

Abstract
Post-transcriptional modifications have critical roles in tRNA stability and function1–4. In thermophiles, tRNAs are heavily modified to maintain their thermal stability under extreme growth temperatures5,6. Here we identified 2′-phosphouridine (Up) at position 47 of tRNAs from thermophilic archaea. Up47 confers thermal stability and nuclease resistance to tRNAs. Atomic structures of native archaeal tRNA showed a unique metastable core structure stabilized by Up47. The 2′-phosphate of Up47 protrudes from the tRNA core and prevents backbone rotation during thermal denaturation. In addition, we identified the arkI gene, which encodes an archaeal RNA kinase responsible for Up47 formation. Structural studies showed that ArkI has a non-canonical kinase motif surrounded by a positively charged patch for tRNA binding. A knockout strain of arkI grew slowly at high temperatures and exhibited a synthetic growth defect when a second tRNA-modifying enzyme was depleted. We also identified an archaeal homologue of KptA as an eraser that efficiently dephosphorylates Up47 in vitro and in vivo. Taken together, our findings show that Up47 is a reversible RNA modification mediated by ArkI and KptA that fine-tunes the structural rigidity of tRNAs under extreme environmental conditions.
Subject terms: tRNAs, RNA modification
Reversible internal RNA phosphrylation contributes to thermal stability and nuclease resistance of tRNA, and cellular thermotolerance of hyperthermophiles.
Main
Recent advances in epitranscriptomics research have demonstrated the chemical diversity and biological importance of RNA modifications1–4. Thus far, about 150 types of RNA modification have been reported in various RNA molecules from all domains of life7. In particular, tRNAs contain the widest variety and largest number of modified nucleosides, with 80% of RNA modifications identified in tRNA molecules. Diverse RNA modifications are clustered in the anticodon loop, especially at positions 34 and 37 (refs. 1,8). These modifications have critical roles in stabilizing and modulating codon–anticodon interactions on the ribosome, ensuring accurate and efficient protein synthesis. Many RNA modifications are also found in the tRNA body composed of the D-loop, TΨC loop (T-loop) and variable loop (V-loop)9,10 (Fig. 1a). These RNA modifications are required for correct folding and stability of the tRNA core structure. In particular, 2′-O-methyl modifications (Nm) confer conformational rigidity to the tRNA core region by fixing C3′-endo ribose puckering9,11.
Fig. 1. Identification and biochemical characterization of Up at position 47 of S. tokodaii tRNAVal3.

a, Secondary structure of S. tokodaii tRNAVal3 with post-transcriptional modifications. N324 is shown in red. The cleavage sites of RNase T1 and RNase A that generate RNA fragments with N324 are indicated by black and grey triangles, respectively. b, CID spectrum of the N324-containing fragment (m/z 852.59) of S. tokodaii tRNAVal2/3 digested with RNase T1. c- and y-series product ions are indicated. c, CID spectrum of the N324-containing fragment (m/z 1,215.15) of S. tokodaii tRNAVal2/3 digested with RNase A. c- and y-series product ions are indicated. d, RNA-MS of RNA fragments with or without N324 and Um50. The upper panel shows the base peak chromatogram (BPC) of RNase T1-digested fragments. The second, third, fourth and fifth panels represent extracted ion chromatograms (XICs) of the divalent negative ions of the respective fragments, as indicated. e, Chemical structure of 2′-phosphouridine (Up). The 2′-phosphate group is shown in red. f, Melting curves of S. tokodaii tRNAVal3 with (red line) or without (blue line) Up47. Tm values were determined on the basis of the inflection point of the melting curve. Data represent the average values of technical triplicates ± s.d. P < 1.02 × 10–4 by two-sided Student’s t test. g, RNase probing of S. tokodaii tRNAVal3 with (red line) or without (blue line) Up47. Top, PAGE gels showing degradation of 32P-labelled tRNA with or without Up47 by RNase I at the indicated time (min). Intact tRNA is indicated by a triangle. Data represent the average values of technical triplicates ± s.d. The unprocessed gel image is provided in Supplementary Fig. 10.
In thermophilic bacteria and archaea, unique RNA modifications contribute to the thermal adaptation of tRNAs5,6. 5-Methyl-2-thiouridine (m5s2U or s2T) is found at position 54 in the T-loop of tRNAs from thermophiles12. m5s2U54 adopts a rigid conformation with C3′-endo ribose puckering, thereby stabilizing the tRNA body in high-temperature environments11. The 2-thiolation level of m5s2U54 increases as the growth temperature rises13,14. m5s2U54 contributes to the thermotolerance of Thermus thermophilus15. In Pyrococcus furiosus, the relative abundance of N4-acetylcytidine (ac4C) and its 2′-O-methyl derivative (ac4Cm) were markedly increased with rising growth temperature14. ac4C is a prevalent modification that is present in tRNAs, rRNAs and other RNAs in hyperthermophilic archaea14,16. ac4C favours the C3′-endo form and stabilizes tRNAs17,18. Loss of ac4C results in a growth defect in Thermococcus kodakarensis at high temperature, contributing to cellular thermotolerance19. In Bacillus stearothermophilus, 2′-O-methylation in tRNAs increases when the growth temperature rises20. Archaeosine (G+) is a unique 7-deazaguanosine derivative found at position 15 in the D-loop of archaeal tRNAs21. On the basis of quantum mechanics calculations, G+15 stabilizes the Levitt base pair with C48 (ref. 22). In line with this, biochemical and genetic studies have shown that G+ confers thermal stability to tRNAs and contributes to thermotolerance19,23.
Here we report the identification of 2′-phosphouridine (Up) in tRNAs, which, to our knowledge, is the first known instance of internal RNA phosphorylation. Biochemical, structural and genetic studies showed that Up47 is a reversible RNA modification and confers thermal stability to tRNA, thereby contributing to cellular thermotolerance.
Discovery of Up in tRNA
To explore tRNA modifications in hyperthermophilic archaea, we isolated 12 tRNA species from the thermoacidophilic crenarchaeon Sulfurisphaera tokodaii using our original method for RNA isolation by reciprocal circulating chromatography (RCC) (Extended Data Fig. 1)24.
Extended Data Fig. 1. Isolation of S. tokodaii tRNAs.

tRNAs for Val3, Ile2 and Phe were isolated by CCC52. The other tRNAs were isolated by RCC24. The isolated tRNAs and total RNA as a maker were resolved by electrophoresis on a 10% (w/v) polyacrylamide gel containing 7 M urea and 1 × TBE, and stained with SYBR Gold. Smearing of bands is due to the high GC content of tRNAs. The precursor tRNAs for Ile2 and Phe containing an intron are indicated by red triangles. 5S rRNA, Class II tRNAs, and Class I tRNAs are indicated. We confirmed the reproducibility of this result. The unprocessed gel images are provided in Supplementary Fig. 10.
First, we comprehensively analysed all post-transcriptional modifications of tRNAVal3 by mass spectrometry (RNA-MS)25–27 and mapped 13 types of RNA modification at 18 positions in this tRNA molecule (Fig. 1a, Extended Data Fig. 2a–c, Supplementary Note 1, Supplementary Fig. 1, Supplementary Table 1). Among the modifications, we detected an unknown uridine derivative with molecular mass of 324 (tentatively named N324) at position 47 of the RNA fragments digested with RNases (Fig. 1b, c). The relative intensity of the mass chromatograms showed that N324 occurred at a frequency of 96.8% (Fig. 1d), indicating that N324 is an abundant modification. We also detected N324 in the seven other tRNA species (Extended Data Fig. 3a, b, Supplementary Note 1, Supplementary Table 2), indicating that N324 is a prevalent and abundant (82–100%) modification in class I tRNAs bearing U47 in the V-loop, but not in class II tRNAs with a long V-loop (Extended Data Fig. 3a). We also detected N324 in tRNA precursors (Extended Data Fig. 3c, Supplementary Note 1).
Extended Data Fig. 2. Nucleoside analysis and RNA-MS of S. tokodaii tRNAVal2/3.

(a) Nucleosides of S. tokodaii tRNAVal2/3 were subjected to LC/MS analysis. Top panel shows the UV trace at 254 nm. Second to bottom panels show XICs detecting proton adducts of modified nucleosides as indicated. N324 nucleoside was not detected in the bottom panel. m1G is derived from tRNAVal2, which was co-isolated with tRNAVal3 (Supplementary Fig. 1). Asterisks indicate unassigned ions. (b) RNA-MS of RNase T1 digests of the isolated S. tokodaii tRNAVal2/3. The upper panel shows the BPC of RNase T1-digested fragments. The sequence and molecular mass of each RNA fragment numbered in BPC are listed in Supplementary Table 1a. The lower panel shows the XIC of the divalent negative ion of the fragment containing N324. (c) RNA-MS of RNase A digests of isolated S. tokodaii tRNAVal2/3. The upper panel shows the BPC of RNase A-digested fragments. The sequence and molecular mass of each RNA fragment numbered in BPC are listed in Supplementary Table 1b. The lower panel shows the XIC of the divalent negative ion of the fragment containing N324.
Extended Data Fig. 3. N324 is present in various class I tRNAs.

(a) RNA-MS of RNase T1 digests of the isolated S. tokodaii tRNAs. XICs show negative ions of RNA fragments derived from V-loop containing N324 (red line) or U (black line) at position 47. Sequence and m/z value of each fragment are provided in Supplementary Table 2. Modification frequency of N324 indicated in each tRNA was calculated from relative peak intensities of the modified and unmodified fragments. Unassigned fragments are indicated by asterisks. (b) CID spectrum of the RNA fragments detected in (a). The negatively-charged ion of each fragment was used as a precursor ion for CID analysis. The product ions in the CID spectrum are assigned on the sequences. N324 is shown in red. (c) N324 is introduced in precursor tRNAs. RNA-MS of RNase T1 digests of the precursor tRNAs for Ile2 and Phe isolated from S. tokodaii (Extended Data Fig. 1). XICs show negative ions of the RNA fragments derived from V-loop containing N324 (red line) or U (black line) at position 47. Modification frequency of N324 indicated in each tRNA was calculated from relative peak intensities of the modified and unmodified fragments.
High-resolution mass analysis of the N324-containing fragment showed that the additional mass of N324 attached to the uridine residue was 79.97067 Da, equivalent to one phosphate group (theoretical mass, 79.96632 Da), with a low error value of 4.4 millimass unit , indicating that N324 is a phosphorylated uridine residue. This prediction explains why the N324 nucleoside was not detected in our nucleoside analysis (Extended Data Fig. 2a), owing to N324 being dephosphorylated during nucleoside preparation. To determine the phosphorylation site of N324, we prepared the N324-containing nucleotide and analysed its chemical structure through collision-induced dissociation (CID) and biochemical approaches (Supplementary Note 2, Extended Data Fig. 4a–h). We found that phosphorylation occurs on the 2′-OH group of the ribose moiety and concluded that N324 is 2′-phosphouridine (denoted Up, where ‘p’ is superscript to discriminate it from 3′-phosphate) (Fig. 1e).
Extended Data Fig. 4. Chemical structure determination of N324.

(a) RNA-MS of the N324-containing fragment of tRNAVal3 digested with RNase T1, with (+) or without (-) BAP treatment. XICs show the divalent negative ions of N324m5Cm5CUGp (m/z 845.59, z = -2) and Um5Cm5CUG-OH (m/z 765.52, z = -2). Two phosphates were removed by this treatment. (b) CID spectrum of the N324-containing fragment treated with BAP. The product ions are assigned on Um5Cm5CUG-OH. (c) Preparation scheme of pN324p. S. tokodaii tRNA fraction is digested with nuclease P1, yielding dinucleotide pN324m5C. The digests were subjected to periodate oxidation and β-elimination to remove the 3′ terminal residue. The resultant pN324p was purified by anion exchange chromatography and subjected to LC/MS/MS analysis. (d) LC/MS nucleotide analysis of the nuclease P1 digest of S. tokodaii tRNA fraction. UV trace at 254 nm (upper panel) and XIC of the negatively charged ion of pN324m5C (m/z 722, z = -1) (lower panel) are shown. (e) CID spectrum of pN324m5C. The product ions were assigned on the predicted chemical structure of pN324m5C. The phosphate group of N324 is shown in red. (f) CID spectrum of the N324 nucleotide (pN324p; m/z 483, z = -1). The product ions are assigned in the predicted chemical structure of pN324p. (g) RNA-MS of the V-loop-containing RNA fragment with (+) or without (-) Tpt1p treatment before RNase T1 digestion. XICs show the divalent negative ions of Upm5Cm5CUGp (m/z 845.59, z = -2) and Um5Cm5CUGp (m/z 805.60, z = -2). (h) CID spectrum of the dephosphorylated fragment by Tpt1p. The Tpt1p-treated tRNAVal3 was digested with RNase T1 and analyzed by RNA-MS. The V-loop containing fragment was selected as a precursor for CID. The product ions are assigned on the sequence as indicated.
Up47 stabilizes tRNA structure
Given that Up47 is a thermophile-specific modification found in the tRNA core region, we investigated whether Up47 stabilizes the tertiary structure of tRNA. To this end, we treated S. tokodaii tRNAVal3 with yeast Tpt1p (2′-phosphotransferase) to remove the 2′-phosphate of Up47. We measured the melting temperature (Tm) of S. tokodaii tRNAVal3 with and without Up47 (Fig. 1f). In the melting curves, the tRNA without Up47 gradually melted at around 65 °C while its hyperchromicity increased with temperature, whereas the tRNA with Up47 remained stable even at 70 °C. The Tm values of the tRNA with and without Up47 were 85.8 ± 0.5 °C and 79.2 ± 0.5 °C, respectively. These observations clearly demonstrate that a single Up47 modification increases the thermal stability of tRNAVal3 by 6.6 °C.
We next performed an RNase probing experiment to examine the nuclease resistance of tRNA with and without Up47. S. tokodaii tRNAVal3 and its Tpt1p-treated form were labelled with 32P at their 3′ termini and were probed with RNase I at 65 °C (Fig. 1g). Over time, the intact tRNAs were gradually degraded into RNA fragments. Compared with the intact tRNA with Up47, the Tpt1p-treated tRNA was degraded more rapidly, within 5 min, indicating that the tRNA without Up47 was highly sensitive to RNase I. This observation demonstrates that Up47 stabilizes and protects tRNAs from nucleolytic degradation.
Structural study of Up47 in native tRNA
To determine the molecular basis for thermal stabilization of tRNA by Up47, we crystalized S. tokodaii tRNAVal3 and determined its atomic structure at a resolution of up to 1.9 Å by X-ray crystallography (Fig. 2a, Extended Data Table 1, Extended Data Fig. 5, Supplementary Note 3, Supplementary Fig. 2). One unit cell contains two tRNA molecules, denoted as molecule A and molecule B. Molecule A formed a canonical tRNA core structure (Fig. 2b, c, Extended Data Fig. 5a), whereas molecule B had an altered core structure with a non-canonical base triple (Fig. 2b, c, Extended Data Fig. 5b). We clearly observed electron densities for tRNA modifications, including Up47 (Fig. 2a, Extended Data Fig. 5c, d). In both molecules, the 2′-phosphate of Up47 was oriented towards the solvent side and did not interact with any residues (Fig. 2a–c). The ribose puckering of Up47 adopted a C2′-endo conformation (Supplementary Table 3), as observed in the synthetic nucleotide28. The O4′ position in the ribose of Up47 formed a hydrogen bond with the N6-amino group of A21 in both molecules (Fig. 2d). The uracil base of Up47 faced the tRNA core (Fig. 2b, d). Because the uracil base at position 47 favours an outward orientation, as observed in well-known structures of yeast tRNAPhe and other class I tRNAs29, we observed backbone rotation of the V-loop at positions 46–48 caused by Up47 (Fig. 2e, Extended Data Fig. 6a, b). When Up47 was virtually introduced to yeast tRNAPhe, the 2′-phosphate clashed with T-stem residues at positions 49 and 50, inducing backbone rotation of the V-loop that orients the uracil base inwards and the 2′-phosphate outwards (Fig. 2e). In this rotation from yeast tRNAPhe to molecule A (Fig. 2b, c, e, Extended Data Fig. 6b), G46 changed its ribose pucker from C2′-endo to C3′-endo with altered torsion angles (δ, ε and ζ were changed by –58°, –28° and 68°, respectively) (Extended Data Fig. 6b, Supplementary Table 3). In addition, the Up47 backbone was substantially rotated with the α and ζ angles changing by –113° and 167°, respectively (Extended Data Fig. 6b, Supplementary Table 3). The m5C48 backbone was also rotated, with the α and γ angles changing by –36° and –86°, respectively (Extended Data Fig. 6b, Supplementary Table 3).
Fig. 2. Structural characterization of Up47 in S. tokodaii tRNAVal3.

