

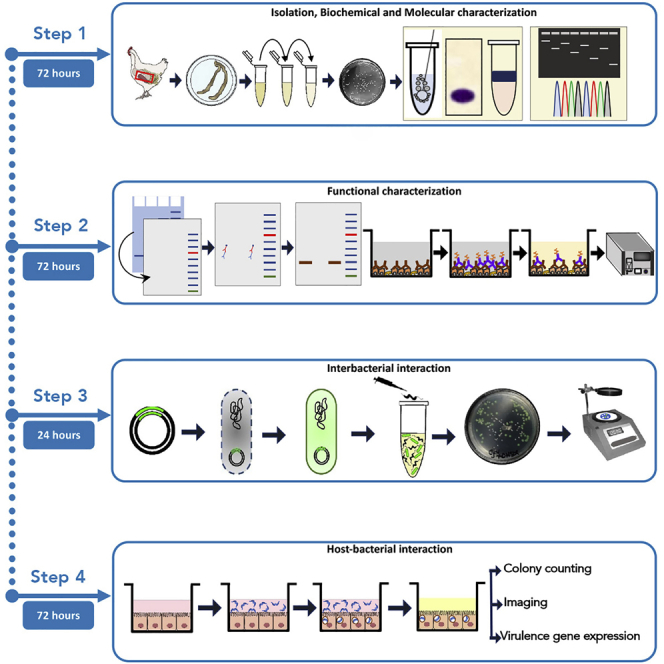
Summary
The bacterial Type VI Secretion System (T6SS) functions as a nanomachine used by many gut pathogens. In the present protocol, we outlined how such molecular activities during interspecies interaction can be demonstrated at a population level. To this end, we first present a comprehensive protocol for isolation, identification, and functional characterization of T6SS-positive Campylobacter jejuni. Further, we developed straightforward techniques for unraveling how the T6SS targets prey populations and host cells when growing with or without environmental stressors.
For complete details on the use and execution of this protocol, please refer to Gupta et al. (2021).
Subject areas: Microbiology, Microscopy, Model Organisms, Molecular Biology
Graphical abstract
Highlights
-
•
A technique for isolation and identification of C. jejuni from its primary host
-
•
Molecular and Functional characterization of C. jejuni T6SS
-
•
Protocol for direct visualization of T6SS-dependent interspecies interaction
The bacterial Type VI Secretion System (T6SS) functions as a nanomachine used by many gut pathogens. In the present protocol, we outlined how such molecular activities during interspecies interaction can be demonstrated at a population level. To this end, we first present a comprehensive protocol for isolation, identification, and functional characterization of T6SS-positive Campylobacter jejuni. Further, we developed straightforward techniques for unraveling how the T6SS targets prey populations and host cells when growing with or without environmental stressors.
Before you begin
Note: Researcher should acquire permissions from the relevant institutions before start of the experiment.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-Rabbit IgG (H+L) (HRP Conjugated) (Antibody Dilution: 1:3,500) | BioBharati Life Science Pvt Ltd, India | Cat# BB-SAB01C |
| Bacterial and virus strains | ||
| E. coli (DH5α) | BioBharati Life Science Pvt Ltd, India | Cat# BB-X0051 |
| Chemicals, peptides, and recombinant proteins | ||
| Blood Free Campylobacter Selectivity Agar Base Medium | HiMedia, India | Cat# M887 |
| CAT Selective Supplement | HiMedia, India | Cat# FD 145 |
| Muller-Hinton Broth | HiMedia, India | Cat# M391 |
| Luria-Bertani Broth | HiMedia, India | Cat# M575-500G |
| Bacteriological Agar powder | HiMedia, India | Cat# GRM026 |
| Trichloro acetic acid | Merck | Cat# 1.94971.0521 |
| 2-Mercaptoethanol | SRL | Cat# 69892 (1327198) |
| Bromophenol blue | HiMedia, India | Cat# MB123-5G |
| 3,3′-Diaminobenzidine | Sigma-Aldrich | Cat# D12384-1G |
| 3, 3′, 5, 5′- Tetramethylebenzidine (TMB) Substrate | Sigma-Aldrich | Cat# T0440-100ML |
| Bovine serum albumin | HiMedia, India | Cat# MB083-100G |
| Ultra-Pure Tris Base | Invitrogen | Cat#15504-020 |
| Glycine | Merck | Cat#8.16013.0521 |
| Tetramethylethylenediamine (TEMED) | Thermo Fisher Scientific | Cat#17919 |
| Methanol | Merck Millipore | Cat#106012 |
| Ammonium persulfate | Merck Millipore | Cat#2300-OP |
| Tween20 | Sigma-Aldrich | Cat# P9416-100ML |
| Acetic acid (Glacial) | Merck | Cat#1.93402.2521 |
| X-gal | HiMedia, India | Cat# MB069 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | BioBharati Life Science Pvt Ltd, India | Cat# BB-C0010 |
| Vecta-Sheild Mounting Media | Vector Laboratories, Inc., Burlingame, CA | Cat# H-1000-10 |
| Glutaraldehyde | Merck | Cat# 8.20603.0521 |
| Paraformaldehyde | Merck | Cat# 8.18715.0100 |
| Penicillin-streptomycin | Thermo Fisher Scientific | Cat# 15140122 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco (Invitrogen) | Cat# 11995-065 |
| Fetal Bovine Serum | Invitrogen | Cat# 10270106 |
| Gentamicin | HiMedia, India | Cat# CMS461-1G |
| Triton X-100 | Sigma-Aldrich | Cat# T8787-100ML |
| DAPI | USB Corporation | Cat# 14564 10M |
| Sodium chloride | Merck | Cat# 1.93606.0521 |
| Potassium chloride | Merck | Cat# 61779205001730 |
| Di-sodium hydrogen phosphate | Merck | Cat# 1.93622.0521 |
| Sodium hydroxide | Merck | Cat# MB095-100G |
| Sulphuric acid | Merck | Cat#1.93000.0521 |
| Hydrochloric acid | Merck | Cat#1.93001.2521 |
| N, N, N′, N′- Tetramethyl-p-phenylenediamine dihydrochloride | HiMedia, India | Cat# GRM445-5G |
| Hydrogen peroxide | Merck | Cat# 1.93407.0521 |
| Ninhydrin | HiMedia, India | Cat# MB234 |
| Acetone | Merck | Cat# 1.94500.2521 |
| Butanol | Merck | Cat# 61775805001730 |
| Hippuric acid sodium salt | HiMedia, India | Cat# RM65232 |
| Citric Acid Monohydrate | Merck | Cat# 1.93011.0521 |
| Ethylenediaminetetraacetic acid (EDTA) | Himedia, India | Cat# MB011-500G |
| Sodium dodecyl sulfate | Merck | Cat# 1.94954.0521 |
| Chloroform | Merck | Cat# 1.94506.0521 |
| Chloroform (Molecular Biology Grade) | Sigma-Aldrich | Cat# C2432-25ML |
| Ethanol (Molecular Biology Grade) | Sigma-Aldrich | Cat# E7023 |
| Tris-EDTA Buffer | Invitrogen | Cat# 8019005 |
| Gram Stains-Kit | HiMedia, India | Cat# K001-1KT |
| Phalloidin-iFluor 647 conjugate | Abcam | Cat# Ab176759 |
| MulV super-transcriptase Kit | BioBharati Life Science Pvt Ltd, India | Cat# BB-E043 |
| TriZOL Reagent | Ambion (Life Technologies) | Cat# 15596026 |
| Iso-propanol | Sigma-Aldrich | Cat# I9516-500ML |
| Quick PCR purification Kit | Invitrogen | Cat# K310001 |
| Experimental models: Organisms/strains | ||
| Chicken (Strains: Rhode Island Red; Age: More than 5 Weeks) | N/A | N/A |
| Other | ||
| Tissue culture plates | Thermo Fisher Scientific (Nunc) | Cat#144530 |
| Petri plates | Tarsons | Cat#460020 |
| Glass slides | Riviera | Cat# 72910135 |
| 96-well ELISA Plate | Nunc, Thermo Fisher | Cat# 44-2404-21 |
| Supra 55 Carl Zeiss Scanning Electron Microscope | Carl ZEISS, Germany | https://www.zeiss.com/microscopy/int/products/scanning-electron-microscopes/geminisem.html |
| Epoch2 Micro-plate Reader | BioTek | https://www.biotek.com/products/detection-microplate-readers/epoch-2-microplate-spectrophotometer/ |
| Spectramax M2e Multi Detection Microplate Readers | Molecular Devices LLC, USA | https://www.moleculardevices.com/products/microplate-readers/multi-mode-readers/spectramax-m-series-readers#gref |
| Software and algorithms | ||
| Zen | Carl ZEISS, Germany | https://www.zeiss.com/microscopy/int/products/microscope-software/zen-lite.html |
| ImageLab | Bio-Rad Laboratories, Inc. | https://www.bio-rad.com/en-in/product/image-lab-software?ID=KRE6P5E8Z |
| SoftMax® Pro Microplate Data Acquisition and analysis software | Molecular Devices, LLC. | https://www.moleculardevices.com/products/microplate-readers/acquisition-and-analysis-software/softmax-pro-software#gref |
| ImageJ | Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA | https://imagej.nih.gov/ij/, 1997–2018 |
| Graphpad Prism 8.0 | GraphPad, USA | http://www.graphpad.com/ |
| Inkscape, (Version 0.92.5) | Inkscape Project, 2020. | https://inkscape.org/ |
| Molecular Evolutionary Genetics Analysis (MEGA X) | Institute of Molecular Evolutionary Genetics, The Pennsylvania State University, University Park, PA 16802, USA |
https://www.megasoftware.net/ |
Materials and equipment
Timing: 5 h
Preparation of 10× PBS Solution
| Reagents | Amount |
|---|---|
| Di-Sodium hydrogen phosphate | 25.6 g |
| Sodium chloride | 80 g |
| Potassium chloride | 2 g |
| Potassium dihydrogen phosphate | 2 g |
| Distilled water | 1,000 mL |
Note: Adjust the pH∼7.4. Autoclave the solution at 121°C and 15 lb pressure for 15 min.
