

Abstract
At present, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spread worldwide, which has emerged multiple variants and brought a threat to global public health. To analyze the genomic characteristics and variations of SARS-CoV-2 imported into Beijing, we collected the respiratory tract specimens of 112 cases of coronavirus disease 2019 (COVID-19) from January to September 2021 in Beijing, China, including 40 local cases and 72 imported cases. The whole-genome sequences of the viruses were sequenced by the next-generation sequencing method. Variant markers and phylogenic features of SARS-CoV-2 were analyzed. Our results showed that in all 112 sequences, the mutations were concentrated in spike protein. D614G was found in all sequences, and mutations including L452R, T478K, P681R/H, and D950N in some cases. Furthermore, 112 sequences belonged to 23 lineages by phylogenetic analysis. B.1.1.7 (Alpha) and B.1.617.2 (Delta) lineages were dominant. Our study drew a variation image of SARS-CoV-2 and could help evaluate the potential risk of COVID-19 for pandemic preparedness and response.
Keywords: Severe acute respiratory syndrome coronavirus 2, Variation, Genome, Phylogenetic analysis
1. Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in December 2019, which was highly transmissible and pathogenic and spread rapidly around the world [1], [2]. To date, over 481 million coronavirus disease (COVID-19) confirmed cases and over −6.1 million deaths globally had been reported in total by World Health Organization (WHO) [3]. The COVID-19 global pandemic has brought a massive disaster to humans.
SARS-CoV-2 is a member of the family Coronaviridae, subfamily Orthocoronavirinae, and betacoronavirus, the enveloped viruses with genome sizes of 29,903 bases in length [4]. The SARS-CoV-2 genome contains four structural proteins, including nucleocapsid protein (N), envelop protein (E), membrane protein (M), and spike protein (S) [5]. Besides, the SARS-CoV-2 genome was similar to that of a typical coronavirus and contained at least ten open reading frames (ORFs) [5]. Furthermore, the S protein played an essential role in identifying receptor binding sites and subsequent viral entry [6]. Furthermore, some significant amino acid substitutions in S protein might increase the infectivity and escape immune [7], [8]. Therefore, the emergence variants of SARS-CoV-2 posed an increased risk to global public health. There are five variants of concern (VOC) worldwide, including Alpha, Beta, Gamma, Delta, and Omicron, which could increase the transmissibility, virulence, or the risk of reinfection [9].
Soon after the COVID-19 outbreak, China quickly took strict measures to control the local spread and the import of SARS-CoV-2. China has entered the stage of regular COVID-19 prevention and control, implementing a “dynamic COVID-19 case clearing policy” strategy. As the capital of China, Beijing has a high population density, frequent international exchanges and population flows, and a high risk of local outbreaks caused by domestic and abroad imports. In January 2020, Beijing reported the first case of COVID-19 infection [10]. And then, there have been several imported outbreaks caused by human and cargo transmission [10], [11]. Therefore, it is necessary to monitor the genomic variation and genetic evolution characteristics of SARS-CoV-2.
In response to the COVID-19 outbreak, we have carried out long-term genetic surveillance. This study analyzed 112 whole genomes of SARS-CoV-2 collected from January to September 2021. Based on the presence or absence of specific variation of SARS-CoV-2, we can assess the transmissibility and virulence of SARS-CoV-2. In addition, genomic variation and genetic evolution characteristics of SARS-CoV-2 were shown, providing a reference for risk assessment, prevention, and control measures of COVID-19.
2. Materials and methods
2.1. Sample collection
The samples of nasopharyngeal swabs and oropharyngeal swabs from confirmed and asymptomatic cases of COVID-19 were obtained from January 2021 to September 2021 in Beijing. In addition, the general information was collected through epidemiological investigation.
2.2. Sample nucleic acid extraction
The viral RNA was extracted by the Nucleic Acid Exaction System (Xi’an Tianlong Science and Technology CO., Ltd., Xi’an, China). According to the 200 μl nasopharyngeal swabs or oropharyngeal swabs, samples were transferred and used for RNA extraction according to the instructions.
2.3. Whole-genome sequencing
The ULSEN SARS-CoV-2 whole-genome capture kit (Beijing Microfuture Technology CO., Ltd., Beijing, China) was used for polymerase chain reaction (PCR) amplification. The PCR amplification process was carried out according to the kit instructions. Nextera XT DNA library preparation kit (Illumina, San Diego, CA, USA) was used to construct a DNA sequencing library. DNA libraries were sequenced on a MiniSeq platform (Illumina) using a MiniSeq reagent kit (300-cycles). The SARS-CoV-2 Wuhan-Hu-1 (accession number NC_045512) was regarded as a reference. The CLC software was used for genome sequence assembly and alignment.