a, Overview of the crystal structures of S. tokodaii tRNAVal3. Molecules A and B are shown in stick representation in white and light green, respectively. Up47 is shown in red. The 2Fo–Fc electron density map contoured at 1.0σ around Up47 is shown in the lower right box for each molecule. 2′p, 2′-phosphate. b, Close-up views of the core structures of molecules A (left) and B (right). Up47 (red) and the top (15–48; black), second (8–14–21; green), third (13–22–46; blue), fourth (9–12–23; yellow), fifth (11–24; orange), sixth (10–25–45; pink) and seventh (26–44; light purple) layers are shown in stick representation. c, Schematic views of the core structures of molecules A (left) and B (right). d, f, g, Atomic structures of the base triples s4U8–A14–A21 (d), Ψ13–G22–G46 (f) and C12–G23–C9 (g) in the core region of molecules A (left) and B (right). Dashed lines indicate predicted interactions with bond length (Å). Up47 is shown in red. e, The V-loop structures of molecule (mol.) A (blue), molecule B (green) and Saccharomyces cerevisiae tRNAPhe (PDB, 1EHZ) (gold) are overlaid. The residues at position 47 are shown in stick representation. The 2′-phosphate of Up47 is indicated. h, The V-loop structures of intact tRNA molecule A (blue), Tpt1p-treated tRNA molecule A (magenta) and Tpt1p-treated tRNA molecule B (orange) are overlaid. The residues at position 47 are shown in stick representation. The 2′-phosphate of Up47 is indicated.
Extended Data Table 1.
Data collection and refinement statistics.

*Values in parentheses are for the highest-resolution shells.
Extended Data Fig. 5. Crystal structures of S. tokodaii tRNAVal3.

(a,b) Schematic views of Mol. A (a) and B (b) of S. tokodaii tRNAVal3. Each residue is shown as a box. Color codes for the base pairs and base triples are the same as those in Fig. 2b,c. Tertiary interactions are shown as blue dashed lines. (c,d) Simulated annealing-omit Fo-Fc map contoured at 3.0 sigma around 2′-phosphate of Mol. A (c) and Mol. B (d). (e) Levitt base pair of G+15 and m5C48 with neighboring residues shown in stick representation with electron density map. 2Fo-Fc electron density map contoured at 0.76 sigma around G+15–m5C48 is shown in the lower panel. (f) Close-up view of the tRNA core around the G+15–m5C48 base pair. G+15, m5C48, and neighboring residues are shown in stick representation. Other residues are indicated as lines. Backbones are shown as cartoons. Hydrogen bonds are indicated by yellow dash lines. (g) Base pair of ac4C6 with G67 in stick representation with electron density map. 2Fo-Fc electron density map contoured at 0.76 sigma around ac4C6–G67 is shown in the lower panel. (h) Close-up view of the acceptor stem including ac4C6, G67, and neighboring base-pairs and nucleotides. (i) Electron density map of m1I57 and m1A58. 2Fo-Fc electron density maps contoured at 0.76 sigma for m1I57 and 1.02 sigma for m1A58 are shown. (j) Close-up view of T-loop including m1I57, s2U54, m1A58, and neighboring base-pairs and nucleotides. (k) Base pair of m2,2G26 with A44 in stick representation with electron density map. 2Fo-Fc electron density map contoured at 0.76 sigma around m2,2G26–A44 is shown in the lower panel. (l) Close-up view of D- and anticodon-stems including m2G10, m2,2G26, and neighboring base-pairs and nucleotides.
Extended Data Fig. 6. Torsion angles of nucleotides around position 47 of tRNAs.

(a) Key to torsion angles of nucleic acid backbone. Nomenclature of each angle is shown next to its direction, depicted as a black curved arrow. (b) Comparison of torsion angles at positions 44–48 of S. cerevisiae tRNAPhe (PDB: 1EHZ) (leftmost), and S. tokodaii tRNAVal3 Mol. A (left), Mol. B (right), and Tpt1p-treated tRNAVal3 (rightmost). Torsion angles of each tRNA are listed in Supplementary Table 3. Torsion angle changes from yeast tRNAPhe to Mol. A are shown on Mol. A as curved arrows. Torsion angle changes from Mol. A to Mol. B are shown on Mol. B as curved arrows. Torsion angle changes from Mol. A to Tpt1p-treated tRNA (Mol. A) are shown on Tpt1p-treated tRNA as curved arrows. The lower panels show the 90 degree-rotated models.
Although molecule A had a canonical tRNA core structure stabilized by multiple tertiary interactions between the D-arm and V-loop, including the base triples s4U8– A14–A21, Ψ13–G22–G46, C12–G23–C9 and m2G10–C25–G45 (Fig. 2b, c), molecule B unexpectedly had a non-canonical core structure (Fig. 2b, c). In molecule B, G46 was dissociated from the base triple Ψ13–G22–G46 and stacked with Up47 (Fig. 2f, Supplementary Video 1). The N2-amino group of G46 formed hydrogen bonds with A21 by inserting itself between the base triple and Up47 (Fig. 2d). This interaction pushes A21 towards A14 to make additional hydrogen bonds that stabilize the s4U8–A14–A21 triple (Fig. 2d). Because Up47 does not substantially change its backbone conformation (Extended Data Fig. 6b, Supplementary Table 3, Supplementary Video 1), the G46 base was stably trapped by Up47 in molecule B (Fig. 2d, f). To compensate for this conformational change, C9 comes up from the lower layer (C12–G23–C9) (Fig. 2g, Supplementary Video 1) to form the non-canonical base triple Ψ13–G22–C9 (Fig. 2f, Supplementary Video 1). Thus, the molecule B structure has a non-canonical base triple that might be stabilized by Up47. In this structural alteration, the torsion angles of A44 and G45 were slightly changed to make the backbone bulge outwards, flipping the G46 base out with the χ angle altered by –70° (Extended Data Fig. 6b, Supplementary Table 3). C9 changes its backbone, altering the α, β, γ and χ angles by 171°, –37°, –180° and 26°, respectively (Supplementary Table 3).
To further investigate the structural role of Up47, we also solved a crystal structure for Tpt1p-treated S. tokodaii tRNAVal3 (Extended Data Fig. 7a). Both molecules A and B of the Tpt1p-treated tRNA showed the canonical structure with the standard core (Extended Data Fig. 7b–f). In both molecules, U47 was dissociated from the s4U8– A14–A21 base triple (Extended Data Fig. 7b–e) with backbone angles α, γ and ε altered by 153°, –109° and –37°, respectively (molecule A) (Extended Data Fig. 6b, Supplementary Table 3), thereby placing the uracil base of U47 outwards (Fig. 2h, Extended Data Fig. 7b–d). In another aspect of the Tpt1p-treated tRNA, C9 was detached from the C12–G23–C9 base triple in both molecules (Extended Data Fig. 7f). These findings imply that Up47 stabilizes the metastable tRNA core structure with a non-canonical base triple during thermal denaturation.
Extended Data Fig. 7. Crystal structure of Tpt1p-treated S. tokodaii tRNAVal3.

(a) Overviews of crystal structure of Tpt1p-treated S. tokodaii tRNAVal3 with stick representation. Molecules A (left) and B (right) are shown in stick representation in pink and orange, respectively. U47 is colored in red. (b) Close-up views of the core structure of Mol. A (left) and B (right). Color code is the same as in Fig. 2b. (c) Schematic views of the core structure of Mol. A (left) and B (right). (d, e, f) Atomic structures of the base triples s4U8–A14–A21 (top), Ψ13–G22–G46 (middle) and C12–G23–C9 (bottom), in the core region of Mol. A (left) and B (right). Dashed lines indicate predicted interactions, with bond length in Å. U47 is shown in red.
Identification of an RNA kinase for Up47
To identify a gene responsible for Up47 formation, we narrowed down the candidate genes in the S. tokodaii genome by performing a comparative genomic analysis of sequenced genomes using RECOG (http://mbgd.genome.ad.jp/RECOG/). According to our analysis of Up47 distribution in archaeal species (Supplementary Note 4, Extended Data Fig. 8a–d), Up47 is present in seven archaeal species, including in S. tokodaii, but is absent in two species (Fig. 3a). Among the 2,826 genes encoded in the S. tokodaii genome, only nine genes (Supplementary Table 4) were commonly found in all seven archaeal species with Up47 (Fig. 3b). Among them, five genes (Supplementary Table 4) were of uncharacterized function (Fig. 3b). We chose one gene encoding a putative protein kinase, STK_09530 (hypothetical serine/threonine kinase, COG2112), as a strong candidate (Fig. 3b). STK_09530 resides in an operon containing a gene for a tRNA nucleotidyltransferase (STK_09520), implying that it encodes an enzyme related to tRNA maturation. We then constructed a strain of T. kodakarensis lacking tk2051, an orthologue of STK_09530. The tRNA fraction obtained from the Δtk2051 strain was subjected to liquid chromatography followed by MS (LC–MS) nucleotide analysis. A pUpm5C dimer was clearly observed in the parental strain (wild type) of T. kodakarensis (KU216), but was absent in the Δtk2051 strain (Fig. 3c). Therefore, tk2051 is the gene responsible for Up47 formation in cells. We designated the gene arkI (archaeal RNA kinase).
Extended Data Fig. 8. Phylogenetic distribution of Up47 in archaeal species.

