydroxychloroquine on SARS-CoV-2: a molecular docking study. J. Biomol. Struct. Dyn. 2021;39(15):5792–5798. doi: 10.1080/07391102.2020.1792993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Navya V.B., Hosur M.V. A computational study on hydroxychloroquine binding to target proteins related to SARS-COV-2 infection. Inform. Med. Unlocked. 2021;26 doi: 10.1016/j.imu.2021.100714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Celik I., Onay-Besikci A., Ayhan-Kilcigl L.G. Approach to the mechanism of action of hydroxychloroquine on SARS-CoV-2: a molecular docking study. J. Biomol. Struct. Dyn. 2021;39(15):5792–5798. doi: 10.1080/07391102.2020.1792993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chai J., Cai Y., Pang C., Wang L., McSweeney S., Shanklin J., et al. Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction protein PALS1. Nat. Commun. 2021;12(1):3433. doi: 10.1038/s41467-021-23533-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chernyshev A. 2020. Pharmaceutical Targeting the Envelope Protein of SARS-CoV-2: The Screening for Inhibitors in Approved Drugs. [Google Scholar]
- 54.Sandberg J. The State University of New Jersey-Camden; 2021. SARS-CoV-2 Envelope Protein Induces Membrane Curvature through Asymmetric Hydrophobic Mismatch: Rutgers. [Google Scholar]
- 55.Caillet-Saguy C., Durbesson F., Rezelj V.V., Gogl G., Tran Q.D., Twizere J.C., et al. Host PDZ-containing proteins targeted by SARS-CoV-2. FEBS J. 2021;288(17):5148–5162. doi: 10.1111/febs.15881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dey D., Borkotoky S., Banerjee M. In silico identification of tretinoin as a SARS-CoV-2 envelope (E) protein ion channel inhibitor. Comput. Biol. Med. 2020;127 doi: 10.1016/j.compbiomed.2020.104063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chai J., Cai Y., Pang C., Wang L., McSweeney S., Shanklin J., et al. Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction protein PALS1. Nat. Commun. 2021;12(1):1–6. doi: 10.1038/s41467-021-23533-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Toto A., Ma S., Malagrinò F., Visconti L., Pagano L., Stromgaard K., et al. Comparing the binding properties of peptides mimicking the envelope protein of SARS-CoV and SARS-CoV-2 to the PDZ domain of the tight junction-associated PALS1 protein. Protein Sci. 2020;29(10):2038–2042. doi: 10.1002/pro.3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mukherjee S., Bhattacharyya D., Bhunia A. Host-membrane interacting interface of the SARS coronavirus envelope protein: immense functional potential of C-terminal domain. Biophys. Chem. 2020;266 doi: 10.1016/j.bpc.2020.106452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fedorowski J.J. Could amantadine possibly interfere with COVID-19 vaccines based on LNP-mRNA platform? Arch. Med. Sci. 2021;17(3):827–828. doi: 10.5114/aoms/134716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilson L., McKinlay C., Gage P., Ewart G. SARS coronavirus E protein forms cation-selective ion channels. Virology. 2004;330(1):322–331. doi: 10.1016/j.virol.2004.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fink K., Nitsche A., Neumann M., Grossegesse M., Eisele K.-H., Danysz W. Amantadine inhibits SARS-CoV-2 in vitro. Viruses. 2021;13(4):539. doi: 10.3390/v13040539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jeppesen M.G., Toft-Bertelsen T.L., Kledal T.N., Rosenkilde M.M. 2020. Amantadin Has Potential for the Treatment of COVID-19 because it Targets Known and Novel Ion Channels Encoded by SARS-CoV-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baig A.M., Khaleeq A., Syeda H. Docking prediction of amantadine in the receptor binding domain of spike protein of SARS-CoV-2. ACS Pharmacol. Transl. Sci. 2020;3(6):1430–1433. doi: 10.1021/acsptsci.0c00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singh Tomar P.P., Arkin I.T. SARS-CoV-2 E protein is a potential ion channel that can be inhibited by Gliclazide and Memantine. Biochem. Biophys. Res. Commun. 2020;530(1):10–14. doi: 10.1016/j.bbrc.2020.05.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adil M.S., Narayanan S.P., Somanath P.R. Is amiloride a promising cardiovascular medication to persist in the COVID-19 crisis? Drug Discov. Ther. 2020;14(5):256–258. doi: 10.5582/ddt.2020.03070. [DOI] [PubMed] [Google Scholar]