2.4. Sequence alignment and phylogenetic analysis
All available SARS-CoV-2 genomic sequences were aligned with MAFFT v7.222 [12]. Phylogenetic trees were generated using the Neighbour-Joining (NJ) method with the Kimura 2-parameter model. 1,000 bootstrap replicates determined the robustness of N.J. topology.
3 Results.
3.1. The descriptive analysis of the cases of COVID-19 in Beijing
From January to September 2021, we obtained 112 SARS-CoV-2 whole-genome sequences from 112 cases. In all 112 infections, 40 cases were linked with the local cluster, with 72 imported ones. Among them, 94 infections had case information and were available for analysis. 55% (22/40) of local cases were male. However, 63% (34/54) of imported cases were male (Table 1 ). There was no significant difference in the virus infection by gender. Local cases and imported cases infected with SARS-CoV-2 were mainly in the 30–39 age group, accounting for 25% (10/40) and 25.9% (14/54), respectively (Table 1).
Table 1.
General information of the cases of COVID-19 in Beijing.
| Characteristic of cases | General information | Count | Percentage (%) * |
|---|---|---|---|
| Local cases (40 cases) | Gender | ||
| male | 22 | 55 | |
| female | 18 | 45 | |
| Age | |||
| 0~9 | 4 | 10 | |
| 10~19 | 0 | 0 | |
| 20~29 | 1 | 2.5 | |
| 30~39 | 10 | 25 | |
| 40~49 | 9 | 22.5 | |
| 50~59 | 4 | 10 | |
| >60 | 12 | 30 | |
| Imported cases (54 cases) | Gender | ||
| male | 34 | 63 | |
| female | 20 | 37 | |
| Age | |||
| 0~9 | 9 | 16.7 | |
| 10~19 | 6 | 11.1 | |
| 20~29 | 12 | 22.2 | |
| 30~39 | 14 | 25.9 | |
| 40~49 | 6 | 11.1 | |
| 50~59 | 2 | 3.7 | |
| >60 | 5 | 9.3 |
*The general information of imported cases collected were from the 54 cases of coronavirus disease 2019 (COVID-19).
3.2. Genetic variations of SARS-CoV-2
A total of 112 SARS-CoV-2 whole-genome sequences were obtained by sequences alignment. We analyzed the molecular characteristics associated with the infectivity, immune escape, and unknown functional impact of all SARS-CoV-2 (Table 2 ). Compared with the Wuhan-Hu-1 strain, the amino acid mutation sites of analyzed sequences mainly concentrated on ORF1a, ORF1b, N, and S genes, among which the number of mutations in the S gene was the largest (Table 2). It showed that the numbers of amino acid mutations in spike protein ranged from 1 to 15, and all analyzed sequences shared the D614G mutation. The majority of SARS-CoV-2 contained P681R/H (84.8%) in spike protein. We found that the variant of Alpha was H at position 681 in spike protein, and the variant of Delta was R at position 681 in spike protein. The L452R and T478K variations in spike protein receptor-binding domains accounted for 44.6%. Some other mutations included T19R (44.6%), R158G (44.6%), and D950N (42.9%) in spike protein, accounting for nearly half. The mutations T19R, R158G, L452R, T478K, and D950N sites were characteristic variation sites of the Delta variant compared to the Alpha and Beta variants. The percentage of R203K and G240R in nucleocapsid protein of SARS-CoV-2 were 44.6% and 44.6%, respectively (Table 3 ). The mutations in nucleocapsid protein of SARS-CoV-2 were D3L (35.7%), D63G (43.8%), G215C (33%), S235F (35.7%), and D377Y (43.8%) (Table 3). Some mutations in membrane protein were observed, such as V23L, M64F, I82T, and L87F. The mutation I82T accounted for 49.1%. The percentage of T1001I, A1708D, and I2230T in ORF1a protein of SARS-CoV-2 were 35.7%, 35.7%, and 35.7%, respectively. The P314L mutation in the ORF1b protein was already widespread in SARS-CoV-2, which accounted for 99.1%. The percentage of T60A in the ORF9b protein was 43.8%.
Table 2.
The mutations in spike protein from the whole genome sequence.
 |
*The representative strains were selected, which belonged to 23 lineages. For multiple strains in the same lineage, we selected the strains with the largest number of mutations in spike protein.
Table 3.
The mutations in nucleocapsid protein from the whole genome sequence.
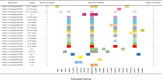 |
*The representative strains were selected, which belonged to 23 lineages. For multiple strains in the same lineage, we selected the strains with the largest number of mutations in spike protein.