(a) LC/MS nucleotide analyses of tRNA fractions from S. tokodaii, S. acidocaldarius, S. solfataricus, A. pernix, T. kodakarensis, and M. acetivorans. UV trace at 254 nm (upper panels) and XICs of the proton adducts of pN324m5C (m/z 724, z = 1) (lower panels) are shown. Asterisks indicate unassigned ions. (b) RNA-MS shotgun analysis of N. viennensis tRNA fraction treated with (right panel) or without (left panel) Tpt1p before RNase T1 digestion. XICs show the RNA fragments containing Up47 (UpCGp; m/z 1,053.08, z = -1). The product ions in the CID spectrum are assigned on UpCGp. (c) RNA-MS shotgun analysis of P. oguniense tRNA fraction treated with (right panel) or without (left panel) Tpt1p before RNase T1 digestion. XICs show the RNA fragments containing Up47 (Upm5Cm5Cac4CGp; m/z 866.10, 1,733.20, z = -2, -1). The product ions in the CID spectrum are assigned on Upm5Cm5Cac4CGp. (d) RNA-MS shotgun analysis of T. acidophilum tRNA fraction digested by RNase T1. XICs show the expected RNA fragments containing Up47 (left panels) or U47 (right panels) as indicated.
Fig. 3. Identification and characterization of the RNA kinase responsible for Up47 and its physiological role.

a, Venn diagram showing unique and shared genes among the Bacteria (E. coli), Eukarya (Homo sapiens, S. cerevisiae and Arabidopsis thaliana) and Archaea (Methanosarcina acetivorans, Thermoplasma acidophilum, S. tokodaii, Sulfolobus acidocaldarius, Saccharolobus solfataricus, Aeropyrum pernix, Pyrobaculum oguniense, T. kodakarensis and Nitrososphaera viennensis) domains possessing (+) or lacking (–) Up47. The pale red area includes genes unique to archaea having Up47. b, Comparative genomic analysis performed to narrow down the candidate genes responsible for Up47 modification. c, LC–MS nucleotide analysis of tRNA fractions from wild-type (WT, KU216) (left) and Δtk2051 (right) strains of T. kodakarensis. The upper panel shows the UV trace at 254 nm. The peaks for pA, pU, pC and pG are marked. The lower panel shows the XIC for the proton adduct of the dimer pUpm5C (m/z 724.1, z = 1). d, Growth measurement (OD600) of wild-type (KU216) (open circles), ΔqueE (squares), ΔarkI (closed circles) and ΔarkIΔqueE (triangles) strains of T. kodakarensis at 83 °C (left), 87 °C (middle) and 91 °C (right). Data represent the average values of technical triplicates ± s.d. e, In vitro reconstitution of Up47 with recombinant TkArkI in the presence (right panels) or absence (left panels) of ATP. XICs show the sum of monovalent and divalent negative ions from RNase T1-digested fragments containing Up47 (upper panels) or U47 (lower panels). f, Kinetic measurements of in vitro Up47 formation by TkArkI. The initial velocity (Vi) of the phosphorylation reaction was measured at the indicated concentrations of tRNA (left) and ATP (right). Data represent the average values of technical triplicates ± s.d. The Km and Vmax values are shown below each graph.
Up47 confers cellular thermotolerance
Next, we investigated the physiological importance of Up47 in T. kodakarensis. The ΔarkI strain grew as well as the wild-type strain (KU216) at the nearly optimal temperature of 83 °C (Fig. 3d), whereas it showed a weak temperature-sensitive phenotype with slower growth than the wild-type strain at 87 °C and 91 °C (Fig. 3d). We considered synthetic effects of Up47 with other tRNA modification, thus constructing a ΔarkIΔqueE double-knockout strain, in which queE is responsible for archaeosine (G+15) formation, because G+15 thermally stabilizes tRNAs and contributes to cellular thermotolerance19. We confirmed the absence of Up47 and G+15 in tRNAs from the double-knockout strain (Supplementary Fig. 3). The ΔqueE strain grew well at 83 °C, slowly at 87 °C and not at all at 91 °C (Fig. 3d), as reported19. The ΔarkIΔqueE strain grew slower than the wild-type, ΔarkI and ΔqueE strains at 83 °C (Fig. 3d). The strain exhibited a severe growth phenotype at 87 °C (Fig. 3d) and was unable to survive at 91 °C (Fig. 3d). This finding indicates that Up47 and G+15 cooperatively stabilize the tRNA core structure at high temperatures, thereby contributing to cellular thermotolerance.
Kinetics of tRNA phosphorylation by ArkI
We prepared recombinant T. kodakarensis ArkI (TkArkI) and examined in vitro Up47 formation. Up47 was efficiently reconstituted only in the presence of ATP (Fig. 3e). We then performed kinetic measurement of Up47 formation catalysed by TkArkI. The Km and Vmax values for tRNA were 97.3 nM and 9.9 nM min–1, respectively (Fig. 3f), showing that TkArkI efficiently recognizes tRNA substrate. By contrast, the Km value for ATP was found to be 1.2 mM (Fig. 3f). This value is extremely high when compared with the values for known protein kinases. This finding indicates that TkArkI-mediated Up47 formation might be regulated by sensing the cellular ATP concentration. We also characterized ArkI homologues from other archaeal and bacterial species (Supplementary Note 5, Supplementary Figs. 4, 5a–c).
Crystal structure of TkArkI
To find the structural basis of Up47 formation, we crystallized TkArkI and determined its atomic structure at a resolution of 1.8 Å using X-ray crystallography (Fig. 4a, Extended Data Table 1). On the basis of its amino acid sequence, TkArkI belongs to a superfamily of eukaryotic protein kinases (ePKs)30. As observed for ePKs, TkArkI also consisted of two lobes, termed the N-terminal and C-terminal lobes, which were connected by a hinge (positions 96–109) (Fig. 4a, Extended Data Fig. 9). ePKs consist of 12 conserved subdomains that fold into the catalytic core. TkArkI had subdomains I–V in the N-terminal lobe and subdomains VIab, VII, IX and XI in the C-terminal lobe, but lacked subdomains VIII and X (Fig. 4b, Extended Data Fig. 9). The conserved motifs of the P-loop (positions 31–38), catalytic loop (positions 128–140) and metal-binding loop (positions 145–153) were present in subdomains I, VIb and VII, respectively (Fig. 4b, Extended Data Fig. 9). Compared with the canonical ePK, the characteristic sequences in the conserved motifs were altered in TkArkI. The HRD triplet in the catalytic loop (VIb) of ePKs was replaced with HGQ in TkArkI (Extended Data Fig. 9). In addition, the DFG triplet in the metal-binding loop (VII) of ePKs was replaced with DFE in TkArkI (Extended Data Fig. 9). In subdomain IX, TkArkI had an α-helix (α6) specific to ArkI homologues. In subdomain XI, TkArkI had a longer α-helix (α7), when compared with the same helix in mouse PRKACA. In the extended C terminus of α7, the YKR motif is conserved in ArkI-family proteins (Extended Data Fig. 9), indicating that this positively charged motif is involved in RNA binding.
Fig. 4. Crystal structure and characterization of TkArkI.
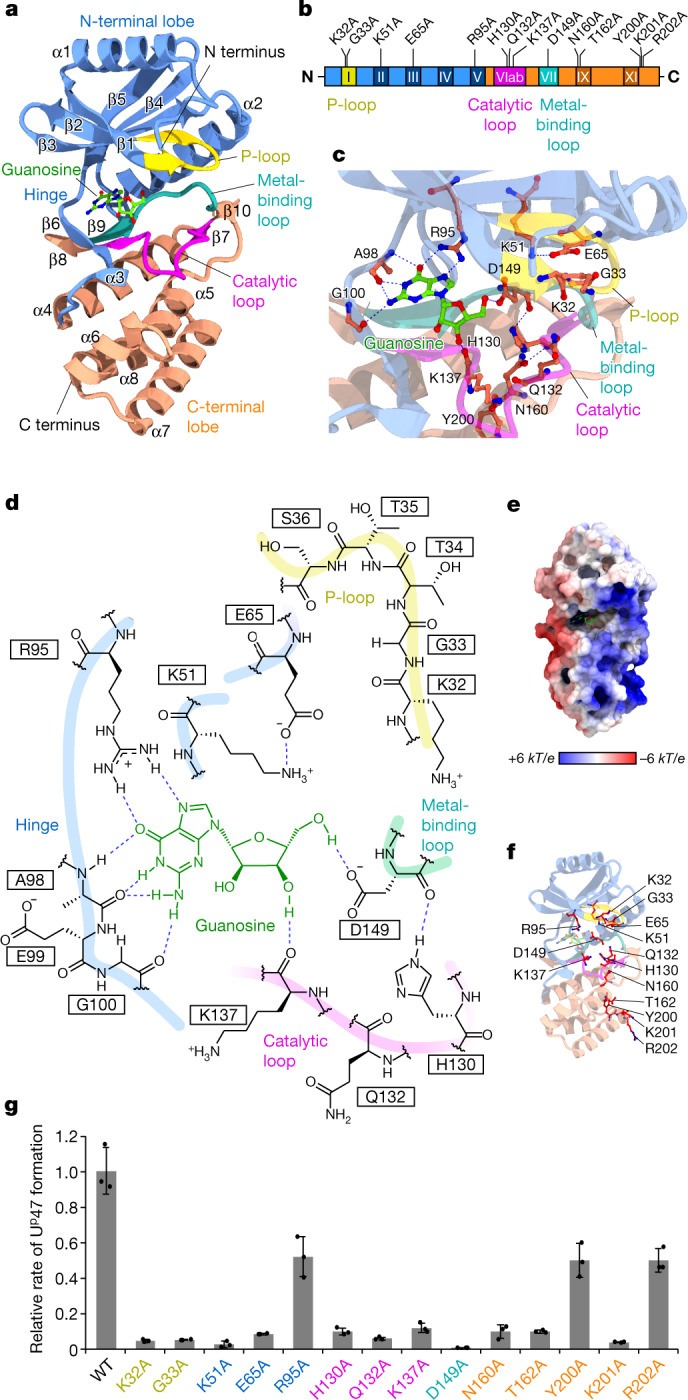
a, Overall structure of TkArkI with five features highlighted: the N-terminal lobe (residues 1–30 and 39–109; blue), P-loop (residues 31–38; yellow), C-terminal lobe (residues 110–127, 141–144 and 154–216; orange), subdomain VIb (catalytic loop, residues 128–140; pink) and subdomain VII (metal-binding loop, residues 145–153; cyan). Guanosine observed in a putative ATP-binding pocket is shown in ball-and-stick representation. b, Subdomains of TkArkI showing the locations of mutations examined in this study. Colour codes for each feature are the same as in a. c, Close-up view of the putative ATP-binding pocket in TkArkI. Residues for which mutations were examined in this study are indicated. Guanosine is shown in ball-and-stick representation. d, Schematic diagram of guanosine binding in the putative ATP-binding pocket. Predicted interactions are indicated with dashed lines. The main chains of the P-loop, hinge, catalytic loop and metal-binding loop are shown with bold lines. e, Electrostatic surface potential of TkArkI. Positively and negatively charged areas are coloured in blue and red, respectively. Guanosine is shown in ball-and-stick representation. The surface potential is described as dimensionless numbers. kT/e refers to the conversion factor (k, proportion constant; T, temperature; e; charge unit). f, Positions of mutation sites indicated in the crystal structure. g, Relative activities of a series of TkArkI mutants, normalized against the activity of wild-type TkArkI. Data represent the average values of technical triplicates ± s.d.
Extended Data Fig. 9. Sequence alignment of ArkI family (COG2112).