Storage: PBS should be stored at 4°C or room temperature (∼25°C). The concentrated solution tends to precipitate when cooled down. It should be kept at room temperature (∼25°C) until the precipitate completely dissolves before use.
PBS-Tween
| Reagents | Amount |
|---|---|
| 1× PBS | 10 mL |
| Tween20 | 0.01 mL |
Note: Do not store the solution. Freshly prepare the solution on the day of the experiment.
Oxidase Reagent
| Reagents | Amount |
|---|---|
| L (+)-Ascorbic acid | 0.01 g |
| N, N, N′, N′-Tetramethyl-p-phenylenediamine dihydrochloride | 0.01 g |
| Sterile distilled water | 10 mL |
Note: Do not store the solution. Prepare the solution on the day of the experiment.
Ninhydrin solution (3.5%, w/v)
| Reagents | Amount |
|---|---|
| Ninhydrin | 0.7 g |
| Acetone | 10 mL |
| Butanol | 10 mL |
Note: Mix the reagents properly to make a uniform solution.
Storage: At 4°C in dark bottles. Aluminum foil-wrapped bottles also can be used as an alternative to avoid direct exposure to light.
Hippuric acid solution (1%, w/v)
| Reagents | Amount |
|---|---|
| Sodium Hippurate | 0.2 g |
| 1× PBS | 19.8 mL |
Note: Solution can be stored at −20°C in a 50 mL tube for maximum 3 days.
50% Glycerol (v/v)
| Reagents | Amount |
|---|---|
| Glycerol | 5 mL |
| Distilled water | 5 mL |
Note: Autoclave the mixture at 121°C and 15 lb pressure and store it at 4°C in a sealed falcon tube to avoid contamination.
Glacial Acetic Acid (30%, v/v)
| Reagents | Amount |
|---|---|
| Glacial Acetic Acid | 30 mL |
| Autoclaved distilled water | 70 mL |
CRITICAL: Glacial acetic acid can cause eye and upper respiratory tract irritation.
Precautionary measures: Use nitrile rubber gloves as glacial acetic acid perforates latex gloves. Double-gloving, as well as apron and face shield, can also be used. Open the chemical in a fume hood.
Note: Do not store. Freshly prepare ethanol gradient on the day of the experiment.
Glutaraldehyde (2.5%, v/v)
| Reagents | Amount |
|---|---|
| 25% glutaraldehyde solution | 1 mL |
| Autoclaved distilled water | 9 mL |
Note: Prepare the homogenous solution of 2.5% glutaraldehyde in autoclaved distilled water. The solution can be stored at 4°C in a 15 mL tube maximum for 3 days.
Ethanol Gradient (in water)
| Concentration (v/v) | Total volume | Water (mL) | Ethanol (mL) |
|---|---|---|---|
| 35% | 10 mL | 6.5 | 3.5 |
| 50% | 10 mL | 5 | 5 |
| 75% | 10 mL | 2.5 | 7.5 |
| 95% | 10 mL | 0.5 | 9.5 |
Note: Do not store. Freshly prepare ethanol gradient on the day of the experiment.
Bacterial lysis buffer for the isolation of genomic DNA
| Reagents | Amount |
|---|---|
| 0.5 M EDTA (pH∼8.0) | 1 mL |
| Tris Base | 2.42 g |
| Glacial Acetic acid | 0.57 mL |
| SDS | 0.1 g |
| Nuclease free water | 8.4 mL |
Note: Do not store. Freshly prepare the buffer on the day of the experiment.
5 M Sodium chloride solution
| Reagents | Amount |
|---|---|
| Sodium chloride | 2.92 g |
| Autoclaved distilled water | 10 mL |
Note: Do not store. Freshly prepare the solution on the day of the experiment. If preparation is not possible on the day of an experiment, do not use more than 2 days old solution.
Note: Commercially available 5 M sodium chloride solution can also be used.
Crystal violet solution (0.1%, v/v)
| Reagents | Amount |
|---|---|
| Crystal violet | 10 μL |
| Autoclaved distilled water | 10 mL |
Note: Do not store. Freshly prepare the staining solution on the day of experiment.
1.5 M Tris-Cl (∼pH 8.8)
| Reagents | Amount |
|---|---|
| Tris base | 9.08 gm |
| Distilled water | 50 mL |
Note: Adjust the pH of the solution up to ∼8.8 adding Hydrochloric acid (HCl) slowly. Store the solution at 4°C.
1 M Tris-Cl (∼pH 6.8)
| Reagents | Amount |
|---|---|
| Tris base | 6.05 gm |
| Distilled water | 50 mL |
Note: Adjust the pH of the solution up to ∼6.8 adding Hydrochloric acid (HCl) slowly. Store the solution at 4°C.
Acrylamide Solution
| Reagents | Amount |
|---|---|
| Acrylamide | 28.5 gm |
| Bis-acrylamide | 1.5 gm |
| Water | 100 mL |
Note: Mix the reagents properly and adjust the pH of the solution up to ∼6.5 adding 1 M hydrochloric acid (HCl) slowly. Store the solution at 4°C.
10% SDS solution (w/v)
| Reagents | Amount |
|---|---|
| SDS | 1 gm |
| Autoclaved Distilled water | 10 mL |
Note: Store the solution at 4°C.
10% Ammonium per-sulfate solution (w/v)
| Reagents | Amount |
|---|---|
| Ammonium persulfate | 1 gm |
| Autoclaved Distilled water | 10 mL |
Note: Store the solution at 4°C.
1× Tris-Buffer Saline (TBS)
| Reagents | Amount |
|---|---|
| Sodium Chloride | 8.76 g |
| Tris base | 6.05 g |
| Autoclaved distilled water | 1,000 mL |
Note: Dissolve the chemicals in the solution and adjust the pH to ∼7.5 using 1 M Hydrochloric acid (HCl). Store at 4°C.
2× Laemmli Buffer
| Reagents | Amount |
|---|---|
| 2-Mercaptoethanol | 20 μL |
| Bromophenol blue | 10 mg |
| Glycerol | 2 mL |
| SDS | 0.2 g |
| Tris-HCl | 76 mg |
Note: Prepare the solution just on the day of the experiment.
SDS-PAGE Running Buffer (1×)
| Reagents | Amount |
|---|---|
| Tris Base | 3 gm |
| Glycine | 14.4 gm |
| SDS | 1 gm |
| Distilled water | 1,000 mL |
Note: Adjust the pH to ∼8.3 using 1 M Hydrochloric acid (HCl) slowly. Prepare the solution just on the day of the experiment.