- 67.Wilson L., Gage P., Ewart G. Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology. 2006;353(2):294–306. doi: 10.1016/j.virol.2006.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pervushin K., Tan E., Parthasarathy K., Lin X., Jiang F.L., Yu D., et al. Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog. 2009;5(7) doi: 10.1371/journal.ppat.1000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Read R. 2020. Flawed Methods in “COVID-19: Attacks the 1-beta Chain of Hemoglobin and Captures the Porphyrin to Inhibit Human Heme Metabolism”. [Google Scholar]
- 70.Fong C. Eigenenergy; Adelaide South Australia Australia: 2021. Inhibition of SARS-CoV-2 Envelope Ion Channel and the Relationship with the Membrane Dipole Potential. [Google Scholar]
- 71.Singh Tomar P.P., Arkin I.T. SARS-CoV-2 E protein is a potential ion channel that can be inhibited by gliclazide and memantine. Biochem. Biophys. Res. Commun. 2020;530(1):10–14. doi: 10.1016/j.bbrc.2020.05.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Selvaraj G., Kaliamurthi S., Peslherbe G.H., Wei D.-Q. Identifying potential drug targets and candidate drugs for COVID-19: biological networks and structural modeling approaches. F1000Res. 2021;10 doi: 10.12688/f1000research.50850.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y., Fang S., Wu Y., Cheng X., Zhang L.-k., Shen X.-r., et al. Discovery of SARS-CoV-2-E channel inhibitors as antiviral candidates. Acta Pharmacol. Sin. 2021:1–7. doi: 10.1038/s41401-021-00732-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Butterworth R.F. Potential for the repurposing of adamantane antivirals for COVID-19. Drugs R&D. 2021;21(3):267–272. doi: 10.1007/s40268-021-00351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bozdaganyan M.E., Orekhov P.S., Bragazzi N.L., Panatto D., Amicizia D., Pechkova E., et al. Docking and molecular dynamics (MD) simulations in potential drugs discovery: an application to influenza virus M2 protein. Am. J. Biochem. Biotech. 2014;10(3):180–188. [Google Scholar]
- 76.Borkotoky S., Banerjee M. A computational prediction of SARS-CoV-2 structural protein inhibitors from Azadirachta indica (Neem) J. Biomol. Struct. Dyn. 2021;39(11):4111–4121. doi: 10.1080/07391102.2020.1774419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tomar P.P.S., Krugliak M., Arkin I.T. Blockers of the SARS-CoV-2 3a channel identified by targeted drug repurposing. Viruses. 2021;13(3) doi: 10.3390/v13030532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Umar H.I., Siraj B., Ajayi A., Jimoh T.O., Chukwuemeka P.O. Molecular docking studies of some selected gallic acid derivatives against five non-structural proteins of novel coronavirus. J. Genet. Eng. Biotechnol. 2021;19(1):1–14. doi: 10.1186/s43141-021-00120-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yan F., Gao F. An overview of potential inhibitors targeting non-structural proteins 3 (PLpro and Mac1) and 5 (3CLpro/Mpro) of SARS-CoV-2. Computat. Struct. Biotechnol. J. 2021;19:4868–4883. doi: 10.1016/j.csbj.2021.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Osipiuk J., Azizi S.-A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12(1):1–9. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587(7835):657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Delre P., Caporuscio F., Saviano M., Mangiatordi G.F. Repurposing known drugs as covalent and non-covalent inhibitors of the SARS-CoV-2 papain-like protease. Front. Chem. 2020;8 doi: 10.3389/fchem.2020.594009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu C.