ArkI homologs and PRKACA (PRKACA_MOUSE, PDB: 1ATP) as a canonical ePK are aligned based on structure comparison using DALI (http://ekhidna2.biocenter.helsinki.fi/dali/). Bacterial and archaeal homologs of ArkI are added using MAFFT (https://mafft.cbrc.jp/alignment/server/). Black and gray boxes indicate the degree of sequence similarity. Residues mutated in TkArkI are indicated as red letters. The alpha helices and beta strands observed in the TkArkI structure are depicted on top of alignments as helices and arrows, respectively. Those observed in PRKACA are depicted under the alignments, as well. Subdomains (I to XI) and representative motifs (P-loop, HRD, DFG, and APE) in ePK are underlined and featured. Abbreviations for organisms: Tk, Thermococcus kodakarensis; PAB, Pyrococcus abyssi; STK, Sulfurisphaera tokodaii; Mefer, Methanocaldococcus fervens; Pogu, Pyrobaculum oguniense; aq, Aquifex aeolicus.
Although we demonstrated that TkArkI is an ATP-dependent RNA kinase involved in the formation of Up47 (Fig. 3e, f), we observed a clear electron density for guanosine in the cleft of the two lobes (Fig. 4a, c, d), which corresponds to the ATP-binding site of ePKs surrounded by the hinge and metal-binding, catalytic and P-loops (Supplementary Fig. 6a, b). We confirmed guanosine (and deoxyguanosine) as a ligand that tightly binds to TkArkI (Supplementary Note 6, Supplementary Fig. 7a–c). These observations indicate that TkArkI has binding affinity for guanosine and deoxyguanosine but uses ATP as a major phosphate donor. In the ATP-binding site of mouse PRKACA (Supplementary Fig. 6a, b), the triphosphate of ATP coordinates two Mn2+ ions and interacts tightly with the conserved motifs, especially the metal-binding loop and P-loop. However, in the guanosine-bound TkArkI structure, the P-loop was dislocated from the ligand-binding site (Fig. 4c, d). Thus, ATP does not bind the ligand-binding site of the observed structure. In homology modelling to ePKs (Supplementary Fig. 6c), ATP virtually bound to the active form of the ligand-binding site of TkArkI. It is likely that the P-loop and other motifs form the active pocket for ATP binding following tRNA binding to TkArkI. Although the biological relevance of guanosine binding to TkArkI is not known, guanosine may compete with ATP to regulate tRNA phosphorylation, similar to the mechanism by which nucleoside derivatives inhibit protein kinases31,32. Judging by its high Km value for ATP (1.2 mM) (Fig. 3f), TkArkI might sense the cellular energy status and guanosine binding to TkArkI might have a regulatory role in Up47 formation. Given that TkArkI was a recombinant protein expressed in Escherichia coli, we cannot rule out the possibility that guanosine was an artificial ligand bound to the inactive form of TkArkI. It is unclear whether guanosine actually binds to TkArkI within archaeal cells at high growth temperatures.
The electrostatic surface potential showed a large positive area on one side of the TkArkI structure (Fig. 4e). The positively charged surface covered the ATP-binding site in the N-terminal lobe and extended to the ArkI-specific elongated α7 helix in the C-terminal lobe (Extended Data Fig. 9). Instead of the missing subdomain VIII involved in recognition of substrate peptide in ePKs (mouse PRKACA), the basic surface in the C-terminal lobe might bind substrate tRNA through electrostatic interaction.
To characterize the conserved residues in TkArkI, we constructed 14 TkArkI mutants in which targeted residues were replaced by alanine (Fig. 4b, f). All mutants were expressed in soluble form and purified. The tRNA phosphorylation activity of each mutant was measured (Fig. 4g). In the ATP-binding site, K32A, G33A, K51A and E65A substitutions markedly reduced activity, whereas the R95A substitution caused a mild reduction in activity. In addition, a severe reduction in activity was observed in the H130A, Q132A and K137A mutants with substitutions in the catalytic loop. No activity was detected for the D149A mutant, in which the mutated residue is in subdomain VII involved in metal binding. These results clearly confirm the critical role of catalytic residues in kinase activity. The N160A and T162A substitutions in subdomain IX led to decreased activity. We mutated the YKR motif in the α7 helix, finding a severe reduction in activity with the K201A substitution and a mild reduction with the Y200A and R202A substitutions. These observations indicate the importance of the conserved residues and positively charged surface in the C-terminal lobe.
KptA acts as an eraser for Up47
Tpt1p removes the 2′-phosphate from tRNA precursors during maturation33. Tpt1/KptA homologues are distributed across all domains of life34,35 (Supplementary Fig. 4). Although Tpt1/KptA homologues are also present in thermophilic archaea and bacteria (Supplementary Fig. 4), natural RNA substrates with 2′-phosphate have not been identified.
Efficient removal of Up47 by yeast Tpt1p prompted us to speculate that archaeal KptA is capable of removing the 2′-phosphate of Up47 from tRNAs in the cell (Fig. 5a). To explore this possibility, we conducted in vitro dephosphorylation of Up47 with T. kodakarensis KptA (TkKptA) in the presence of NAD+, with the results indicating that the 2′-phosphate of Up47 was efficiently removed (Fig. 5b). In the same reaction conditions used for Up47 formation by TkArkI, we measured the kinetic parameters of Up47 dephosphorylation catalysed by TkKptA: the Km and Vmax values for tRNA were 180 nM and 27 nM s–1, respectively (Fig. 5c). The Km value for dephosphorylation by TkKptA is comparable to that of phosphorylation by TkArkI, implying that TkKptA acts as an eraser for Up47 in the cell.
Fig. 5. KptA acts as a potential eraser for Up47.
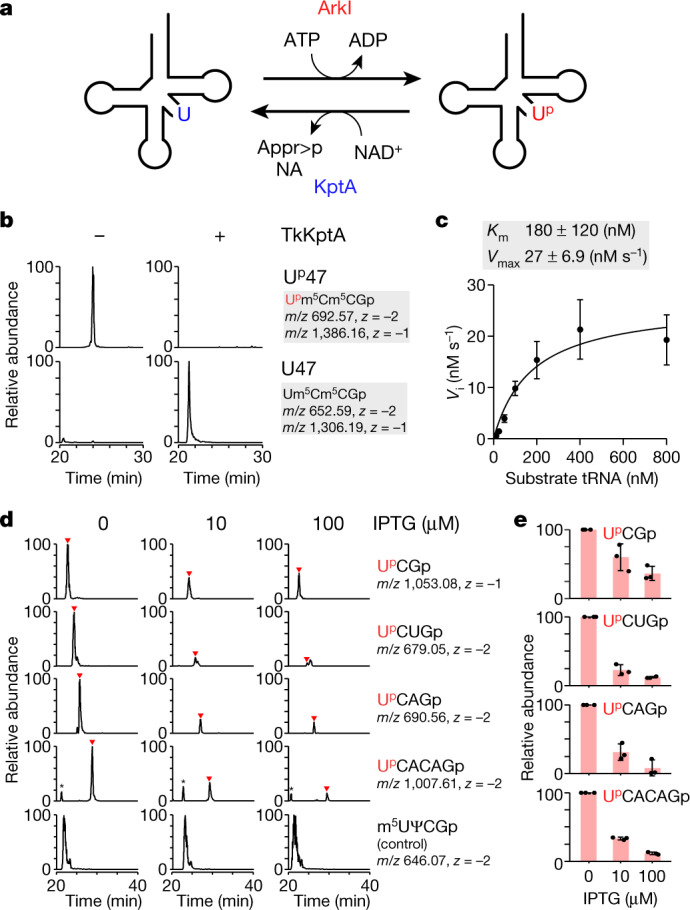
a, Reversibility of Up47 mediated by ArkI and KptA. ArkI phosphorylates U47 of tRNA to form Up47 using ATP as a phosphate donor, producing ADP as a by-product. KptA converts Up47 to U47 by transferring the phosphate group of Up47 to NAD+, producing ADP-ribose 1′′,2′′-cyclic phosphate (Appr>p) and nicotinamide (NA) as by-products. b, In vitro dephosphorylation of Up47 with (+) or without (–) TkKptA. XICs show the sum of monovalent and divalent negative ions from RNase T1-digested fragments containing Up47 (upper panels) or U47 (lower panels). c, Kinetic measurement of in vitro Up47 dephosphorylation by TkKptA. The initial velocity (Vi) of the dephosphorylation reaction was measured at the indicated tRNA concentrations. Data represent the average values of technical triplicates ± s.d. The Km and Vmax values are shown above the graph. d, Dephosphorylation of Up47 by TkKptA in E. coli. XICs show Up47-containing fragments derived from various E. coli tRNA species (Supplementary Table 5) from an E. coli ΔtrmBΔtapT strain expressing N. viennensis ArkI: UpCGp (top panels), UpCUGp (second panels), UpCAGp (third panels), UpCACAGp (fourth panels) and m5UΨCGp as a control fragment (bottom panels). Relative abundance of the Up47-containing fragments was measured in E. coli strains in which TkKptA was not expressed (left panels) or where TkKptA expression was induced with 10 μM (middle panels) or 100 μM (right panels) IPTG. e, Relative peak intensity of each Up47-containing fragment detected in the tRNA fraction from E. coli strains cultured with 0, 10 or 100 μM IPTG. Data represent the average values of technical triplicates ± s.d.
We then examined the in vivo function of Tpt1/KptA homologues in Up47 dephosphorylation, using E. coli as a model organism. Because E. coli tRNAs have m7G46 and acp3U47 modifications, which inhibit Up47 formation in the V-loop, we used the E. coli ΔtrmBΔtapT strain as a host cell in which both of these tRNA modifications are absent and then expressed Nitrososphaera viennensis ArkI (NvArkI), because N. viennensis is a mesophilic archaeon and its ArkI homologue was predicted to have efficient activity in E. coli. The class I tRNA fraction prepared from this strain was subjected to shotgun analysis to detect the Up47 modification. We clearly detected four Up47-containing fragments derived from various E. coli tRNA species (Fig. 5d, Supplementary Table 5). Each fragment was sequenced by higher-energy collision dissociation analysis, confirming the presence of Up at position 47 (Supplementary Fig. 8). Next, we introduced TkKptA under the control of an isopropyl β- d-1-thiogalactopyranoside (IPTG)-inducible promotor and quantified the peak intensity of each Up47-containing fragment when TkKptA expression was induced by addition of 10 or 100 μM IPTG (Fig. 5d, e). All four Up47-containing fragments had decreased abundance as a function of IPTG concentration, demonstrating that TkKptA erases Up47 in E. coli. We obtained similar results with E. coli KptA (Extended Data Fig. 10a, b) and S. cerevisiae Tpt1p (Extended Data Fig. 10c, d). Together, these data demonstrate that Tpt1/KptA homologues dephosphorylate Up47 of tRNAs in vivo.
Extended Data Fig. 10. Dephosphorylation of Up47 by KptA/Tpt1p homologs in E. coli.