Western Blot Transfer Buffer (1×)
| Reagents | Amount |
|---|---|
| Tris Base | 3 gm |
| Glycine | 14.4 gm |
| Methanol | 200 mL |
| Distilled water | 800 mL |
Note: Adjust the pH to ∼8.3 using 1 M Hydrochloric acid (HCl) slowly. Prepare the solution just on the day of the experiment.
TBS-Tween (TBST)
| Reagents | Amount |
|---|---|
| 1× TBS | 10 mL |
| Tween-20 | 0.01 mL |
Note: Do not store. Freshly prepare the solution on the day of the experiment.
Blocking Buffer
| Reagents | Amount |
|---|---|
| 1× TBS | 10 mL |
| Bovine Serum Albumin (BSA) | 0.3 g |
Note: Do not store. Freshly prepare the solution on the day of the experiment.
Primary antibody dilution (1: 5000) for Western blot
| Reagents | Amount |
|---|---|
| 1× TBS | 10 mL |
| BSA | 0.2 g |
| Rabbit anti-Hcp polyclonal antibody | 2 μL |
Note: Freshly prepare the solution before the experiment. However, the diluted solution can be stored at 4°C if intended for use within the next 1–2 days.
Secondary antibody dilution (1: 3500) for Western blot
| Reagents | Amount |
|---|---|
| 1× TBS | 10 mL |
| BSA | 0.2 g |
| Goat anti-rabbit IgG (H+L) HRP conjugated | 3 μL |
Note: Freshly prepare the solution before the experiment. However, the diluted solution can be stored at 4°C if intended for use within the next 1–2 days.
3, 3′-diaminobenzidine (DAB) substrate solution
| Reagents | Amount |
|---|---|
| DAB | 5 mg |
| TBS | 10 mL |
| 30% H2O2 | 10 μL |
Note: Do not store. Freshly prepare the solution on the day of the experiment. Mix the DAB reagents completely into TBS.
Coating buffer solution
| Reagents | Amount |
|---|---|
| Sodium bicarbonate | 16.8 g |
| Sodium carbonate | 22 g |
| Autoclaved distilled water | 1,000 mL |
Note: Dissolve the reagents in 800 mL autoclaved distilled water and adjust the pH to 9.6. Make the volume up to 1,000 mL by adding water. Store the buffer solution at 4°C for a maximum of 3 months.
Blocking buffer (5%, w/v) for ELISA
| Reagents | Amount |
|---|---|
| 1× PBS | 10 mL |
| Bovine serum albumin (BSA) | 0.5 g |
Note: Freshly prepare the solution on the day of the experiment. However, it can be stored at 4°C if intended for use within the next 1–2 days.
Primary antibody dilution (1:2,000) for indirect ELISA
| Reagents | Amount |
|---|---|
| 1× PBS | 10 mL |
| BSA | 0.2 g |
| Rabbit anti-Hcp polyclonal antibody | 5 μL |
Note: Freshly prepare the solution on the day of the experiment. However, it can be stored at 4°C if intended for use within the next 1–2 days.
Secondary antibody dilution (1:3,500) for indirect ELISA
| Reagents | Amount |
|---|---|
| 1× PBS | 10 mL |
| BSA | 0.2 g |
| Goat anti-rabbit IgG (H+L) HRP conjugated | 3 μL |
Note: Freshly prepare the solution on the day of the experiment. However, it can be stored at 4°C if intended for use within the next 1–2 days.
3, 3′, 5, 5′-Tetramethylbenzidine (TMB) substrate solution for ELISA
| Reagents | Amount |
|---|---|
| TMB substrate | 1 mL |
| Autoclaved distilled water | 9 mL |
Note: Do not store. Do not expose to light. Freshly prepare the solution before the experiment and keep it in a 15 mL tube under the dark.
Stopping solution (1 M H2SO4)
| Reagents | Amount |
|---|---|
| Sulphuric acid | 0.54 mL |
| Autoclaved distilled water | 9.46 mL |
Note: H2SO4 is hygroscopic, it will be better to use the freshly prepared solution before the experiment.
Paraformaldehyde solution (4%, w/v)
| Reagents | Amount |
|---|---|
| Paraformaldehyde | 4 g |
| Sodium hydroxide (NaOH) | 1 mL (1 M) |
| Hydrochloric acid (HCl) | 1 mL (1 M) |
| Autoclaved distilled water | 100 mL |
Note: For preparing 4% paraformaldehyde solution, add 4 g of paraformaldehyde to 50 mL of distilled H2O. Add 1 mL of 1 M NaOH and stir the heating block gently at ∼60°C until the paraformaldehyde is dissolved. Add 10 mL of 10× PBS and allow the mixture to cool at room temperature (∼25°C). Adjust the pH to 7.4 and then make the final volume 100 mL with H2O. The solution can be stored for one month at 4°C.
Standard growth media for human INT407 cells
| Reagents | Final concentration | Amount |
|---|---|---|
| Dulbecco’s Modified Eagle Medium (DMEM) | 89% | 44.5 mL |
| Fetal Bovine Serum | 10% | 5 mL |
| Penicillin-Streptomycin | 1% | 0.5 mL |
Note: Cell culture medium for human INT407 cells can be stored at 4°C for 1 month.
Note: Optimal medium composition can vary considerably for different cell types.
Triton-X 100 (1%, v/v)
| Reagents | Amount |
|---|---|
| TritonX-100 | 0.1 mL |
| 1× PBS | 10 mL |
Note: Do not store. Freshly prepare the solution on the day of the experiment.
4′, 6-diamidino-2-phenylindole (DAPI) stock solution
| Reagents | Amount |
|---|---|
| DAPI powder | 50 mg |
| 1× PBS | 1 mL |
Note: Prepare the solution in the dark. To avoid direct light exposure, store the solution at −20°C in an aluminum foil-wrapped 500 μL micro-centrifuge tube.
DAPI working solution
| Reagents | Amount |
|---|---|
| DAPI stock solution | 50 μL |
| 1× PBS | 1 mL |
Note: Do not store. Freshly prepare the solution on the day of the experiment.
2′, 7′-Dichlorodihydrofluorescein diacetate (H2DCFDA) Stock solution
| Reagents | Amount |
|---|---|
| H2DCFDA reagent | 485 mg |
| 1× PBS | 1 mL |
Note: Dissolve the chemical in 1× PBS. Aliquot the solution in a dark micro-centrifuge tube. Store it at −20°C.
H2DCFDA working solution
| Reagents | Amount |
|---|---|
| H2DCFDA stock solution | 4.8 μL |
| 1× PBS | 10 mL |
Note: Do not store. Freshly prepare the solution on the day of the experiment.
Step-by-step method details
Isolation of Campylobacter spp. from the chicken cecal contents
-
1.
Sample collection and processing:
Place freshly collected fecal contents from broiler chickens in sterile 1× PBS (pH 7.4). Samples should be kept on ice and processed immediately (Singh and Mallick, 2019) (Figure 1A).-
a.Place the chicken on top of the cage.
-
b.Humanely kill the chickens in accordance with the approved animal ethics protocol.
-
c.Lay the chicken on its back on tissue paper and clean the surface of the chicken thoroughly in 70% ethanol.Note: This important step reduces the risk of contaminating the tissue sample.
-
d.Pin the sacrificed chicken down to secure the chicken to a tray at the wings, shoulders, and legs.
-
e.Use a pair of sterile scissors to remove the skin of the chicken, starting from the lower abdomen and to the top of the thorax to expose the digestive tract.
-
f.Cut the cecum pair and gently place it in a sterile petri-plate.
-
g.Carefully clean the outer surface of the cecum using a sterile scalpel to remove the mucous or blood clots.
-
h.Cut the cecum longitudinally.
-
i.Use a sterile scalpel to scrape and gently rub the inner epithelial surface of the cecum and take the cecal content into the sterile micro-centrifuge tube.
-
j.Suspend 500 mg cecal content in 1 mL of sterile 1× PBS. Mix the cecum content by vortexing for 10 min to make a homogenous mixture.
-
k.Serially dilute homogenized samples up to 10-3 in Mueller Hinton broth (MH broth).
-
l.Inoculate 100 μL sample from 10-3 dilution and spread onto Blood Free Campylobacter Selectivity Agar Base media supplemented with CAT Selective Supplement (cefoperazone 8 mg/L, amphotericin 10 mg/L, and teicoplanin 4 mg/L).