-H., Chou C.-C., Tu H.-F., Huang W.-C., Ho Y.-Y., Khoo K.-H., et al. Antibody-assisted target identification reveals afatinib, an EGFR covalent inhibitor, down-regulating ribonucleotide reductase. Oncotarget. 2018;9(30):21512–21529. doi: 10.18632/oncotarget.25177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hosseini-Zare M.S., Thilagavathi R., Selvam C. Targeting severe acute respiratory syndrome-coronavirus (SARS-CoV-1) with structurally diverse inhibitors: a comprehensive review. RSC Adv. 2020;10(47):28287–28299. doi: 10.1039/d0ra04395h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen X., Chou C.Y., Chang G.G. Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir. Chem. Chemother. 2009;19(4):151–156. doi: 10.1177/095632020901900402. [DOI] [PubMed] [Google Scholar]
- 86.Ma C., Wang J. Validation and invalidation of SARS-CoV-2 papain-like protease inhibitors. ACS Pharmacol. Transl. Sci. 2021;5(2):102–109. doi: 10.1021/acsptsci.1c00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Klemm T., Ebert G., Calleja D.J., Allison C.C., Richardson L.W., Bernardini J.P., et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020;39(18) doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chou C.-Y., Chien C.-H., Han Y.-S., Prebanda M.T., Hsieh H.-P., Turk B., et al. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem. Pharmacol. 2008;75(8):1601–1609. doi: 10.1016/j.bcp.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou P., Tian F., Lv F., Shang Z. Geometric characteristics of hydrogen bonds involving sulfur atoms in proteins. Proteins. 2009;76(1):151–163. doi: 10.1002/prot.22327. [DOI] [PubMed] [Google Scholar]
- 90.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 91.Zhu L., George S., Schmidt M.F., Al-Gharabli S.I., Rademann J., Hilgenfeld R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antivir. Res. 2011;92(2):204–212. doi: 10.1016/j.antiviral.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S.J., et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti–COVID-19 drug design. Sci. Adv. 2020;6(42) doi: 10.1126/sciadv.abd4596. eabd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang H., Rao Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021;19(11):685–700. doi: 10.1038/s41579-021-00630-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gradišar H., Keber M.M., Pristovšek P., Jerala R. MD-2 as the target of curcumin in the inhibition of response to LPS. J. Leukoc. Biol. 2007;82(4):968–974. doi: 10.1189/jlb.1206727. [DOI] [PubMed] [Google Scholar]
- 95.Kouznetsova V.L., Zhang A., Tatineni M., Miller M.A., Tsigelny I.F. Potential COVID-19 papain-like protease PL(pro) inhibitors: repurposing FDA-approved drugs. PeerJ. 2020;8:e9965. doi: 10.7717/peerj.9965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Whyte G.F., Vilar R., Woscholski R. Molecular recognition with boronic acids-applications in chemical biology. J. Chem. Biol. 2013;6(4):161–174. doi: 10.1007/s12154-013-0099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Whyte G.F., Vilar R., Woscholski R. Molecular recognition with boronic acids-applications in chemical biology. J. Chem. Biol. 2013;6(4):161–174. doi: 10.1007/s12154-013-0099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Groves M., Domling A., Moreno A.J.R., Romero A.R., Neochoritis C., Velasco-Velázquez M. 2020. Gliptin Repurposing for COVID-19. [Google Scholar]