(a, c) In vivo dephosphorylation of Up47 by EcKptA (a) or ScTpt1p (c). XICs show Up47-containing fragments from various E. coli tRNA species (Supplementary Table 5) isolated from E. coli ΔtrmB/ΔtapT strain expressing N. viennensis ArkI; UpCGp (top panels), UpCUGp (second panels), UpCAGp (third panels), UpCACAGp (fourth panels), and m5UΨCGp as a control fragment (bottom panels). Relative abundance of the Up47-containing fragments was measured in E. coli strain in which EcKptA (a) or ScTpt1p (c) is not expressed (left panels) or induced by 10 μM (middle panels) or 100 μM IPTG (right panels). (b, d) Quantification of Up47 dephosphorylation in E. coli by EcKptA (b) or ScTpt1p (d). Peak intensity is shown for each Up47-containing fragment detected in tRNA fractions from E. coli strain cultured with 0, 10, or 100 μM IPTG. Data represent average values of technical triplicates ± s.d.
Discussion
Up47 is, to our knowledge, the first known instance of internal phosphorylation as a stable RNA modification (Supplementary Note 7). 2′-Phosphate at an internal residue appears transiently during tRNA splicing in fungi and plants36,37. However, this moiety is not formed by phosphorylation but rather through hydrolysis of 2′,3′-cyclic phosphate via the healing and sealing pathway36,38. Because the 2′-phosphate is removed by Tpt1p33, it is not present in mature tRNAs.
In S. tokodaii tRNAs isolated in this study, Up47 was detected in nine class I tRNA species with high frequency (82–100%) (Fig. 1d, Extended Data Fig. 3a) but was absent in two class I tRNAs (tRNAGln2 and tRNACys) and two class II tRNAs (tRNALeu4 and tRNASer3) (Extended Data Fig. 3a). Judging from the primary sequences of these species (Supplementary Fig. 9), it is likely that ArkI introduces Up47 in tRNAs bearing a V-loop with five bases, as tRNAGln2 and tRNACys have four and six bases in the V-loop, respectively. Supporting this finding, only the class I tRNA fraction was phosphorylated in total RNA by in vitro reaction (Supplementary Fig. 5b, c).
RNA hydrolysis is mediated by the 2′-OH group in the presence of divalent metal ions such as Mg2+. Especially at high temperatures, RNA is rapidly degraded. Similarly to 2′-O-methylation, the 2′-phosphorylation of Up47 also serves to prevent tRNA degradation. This property partly explains the RNase resistance of tRNA conferred by Up47 (Fig. 1g). It is known that Up adopts C2′-endo ribose puckering28, which confers flexibility to the RNA strand by extending the backbone structure39. Hence, Up47 presumably acts as a defining mark for single-stranded RNA. In the process of tRNA folding, Up47 might have a role in preventing the V-loop from being accidentally incorporated into stem structures, ensuring correct folding of the tRNA L-shape structure. Especially in thermophiles, tRNA might frequently misfold owing to its high G+C content. Thus, Up47 deposition in the tRNA precursor might be required to loop out the V-loop region to ensure correct folding of the tRNA. Other modifications at position 47, acp3U40 and dihydrouridine7, are used in bacteria and eukaryotes, respectively. acp3U directly prevents the V-loop from being incorporated into stem structures by inhibiting base pairing. Dihydrouridine also adopts the C2′-endo conformation41 and confers flexibility to the V-loop. It is interesting that similar functions are evolutionarily conserved in different V-loop modifications across the domains of life.
Intriguingly, S. tokodaii tRNAVal3 was present as two isomers (molecules A and B) with different conformations in the core region (Fig. 2a). Molecule A has a standard core structure found in many tRNAs, whereas molecule B has a non-standard core structure. Because the Tpt1p-treated tRNA has the canonical structure with the standard core (Extended Data Fig. 7a–f), it is likely that the structural alteration is caused by Up47. During thermal denaturation of tRNAs, the core region and D-arm are unwound first42,43. In molecule B, G46 is released from the base triple Ψ13–G22–G46 and stacks with the uracil base of Up47 (Fig. 2d, f). Presumably, this unique conformation is a metastable structure of tRNA during heat denaturation. Curiously, in the structural transition from molecule A to molecule B (Supplementary Video 1), the torsion angle of G46 changes substantially, whereas that of Up47 does not (Extended Data Fig. 6b). Up47 catches the G46 base that is dissociated from the base triple to restrict further rotation of the V-loop, thereby stabilizing the metastable core structure of the tRNA to prevent its heat denaturation. In addition, C9 comes up from the lower layer (C12–G23–C9) to fill in for the missing G46, forming the non-canonical base triple Ψ13–G22–C9 (Fig. 2f). Up47 does not fix the tRNA rigidly but rather maintains a metastable structure when the tRNA core thermally fluctuates, thereby preventing further collapse of the core structure, as well as increasing the chance of return to the canonical structure.
ArkI homologues are mainly distributed in thermophilic archaea but are also present in some bacteria (Supplementary Fig. 4). We confirmed the activity of tRNA phosphorylation for bacterial ArkI homologues (Supplementary Fig. 5a, c). In silico analysis of protein kinases suggested that ArkI-family proteins were originally classified as members of the AQ578 family found in bacterial and archaeal genomes44; the AQ578 family was proposed to have emerged by gene duplication in the early archaeal lineage. The bacterial AQ578 family might have been acquired by horizontal gene transfer of the archaeal homologue, suggesting that the strategy of stabilizing tRNA by internal phosphorylation might have spread across the domains of life.
The ΔarkI strain of T. kodakarensis exhibited weak temperature sensitivity (Fig. 3d), demonstrating that Up47 by itself contributes to cellular thermotolerance. Because multiple tRNA modifications cooperatively stabilize the tRNA structure, we chose to analyse the G+15 modification, showing a synthetic phenotype with Up47 loss. We found that the ΔarkIΔqueE double-knockout strain was extremely susceptible to high temperature (Fig. 3d), suggesting that Up47 and G+15 cooperatively stabilize the tRNA core structure and contribute to cellular thermotolerance. Up47 flexibly deals with the structural change due to thermal denaturation of the core structure, like a padlock, whereas G+15 tightly fixes the core structure, like a screw bolt (Supplementary Note 3). On the basis of these findings, we propose a new mechanism of tRNA stabilization mediated by two distinct but concerted actions of tRNA modification.
In eukaryotic mRNAs and non-coding RNAs, N6-methyladenosine (m6A) has a critical role in RNA metabolism and function as a reversible RNA modification45. If Up47 is a reversible modification, it is expected that tRNA function and stability are dynamically regulated by a writer and eraser, raising the possibility of epitranscriptomic regulation of tRNAs in translation. The mechanism closely resembles post-translational modification of proteins. Phosphorylation and dephosphorylation rapidly and dynamically control protein function46–48. Because tRNA is a stable molecule with a low turnover rate and long lifetime in the cell, it would be reasonable for tRNA function to be regulated by Up47 modification. We found efficient dephosphorylation of Up47 by TkKptA in vitro (Fig. 5b) and confirmed the in vivo activity of Tpt1/KptA homologues in E. coli cells (Fig. 5d, e and Extended Data Fig. 10a–d). In fact, tRNA stability is regulated by thermophile-specific tRNA modifications including m5s2U and ac4C, which become much more abundant as the growth temperature increases14,49 but are not reversible. Reversible Up47 modification would be beneficial for hyperthermophilic organisms in extremely harsh environments. Future studies will be necessary to investigate Up47 frequency and the expression levels of ArkI and KptA under various growth conditions, including during rapid changes in growth temperature and introduction of environmental stresses.
Methods
Archaeal strains and media
S. tokodaii str. 7, Methanosarcina acetivorans C2A and Thermoplasma acidophilum were kindly provided by T. Oshima (Kyowa Kako Co., Ltd), T. Yokogawa (Gifu University) and H. Hori (Ehime University), respectively. Sulfolobus acidocaldarius (JCM no. 8929), Saccharolobus solfataricus (JCM no. 8930), Aeropyrum pernix (JCM no. 9820), Pyrobaculum oguniense (JCM no. 10595) and N. viennensis (JCM no. 19564) were obtained from Japan Collection of Microorganisms, RIKEN BRC which is participating in the National BioResource Project of the MEXT, Japan.
S. tokodaii and S. acidocaldarius were cultured at 80 °C in JCM medium no. 165 consisting of 1 g l–1 yeast extract, 1 g l–1 casamino acids, 1.3 g l–1 (NH4)2SO4, 0.28 g l–1 KH2PO4, 0.25 g l–1 MgSO4·7H2O, 0.07 g l–1 CaCl2·2H2O, 2.0 mg l–1 FeCl3·6H2O, 1.8 mg l–1 MnCl2·4H2O, 4.5 mg l–1 Na2B4O7·10H2O, 0.22 mg l–1 ZnSO4·7H2O, 0.05 mg l–1 CuCl2·2H2O, 0.03 mg l–1 Na2MoO4·2H2O, 0.03 mg l–1 VOSO4·H2O and 0.01 mg l–1 CoSO4·7H2O (adjusted to pH 2.5 with H2SO4). S. solfataricus was cultured at 80 °C in JCM medium no. 171 consisting of 1 g l–1 yeast extract, 2.5 g l–1 (NH4)2SO4, 3.1 g l–1 KH2PO4, 0.2 g l–1 MgSO4·7H2O, 0.25 g l–1 CaCl2·2H2O, 1.8 mg l–1 MnCl2·4H2O, 4.5 mg l–1 Na2B4O7·10H2O, 0.22 mg l–1 ZnSO4·7H2O, 0.05 mg l–1 CuCl2·2H2O, 0.03 mg l–1 Na2MoO4·2H2O, 0.03 mg l–1 VOSO4·H2O and 0.01 mg l–1 CoSO4·7H2O (adjusted to pH 4.0 with H2SO4). A. pernix was cultured at 90 °C in JCM medium no. 224 consisting of 1 g l–1 yeast extract, 1 g l–1 peptone, 1 g l–1 Na2S2O3·5H2O, 24.0 g l–1 NaCl, 7.0 g l–1 MgSO4·7H2O, 5.3 g l–1 MgCl2·6H2O, 0.7 g l–1 KCl and 0.1 g l–1 CaCl2·2H2O (adjusted to pH 7.0 with NaOH). P. oguniense was cultured at 90 °C in JCM medium no. 165 with addition of 1.0 g l–1 Na2S2O3·5H2O (adjusted to pH 7.25 with NaOH). N. viennensis was cultured at 42 °C in JCM medium no. 1004 consisting of 1 g l–1 NaCl, 0.5 g l–1 KCl, 0.4 g l–1 MgCl2·6H2O, 0.2 g l–1 KH2PO4, 0.1 g l–1 CaCl2·2H2O, 1.0 ml l–1 modified trace element mixture (30 mg l–1 H3BO3, 100 mg l–1 MnCl2·4H2O, 190 mg l–1 CoCl2·6H2O, 24 mg l–1 NiCl2·6H2O, 2 mg l–1 CuCl2·2H2O, 144 mg l–1 ZnSO4·7H2O, 36 mg l–1 Na2MoO4·2H2O and 0.3% HCl), 1.0 ml l–1 vitamin solution (20 mg l–1 biotin, 20 mg l–1 folic acid, 100 mg l–1 pyridoxine·HCl, 50 mg l–1 thiamine·HCl, 50 mg l–1 riboflavin, 50 mg l–1 nicotinic acid, 50 mg l–1 DL-calcium pantothenate, 1 mg l–1 vitamin B12, 50 mg l–1 p-aminobenzoic acid and 2 g l–1 choline chloride (adjusted to pH 7.0 with KOH)), 1.0 ml l–1 7.5 mM EDTA·Na·Fe(III) solution (pH 7.0), 2.0 ml l–1 1 M NaHCO3 solution, 10 ml l–1 HEPES solution (238.4 g l–1 HEPES (free acid) and 24 g l–1 NaOH), 1.0 ml l–1 1 M NH4Cl solution and 1.0 ml l–1 1 M sodium pyruvate solution (adjusted to pH 7.6 with NaOH).
T. kodakarensis was cultured at 83 °C, 87 °C or 91 °C, in nutrient-rich medium (ASW-YT-S0 or MA-YT-Pyr) or synthetic medium containing amino acids (ASW-AA-S0), under strict anaerobic conditions. ASW-YT-S0 medium contains 0.8× artificial sea water (ASW)50, 10 g l–1 yeast extract, 5.0 g l–1 tryptone, 2.0 g l–1 elemental sulfur and 0.1% (wt/vol) resazurin. MA-YT-Pyr medium contains 30.5 g l–1 Marine Art SF-1 (Osaka Yakken), 10 g l–1 yeast extract, 5.0 g l–1 tryptone, 5.0 g l–1 pyruvate sodium and 0.1% (wt/vol) resazurin. ASW-AA-S0 medium contains 0.8× ASW, 0.5× amino acid solution50, modified Wolfe’s trace minerals (0.5 g l–1 MnSO4·2H2O, 0.1 g l–1 CoCl2, 0.1 g l–1 ZnSO4, 0.01 g l–1 CuSO4·5H2O, 0.01 g l–1 AlK(SO4)2, 0.01 g l–1 H3BO3 and 0.01 g l–1 NaMoO4·2H2O), 5.0 ml l–1 vitamin mixture51, 2.0 g l–1 elemental sulfur and 0.1% (wt/vol) resazurin. For plate cultivation, 2.0 ml l–1 polysulfide solution (20% elemental sulfur in 67% Na2S·9H2O solution) was added instead of elemental sulfur, and the media were solidified with 1.0% Gelrite (Fujifilm Wako Pure Chemical Corporation). When pyrF-negative transformants were selected0, 75% 5-fluoroorotic acid (5-FOA) was added. We used ASW-YT-S0 medium for standard cultivation, MA-YT-Pyr medium for growth comparisons and ASW-AA-S0 medium for construction of the gene knockout strain.
Preparation of tRNA fractions
For small-scale preparation (~100-ml culture), archaeal cells were resuspended in 3 ml solution D (4 M guanidine thiocyanate, 25 mM citrate–NaOH (pH 7.0), 0.5% (wt/vol) N-lauroylsarcosine sodium salt and 1 mM 2-mercaptoethanol) and mixed with an equal volume of water-saturated phenol and 1/10 volume of 3 M sodium acetate (pH 5.3). The mixture was shaken for 1 h on ice and mixed with 1/5 volume of chloroform, followed by centrifugation at 8,000g for 10 min at 4 °C. The supernatant was collected and mixed with an equal volume of chloroform, followed by centrifugation at 8,000g for 10 min at 4 °C. Total RNA was obtained from the resultant supernatant by isopropanol precipitation. The total RNA prepared in this manner was separated by 10% denaturing PAGE, followed by staining with SYBR Gold or toluidine blue. The visualized tRNA fraction including class I and class II tRNAs was cut out and eluted from the gel slice with elution buffer (0.3 M sodium acetate (pH 5.3) and 0.1% (wt/vol) SDS), followed by filtration to remove the gel pieces and ethanol precipitation for RNA-MS analysis of the tRNA fraction.
For large-scale preparation of tRNA fractions from S. tokodaii, cell pellets (53 g) were resuspended in 530 ml solution D and then mixed with 53 ml of 3 M sodium acetate (pH 5.3) and 425 ml neutralized phenol. The mixture was shaken for 1 h on ice to which 106 ml chloroform/isoamyl alcohol (49:1) was added, followed by centrifugation at 4,500g for 20 min at 4 °C. The supernatant was collected and mixed with 106 ml chloroform/isoamyl alcohol (49:1), followed by centrifugation at 4,500g for 15 min at 4 °C. The aqueous phase was collected and then subjected to isopropanol precipitation. The collected RNA was resuspended in 53 ml water and mixed with 80 ml TriPure Isolation Reagent (Roche), followed by centrifugation at 10,000g for 20 min at 4 °C. The supernatant was collected and mixed with 36 ml chloroform/isoamyl alcohol (49:1), followed by centrifugation at 10,000g for 10 min at 4 °C. The aqueous phase was collected and precipitated with isopropanol. The prepared total RNA (608 mg) was dissolved in 250 ml of buffer consisting of 20 mM HEPES-KOH (pH 7.6), 200 mM NaCl and 1 mM DTT and then loaded on a DEAE Sepharose Fast Flow column (320-ml beads) and fractionated with a gradient of NaCl from 200 to 500 mM. Fractions containing tRNA were collected by isopropanol precipitation.
Isolation of individual tRNAs