-
m.Incubate the agar plates at 42°C for 48 h under microaerophilic conditions (10% CO2, 5% O2, and 85% N2).
-
n.Colonies that grow on the plate can be subjected to further analysis.
-
a.
Figure 1.

Identification of T6SS+ C. jejuni from cecal content of chickens
(A) Schematic of sample collection, processing, and identification of C. jejuni. Cecal content from commercial broilers was collected, serially diluted in MH broth, and plated onto Blood free Campylobacter selective agar media. After overnight incubation (∼18 h), bacterial colonies were subjected to further characterization.
(B) Morphological features and differential staining characteristics of the isolated bacteria. Flat, gray, glistening colonies with rough edges appeared on the plate. Gram-negative spiral-shaped bacteria were visualized under a light microscope (Scale bar: 20 μm) and scanning electron microscopy (Scale bar: 4 μm).
(C) Diagrammatic representation bacterial motility assessment: Following spot inoculation of single colony onto a soft agar plate, the bacteria were grown for 24–48 h. The swarming ability of C. jejuni was assessed by measuring the diameter of hallow formation around the colonies at different time points.
(D) A catalase test for the identification of Campylobacter sp. The individual colony was picked and added into a 4% H2O2 solution. Typical bubble formation (red arrow) in H2O2 solution indicates rapid liberation of oxygen bubbles due to the presence of catalase specific to the Campylobacter sp.
(E) An oxidase test for the identification of Campylobacter sp. Bacterial inoculum was added to a blotting paper soaked with an oxidase solution. The inoculated area turned blue (red arrow) due to the production of bluish indophenol, confirming the presence of cytochrome c oxidase in Campylobacter sp.
(F) A hippurate hydrolysis test for the identification of Campylobacter jejuni. The individual colony was incubated with 1% Hippurate solution, and then a ninhydrin solution was overlaid on the top. The appearance of purple (red arrow) confirms the presence of hippuricase enzyme specific for C. jejuni.
Identification of Campylobacter jejuni (C. jejuni)
Identification of C. jejuni from mixed culture can be achieved by colony morphology, bacterial morphology, biochemical character, and bacterial motility assay.
-
2.
Colony morphology.
Colony morphology can be useful to identify the bacteria present in a mixed culture as the colony of bacteria is distinguishable from each other based on their appearance, shape, and color.-
a.For morphological characterization, examine the colonies that appeared on selective media for size, shape, elevation, color, surface type, odor, etc.
-
b.Examine the subjected colonies for further characterization, such as morphological, biochemical and molecular characterization (Figure 1B).Note: Usually, C. jejuni forms a milky-white, glistening colony with rough edges on Blood Free Campylobacter Selective Media.
-
a.
-
3.
Morphological characterization.
The size and shape of bacteria are relatively stable under suitable conditions. It is important to know the morphological structure of bacteria, as morphology provides a better understanding of bacterial physiology and allows to characterize them by species. The Gram Staining procedure and Field Emission Scanning Electron Microscopy (FESEM) can be used as key techniques to characterize bacterial morphology.-
a.Gram staining.Conventional Gram Staining protocol (Coico, 2005) can be followed to identify C. jejuni by its Gram-negative and spiral shape character (Figure 1B).
-
i.Take bacterial samples from an overnight grown (18–24 h) culture and prepare a smear on a glass slide.Note: To prepare a good smear, bacteria cells from a culture should be laid out using a sterile loop in circular motions to make a thin film over a small area of a microscope slide.
-
ii.Air-dry cell smear. Heat fix the slide by passing it over a heat source (flame) several times to kill and firmly adhere bacteria on the glass slide (Madani, 2003).Note: Smear quality affects gram staining results. Pass the slide over the heat source quickly to avoid excessive heating during heat fixation.
-
iii.Flood the dried smear with crystal violet staining reagent for 1 min.
-
iv.Discard the flooded crystal violet stain and wash the slide under a slow stream of tap water for 5 s.
-
v.Cover the smear with Gram’s iodine and wait for 1 min.
-
vi.Discard the residual Gram’s iodine and wash the slide under a slow stream of tap water for 5 s.
-
vii.Add decolorizing agent (95% ethanol) and keep it for 15 s.
-
viii.Rinse the slide under a slow stream of water for 10 s.
-
ix.After completely drying, flood the slide with a safranin counterstain and wait for another 30 s.
-
x.Wash the slide under a slow stream of tap water until no color appears in the effluent.
-
xi.Observe the results of the staining procedure under oil immersion using a brightfield microscope (Objective magnification 60×) (Figure 1B).Note: As C. jejuni is a Gram-negative bacterium with less peptidoglycan in the cell wall, it cannot retain the crystal violet stain and hence acquire the counterstain (safranin) to shade pink color.
-
i.
-
b.Field Emission Scanning Electron microscope (FESEM).To visualize the cell morphology, cell integrity, and cell-to-cell adherent properties at a higher resolution (14,990×), FESEM can be performed (Relucenti et al., 2021) (Figure 1B).
-
i.Take the bacterial culture and adjust the OD600 to ∼0.6 by adding a fresh medium. Further, take 1 mL of the culture and centrifuge it at 5,000 × g for 6 min at room temperature (∼25°C).
-
ii.Resuspend the cells with 300 μL of 1× PBS and centrifuge at 5,000 × g for 6 min. Repeat this step for two more rounds.
-
iii.To fix the cells, add 300 μL of 2.5% (v/v) glutaraldehyde to the cell mixture and incubate at room temperature (∼25°C) for 1 h. Further, centrifuge the cells at 5,000 × g for 6 min.
-
iv.Wash the cell pellet thrice with 300 μL of 1× PBS for 10 min each.
-
v.Resuspend the cells using 300 μL of 35% ethanol and incubate for 10 min at room temperature (∼25°C). Further, centrifuge the cells at 5,000 × g for 6 min.
-
vi.Dehydrate the fixed cells sequentially at 50%, 70%, and 95% ethanol as mentioned in the previous step (Sub-step 3; b; v).
-
vii.After that, resuspend the cells using 500 μL of 100% ethanol followed by 1 h incubation for complete dehydration.
-
viii.Cast 10 μL of the sample on a small cover slip or silicon wafer.
-
ix.Dry the cover slip at room temperature (∼25°C) for 2 h.
-
x.Finally, vacuum-dry the fixed and dehydrated samples for 1 h and fix them to aluminum stubs.
-
xi.Thereafter, put the aluminum stub with the sample inside the coating instrument.
-
xii.Then sputter-coat the sample with platinum (Figure 1B) (Methods video S1: Method video of FESEM sample fixation to aluminum stubs with silver conductive paint and sputter-coating with gold, related to step 3b).
-
xiii.Capture the images of the Platinum-coated samples using the secondary electron detector under a high vacuum at 6 kV, with an 8 mm distance and 30 μM objective lens apertures, magnifications ranging from 3,000× to 20,000×.Note: We used Supra 55 Carl Zeiss scanning electron microscopes for our experimental purposes. We observed the cell morphology (spiral-shaped) and integrity of C. jejuni.
-
i.
-
a.
-
4.
Bacterial motility assay.
Motility is the ability of some bacteria to move. Using metabolic energy, bacterial flagella facilitate this movement in the culture or inside the hosts and is considered one of the key virulence phenotypes. As C. jejuni is a flagellated bacterium, motility phenotypes represent a critical virulence determinant for C. jejuni.CRITICAL: Do not use high agar concentration to study bacterial motility. A higher agar concentration in the medium may cause inhibition of bacterial motility.-
a.Pour 0.4% Mueller Hinton Agar (MH agar) into each well of a 6-well tissue culture plate.
-
b.Adjust the absorbance (OD600) of the culture to 1.00.
-
c.Inoculate 5 μL of the culture as a spot in the center of the agar and let it dry.
-
d.Incubate the inoculated plate for 24 h at 42°C under micro-aerophilic conditions.
-
e.After 24 h, measure the diameter (in a millimeter-scale) of the spread zone with a ruler throughout the area of growth around the central point of the plate (Figure 1C).Note: As C. jejuni are motile and flagellated bacteria, bacterial spots on the agar surface should be outspread (hallow) from the center to the periphery (Figure 1C). Hallow formation is absent in the case of non-motile bacteria.