- 99.Báez-Santos Y.M., St. John SE, Mesecar AD. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bhati S. Structure-based drug designing of naphthalene based SARS-CoV PLpro inhibitors for the treatment of COVID-19. Heliyon. 2020;6(11):e05558. doi: 10.1016/j.heliyon.2020.e05558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sargsyan K., Lin C.-C., Chen T., Grauffel C., Chen Y.-P., Yang W.-Z., et al. Multi-targeting of functional cysteines in multiple conserved SARS-CoV-2 domains by clinically safe Zn-ejectors. Chem. Sci. 2020;11(36):9904–9909. doi: 10.1039/d0sc02646h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Menéndez J.C. Approaches to the potential therapy of COVID-19: a general overview from the medicinal chemistry perspective. Molecules. 2022;27(3):658. doi: 10.3390/molecules27030658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 104.Mengist H.M., Dilnessa T., Jin T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front. Chem. 2021;9 doi: 10.3389/fchem.2021.622898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Keillor J., Brown R. Attack of zwitterionic ammonium thiolates on a distorted anilide as a model for the acylation of papain by amides. A simple demonstration of a bell-shaped pH/rate profile. J. Am. Chem. Soc. 1992;114(21):7983–7989. [Google Scholar]
- 106.Du Q.-S., Wang S.-Q., Zhu Y., Wei D.-Q., Guo H., Sirois S., et al. Polyprotein cleavage mechanism of SARS CoV Mpro and chemical modification of the octapeptide. Peptides. 2004;25(11):1857–1864. doi: 10.1016/j.peptides.2004.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chou K.-C. Prediction of human immunodeficiency virus protease cleavage sites in proteins. Anal. Biochem. 1996;233(1):1–14. doi: 10.1006/abio.1996.0001. [DOI] [PubMed] [Google Scholar]
- 108.Robello M., Barresi E., Baglini E., Salerno S., Taliani S., Settimo F.D. The alpha keto amide moiety as a privileged motif in medicinal chemistry: current insights and emerging opportunities. J. Med. Chem. 2021;64(7):3508–3545. doi: 10.1021/acs.jmedchem.0c01808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science (New York, N.Y.) 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., et al. α-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020;63(9):4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 111.Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kuo C.J., Liang P.H. Characterization and inhibition of the main protease of severe acute respiratory syndrome coronavirus. ChemBioEng Rev. 2015;2(2):118–132. [Google Scholar]
- 113.Mellott D.M., Tseng C.-T., Drelich A., Fajtová P., Chenna B.C., Kostomiris D.H., et al. A cysteine protease inhibitor blocks SARS-CoV-2 infection of human and monkey cells. bioRxiv Preprint Server Biol. 2020 doi: 10.1021/acschembio.0c00875. 2020.10.23.347534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. [DOI] [PMC free article] [PubMed]
- 115.Cremer D., Gauss J. Theoretical determination of molecular structure and conformation. 20. Reevaluation of the strain energies of cyclopropane and cyclobutane carbon-carbon and carbon-hydrogen bond energies, 1, 3 interactions, and sigma-aromaticity. J. Am. Chem. Soc. 1986;108(24):7467–7477. doi: 10.1021/ja00284a004. [DOI] [PubMed] [Google Scholar]
- 116.Pillaiyar T., Wendt L.L., Manickam M., Easwaran M. The recent outbreaks of human coronaviruses: a medicinal chemistry perspective. Med. Res. Rev. 2021;41(1):72–135. doi: 10.1002/med.21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Amin S.A., Banerjee S., Singh S., Qureshi I.A., Gayen S., Jha T. First structure-activity relationship analysis of SARS-CoV-2 virus main protease (Mpro) inhibitors: an endeavor on COVID-19 drug discovery. Mol. Divers. 2021;25(3):1827–1838. doi: 10.1007/s11030-020-10166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Citarella A., Gentile D., Rescifina A., Piperno A., Mognetti B., Gribaudo G., et al. Pseudo-dipeptide bearing α,α-difluoromethyl ketone moiety as electrophilic warhead with activity against coronaviruses. Int. J. Mol. Sci. 2021;22(3):1398. doi: 10.3390/ijms22031398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dai W., Zhang B., Jiang X.-M., Su H., Li J., Zhao Y., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science (New York, N.Y.) 2020;368(6497):1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Goetz D.H., Choe Y., Hansell E., Chen Y.T., McDowell M., Jonsson C.B., et al. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry. 2007;46(30):8744–8752. doi: 10.1021/bi0621415. [DOI] [PubMed] [Google Scholar]