Isolation of individual tRNAs from thermophilic organisms is extremely difficult owing to their high melting temperatures, which are the consequence of their high G+C content and complex modifications. We thus optimized our original method for RNA isolation by RCC24 or chaplet column chromatography (CCC)52. Approximately 200 absorbance at 260 nm (A260) units of the S. tokodaii tRNA fraction was subjected to RCC. The isolation procedure was carried out as follows: hybridization at 66 °C in 6× NHE buffer (30 mM HEPES-KOH (pH 7.5), 15 mM EDTA (pH 8.0), 1.2 M NaCl, 1 mM DTT), washing at 50 °C with 0.1× NHE buffer (0.5 mM HEPES-KOH (pH 7.5), 0.25 mM EDTA (pH 8.0), 20 mM NaCl, 0.5 mM DTT) and elution at 72 °C with 0.1× NHE buffer. Eluted tRNAs were recovered by ethanol precipitation. Mature and precursor tRNAs were separated by 10% denaturing PAGE and stained with SYBR Gold. Visualized bands of mature and precursor tRNAs were cut out and eluted from the gel slices with elution buffer, followed by filtration to remove the gel pieces and precipitation with ethanol.
To crystalize native tRNA bearing Up47, we conducted large-scale isolation of S. tokodaii tRNAVal3 using CCC52. The S. tokodaii tRNA fraction (2,000 A260 units) was subjected to CCC with tandem affinity chaplet columns for tRNAVal3, tRNAIle2 and tRNAPhe. The isolation procedure was carried out as follows: hybridization at 66 °C in 6× NHE buffer, washing separately at 50 °C with 0.1× NHE buffer and elution at 72 °C with 0.1× NHE buffer. The eluted tRNAs were recovered by isopropanol precipitation. The sequences of the DNA probes are shown in Supplementary Table 6. The isolated tRNAVal3 was further purified by anion exchange chromatography to completely remove tRNAVal2, as described below.
RNA mass spectrometry
For tRNA fragment analysis by RNA-MS, 30 ng (900 fmol) of the isolated tRNA or 150 ng (4.5 pmol) of tRNA mixture was digested with RNase T1 (Epicentre or Thermo Fisher Scientific) or RNase A (Ambion) and analysed with a linear ion trap–Orbitrap hybrid mass spectrometer (LTQ Orbitrap XL, Thermo Fisher Scientific) equipped with a custom-made nanospray ion source and a splitless nanoHPLC system (DiNa, KYA Technologies) as described previously26,27. To analyse Ψ sites, tRNA was treated with acrylonitrile to cyanoethylate Ψ53 and subjected to RNA-MS. For dephosphorylation of the Up47-containing fragment (Extended Data Fig. 4a, b), RNase T1 digestion was performed in the presence of 0.01 U μl–1 bacterial alkaline phosphatase (BAP C75, Takara Bio). To precisely map tRNA modifications, RNA fragments were decomposed by CID in the instrument. The normalized collision energy of LTQ Orbitrap XL was set to 40%. Mongo Oligo Mass Calculator v2.08 (https://mods.rna.albany.edu/masspec/Mongo-Oligo) was used for assignment of the product ions in CID spectra.
For nucleoside analysis, 800 ng (24 pmol) of the isolated tRNAVal3 was digested with 0.09 U nuclease P1 (Fujifilm Wako Pure Chemical Corporation) in 20 mM ammonium acetate (pH 5.2) at 50 °C for 1 h and mixed with 1/8 volume of 1 M trimethylamine-HCl (TMA-HCl) (pH 7.2) and 0.06 U phosphodiesterase I (Worthington Biochemical Corporation), followed by incubation at 37 °C for 1 h. To this mixture, 0.08 U BAP was added, and the sample was incubated at 50 °C for 1 h. After that, 9 volumes of acetonitrile were added, followed by LC–MS/MS analysis as described in refs. 25,54 with some modifications as follows. The samples were chromatographed with a ZIC-cHILIC column (3-μm particle size, 2.1 × 150 mm; Merck) and eluted with 5 mM ammonium acetate (pH 5.3) (solvent A) and acetonitrile (solvent B) at a flow rate of 100 μl min–1 with a multistep linear gradient: 90–50% solvent B for 30 min, 50% solvent B for 10 min, 50–90% solvent B for 5 min and then initialization with 90% solvent B. The chromatographed eluent was directly introduced into the electrospray ionization source of the Q Exactive Hybrid Quadrupole–Orbitrap mass spectrometer (Thermo Fisher Scientific).
For nucleotide analysis, 800 ng (24 pmol) of the tRNA fraction or individual tRNA was digested with 0.09 U nuclease P1 in 20 mM ammonium acetate (pH 5.2) at 50 °C for 1 h and then mixed with 9 volumes of acetonitrile for LC–MS. The digests were chromatographed with a ZIC-cHILIC column and analysed by Q Exactive Hybrid Quadrupole–Orbitrap mass spectrometer (Thermo Fisher Scientific) or LTQ Orbitrap XL (Thermo Fisher Scientific) with a multistep linear gradient: 90–50% solvent B for 30 min, 50% solvent B for 10 min, 50–90% solvent B for 5 min and then initialization with 90% solvent B.
The acquired LC–MS data were analysed using Xcalibur 4.1 (Thermo Fisher Scientific) and were visualized with Canvas X (Nihon poladigital k.k).
Isolation and detection of pN324p
Five A260 units of the S. tokodaii tRNA fraction was completely digested with nuclease P1. Digests containing pN324m5C dinucleotide were subjected to periodate oxidation with 10 mM NaIO4 for 1 h on ice in the dark. The reaction was stopped by addition of 1 M l-rhamnose and incubation for 30 min. For β-elimination, an equal volume of 2 M lysine-HCl (pH 8.5) was added, and the sample was incubated at 45 °C for 90 min. The product containing pN324p was then subjected to anion exchange chromatography with a Q Sepharose Fast Flow column (GE Healthcare) equilibrated with 20 mM triethylammonium bicarbonate (TEAB) (pH 8.2). The eluate with 2 M TEAB was collected and dried by evaporation in vacuo. The pellet was dissolved with water and mixed with an equal volume of chloroform, followed by centrifugation at 20,000g for 5 min at 4 °C. The supernatant was recovered and dried again. This process was repeated five times. The resultant digest was mixed with 9 volumes of acetonitrile and subjected to LC–MS/MS using an LCQ-Advantage ion trap mass spectrometer (Thermo Scientific), equipped with an electrospray ionization source and an HP1100 LC system (Agilent Technologies). For LC, the digest was chromatographed with a ZIC-HILIC column (3.5 μm; pore size, 100 Å; internal diameter, 2.1 × 150 mm; Merck) and eluted with 5 mM formic acid (pH 3.4) (solvent A) and acetonitrile (solvent B) at a flow rate of 100 μl min–1 with a multistep gradient: 90–70% solvent B for 25 min, 70–10% solvent B for 15 min, 10% solvent B for 5 min and then initialized with 90% solvent B.
Expression and purification of recombinant proteins
Synthetic genes for arkI from T. kodakarensis, Methanocaldococcus fervens, P. oguniense, Aquifex aeolicus, Nautilia profundicola and Leptolyngbya sp. PCC7376 were designed with codons optimized for E. coli expression and synthesized by GENEWIZ or Thermo Fisher Scientific. Each gene was cloned into the pE-SUMO-TEV vector by the SLiCE method55. N. viennensis arkI was PCR amplified from genomic DNA with a set of primers (Supplementary Table 6) and cloned into the BamHI and NotI sites of pE-SUMO-TEV.
E. coli BL21(DE3) or Rosetta2(DE3) cells transformed with the pE-SUMO-TEV vector carrying each arkI gene were cultured in 250 ml or 1 l of LB containing 50 μg ml–1 kanamycin and 20 μg ml–1 chloramphenicol when necessary. His6–SUMO-tagged recombinant protein was expressed at 37 °C for 3–4 h by induction with 0.1 or 1 mM IPTG or 2% (wt/vol) lactose when the cells reached OD610 = 0.4–0.6. P. oguniense ArkI was expressed in cells cultured overnight at 18 °C. The collected cells were resuspended in lysis buffer (50 mM HEPES-KOH (pH 8.0), 150 mM KCl, 2 mM MgCl2, 20 mM imidazole, 12% (vol/vol) glycerol, 1 mM 2-mercaptoethanol and 1 mM PMSF) and disrupted by sonication, followed by centrifugation at 15,000g for 15 min at 4 °C. The supernatant was boiled at 60 °C for 20 min (for ArkI homologues from T. kodakarensis, M. fervens, P. oguniense and A. aeolicus) and centrifuged at 15,000g for 15 min at 4 °C. The recombinant protein was affinity captured on an Ni-Sepharose 6 Fast Flow column (GE Healthcare) and then eluted with lysis buffer containing 300 mM imidazole, followed by gel filtration with a PD-10 column (GE Healthcare) to remove the imidazole. The recombinant protein for N. viennensis ArkI was purified using a HisTrap column (GE Healthcare) with a linear gradient of 0–500 mM imidazole, followed by dialysis using a Slide-A-Lyzer Dialysis Cassette (Thermo Fisher Scientific) to remove imidazole. The purified protein was subjected to Ulp1 digestion at 4 °C overnight to cleave the His6–SUMO tag and then passed through a Ni-Sepharose 6 Fast Flow column to remove the tag. Because ArkI homologues from M. fervens (MfArkI) and Leptolyngbya sp. PCC7376 (LeArkI) aggregated following tag removal, His6–SUMO tag-fused proteins of these homologues were used for the phosphorylation assay. Purified protein was quantified by the Bradford method using BSA as a standard.
For large-scale preparation of T. kodakarensis ArkI for crystallization, the E. coli BL21(DE3) strain carrying pE-SUMO-TkArkI was cultured in 2 l of LB containing 50 μg ml–1 kanamycin and TkArkI was expressed at 25 °C overnight by induction with 0.1 mM IPTG when the cells reached OD610 = 0.4. The cells were collected and disrupted by sonication in lysis buffer (50 mM HEPES-KOH (pH 8.0), 150 mM KCl, 2 mM MgCl2, 20 mM imidazole, 12% (vol/vol) glycerol, 1 mM 2-mercaptoethanol and 1 mM PMSF). The protein was purified using a HisTrap column with a linear gradient of 20–520 mM imidazole. Fractions containing TkArkI were pooled and subjected to Ulp1 digestion at 4 °C overnight to cleave the tag, followed by passage through a Ni-Sepharose 6 Fast Flow column to remove the tag fragment. The flow-through fraction was filtered through a 0.45-μm PVDF membrane to remove the resin. The protein was further purified by affinity chromatography with a HiTrap Heparin HP column (GE Healthcare) using a linear gradient of 150–1,150 mM KCl. TkArkI was further purified by size exclusion chromatography using a Superdex 75 10/300 GL column (GE Healthcare) with buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl and 10 mM 2-mercaptoethanol and then concentrated to 5.74 mg ml–1 and stored at –80 °C.
The T. kodakarensis kptA gene was PCR amplified from genomic DNA from T. kodakarensis with the primers listed in Supplementary Table 6 and cloned into pE-SUMO-TEV to give pE-SUMO-TEV-tkkptA. The E. coli Rosetta2(DE3) strain carrying pE-SUMO-TEV-tkkptA was cultured in 1 l LB containing 50 μg ml–1 kanamycin and 20 μg ml–1 chloramphenicol, and TkKptA was expressed at 37 °C for 3 h by induction with 0.1 mM IPTG when the cells reached OD610 = 0.6. The recombinant TkKptA was purified as described above. The gene encoding Tpt1p was PCR amplified from the genomic DNA of S. cerevisiae BY4742 with the set of primers listed in Supplementary Table 6 and was cloned into pET21b (Merck) between the NdeI and XhoI sites. Recombinant Tpt1p was purified as described above.
Removal of the 2′-phosphate of Up47 by Tpt1p
Removal of the 2′-phosphate of Up47 by yeast Tpt1p was performed as described33. Individual tRNAs or the tRNA fraction was incubated for 3 h at 30 °C in a reaction mixture (25 μl) consisting of 20 mM Tris-HCl (pH 7.4), 0.5 mM EDTA (pH 8.0), 1 mM NAD+, 2.5 mM spermidine, 0.1 mM DTT, 0.9 μM tRNA and 0.1 μg μl–1 recombinant Tpt1p. The tRNA was extracted by phenol/chloroform treatment and recovered by ethanol precipitation, followed by desalting with Centri-Sep spin columns (Princeton Separations). For crystallization of Tpt1p-treated tRNA, S. tokodaii tRNAVal3 (202.5 μg) was dephosphorylated by yeast Tpt1p in a 200-μl reaction mixture.
Measurement of the thermal stability of tRNA
S. tokodaii tRNAVal3 (25 pmol) with or without Up47 was dissolved in degassed buffer consisting of 50 mM Tris-HCl (pH 7.4), 100 mM NaCl and 1 mM MgCl2 and incubated at 80 °C for 5 min, followed by cooling to 25 °C at a rate of 0.1 °C s–1. The samples were placed onto a Type 8 multi-micro UV quartz cell (path length, 10 mm). The hyperchromicity of tRNA was monitored on a UV–visible light spectrophotometer (V-630, JASCO). The gradients were as follows: 25 °C for 30 s, 25–40 °C at 5 °C min–1, 40 °C for 5 min and 40–105 °C at 0.5 °C min–1. The Tm was calculated using Spectra Manager v2 (JASCO). Melting curves were generated using Microsoft Excel.
RNase probing of tRNA
S. tokodaii tRNAVal3 (25 pmol) with or without Up47 was labelled with 32P at the 3′ terminus by ligation with [5′-32P]cytidine 3′,5′-bisphosphate (PerkinElmer). The labelled tRNA was separated on a 7.5% (wt/vol) polyacrylamide gel containing 7 M urea, 1× TBE and 10% (vol/vol) glycerol and was purified by gel extraction. Labelled tRNA was mixed with the S. tokodaii tRNA fraction as a carrier to a concentration of 100,000 counts per minute (c.p.m.) per A260 unit and was precipitated with ethanol. The pellet was dissolved in water to a concentration of 0.1 A260 units per μl. For the RNase degradation assay, the labelled tRNA (0.1 A260 units, 10,000 c.p.m.) was incubated at 65 °C in a reaction mixture consisting of 10 mM HEPES-KOH (pH 7.6), 0.5 mM MgCl2, 100 mM NaCl and 0.1 U μl–1 RNase I (Promega). At time points of 1, 3, 5, 10, 15 and 30 min after starting the reaction, aliquots were taken from the mixture and mixed well with chilled phenol/chloroform/isoamyl alcohol (25:24:1, pH 7.9) to stop the reaction, followed by centrifugation at 15,000g for 15 min at 4 °C. The supernatant was collected and treated with an equal volume of chloroform, followed by centrifugation at 15,000g for 5 min at 4 °C. The supernatant was mixed with 2× loading solution (2× TBE, 7 M urea, 13.33% (wt/vol) sucrose, 0.05% (wt/vol) xylene cyanol and 0.05% (wt/vol) bromophenol blue) and subjected to 10% denaturing PAGE. The gel was exposed to an imaging plate, and radioactivity was visualized by using an FLA-7000 imaging analyser (Fujifilm). Graphs were generated using Microsoft Excel.
Crystallization of S. tokodaii tRNAVal3
S. tokodaii tRNAVal3 (500 μg), isolated as described above, was refolded in annealing buffer (50 mM HEPES-KOH (pH 7.6), 5 mM MgCl2 and 1 mM DTT) by incubation for 5 min at 80 °C and cooling to 25 °C with a rate of 0.1 °C s–1. tRNAVal3 was further purified by anion exchange chromatography using a Mono Q 5/50 GL column (GE Healthcare) with a linear gradient of 200–1,000 mM NaCl. The major peak was collected, precipitated with isopropanol, dissolved in water and precipitated with ethanol. Tpt1p-treated tRNAVal3 was prepared with the same procedure as described above. The purified tRNA was dissolved in buffer consisting of 10 mM Tris-HCl (pH 7.1) and 5 mM MgCl2 to a concentration of 50 μM. One microlitre of tRNA solution was mixed with 1 μl Natrix 2 no. 32 (80 mM NaCl, 12 mM spermine-4HCl, 40 mM sodium cacodylate·3H2O (pH 7.0) and 30% (vol/vol) MPD) (Hampton Research) on silicon-coated glass and crystalized by the hanging drop vapor diffusion method at 20 °C.