-
a.
-
5.
Biochemical characterization.
Biochemical tests are commonly used to accurately identify the bacterial genus or species based on differences in their biochemical activities. The standard biochemical tests rely on nutrient utilization (carbohydrate utilization, amino acid degradation, lipid degradation), resistance to inhibitory substances (high salt, antibiotics, etc.), enzyme production (catalase, oxidase, hippuricase, etc.) of particular bacteria. To identify and characterize C. jejuni, biochemical tests such as Catalase, Oxidase, and Hippurate hydrolysis test can be performed. Bacteria can be grown either on agar plates or in broth culture, depending on the type of the biochemical test. Bacteria can be incubated for 18–24 h in microaerophilic conditions (85% N2, 10% CO2, and 5% O2) at 37°C to obtain bacterial growth on agar plates (distinguishable colony) as well as in broth (OD600∼0.6).-
a.Catalase test.The catalase test helps identify the bacteria that have catalase enzymes. As C. jejuni is a catalase-positive bacterial species, this assay can be helpful in the species-level identification of C. jejuni. A catalase enzyme in a C. jejuni can be scrutinized when bacterial inoculum is mixed with hydrogen peroxide (H2O2) solution.CRITICAL: To limit catalase aerosols, which have been shown to carry viable bacterial cells, petri dish is strongly recommended. Be careful not to pick up any agar.
-
i.Place a microscope slide inside a petri dish.
-
ii.Keep the petri dish cover available.
-
iii.Place one drop of 4% H2O2 onto the bacteria on the microscope slide using a dropper.
-
iv.Using a sterile inoculating loop or wooden stick, collect a small amount of well-isolated colony and place it onto the microscope slide.
-
v.Immediately cover the petri dish with a lid to limit aerosols and observe immediate bubble formation.
-
vi.Observing the formation of bubbles against a dark background enhances readability.Alternative Protocol:
-
vii.Take 1 mL of well-grown (18–24 h after primary culture) culture in a 1.5 mL microcentrifuge tube.
-
viii.Centrifuge the cells at 6,000 × g for 5 min and decant the supernatant.
-
ix.Resuspend the cells in 1 mL of 1× PBS.
-
x.Add 150 μL of 30% H2O2 solution.
-
xi.Observing the formation of bubbles against a dark background enhances readability (Figure 1D).Note: As C. jejuni is catalase-positive, it can facilitate the breakdown of H2O2 into oxygen and water. This is exhibited by the rapid release of oxygen and forms bubbles.
-
i.
-
b.Oxidase test.The Oxidase test determines the ability of bacteria to synthesize cytochrome oxidase and is used to identify Neisseria, Moraxella, Campylobacter, and Pasteurella spp (oxidase positive). Cytochrome oxidase catalyzes electron transport from donor compounds (NADH) to acceptors (usually oxygen). The test reagent, N, N, N′, N′-tetramethyl-p-phenylenediamine dihydrochloride (TMPD), serves as an electron acceptor. The oxidized reagent forms the colored compound indophenol blue, suggesting positive test results.
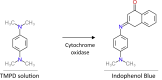 CRITICAL: Let the filter paper dry after adding oxidase reagent.
CRITICAL: Let the filter paper dry after adding oxidase reagent.-
i.Soak a small filter paper in an oxidase reagent and let it dry.
-
ii.Using a loop, pick a well-isolated colony from a fresh bacterial plate (grown overnight for 18–24 h), rub it onto treated filter paper, and wait for 10–15 min.
-
iii.Observe for color changes to blue, indicating oxidase-positive (Figure 1E).Note: As C. jejuni is oxidase-positive, blue color forms in the presence of N, N, N′, N′-tetramethyl-p-phenylenediamine dihydrochloride.
-
i.
-
c.Hippurate hydrolysis test.The hippurate hydrolysis test aims to confirm the presence of the hippuricase enzyme in bacteria. It is primarily used to identify C. jejuni, Listeria monocytogenes, Gardnerella vaginalis, and Streptococcus agalactiae. In addition, the hippurate hydrolysis test is a reliable assay to differentiate C. jejuni from the other Campylobacter spp. such as C. coli and C. upsaliensis since the latter does not produce hippuricase enzymes. This biochemical assay is based on the ability of C. jejuni to hydrolyze sodium hippurate to benzoic acid and glycine using the hippuricase enzyme. Further glycine is deaminated by the oxidizing action of ninhydrin which forms reduced ninhydrin, resulting in a purple-colored complex.
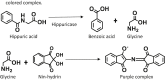 CRITICAL: The bacteria wash step (steps 2 and 3) is essential to remove the content of the medium that may give false-positive results.
CRITICAL: The bacteria wash step (steps 2 and 3) is essential to remove the content of the medium that may give false-positive results.-
i.Suspend a loop full of the well-isolated colonies from a fresh (18–24 h) bacterial plate in 5 mL MH broth and grow the culture for 18–24 h (OD600∼0.6) at 37°C in microaerophilic condition (85% N2, 10% CO2, 5% O2).
-
ii.Centrifuge the grown culture at 5,000 × g for 6 min and discard the supernatant.
-
iii.To wash the bacterial cells, resuspend the cell pellet in 1 mL 1× PBS (pH∼7.4). Centrifuge the suspension at 5,000 × g for 6 min. Repeat this step three times.
-
iv.Discard the supernatant.
-
v.Add 400 μL of 1%-hippurate solution and incubate the mixture for 2 h at 37°C.
-
vi.Then slowly add 200 μL of the 3.5%-ninhydrin solution to the side of the tube to form an overlay and re-incubate at 37°C for 20 min.
-
vii.See the formation of purple color (Figure 1F).Note: Identified bacteria, which are characterized as Gram-negative, spiral in shape, motile and positive for catalase, oxidase, and hippurate hydrolysis test, can be performed for further molecular and phenotypic characterization.
-
i.
-
a.
Characterization of C. jejuni
-
6.
Molecular characterization.
To identify and characterize bacteria at the species level, molecular, biochemical and morphological characterization are essential.-
a.Isolation of Genomic DNA.Bacterial genomic DNA (gDNA) isolation protocol (Chen and Kuo, 1993) can be performed for molecular characterization by observing the presence or absence and sequence of individual genes, modifying sections of DNA (isogenic mutant generation), and more.
-
i.Centrifuge 5 mL well grown (16–18 h, OD600∼0.6) C. jejuni culture at 10,000 × g for 3 min.
-
ii.Resuspend the pellet in lysis buffer (200 μL) and mix it with vigorous pipetting.
-
iii.Incubate it at 37°C for 30 min.
-
iv.Add 80 μL of 5 M NaCl solution and mix well.
-
v.Centrifuge the viscous solution at 10,000 × g for 8 min at 4°C.
-
vi.Transfer the clear supernatant into a new micro-centrifuge tube.
-
vii.Add 200 μL of chloroform.CRITICAL: As chloroform is a volatile hazardous chemical, perform this step in the fume hood.
-
viii.Invert it at least 80 times to form a milky solution or rotate the tube by placing it on a spinning wheel for at least 1 min.
-
ix.Centrifuge it at 10,000 × g for 3 min.
-
x.Pipette the supernatant into a new vial without touching the second layer.
-
xi.Add 150 μL of 100% molecular grade ethanol to the supernatant and tap the tube gently for proper mixing.
-
xii.Centrifuge it at 10,000 × g for 5 min and decant the supernatant.
-
xiii.Add 150 μL of 70% ethanol and tap gently for proper mixing.
-
xiv.Centrifuge it at 10,000 × g for 5 min and decant the supernatant.
-
xv.Repeat steps xiii–xiv twice.
-
xvi.Dry the pellet inside a laminar air-flow for 15 min to minimize ethanol contamination which may hinder the downstream applications.
-
xvii.Dissolve the pellet with 30 μL TE buffer.
-
i.
-
b.Identification of major genes of T6SS +ve C. jejuni by PCR.
-
c.Sequencing of 16S rRNA and hcp genes of C. jejuni isolate, BLAST analysis, and construction of Phylogenetic tree:As 16S rRNA gene sequencing is used to identify bacteria at the species level and assist with differentiating between closely related bacterial species, 16S rRNA gene sequencing can be performed as a confirmatory test to identify 16S rRNA sequencing. As hcp is a hallmark gene of T6SS machinery, determining the hcp gene sequence can also be performed by the Sanger Sequencing technique.