- 121.Mandal A., Jha A.K., Hazra B. Plant products as inhibitors of coronavirus 3CL protease. Front. Pharmacol. 2021;12(167) doi: 10.3389/fphar.2021.583387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Alamri M.A., Tahir ul Qamar M., Mirza M.U., Bhadane R., Alqahtani S.M., Muneer I., et al. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2021;39(13):4936–4948. doi: 10.1080/07391102.2020.1782768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chataigner I., Panel C., Gérard H., Piettre S.R. Sulfonyl vs. carbonyl group: which is the more electron-withdrawing? Chem. Commun. 2007;31:3288–3290. doi: 10.1039/b705034h. [DOI] [PubMed] [Google Scholar]
- 124.Nguyen D.D., Gao K., Chen J., Wang R., Wei G.W. Potentially highly potent drugs for 2019-nCoV. bioRxiv. 2020 [Google Scholar]
- 125.Alamri M.A., ul Qamar M.T., Alqahtani S.M. 2020. Pharmacoinformatics and molecular dynamic simulation studies reveal potential inhibitors of SARS-CoV-2 main protease 3CLpro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Abdusalam A.A.A., Murugaiyah V. Identification of potential inhibitors of 3CL protease of SARS-CoV-2 from ZINC database by molecular docking-based virtual screening. Front. Mol. Biosci. 2020;7(419) doi: 10.3389/fmolb.2020.603037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mahmoud A., Mostafa A., Al-Karmalawy A.A., Zidan A., Abulkhair H.S., Mahmoud S.H., et al. Telaprevir is a potential drug for repurposing against SARS-CoV-2: computational and in vitro studies. Heliyon. 2021;7(9) doi: 10.1016/j.heliyon.2021.e07962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ghahremanpour M.M., Tirado-Rives J., Deshmukh M., Ippolito J.A., Zhang C.-H., Cabeza de Vaca I., et al. Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020;11(12):2526–2533. doi: 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Araújo J.L., de Oliveira Sousa A., Santos G.T., de Sousa L.A. Analysis of the molecular activity of atazanavir against SARS-COV-2 by molecular docking studies. Acta Scient. Anat. 2021;1(Suppl. 2):25–26. [Google Scholar]
- 130.Beck B.R., Shin B., Choi Y., Park S., Kang K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Computat. Struct. Biotechnol. J. 2020;18:784–790. doi: 10.1016/j.csbj.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ray A.K., Gupta P.S.S., Panda S.K., Biswal S., Bhattacharya U., Rana M.K. Repurposing of FDA-approved drugs as potential inhibitors of the SARS-CoV-2 main protease: molecular insights into improved therapeutic discovery. Comput. Biol. Med. 2022;142 doi: 10.1016/j.compbiomed.2021.105183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Teli D.M., Shah M.B., Chhabria M.T. In silico screening of natural compounds as potential inhibitors of SARS-CoV-2 Main protease and spike RBD: targets for COVID-19. Front. Mol. Biosci. 2021;7 doi: 10.3389/fmolb.2020.599079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kandeel M., Abdelrahman A.H.M., Oh-Hashi K., Ibrahim A., Venugopala K.N., Morsy M.A., et al. Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. J. Biomol. Struct. Dyn. 2021;39(14):5129–5136. doi: 10.1080/07391102.2020.1784291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ramos-Guzmán C.A., Ruiz-Pernía J.J., Tuñón I. Computational simulations on the binding and reactivity of a nitrile inhibitor of the SARS-CoV-2 main protease. Chem. Commun. 2021;57(72):9096–9099. doi: 10.1039/d1cc03953a. [DOI] [PubMed] [Google Scholar]