Crystallization of T. kodakarensis ArkI
The concentration of TkArkI was adjusted to 5 mg ml–1 before crystallization. One microlitre of the protein solution was mixed with 0.5 μl reservoir solution, containing 25% (vol/vol) ethylene glycol. TkArkI was crystallized by the hanging drop vapor diffusion method at 20 °C.
Data collection and crystal structure determination
The datasets were collected at beamline BL-17A at the Photon Factory at KEK, Japan. For data collection for the tRNAVal3 crystals, the crystals were cryoprotected with a portion of the reservoir solution. For data collection for the native TkArkI crystal, the crystal was cryoprotected with solution containing 25% (vol/vol) ethylene glycol, 2 mM MgCl2 and 1 mM ATP. For data collection for the iodide-derivative TkArkI crystal, the crystal was briefly soaked in and cryoprotected with solution containing 300 mM potassium iodide and 22.5% (vol/vol) ethylene glycol, and the diffraction dataset was collected at a wavelength of 1.5 Å. The datasets were indexed, integrated and scaled using xds56. The initial phase of tRNAVal3 was determined by molecular replacement with Phaser57. The structure of T. thermophilus tRNAVal (PDB, 1IVS)58 was used for the model. The initial phase of TkArkI was determined by the SAD method using the anomalous signal of iodide ions. The iodine sites were located by SHELX59, and the initial phase was calculated by Phaser. Subsequent density modification and initial model building were performed with RESOLVE60. The model was further modified with Coot61 and refined with Phenix62. Crystal structures and their electron density maps were visualized using PyMOL, Cuemol or Coot. Torsion angles of the tRNAs were analysed with DSSR software63.
Analysis of ligands bound to TkArkI
TkArkI purified by affinity chromatography with a HiTrap Heparin HP column (GE Healthcare) (100 pmol) was mixed with [15N]adenosine (10 pmol) and [15N]guanosine (10 pmol) as tracer molecules, followed by addition of 4 volumes of methanol, an equal volume of chloroform and 3 volumes of water and vigorous mixing. The denatured protein was removed by centrifugation at 15,000g for 1 min at 4 °C. The supernatant was dried in vacuo and dissolved in 20 μl water. Half of the extract was analysed by LC–MS. The tracer molecules were prepared by dephosphorylation of [15N]ATP and [15N]GTP as follows: 1,000 pmol each of [15N]ATP (Silantes) and [15N]GTP (Silantes) was treated with 0.04 U alkaline phosphatase (PAP, from Shewanella sp. SIB1, BioDynamics Laboratory) in 20 mM ammonium acetate (pH 8.0) at 60 °C for 30 min. After dephosphorylation, PAP was heat denatured at 95 °C for 5 min.
Construction of gene knockout strains of T. kodakarensis
Knockout strains of T. kodakarensis were constructed by pop-in/pop-out recombination as described previously64. The 5′ and 3′ flanking regions (about 1,000 bp) of T. kodakarensis arkI and kptA were PCR amplified from genomic DNA with a set of primers (Supplementary Table 6) and inserted into the pUD3 vector bearing the pyrF marker65 to yield pUD3-arkI and pUD3-kptA. The T. kodakarensis KU216 strain (ΔpyrF) was transformed with pUD3-arkI or pUD3-kptA, and the uracil-prototrophic transformants generated by pop-in recombination were selected on an ASW-AA-S0 plate without uracil. The selected strains were then cultured on an ASW-AA-S0 plate supplemented with 5-FOA to obtain uracil-auxotrophic, 5-FOA-resistant transformants formed by pop-out recombination. The knockout strains of arkI or kptA were selected among the transformants by genomic PCR with a set of primers (Supplementary Table 6). The double-knockout strain of arkI and queE (ΔarkI/queE::Tn) was constructed by deletion of arkI from FFH05 (queE::Tn) isolated from a random mutagenesis library19. T. kodakarensis strains used in this study are listed in Supplementary Table 7.
Growth phenotype analysis
T. kodakarensis KU216 (wild type), FFH05 (queE::Tn), ΔarkI and ΔarkI/queE::Tn strains were precultured in MA-YT-Pyr medium at 83 °C overnight and inoculated into 8 ml fresh MA-YT-Pyr medium with an initial OD600 of 0.01. The cells were cultured at 83 °C, 87 °C or 91 °C, and cell growth was monitored every 2 h by measuring OD600 with an S1200 diode array spectrophotometer. Graphs were generated using Microsoft Excel.
In vitro transcription of tRNA
For in vitro transcription of T. kodakarensis tRNAVal3 and its G5–C68 variants by T7 RNA polymerase66, template DNAs were constructed by PCR using synthetic DNA (Supplementary Table 6). The tRNAs were transcribed at 37 °C overnight in a reaction mixture consisting of 40 mM Tris-HCl (pH 7.5), 24 mM MgCl2, 5 mM DTT, 2.5 mM spermidine, 0.01% (vol/vol) Triton X-100, 0.8 μg ml–1 T7 RNA polymerase, 1 μg ml–1 pyrophosphatase, 30 nM DNA template, 2 mM ATP, 2 mM CTP, 2 mM UTP, 2 mM GTP and 10 mM GMP, followed by extraction with phenol/chloroform treatment and desalting with PD-10 columns (GE Healthcare). In vitro transcripts prepared in this way were separated by 10% denaturing PAGE, followed by staining with toluidine blue. The stained bands were cut out and eluted from the gel slice with elution buffer, followed by filtration to remove the gel pieces and ethanol precipitation.
In vitro phosphorylation of tRNA by ArkI
Up47 formation by TkArkI was carried out at 70 °C for 20 min in a reaction mixture (30 μl) containing 50 mM HEPES-KOH (pH 7.5), 1 mM MgCl2, 1 mM MnCl2, 1 mM DTT, 10% (vol/vol) glycerol, 0.5 mM ATP, 0.9 μM tRNA fraction (from the T. kodakarensis ΔarkI strain) and 1 μM TkArkI. After the reaction, the tRNA was extracted by acidic phenol/chloroform, desalted on a NAP-5 column (GE Healthcare) and precipitated with isopropanol. For RNA-MS, the prepared tRNA was dialysed against water on a nitrocellulose membrane (0.025-μm VSWP, MF-Millipore, Merck) for 2 h (drop dialysis). To examine GTP as a phosphate donor, 0.5 mM ATP or GTP was added to the reaction mixture and Up47 formation was performed with 0.5 μM TkArkI for 5 min, followed by RNA-MS analysis. The activities of TkArkI variants were measured by γ-phosphate transfer from [γ-32P]ATP to tRNA similarly to the kinetic studies of TkArkI (see below). tRNA phosphorylation was performed at 70 °C for 15 min in an 8-μl reaction mixture. For PAGE analysis, 4 μl of the reaction mixture was mixed with 4 μl of 2× loading solution, resolved by 10% denaturing PAGE and exposed to an imaging plate to visualize radiolabelled RNA with an FLA-9000 imaging analyser (Fujifilm). The gel image was analysed using Multi Gauge (Fujifilm). Bar graphs with independent plots were prepared with R (R Foundation). For phosphorylation of total RNA, the reaction was performed at 70 °C for 30 min in an 8-μl reaction mixture consisting of 50 mM HEPES-NaOH (pH 7.5), 1 mM MgCl2, 1 mM MnCl2, 1 mM DTT, 10% (vol/vol) glycerol, 100 μM [γ-32P]ATP (3,000 mCi mmol–1; PerkinElmer), 1.8 μM TkArkI and 50 ng μl–1 total RNA fraction (from the T. kodakarensis ΔarkI strain). Then, 0.5 μl of 50 mM EDTA (pH 8.0) was added, and 4 μl of reaction mixture was mixed with 2× loading solution, resolved by 10% denaturing PAGE and visualized as described above.
Formation of Up47 by other ArkI homologues was carried out at 70 °C for 30 min in a reaction mixture (30 μl) containing 50 mM PIPES-NaOH (pH 6.9), 125 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 1 mM DTT, 10% (vol/vol) glycerol, 500 μM ATP, 0.05 mg ml–1 BSA (Takara), 1 μM tRNA transcript and 0.5 μM ArkI protein. For NvArkI, the reaction temperature was set to 45 °C. For ArkI homologue from N. profundicola (NpArkI), the reaction was carried out at 50 °C for 60 min. After the reaction, tRNA was prepared as described above. For PAGE analysis, Up47 formation was carried out in a reaction mixture (8 μl) containing 50 mM PIPES-NaOH (pH 6.9), 125 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 1 mM DTT, 10% (vol/vol) glycerol, 100 μM [γ-32P]ATP (3,000 mCi mmol–1; PerkinElmer), 0.1 mg ml–1 BSA (Takara), 0.75 μM recombinant ArkI homologue (NpArkI, NvArkI or LeArkI) and 50 ng μl–1 E. coli total RNA. Then, the reaction mixture was mixed with 2× loading solution, resolved by 10% denaturing PAGE and visualized as described above.
In vitro dephosphorylation of tRNA by T. kodakarensis KptA
Dephosphorylation of Up47 by TkKptA was carried out at 60 °C for 1 h in a reaction mixture (30 μl) containing 20 mM Tris-HCl (pH 7.4), 0.5 mM EDTA (pH 8.0), 1 mM NAD+, 2.5 mM spermidine, 0.1 mM DTT, 0.9 μM T. kodakarensis tRNA fraction and 0.1 μg μl–1 recombinant TkKptA. After the reaction, the tRNA was extracted by acidic phenol/chloroform, desalted on a NAP-5 column (GE Healthcare) and precipitated with isopropanol. For RNA-MS, the prepared tRNA was desalted by drop dialysis as described above.
Kinetic studies of T. kodakarensis ArkI and KptA
TkArkI-mediated Up47 formation was quantified by γ-phosphate transfer from [γ-32P]ATP to tRNA. For kinetic measurement of the tRNA substrate, tRNA phosphorylation was performed at 70 °C in a reaction mixture (25 μl) consisting of 50 mM PIPES-NaOH (pH 6.9), 125 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 1 mM DTT, 10% (vol/vol) glycerol, 100 μM [γ-32P]ATP (1,500 mCi mmol–1; PerkinElmer), 0.05 mg ml–1 BSA (Takara), 0.05 μM TkArkI and 0.1–5.0 μM of in vitro-transcribed T. kodakarensis tRNAVal3. For kinetic measurement of the ATP substrate, the ATP concentration was altered from 15.6 to 1,000 μM [γ-32P]ATP (750 mCi mmol–1; PerkinElmer) and the tRNA concentration was increased to 1.0 μM. At each time point (2 and 5 min), 8-μl aliquots were taken and mixed with an equal volume of 2× loading solution (7 M urea, 0.2% (wt/vol) bromophenol blue, 0.2% (wt/vol) xylene cyanol and 50 mM EDTA (pH 8.0)) to quench the reaction. Each sample was subjected to 10% denaturing PAGE. The gel was exposed on an imaging plate to measure radiolabelled tRNAs using an FLA-9000 imaging analyser. Kinetic parameters were calculated using Prism 7 (GraphPad).
TkKptA-mediated dephosphorylation of Up47 was quantified by measuring the reduction in radioactivity for tRNA. In vitro-transcribed T. kodakarensis tRNAVal3 was phosphorylated by TkArkI with [γ-32P]ATP as described above and then purified by gel extraction and isopropanol precipitation. In addition, the same tRNA was phosphorylated by TkArkI with unlabelled ATP. By mixing labelled and unlabelled tRNAs, the specific activity of the labelled tRNA was adjusted to 6,250 c.p.m. per pmol in buffer consisting of 50 mM HEPES-KOH (pH 7.6), 5 mM MgCl2 and 1 mM DTT. The labelled tRNA was incubated at 80 °C for 5 min and then cooled at room temperature, followed by isopropanol precipitation. The labelled tRNA was dissolved in water to a concentration of 8 μM (50,000 c.p.m. per μl). Dephosphorylation of the labelled tRNA by TkKptA was performed at 70 °C in a reaction mixture (30 μl) consisting of 50 mM PIPES-NaOH (pH 6.9), 125 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 1 mM DTT, 10% (vol/vol) glycerol, 1 mM NAD+, 0.05 mg ml–1 BSA (Takara), 1 nM TkKptA and 12.5–800 nM 32P-labelled tRNA. At each time point (2 and 5 min), 8-μl aliquots were spotted on Whatman 3MM filter paper, which was immediately soaked in 5% (wt/vol) trichloroacetic acid. The filter paper was washed three times for 15 min with ice-cold 5% (wt/vol) trichloroacetic acid, rinsed for 5 min with ice-cold ethanol and dried in air. Radioactivity on the filter paper was measured by liquid scintillation counting (Tri-Carb 2910TR, PerkinElmer). Kinetic parameters were calculated using Prism 7.
In vivo dephosphorylation of Up47 by KptA
N. viennensis arkI was PCR amplified and cloned into pMW118 (Invitrogen) under the control of the synthetic constitutive J23106 promoter67,68, followed by insertion of sequences encoding a His6 tag and a 3×Flag tag at the C terminus of the N. viennensis arkI gene, yielding pMW-J23106-nvarkI (Supplementary Table 7). T. kodakarensis kptA, E. coli kptA and S. cerevisiae tpt1 were PCR amplified and cloned into pQE-80L (Qiagen). The ampicillin resistance cassette (Ampr) was replaced with a chloramphenicol resistance cassette (Camr), yielding pQE-80LC-tkkptA, pQE-80LC-eckptA and pQE-80LC-sctpt1, respectively (Supplementary Table 7). The E. coli ΔtrmBΔtapT (Kanr) strain was transformed with pMW-J23106-nvarkI and further transformed with pQE-80LC-tkkptA, pQE-80LC-eckptA or pQE-80LC-sctpt1. The transformants were inoculated in 3 ml LB supplemented with 20 μg ml–1 chloramphenicol, 50 μg ml–1 kanamycin and 100 μg ml–1 ampicillin and cultured at 37 °C until mid-log phase. When the OD610 reached 0.6, IPTG was added to a final concentration of 10 or 100 μM to induce expression of the KptA/Tpt1p homologue and cells were cultured for 3.5 h. A 1.5-ml aliquot of the culture was taken, and the tRNA fraction was extracted and analysed by shotgun RNA-MS as described above. Primers, E. coli strains and plasmids used are listed in Supplementary Tables 6, 7. Bar graphs with independent plots were prepared with R (R Foundation).
Drawing of chemical structures
Chemical structures were drawn with chemical structure drawing tools, including ACD/ChemSketch (ACD/Labs) or ChemDraw (PerkinElmer).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-022-04677-2.
Supplementary information
This file contains Supplementary Notes 1–7, Supplementary Figs. 1–10, Supplementary Tables 1–7 and the legend for Supplementary Video 1.
Structural switch of the core region in S. tokodaii tRNAVal3.
Source data
Acknowledgements
We thank the members of the Suzuki laboratory for their continuous technical assistance and fruitful discussion. We also thank the beamline staff at BL-17A of the Photon Factory for technical assistance during data collection. S. tokodaii, M. acetivorans C2A, T. acidophilum and the E. coli ΔtrmBΔtapT strain were kindly provided by T. Ohshima (Kyowa Kako Co., Ltd), T. Yokogawa (Gifu University), H. Hori (Ehime University) and M. Takakura (Suzuki laboratory), respectively. This work was carried out with the support of the Isotope Science Center, University of Tokyo. This work was supported by Grants-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports and Culture of Japan; Research Fellowships for Young Scientists from the Japan Society for the Promotion of Science (26113003, 26220205 and 18H05272 to T.S.; 26113002 and 18H03980 to K.T.; 26840005 and 17H04997 to T.O.; and 19J20723 to K. Minowa); and Exploratory Research for Advanced Technology (ERATO, JPMJER2002 to T.S.) from the Japan Science and Technology Agency (JST).