- i.
-
ii.PCR products can be purified by a PCR purification kit (Thermo Scientific) using the manufacturer’s protocol (https://www.thermofisher.com/document-connect/documentconnect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2Fpurelink_pcr_man.pdf) and subjected to Sanger sequencing.
- iii.
-
iv.Perform BLAST analysis of the DNA sequence. To analyze the nucleotide sequence in BLAST, open the histogram of the Sanger sequencing data and copy the sequence. After that go to browser and open search NCBI BLAST and select Nucleotide BLAST. Then paste the copied sequence and click on BLAST option. (Methods video S2: BLAST analysis user method video to compare nucleotide sequence of the desired gene to standard sequence available in a database and calculate the statistical significance of the matches, related to step 6c).
-
v.Create a phylogenetic tree by the MEGA X software. To generate a phylogenetic tree among the 16S rRNA gene of C. jejuni using MEGA software, download a few sequences of C. jejuni 16S rRNA gene from the NCBI website in the FASTA format. Then create a text file, and merge all the sequences including the sequence of C. jejuni isolate. After that in MEGA software open a file and go to align, to build and create a new alignment. Next, select the DNA option and select the same text file to get the alignment data. Then, export the alignment in MEGA format. After that from the Phylogeny, option select the Maximum Likelihood Tree method to construct the phylogeny tree. (Methods video S3: MEGA X software user method video to construct a phylogenetic tree of 16S rRNA gene of C. jejuni isolate and to reflect evolutionary relationships among 16S rRNA gene of C. jejuni, related to step 6c).Note: The evolutionary relatedness can be inferred using the Maximum Likelihood method and Tamura-Nei model (Tamura and Nei, 1993). The tree with the highest log-likelihood is shown (Figures 2B and 2C).Note: After confirmation, prepare 1 mL of 25% (v/v) bacterial glycerol stock (500 μL MH broth and 500 μL 50% glycerol) to store at −80°C the bacterial strain for future experimental purposes. To prepare single-use glycerol stock, reduce the final volume up to 100 μL (50 μL MH broth and 50 μL 50% glycerol) and store at −80°C.
-
a.
-
7.Phenotypic virulence characterization.
-
a.Biofilm formation assay.Similar to other bacterial pathogens, the ability to form biofilms is an important virulence mechanism concerning the transmission of disease-causing Campylobacter spp. to humans (Mah, 2012; Bridier et al., 2015). As we are looking into virulent T6SS+ve C. jejuni, a biofilm assay can be helpful to identify and characterize C. jejuni.CRITICAL: Glacial acetic acid can cause eye and upper respiratory tract irritation.Precautionary measures: Use nitrile rubber gloves as glacial acetic acid perforates latex gloves. Double-gloving can be a reasonable precaution. Use complete protection in addition to non-latex gloves: apron, face shield, etc. Open the chemical under a fume hood.
-
i.Take single-use glycerol stocks of the C. jejuni isolates and grow them to obtain a pure culture on MH plates overnight (∼18 h) at 42°C under microaerophilic conditions.
-
ii.Take the bacterial cells from the MH plate to inoculate into the MH broth and incubate overnight (∼12 h) under continuous shaking conditions (42°C, microaerophilic conditions).
-
iii.Adjust the absorbance (OD600) of the culture to 0.3 by dilution with MH broth.
-
iv.To allow biofilm formation, add 1 mL of the C. jejuni culture to the wells of 24-well cell culture plates and incubate at 42°C under microaerophilic conditions for 24 h.
-
v.Remove the planktonic cell suspension using a pipette.
-
vi.Wash the remaining biofilm with 500 μL of 1× PBS.
-
vii.Add 1 mL of 0.1% (w/v) Crystal Violet (CV) staining solution and incubate for 30 min at room temperature (∼25°C).
-
viii.Remove the unbound dye and wash the plate three times with 1× PBS and allow it to dry.
-
ix.Further, dissolve the bound dye in 30% glacial acetic acid (Merck).
-
x.Incubate for 15 min at room temperature (∼25°C).
-
xi.Finally, measure the absorbance at 595 nm using a microplate reader. To measure the absorbance value of a multi-well plate using a plate reader equipped with Gen5 software, first open the software and go to the ‘New’ option. Then, select the 'Plate type', 'Read' option, 'Absorbance', 'Endpoint', and 'Monochromators' option. Then depending on the experimental purposes set the wavelength and select the ‘Read’ option. After that, to proceed click on ‘OK’ in the temperature dialogue box (Methods video S4: Gen5 software user method video to read ELISA/tissue culture plate in a microplate reader, related to step 7a).Note: We used Epoch2 micro-plate reader for our experimental purposes.
-
xii.To visualize biofilm formation, prepare the bacterial biofilm on the coverslip in a 35 mm petri plate (as described in step 4) and wash it as mentioned earlier (Sub-step 7; a; vi).
-
xiii.Add 0.005% acridine orange to the coverslip and wait for 5 min.
-
xiv.After that, thoroughly wash the stain with 500 μL 1× PBS thrice.
-
xv.Mount the coverslips using 5 μL Vecta-shield mounting media for imaging.
-
xvi.To visualize acridine orange stained biofilm, use a FITC filter with an excitation wavelength of 400–520 nm and an emission wavelength of 480–580 nm (Figure 3A).Note: For our experimental purposes, image acquisition was made under an Axio observer microscope equipped with an ApoTome module (Carl Zeiss) at an objective magnification of 60×. The images were processed in Zen software.Note: Here we demonstrated the quantification and visualization of sub-merged type biofilm and observed the same for C. jejuni (Figure 3A).Note: Identified and characterized T6SS positive (T6SS+ve) C. jejuni bacterial isolate can be analyzed further for its functional characterization.
-
i.
-
a.
Table 1.
Primer sequence used in this study
| Target gene | Primer sequence | References |
|---|---|---|
| 16S rRNA (C. jejuni) | F- 5′ AATCTAATGGCTTAACCATTA 3′ | (Singh and Mallick, 2019) |
| R- 5′ GTAACTAGTTTAGTATTCCGC 3′ | ||
| ciaB (C. jejuni) | F- 5′ TTTCCAAATTTAGATGATGC 3′ | (Singh and Mallick, 2019) |
| R- 5′ GTTCTTTAAATTTTTCATAATGC 3′ | ||
| hipO (C. jejuni) | F- 5′ CTCCTATGCTTACAACTGCTG 3′ | (Singh and Mallick, 2019) |
| R- 5′ GGTGGTCATGGAAGTGCT 3′ | ||
| gltA (C. jejuni) | F-5′ GCCCAAAGCCCATCAAGCGGA 3′ | (Singh and Mallick, 2019) |
| R-5′ GCGCTTTGGGGTCATGCACA 3′ | ||
| cadF (C. jejuni) | F- 5′CGGCGCGCATGCATGGCTGATAACAATGTAAAATTTG 3′ | (Gupta et al., 2021) |
| R- 5′ GCCGCCGCGGTACCTTATCTTAAAATAAATTTAGCATCC 3′ | ||
| jlpA (C. jejuni) | F- 5′ CGCGGCATGCATGTGCGGAAATTCCATAGATG 3′ | (Gupta et al., 2021) |
| R-5′ CGATGGTACCTTAAAATAACGCTCCGCCC 3′ | ||
| vasC (C. jejuni) | F- 5′ CCAATGCTTTGATGGTAAG 3′ | (Gupta et al., 2021) |
| R- 5′ GGAATTGCTATTGAGAATTATGACG 3′ | ||
| vasK (C. jejuni) | F- 5′ ATAATATTCGGGTATTTCATCGCT 3′ | (Gupta et al., 2021) |
| R- 5′ TTCAGTAGATGCACCGCTTGA 3′ | ||
| vasD (C. jejuni) | F- 5′ GTCCATCAAACCAAGCAAC 3′ | (Gupta et al., 2021) |
| R- 5′ GACATCTCATCTTCAAGTAACTG 3′ | ||
| vasE (C. jejuni) | F- 5′ TCATTGAAATACCGCCCACA 3′ | (Gupta et al., 2021) |
| R- 5′ GATGCAAATGGGTTTGGGAAG 3′ | ||
| hcp (C. jejuni) | F-5′ ATAGGATCCATGGCTGAACCAGCGTTTATA 3′ | (Gupta et al., 2021) |
| R-5′ CGCGAATTCTAGCAAAGGCACAGA 3′ | ||
| 16S rRNA (E. coli) | F- 5′ GTTAATACCTTTGCTCATTGA 3′ | (Gupta et al., 2021) |
| R- 5′ ACCAGGGTATCTAATCCTGTT 3′ | ||
| ftsZ (E. coli) | F- 5′ AGCAGAAGCCGGTTGCTAAA 3′ | (Gupta et al., 2021) |
| R- 5′ TCCGGCTCTTTCGCAGTT 3′ |
Table 2.