3.3. Phylogenetic analysis of SARS-CoV-2
The phylogenetic analyses of SARS-CoV-2 were performed to determine the evolution of SARS-CoV-2 in Beijing in 2021. All sequences of SARS-CoV-2 were classified by the Pangolin COVID-19 Lineage Assigner Web application (https://pangolin.cog-uk.io/). We found that the whole genome sequence belonged to 23 lineages. The 40 SARS-CoV-2 strains were grouped into B.1.1.7 lineage, designated as a concern alpha variant by World Health Organization (WHO). The 49 SARS-CoV-2 strains were classified as B.1.617.2 lineage, belonging to a concern delta variant. There were four strains from the B.1.351 lineage, which belonged to a variant of concern beta. The phylogenetic tree showed that the strain sequences belonged to different lineage, which showed that the strains were of different countries of origin (Fig. 1 ).
Fig. 1.

The phylogenetic tree based on the whole genome sequences of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). A total of 112 SARS-CoV-2 sequences were analyzed. All sequences belonged to 23 different lineages. The strains from the local cluster were indicated with red font; while the strains from the imported were indicated with green font. The phylogenetic tree was generated by MEGA7.0 with neighbor-joining method. The bootstrap was 1,000.
4. Discussion
SARS-CoV-2 has extensive genetic variation during transmission, with a mutation rate of ∼10−6 in each round of replication [13]. As the SARS-CoV-2 continues to evolve, many variants of SARS-CoV-2 emerge around the world. Comparative assessment of variant characteristics and public health risks by WHO was designated variants of concern, variants of interest, and variants under monitoring. They could cause great concern for the variant of SARS-CoV-2. Furthermore, the large RNA genome in coronavirus allows for extra plasticity in genome modification by mutations and recombinations, thereby increasing the probability for intraspecies variability and novel variants to emerge under the right conditions [14].
In our study, we analyzed 112 whole sequences of SARS-CoV-2. They belonged to 23 different lineages and contained different mutation sites. The variants of concern included Alpha, Beta, and Delta were found. The Delta variants were dominant. In terms of epidemic trends, the B.1.1.7 lineage spread widely worldwide, and then the B.1.617.2 lineage gradually replaced B.1.1.7 to take the dominant. It was also consistent with the popularity of Beijing. On the other hand, some lineages appeared less frequently, like AL.1, B.1.1.317, B.1.2, and B.1.36.31 lineage, which appeared only once and was also in line with the global pandemic SARS-CoV-2.
Some necessary molecular signatures were analyzed. Spike protein is an important structural protein in SARS-CoV-2. Its primary function is to promote the viral receptor binding domain to bind to angiotensin-converting enzyme 2 of host cells, which fuses the host cell with the virus. Spike protein contained receptor-binding domains at amino acid 319–541. It was noted that the majority of SARS-CoV-2 posed one to three mutations at the receptor-binding domains, which might increase the infectivity and immune escape [15]. The D614G variant was the earliest mutation that has been recognized and attracted attention, which could enhance the infectivity [16]. We found that all SARS-CoV-2 contained the D614G mutation. Furin protease cleavage sites (amino acid sites 681–685) were located in the middle of the two subunits of spike protein, which was the key for the virus to enter human host cells and could enhance the viral infectivity [17]. The Delta variation carried the P681R mutation at the furin cleavage site. Therefore, the Alpha variation beard the P681H mutation. The substitution of R203K and G204R in nucleocapsid protein could also enhance the infectivity, fitness, and virulence of SARS-CoV-2 [18]. Other amino acid variations were also found in spike protein, such as T29I, R102S, and A222V, but whether these variations affect the structure and function of spike protein needs further study. Some mutations located in ORFs are rarely studied, and their molecular mechanism and biological significance remain unclear. Our study analyzed the percentage of the critical mutations in SARS-CoV-2. Some mutations still account for a tiny proportion of SARS-CoV-2. Therefore, continuous surveillance needs to be implemented.
Our finding suggested that the SARS-CoV-2 contained the reported mutations, which could increase the viral transmission, infectivity, and immune escape. Therefore, we should continue to pay attention to the genetic evolution and variations and strengthen the monitoring, which could help evaluate the risk of SARS-CoV-2 for pandemic preparedness and response.