Extended data figures and tables
Author contributions
K. Minowa, T.O. and K.S. mainly performed the series of experiments. K.S. and T.O. conducted LC–MS analyses and biochemical and thermodynamic analyses of S. tokodaii tRNA assisted by K. Miyauchi and Y.S. Crystal structure analysis of tRNA was performed by K.S., T.O., S.Y. and K. Minowa with support from K.T. Gene identification by comparative genomics was conducted by K. Minowa and T.O. Biochemical characterization of ArkI proteins was performed by K. Minowa and R.N. K. Minowa performed genetic work assisted by A.K., I.O. and T.F. K. Minowa performed structural studies of ArkI assisted by S.Y. and K.T. K. Minowa conducted E. coli experiments assisted by K. Miyauchi and Y.S. All authors discussed the results and revised the manuscript. T.O., K. Minowa and T.S. designed the studies and wrote the manuscript. T.S. supervised the project.
Peer review
Peer review information
Nature thanks Mark Helm, Kathy Fange Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 7VNV, 7VNW and 7VNX. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Takayuki Ohira, Keiichi Minowa
Contributor Information
Takayuki Ohira, Email: ohira_t@chembio.t.u-tokyo.ac.jp.
Kozo Tomita, Email: kozo-tomita@edu.k.u-tokyo.ac.jp.
Tsutomu Suzuki, Email: ts@chembio.t.u-tokyo.ac.jp.
Extended data
is available for this paper at 10.1038/s41586-022-04677-2.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-022-04677-2.
References
- 1.Suzuki T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol. 2021;22:375–392. doi: 10.1038/s41580-021-00342-0. [DOI] [PubMed] [Google Scholar]
- 2.Wiener D, Schwartz S. The epitranscriptome beyond m6A. Nat. Rev. Genet. 2021;22:119–131. doi: 10.1038/s41576-020-00295-8. [DOI] [PubMed] [Google Scholar]
- 3.Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361:1346–1349. doi: 10.1126/science.aau1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frye M, Jaffrey SR, Pan T, Rechavi G, Suzuki T. RNA modifications: what have we learned and where are we headed? Nat. Rev. Genet. 2016;17:365–372. doi: 10.1038/nrg.2016.47. [DOI] [PubMed] [Google Scholar]
- 5.Hori H, et al. Transfer RNA modification enzymes from thermophiles and their modified nucleosides in tRNA. Microorganisms. 2018;6:110. doi: 10.3390/microorganisms6040110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lorenz C, Lunse CE, Morl M. tRNA modifications: impact on structure and thermal adaptation. Biomolecules. 2017;7:35. doi: 10.3390/biom7020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boccaletto P, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46:D303–D307. doi: 10.1093/nar/gkx1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki, T. Fine-tuning of RNA functions by modification and editing. in Topics in Current Genetics (ed. Grosjean, H.) Vol. 12, 23–69 (Springer-Verlag, 2005).
- 9.Motorin Y, Helm M. tRNA stabilization by modified nucleotides. Biochemistry. 2010;49:4934–4944. doi: 10.1021/bi100408z. [DOI] [PubMed] [Google Scholar]
- 10.Roovers M, Droogmans L, Grosjean H. Post-transcriptional modifications of conserved nucleotides in the T-loop of tRNA: a tale of functional convergent evolution. Genes. 2021;12:140. doi: 10.3390/genes12020140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokoyama S, Watanabe K, Miyazawa T. Dynamic structures and functions of transfer ribonucleic acids from extreme thermophiles. Adv. Biophys. 1987;23:115–147. doi: 10.1016/0065-227x(87)90006-2. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe K, Oshima T, Saneyoshi M, Nishimura S. Replacement of ribothymidine by 5-methyl-2-thiouridine in sequence GTψC in tRNA of an extreme thermophile. FEBS Lett. 1974;43:59–63. doi: 10.1016/0014-5793(74)81105-1. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe K, Shinma M, Oshima T, Nishimura S. Heat-induced stability of tRNA from an extreme thermophile, Thermus thermophilus. Biochem. Biophys. Res. Commun. 1976;72:1137–1144. doi: 10.1016/s0006-291x(76)80250-1. [DOI] [PubMed] [Google Scholar]
- 14.Kowalak JA, Dalluge JJ, McCloskey JA, Stetter KO. The role of posttranscriptional modification in stabilization of transfer RNA from hyperthermophiles. Biochemistry. 1994;33:7869–7876. doi: 10.1021/bi00191a014. [DOI] [PubMed] [Google Scholar]
- 15.Shigi N, Sakaguchi Y, Suzuki T, Watanabe K. Identification of two tRNA thiolation genes required for cell growth at extremely high temperatures. J. Biol. Chem. 2006;281:14296–14306. doi: 10.1074/jbc.M511675200. [DOI] [PubMed] [Google Scholar]
- 16.Sas-Chen A, et al. Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping. Nature. 2020;583:638–643. doi: 10.1038/s41586-020-2418-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai G, Hashizume T, Miyazawa T, McCloskey JA, Yokoyama S. Conformational characteristics of 4-acetylcytidine found in tRNA. Nucleic Acids Symp. Ser. 1989;12:61–62. [PubMed] [Google Scholar]
- 18.Dewe JM, Whipple JM, Chernyakov I, Jaramillo LN, Phizicky EM. The yeast rapid tRNA decay pathway competes with elongation factor 1A for substrate tRNAs and acts on tRNAs lacking one or more of several modifications. RNA. 2012;18:1886–1896. doi: 10.1261/rna.033654.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orita I, et al. Random mutagenesis of a hyperthermophilic archaeon identified tRNA modifications associated with cellular hyperthermotolerance. Nucleic Acids Res. 2019;47:1964–1976. doi: 10.1093/nar/gky1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agris PF, Koh H, Soll D. The effect of growth temperatures on the in vivo ribose methylation of Bacillus stearothermophilus transfer RNA. Arch. Biochem. Biophys. 1973;154:277–282. doi: 10.1016/0003-9861(73)90058-1. [DOI] [PubMed] [Google Scholar]
- 21.Gregson JM, et al. Structure of the archaeal transfer RNA nucleoside G*-15 (2-amino-4,7-dihydro-4-oxo-7-β-d-ribofuranosyl-1H-pyrrolo[2,3-d]pyrimidine-5-carboximidamide (archaeosine)) J. Biol. Chem. 1993;268:10076–10086. [PubMed] [Google Scholar]
- 22.Oliva R, Tramontano A, Cavallo L. Mg2+ binding and archaeosine modification stabilize the G15 C48 Levitt base pair in tRNAs. RNA. 2007;13:1427–1436. doi: 10.1261/rna.574407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner B, et al. Archaeosine modification of archaeal tRNA: role in structural stabilization. J. Bacteriol. 2020;202:e00748-19. doi: 10.1128/JB.00748-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyauchi K, Ohara T, Suzuki T. Automated parallel isolation of multiple species of non-coding RNAs by the reciprocal circulating chromatography method. Nucleic Acids Res. 2007;35:e24. doi: 10.1093/nar/gkl1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakaguchi Y, Miyauchi K, Kang BI, Suzuki T. Nucleoside analysis by hydrophilic interaction liquid chromatography coupled with mass spectrometry. Methods Enzymol. 2015;560:19–28. doi: 10.1016/bs.mie.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 26.Ohira T, Suzuki T. Precursors of tRNAs are stabilized by methylguanosine cap structures. Nat. Chem. Biol. 2016;12:648–655. doi: 10.1038/nchembio.2117. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki T, Ikeuchi Y, Noma A, Suzuki T, Sakaguchi Y. Mass spectrometric identification and characterization of RNA-modifying enzymes. Methods Enzymol. 2007;425:211–229. doi: 10.1016/S0076-6879(07)25009-8. [DOI] [PubMed] [Google Scholar]
- 28.Tsuruoka H, Shohda K, Wada T, Sekine M. Synthesis and conformational properties of oligonucleotides incorporating 2′-O-phosphorylated ribonucleotides as structural motifs of pre-tRNA splicing intermediates. J. Org. Chem. 2000;65:7479–7494. doi: 10.1021/jo991097e. [DOI] [PubMed] [Google Scholar]
- 29.Shi H, Moore PB. The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: a classic structure revisited. RNA. 2000;6:1091–1105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 31.Tsuyuguchi M, Nakaniwa T, Sawa M, Nakanishi I, Kinoshita T. A promiscuous kinase inhibitor delineates the conspicuous structural features of protein kinase CK2a1. Acta Crystallogr. F. 2019;75:515–519. doi: 10.1107/S2053230X19008951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massillon D, Stalmans W, van de Werve G, Bollen M. Identification of the glycogenic compound 5-iodotubercidin as a general protein kinase inhibitor. Biochem. J. 1994;299:123–128. doi: 10.1042/bj2990123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Culver GM, McCraith SM, Consaul SA, Stanford DR, Phizicky EM. A 2′-phosphotransferase implicated in tRNA splicing is essential in Saccharomyces cerevisiae. J. Biol. Chem. 1997;272:13203–13210. doi: 10.1074/jbc.272.20.13203. [DOI] [PubMed] [Google Scholar]
- 34.Spinelli SL, Malik HS, Consaul SA, Phizicky EM. A functional homolog of a yeast tRNA splicing enzyme is conserved in higher eukaryotes and in Escherichia coli. Proc. Natl Acad. Sci. USA. 1998;95:14136–14141. doi: 10.1073/pnas.95.24.14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang S, et al. The function of KptA/Tpt1 gene—a minor review. Funct. Plant Biol. 2020;47:577–591. doi: 10.1071/FP19159. [DOI] [PubMed] [Google Scholar]
- 36.Yoshihisa T. Handling tRNA introns, archaeal way and eukaryotic way. Front. Genet. 2014;5:213. doi: 10.3389/fgene.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Popow J, Schleiffer A, Martinez J. Diversity and roles of (t)RNA ligases. Cell Mol. Life Sci. 2012;69:2657–2670. doi: 10.1007/s00018-012-0944-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stout CD, et al. Atomic coordinates and molecular conformation of yeast phenylalanyl tRNA. An independent investigation. Nucleic Acids Res. 1976;3:1111–1123. doi: 10.1093/nar/3.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takakura M, Ishiguro K, Akichika S, Miyauchi K, Suzuki T. Biogenesis and functions of aminocarboxypropyluridine in tRNA. Nat. Commun. 2019;10:5542. doi: 10.1038/s41467-019-13525-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dalluge JJ, Hashizume T, Sopchik AE, McCloskey JA, Davis DR. Conformational flexibility in RNA: the role of dihydrouridine. Nucleic Acids Res. 1996;24:1073–1079. doi: 10.1093/nar/24.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilkinson KA, Merino EJ, Weeks KM. RNA SHAPE chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in tRNAAsp transcripts. J. Am. Chem. Soc. 2005;127:4659–4667. doi: 10.1021/ja0436749. [DOI] [PubMed] [Google Scholar]
- 43.Hilbers CW, et al. Thermal unfolding of yeast glycine transfer RNA. Biochemistry. 1976;15:1874–1882. doi: 10.1021/bi00654a013. [DOI] [PubMed] [Google Scholar]
- 44.Leonard CJ, Aravind L, Koonin EV. Novel families of putative protein kinases in bacteria and archaea: evolution of the “eukaryotic” protein kinase superfamily. Genome Res. 1998;8:1038–1047. doi: 10.1101/gr.8.10.1038. [DOI] [PubMed] [Google Scholar]
- 45.Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wurzenberger C, Gerlich DW. Phosphatases: providing safe passage through mitotic exit. Nat. Rev. Mol. Cell Biol. 2011;12:469–482. doi: 10.1038/nrm3149. [DOI] [PubMed] [Google Scholar]
- 47.Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 48.Cohen P. The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem. Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe K, Himeno H, Ohta T. Selective utilization of 2-thioribothymidine- and ribothymidine-containing tRNAs by the protein synthetic systems of Thermus thermophilus HB 8 depending on the environmental temperature. J. Biochem. 1984;96:1625–1632. doi: 10.1093/oxfordjournals.jbchem.a134993. [DOI] [PubMed] [Google Scholar]
- 50.Sato T, Fukui T, Atomi H, Imanaka T. Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 2003;185:210–220. doi: 10.1128/JB.185.1.210-220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robb, F. T. & Place, A. R. Media for Thermophiles (Cold Spring Harbor Laboratory Press, 1995).
- 52.Suzuki T, Suzuki T. Chaplet column chromatography: isolation of a large set of individual RNAs in a single step. Methods Enzymol. 2007;425:231–239. doi: 10.1016/S0076-6879(07)25010-4. [DOI] [PubMed] [Google Scholar]
- 53.Mengel-Jorgensen J, Kirpekar F. Detection of pseudouridine and other modifications in tRNA by cyanoethylation and MALDI mass spectrometry. Nucleic Acids Res. 2002;30:e135. doi: 10.1093/nar/gnf135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyauchi K, Kimura S, Suzuki T. A cyclic form of N6-threonylcarbamoyladenosine as a widely distributed tRNA hypermodification. Nat. Chem. Biol. 2013;9:105–111. doi: 10.1038/nchembio.1137. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 2012;40:12. doi: 10.1093/nar/gkr1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kabsch W. XDS. Acta Crystallogr. D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCoy AJ, et al. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fukai S, et al. Mechanism of molecular interactions for tRNAVal recognition by valyl-tRNA synthetase. RNA. 2003;9:100–111. doi: 10.1261/rna.2760703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheldrick GM. A short history of SHELX. Acta Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 60.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr. D. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu XJ, Bussemaker HJ, Olson WK. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 2015;43:e142. doi: 10.1093/nar/gkv716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sato T, Fukui T, Atomi H, Imanaka T. Improved and versatile transformation system allowing multiple genetic manipulations of the hyperthermophilic archaeon Thermococcus kodakaraensis. Appl. Environ. Microbiol. 2005;71:3889–3899. doi: 10.1128/AEM.71.7.3889-3899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kobori H, et al. Characterization of NADH oxidase/NADPH polysulfide oxidoreductase and its unexpected participation in oxygen sensitivity in an anaerobic hyperthermophilic archaeon. J. Bacteriol. 2010;192:5192–5202. doi: 10.1128/JB.00235-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sampson JR, Uhlenbeck OC. Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc. Natl Acad. Sci. USA. 1988;85:1033–1037. doi: 10.1073/pnas.85.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anderson, C. J. Anderson promoter collection (2007).
- 68.Kelly JR, et al. Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 2009;3:4. doi: 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains Supplementary Notes 1–7, Supplementary Figs. 1–10, Supplementary Tables 1–7 and the legend for Supplementary Video 1.
Structural switch of the core region in S. tokodaii tRNAVal3.
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 7VNV, 7VNW and 7VNX. Source data are provided with this paper.