PCR cycling conditions of 16S rRNA, major virulence genes, including core genes of functional T6SS
| Steps | Temperature | Time | Cycles | ||
|---|---|---|---|---|---|
| Initial denaturation | 94°C | 3 min | 1 | ||
| Denaturation | 94°C | 1 min | 30 cycles | ||
| Annealing temp. of the primer set for each gene | Housekeeping gene | 16S rRNA (814 bp) | 56°C | 1 min | |
| Identification gene | hipO (130 bp) | 48°C | |||
| Virulence genes | ciaB (1,165 bp) | 41°C | |||
| cadF (912 bp) | 60°C | ||||
| jlpA (1,024 bp) | 60°C | ||||
| gltA (140 bp) | 48°C | ||||
| Major T6SS genes | hcp (243 bp) | 58°C | |||
| vasC (854 bp) | 46°C | ||||
| vasD (692 bp) | 47°C | ||||
| vasK (799 bp) | 50°C | ||||
| vasE (733 bp) | 48°C | ||||
| Extension | 72°C | 1 min | |||
| Final extension | 72°C | 5 min | 1 | ||
| Hold | 4°C | Indefinite | |||
Figure 2.

Molecular identification, virulence gene profiling, and phylogenetic relatedness of T6SS+ve C. jejuni
(A) PCR amplification of 16S rRNA, major virulence genes, including core genes of functional T6SS. The genomic DNA was extracted from C. jejuni isolates and the presence of major virulence gene were identified using genes specific primers sets. Lane 1: DNA ladder (HMW); Lane 2: 16S rRNA; Lane 3: hipO; Lane 4: ciaB; Lane 5: cadF; Lane 6: jlpA; Lane 7: gltA; Lane 8: hcp; Lane 9: vasC; Lane 10: vasD; Lane 11: vasK; Lane 12: vas E; Lane 13: DNA ladder LMW. Next, purified PCR products were subjected to DNA sequencing and checked for sequence homology with previously reported sequences.
(B) The predictive relationship (Phylogenetic tree) of the isolated bacterial species with available C. jejuni sequences was determined based on the sequence similarity of the 16S rRNA gene of C. jejuni. The highest log-likelihood of the tree is -3607.23. Red and bold indicate the 16S rRNA gene sequence of C. jejuni strains that were isolated in our laboratory.
(C) The predictive relationship (Phylogenetic tree) of the isolated bacterial species with available C. jejuni sequences was determined based on the sequence similarity of hcp gene of T6SS harboring C. jejuni and other Gram-ve bacteria. The highest log-likelihood of the tree is -4793.71. Red and bold indicate the hcp gene sequence of the C. jejuni strain isolated in our laboratory.
Figure 3.

Phenotypic virulence characterization (biofilm formation) and functional exhibition of T6SS+ve C. jejuni
(A) Quantification and visualization of the biofilm formation by C. jejuni isolates. A pictorial depiction of biofilm formation and its quantification was performed by growing the bacteria in a 24-well tissue culture plate. The amount of biofilm formation was measured after removing the planktonic bacteria from each well and stained with crystal violet, followed by measuring the OD at 595 nm. To visualize biofilm formation, biofilm was grown on the coverslip. After washing, the grown biofilm was stained with 0.005% acridine orange dye and observed under an Axio observer microscope equipped with an ApoTome module (Carl Zeiss) (Objective magnification: 60×) (Scale Bar: 50μm).
(B) Detection and quantification of Hcp protein in the culture supernatant of T6SS+ve C. jejuni: Western blot analysis of TCA precipitated protein from C. jejuni was probed with an anti-Hcp polyclonal antibody (raised in rabbit) detected a protein band corresponding to the size of Hcp (∼20 kDa) (Lane 1:T6SS+ve C. jejuni; Lane 2: T6SS-ve C. jejuni; Lane 3: rHcp; M: Marker). C. Schematic of indirect ELISA to quantify Hcp protein secreted by T6SS+ve C. jejuni. The supernatant (in serial dilution) from C. jejuni was probed with an anti-Hcp polyclonal antibody (raised in rabbit). After incubating with HRP-conjugated goat anti-rabbit IgG secondary antibody, the plates were developed with TMB substrate. The absorbance was measured at 450 nm in a UV-VIS multi-plate reader.
Functional characterization of T6SS+ve C. jejuni
The T6SS has recently emerged as a new pattern of protein secretions in C. jejuni. Within the T6SS cluster, hemolysin coregulated protein (Hcp or TssD) is considered a hallmark of functional T6SS and holds a key role in bacterial virulence. Hcp is often detected in the culture supernatant of major Gram-negative bacteria with functional T6SS (Peng et al., 2016). Thus, the presence of functional T6SS can be verified by detecting Hcp protein in the bacterial culture supernatant.
-
8.Detection of Hcp protein secretion from T6SS+ve C. jejuni by Western Blot.
-
a.Bacteria culture and TCA precipitation:CRITICAL: Trichloroacetic acid (TCA) is a corrosive chemical. Direct contact can cause skin burn and irritation with the risk of permanent eye damage. Breathing TCA can cause nose and throat irritation.CRITICAL: Ice-chill the culture supernatant before adding TCA to prevent protein degradation. After acetone wash, air-dry the pellet; incomplete drying may cause smear formation and affect the sample mobility during SDS-PAGE. Charge the PVDF membrane with 100% methanol before transfer. After that, wash it with 1× transfer buffer and place the membrane on the polyacrylamide gel.
-
i.Take a freshly grown culture (500 mL) of T6SS+ve C. jejuni and adjust the absorbance (OD600) to 1.00.
-
ii.Centrifuge the culture at 6,000 × g for 8 min at 4°C.
-
iii.Take the culture supernatant and chill it on ice for 30 min.
-
iv.Add 15% TCA to the culture supernatant and keep it on ice for 90 min to 120 min.
-
v.Centrifuge the TCA mixed supernatant at 5,000 × g for 20 min at 4°C.
-
vi.Discard the supernatant and resuspend the pellet into ice-chilled acetone.
-
vii.Centrifuge at 5,000 × g for 20 min at 4°C.
-
viii.Let it dry in the air for 15–30 min.
-
ix.Add 50 μL 1× Laemmli buffer.
-
i.
-
b.Run the sample in 15% SDS-PAGE.
-
i.Place the thin and thick glass plates together in an SDS-PAGE casting cassette.
-
ii.To make resolving gel mix the required reagents (add TEMED in last) described in Table 3.
-
iii.Pour the mixed solution between the two glass plates fixed in the casting cassette.
-
iv.Fill the rest of the space above the resolving gel with isopropanol to avoid bubble formation. Wait for 15 min to solidify the gel.
-
v.To make stacking gel, mix the required reagents (add TEMED in last) described in Table 3.
-
vi.Discard the isopropanol, pour the stacking gel solution, and place the comb accordingly.
-
vii.Wait for 15 min to solidify the stacking gel.
-
viii.Place the gel in the running cassette such that the wells would face inwards.
-
ix.Place the entire running cassette with the gel inside the running tank.
-
x.Fill the gel running tank with 1× SDS-running buffer.
-
xi.Remove the comb.
-
xii.Mix 15 μL of the sample (prepared with 1× Laemmli buffer) with 4 μL of 5× sample loading dye and load accordingly inside the wells.
-
xiii.Set the voltage to 90 volts and run the gel until the dye front comes out.
-
i.
-
c.Membrane transfer.
-
i.Cut the gel and measure the size of the gel and keep it to 1× transfer buffer (in shaking condition) for 10 min at RT.
-
ii.Cut the polyvinylidene fluoride (PVDF) membrane according to the gel size.