Acknowledgements
This study was funded by the National Key R&D Program of China (2021ZD0114100 and 2021ZD0114103), the Capital’s Funds for Health Improvement and Research (2021-1G-3012 and 2022-4G-30117), and the Beijing Science and Technology Planning Project of Beijing Science and Technology Commission (Z211100002521015). In addition, we thank the health workers who contributed to the epidemiological survey, sample collection, and transportation.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Author contributions
Zhaomin Feng: Investigation, Data Curation, Writing – Original Draft. Shujuan Cui: Investigation, Data Curation. Bing Lyu: Investigation, Data Curation. Zhichao Liang: Investigation, Data Curation. Fu Li: Investigation, Data Curation. Lingyu Shen: Investigation, Data Curation. Hui Xu: Data Curation. Peng Yang: Supervision, Conceptualization. Quanyi Wang: Supervision, Funding Acquisition. Daitao Zhang: Supervision, Conceptualization. Yang Pan: Supervision, Conceptualization, Writing – Original Draft, Validation.
References
- 1.Zhu N.a., Zhang D., Wang W., Li X., Yang B.o., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P., Yang X., Wang X., Hu B., Zhang L., Zhang W., Zhan F., Wang Y., Xiao G., Shi Z., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO, Coronavirus disease (COVID-19) weekly epidemiological update and weekly operational update, https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports, 2020 (accesed 10 April 2022).
- 4.Wu F., Zhao S., Yu B., Chen Y., Wang W., Song Z., Hu Y., Tao Z., Tian J., Pei Y., Yuan M., Zhang Y., Dai F., Liu Y., Wang Q., Zheng J., Xu L., Holmes E.C., Zhang Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim D., Lee J.-Y., Yang J.-S., Kim J.W., Kim V.N., Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914–921. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92:418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Q., Nie J., Wu J., Zhang L., Ding R., Zhang L., Qu X., Xu W., Huang W., Wang Y., et al. SARS-CoV-2 501Y.V2 variants lack higher infectivity but do have immune escape. Cell. 2021;184:2362–2371. doi: 10.1016/j.cell.2021.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rees-Spear C., Muir L., Griffith S.A., Heaney J., Aldon Y., Snitselaar J.L., Nastouli E., Doores K.J., Gils M.J.V., McCoy L.e., et al. The effect of spike mutations on SARS-CoV-2 neutralization. Cell Rep. 2021;34:108890. doi: 10.1016/j.celrep.2021.108890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.WHO, Tracking SARS-CoV-2 variants. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/, 2022 (accessed 10 April 2022).
- 10.Pan Y., Feng Z., Li F., Cui S., Lv B., Liang Z., Chen L., Wang Q., Zhang D. Genomic characteristics of SARS-CoV-2 from the first outbreak in clusters caused by VOC202012/01-like variant in China. Int. J. Virol. 2021;28(3):182–186. doi: 10.3760/cma.j.issn.1673-402.2021.03.002. [DOI] [Google Scholar]
- 11.Wu S., Pan Y., Duan W., Ma C., Sun Y., Zhang L., Dou X., Wang X., Jia L., Yang P., Wang Q., Pang X. Tracing infection source of an outbreak in Beijing caused by an imported asymptomatic case of COVID-19. Int. J. Virol. 2021;28(3):187–191. doi: 10.3760/cma.j.issn.1673-402.2021.03.003. [DOI] [Google Scholar]
- 12.Kazutaka K., Standley D.M. MAFFT Multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bar-On Y.M., Flamholz A., Phillips R., Milo R. SARS-CoV-2 (COVID-19) by the numbers. eLife. 2020;9 doi: 10.7554/eLife.57309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su S., Wong G., Shi W., Liu J., Lai A.C.K., Zhou J., Liu W., Bi Y., Gao G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24(6):490–502. doi: 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilhelm A., Toptan T., Pallas C., Wolf T., Goetsch U., Gottschalk R., Vehreschild M.J.G.T., Ciesek S., Widera M. Antibody-mediated neutralization of authentic SARS-CoV-2 B.1.617 variants harboring L452R and T478K/E484Q. Viruses. 2021;13(9):1693. doi: 10.3390/v13091693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou B., Thao T.T.N., Hoffmann D., Taddeo A., Ebert N., Jores J., Benarafa C., Wentworth D.E., Thiel V., Beer M., et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature. 2021;592(7852):122–127. doi: 10.1038/s41586-021-03361-1. [DOI] [PubMed] [Google Scholar]
- 17.Lubinski B., Fernandes M.H.V., Frazier L., Tang T., Daniel S., Diel D.G., Jaimes J.A., Whittaker G.R. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike[Preprint] iScience. 2022;25(1):103589. doi: 10.1016/j.isci.2021.103589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H., Xing N., Meng K., Fu B., Xue W., Dong P., Tang W., Xiao Y., Liu G., Luo H., Zhu W., Lin X., Meng G., Zhu Z. Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host Microbe. 2021;29(12):1788–1801. doi: 10.1016/j.chom.2021.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]