- 135.Aftab S.O., Ghouri M.Z., Masood M.U., Haider Z., Khan Z., Ahmad A., et al. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J. Transl. Med. 2020;18(1):1–15. doi: 10.1186/s12967-020-02439-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Balmeh N., Mahmoudi S., Fard N.A. Manipulated bio antimicrobial peptides from probiotic bacteria as proposed drugs for COVID-19 disease. Inform. Med. Unlocked. 2021;23 doi: 10.1016/j.imu.2021.100515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hernández-Morales R., Becerra A., Campillo-Balderas J., Cottom-Salas W., Cruz-González A., Jácome R., et al. Biomedical Innovations to Combat COVID-19. Elsevier; 2022. Structural biology of the SARS-CoV-2 replisome: evolutionary and therapeutic implications; pp. 65–82. [Google Scholar]
- 138.Wang Y., Anirudhan V., Du R., Cui Q., Rong L. RNA-dependent RNA polymerase of SARS-CoV-2 as a therapeutic target. J. Med. Virol. 2021;93(1):300–310. doi: 10.1002/jmv.26264. [DOI] [PubMed] [Google Scholar]
- 139.Tian L., Qiang T., Liang C., Ren X., Jia M., Zhang J., et al. RNA-dependent RNA polymerase (RdRp) inhibitors: the current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021;213 doi: 10.1016/j.ejmech.2021.113201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jiang Y., Yin W., Xu H.E. RNA-dependent RNA polymerase: structure, mechanism, and drug discovery for COVID-19. Biochem. Biophys. Res. Commun. 2021;538:47–53. doi: 10.1016/j.bbrc.2020.08.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Muhammed Y., Nadabo A.Y., Pius M., Sani B., Usman J., Garba N.A., et al. SARS-CoV-2 spike protein and RNA dependent RNA polymerase as targets for drug and vaccine development: a review. Biosafety Health. 2021;3(05):249–263. doi: 10.1016/j.bsheal.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang Q., Wu J., Wang H., Gao Y., Liu Q., Mu A., et al. Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell. 2020;182(2):417–428. doi: 10.1016/j.cell.2020.05.034. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Qudsiani K., Sutriyo R. Polyamidoamine-remdesivir conjugate: physical stability and cellular uptake enhancement. Biomed. Pharmacol. J. 2021:2073–2083. [Google Scholar]
- 144.Zhang L., Zhou R. Structural basis of the potential binding mechanism of remdesivir to SARS-CoV-2 RNA-dependent RNA polymerase. J. Phys. Chem. B. 2020;124(32):6955–6962. doi: 10.1021/acs.jpcb.0c04198. [DOI] [PubMed] [Google Scholar]
- 145.Ning S., Yu B., Wang Y., Wang F. SARS-CoV-2: origin, evolution, and targeting inhibition. Frontiers in cellular and infection. Microbiology. 2021;11 doi: 10.3389/fcimb.2021.676451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mishra A., Rathore A.S. Pharmacophore screening to identify natural origin compounds to target RNA-dependent RNA polymerase (RdRp) of SARS-CoV2. Mol. Divers. 2022:1–17. doi: 10.1007/s11030-021-10358-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Song W.-H., Liu M.-M., Zhong D.-W., Zhu Y.-l., Bosscher M., Zhou L., et al. Tetrazole and triazole as bioisosteres of carboxylic acid: discovery of diketo tetrazoles and diketo triazoles as anti-HCV agents. Bioorg. Med. Chem. Lett. 2013;23(16):4528–4531. doi: 10.1016/j.bmcl.2013.06.045. [DOI] [PubMed] [Google Scholar]
- 148.Mahmoudi S., Bayat M., Savari M.A., Nasiri S., Nasiri R., Jafari-Sales A., et al. Epidemiology, virology, clinical features, diagnosis, and treatment of SARS-CoV-2 infection. J. Exp. Clin. Med. 2021;38(4):649–668. [Google Scholar]
- 149.Meng X.-Y., Zhang H.-X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011;7(2):146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Offerhaus J.A., Wilde A.A., Remme C.A. Prophylactic (hydroxy) chloroquine in COVID-19: potential relevance for cardiac arrhythmia risk. Heart Rhythm. 2020;17(9):1480–1486. doi: 10.1016/j.hrthm.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wiwanitkit V. QTc interval prolongation, COVID-19 and chloroquine. Neth. Hear. J. 2020;28(11):621. doi: 10.1007/s12471-020-01506-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Narayanan A., Narwal M., Majowicz S.A., Varricchio C., Toner S.A., Ballatore C., et al. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022;5(1):169. doi: 10.1038/s42003-022-03090-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lv Z., Cano K.E., Jia L., Drag M., Huang T.T., Olsen S.K. Targeting SARS-CoV-2 proteases for COVID-19 antiviral development. Front. Chem. 2022;9 doi: 10.3389/fchem.2021.819165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Valdez I.R.V., Santiago-Quintana J.M., ROSALEZ M., Farfan E., Soriano-Ursua M.A. 2020. Theoretical Evaluation of Bortezomib and Other Boron-Containing Compounds as Inhibitors of SARS-CoV-2 Main Protease. [Google Scholar]
- 155.Kumar N., Kumar A., Pradhan S., Kumar A., Singh K. Painful blisters of left hand following extravasation of remdesivir infusion in COVID-19. Indian J. Critic. Care Med. Peer-rev. Off. Publ. Indian Soc. Critic. Care Med. 2021;25(2):240. doi: 10.5005/jp-journals-10071-23732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kabinger F., Stiller C., Schmitzová J., Dienemann C., Kokic G., Hillen H.S., et al. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021;28(9):740–746. doi: 10.1038/s41594-021-00651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sharov A.V., Burkhanova T.M., Taskın Tok T., Babashkina M.G., Safin D.A. Computational analysis of molnupiravir. Int. J. Mol. Sci. 2022;23(3):1508. doi: 10.3390/ijms23031508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Akbarzadeh-Khiavi M., Torabi M., Rahbarnia L., Safary A. Baricitinib combination therapy: a narrative review of repurposed Janus kinase inhibitor against severe SARS-CoV-2 infection. Infection. 2021;1-14 doi: 10.1007/s15010-021-01730-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Marinho E.M., de Andrade Batista, Neto J., Silva J., Rocha da Silva C., Cavalcanti B.C., Marinho E.S., et al. Virtual screening based on molecular docking of possible inhibitors of Covid-19 main protease. Microb. Pathog. 2020;148:104365. doi: 10.1016/j.micpath.2020.104365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Steiner T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002;41(1):48–76. doi: 10.1002/1521-3773(20020104)41:1<48::aid-anie48>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 161.Pfaff D., Nemecek G., Podlech J. A Lewis acid-promoted pinner reaction. Beilstein J. Org. Chem. 2013;9(1):1572–1577. doi: 10.3762/bjoc.9.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Saha P., Banerjee A.K., Tripathi P.P., Srivastava A.K., Ray U. A virus that has gone viral: amino acid mutation in S protein of Indian isolate of coronavirus COVID-19 might impact receptor binding, and thus, infectivity. Biosci. Rep. 2020;40(5) doi: 10.1042/BSR20201312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Herrmann L., Yaremenko I., Çapcı A., Struwe J., Hodek J., Belyakova Y., et al. 2021. Artemisinin and Quinoline Hybrid Compounds Inhibit Replication of SARS-CoV-2 in Vitro. [Google Scholar]
- 164.Najar A., Taynaz M., Elmarzugi N., Atia A., Moftah S. COVID-19 drug design based on the active Core of well-known anti-malarial: a computational approaches. J. Theor. Comput. Sci. 2020;6:266. [Google Scholar]
- 165.Jankelson L., Karam G., Becker M.L., Chinitz L.A., Tsai M.-C. QT prolongation, torsades de pointes, and sudden death with short courses of chloroquine or hydroxychloroquine as used in COVID-19: a systematic review. Heart Rhythm. 2020;17(9):1472–1479. doi: 10.1016/j.hrthm.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Al-Wahaibi L.H., Hassan H.M., Abo-Kamar A.M., Ghabbour H.A., El-Emam A.A. Adamantane-isothiourea hybrid derivatives: synthesis, characterization, in vitro antimicrobial, and in vivo hypoglycemic activities. Mol. (Basel, Switzerland). 2017;22(5):710. doi: 10.3390/molecules22050710. [DOI] [PMC free article] [PubMed] [Google Scholar]