-
iii.To prepare the PVDF membrane, charge the membrane with methanol for 2 min at RT (∼25°C).
-
iv.After that, discard the methanol and wash the membrane with 1× transfer buffer for 15 min at RT (∼25°C).
-
v.Meanwhile, soak filter papers and sponges in 1× transfer buffer for 10 min at RT (∼25°C) to assemble a ‘sandwich’ of gel and membrane.
-
vi.After preparing the ‘sandwich’, roll a small glass rod over it to prevent any bubble formation between the gel and the membrane.
-
vii.Fix the transfer cassette into the transfer chamber.
-
viii.Run for 65 min at 90 volts to facilitate protein transfer from the gel to the PVDF membrane.Note: To avoid heat generation in the transfer chamber, perform the transfer procedure at 4°C environment.
-
i.
-
d.Membrane processing and development.
-
i.Take out the PVDF membrane from the Western blot cassette and dip the membrane into a blocking buffer for overnight (∼12 h) at 4°C.
-
ii.Wash the membrane twice with TBST solution and three times with TBS at room temperature (∼25°C).
-
iii.Add anti-Hcp primary antibody (1:5,000 dilution), raised in New-Zealand white rabbit and incubate at room temperature (∼25°C) for 1 h.
-
iv.Wash the membrane twice with TBST solution and three times with TBS at room temperature (∼25°C).
-
v.Add Goat anti-rabbit HRP conjugated IgG (H+L) secondary antibody (1:3,500 dilution) and incubate at room temperature (∼25°C) for 1 h.
-
vi.Wash the membrane two times with TBST solution and three times with TBS.
-
vii.Add 10 mL DAB color developing solution and keep in the dark for 5 min.
-
viii.Observe for brown precipitate formation at the corresponding size of Hcp (∼20 kDa) (Figure 3B).
-
i.
-
a.
-
9.
Quantification of Hcp protein secreted from T6SS+ve C. jejuni in culture supernatant by Indirect ELISA.
An indirect ELISA can be used to quantify and detect the secretion of Hcp protein in the culture supernatant by T6SS+ve C. jejuni isolates.-
a.ELISA plate coating.
-
i.Take the freshly grown culture of T6SS+ve C. jejuni and adjust the OD600 to 1.
-
ii.Centrifuge the culture at 5,000 × g for 5 min and take the culture supernatant.
-
iii.Coat the 96-well ELISA plates with bacterial culture supernatant serially diluted (two-fold) in the carbonate-bicarbonate buffer (pH ∼9.6).Note: Use the micropipette to dispense 100 μL of carbonate-bicarbonate buffer (pH∼9.6) to the second, third, and fourth well. Next, use the micropipette to transfer 200 μL of the test sample (undiluted bacterial culture supernatant) to the first well. After that, use the micropipette to mix the test sample (undiluted bacterial culture supernatant) in the well. Then, draw up 100 μL of the supernatant and dispense it to the second well (to make 1:2 dilution in the second well). Now, mix the solution by pipetting, draw up 100 μL of the supernatant from the second well, and dispense it to the third well (to make 1:4 dilution in the third well). Mix it thoroughly by pipetting. After that, draw up 100 μL of the diluted supernatant from the third well and expel it in the fourth well (to make 1:8 dilution in the fourth well). Use the micropipette to mix it by pipetting. Lastly, draw up 100 μL of the diluted supernatant from the fourth well and discard it.
-
iv.Incubate the plate overnight (∼12 h) at 4°C.Note: We used rHcp (purified recombinant 6His-tagged Hcp protein) as a positive control for validation.
-
i.
-
b.Washing and blocking.
-
i.Subsequently, remove the coated sample and wash the wells thrice with 100 μL of PBS-T/well into the well and discard the content by tapping the inverted plate on the absorbent material such as tissue paper.
-
ii.After washing, add 100 μL of 5% blocking solution to each well and incubate for 1 h at 37°C.
-
iii.Following incubation, wash the plate thrice with 100 μL of PBS-T/ well as mentioned earlier (see Sub-step 7; b; i).
-
i.
-
c.Antibody treatment and washing.
-
i.Probe with rabbit polyclonal anti-Hcp hyper-immune sera (1:2,000 dilution) (100 μL in each well) as the primary antibody for 2 h at RT.
-
ii.Following incubation, wash the plate thrice with 100 μL of PBS-T in each well.
-
iii.After thorough washing, add 100 μL HRP-conjugated goat anti-rabbit IgG (H+L) secondary antibody (1:3,500 dilution) in each well and incubate for another 1 h at RT (∼25°C).
-
iv.Following incubation, wash the plate thrice with 100 μL of PBS-T in each well.
-
i.
-
d.Color development.
-
i.After washing, add 100 μL of 1× TMB substrate to develop the reaction.Note: Color development generally takes 2 min for recombinant protein and approximately 5 min for bacterial culture supernatant.
-
ii.After color formation (blue), stop the reaction by adding a stopping solution (color turns yellow).
-
iii.Read the absorbance at 450 nm in a microplate reader (Methods video S4: Gen5 software user method video to read ELISA/tissue culture plate in a microplate reader, related to step 9d).Note: For our experimental purposes, we used an Epoch2 microplate reader (BioTek, USA) (Figure 3B).Note: T6SS+ve C. jejuni can be extensively used to study interspecies interactions such as interbacterial interaction and host-bacteria interaction.
-
i.
-
a.
Table 3.
15% SDS-PAGE reagent list
| Reagents | Amount (mL) |
|---|---|
| Reagents for Resolving Gel | |
| Distilled water | 1.1 |
| 1.5 M Tris-Cl (pH∼8.8) | 1.3 |
| Acrylamide Solution | 2.5 |
| 10% APS | 0.05 |
| 10% SDS | 0.05 |
| TEMED | 0.007 |
| Reagents for Stacking Gel | |
| Distilled water | 1.49 |
| 1 M Tris-Cl (pH∼6.8) | 0.625 |
| Acrylamide Solution | 0.335 |
| 10% APS | 0.025 |
| 10% SDS | 0.025 |
| TEMED | 0.005 |
Interbacterial interaction
Within the same environmental niche, a functional T6SS facilitates host cell adherence, invasion, and bacterial predation, resulting in the selective advantage of the predator over other prey bacteria (Lertpiriyapong et al., 2012). These selective advantages raise the interest to study the nature of interbacterial interactions. Here, we have demonstrated the procedure to establish interbacterial interaction between C. jejuni and E. coli.
-
10.
Generation of GFP expressing E. coli.
To differentiate competing bacteria during inter-bacterial interaction, bacterial cells should possess morphological or chromogenic heterogeneity. For this, we have generated GFP-expressing E. coli which have been effectively used during co-culture experiments to differentiate prey (E. coli, green colonies) and predator bacteria (C. jejuni, milky white colonies).Note: Prepare chemically competent E. coli (DH5α) cells and store them at −80°C.Note: Commercially obtained cells also can be used as an alternative.CRITICAL: Do not thaw the competent cells till other reagents are ready; this may affect the competency of the cells. Do not use more than 500 ng of plasmid DNA for the transformation; a high amount of DNA may cause bacterial lawn formation after plating, which affects the selection of positive transformant cells. Do not give heat shock for more than 45 sec, which may cause cell death.-
a.Take out E. coli (DH5α) competent cell vial from −80°C and thaw in ice for 10 min.
-
b.Add 300 ng plasmid DNA (pTurbo GFP-B plasmid) to the competent cell.
-
c.Gently tap the tube 2–3 times to mix cells and place the vial into ice again for 30 min.Note: Set the heat-block temp to 42°C.
-
d.After that, place the vial in the heat block for 45 s.
-
e.Immediately chill the vial on ice for 10 min.
-
f.Add 1 mL LB media into the vial and incubate it for 1 h at 37°C at 180 rpm shaking.
-
g.After 1 h harvest the cell by centrifugation at 5,000 × g for 5 min and discard the supernatant.
-
h.Add 100 μL fresh LB medium and plate it on LB agar plate containing Ampicillin (100 μg/mL) and incubate overnight (∼14 h) at 37°C.
-
i.Pick the green colony from the agar plate for further study (Figure 4A).
-
a.
Figure 4.

Transformation of E. coli (DH5α) with a plasmid expressing GFP and co-culture with C. jejuni
(A) Schematic of the transformation procedure of chemically competent E. coli (DH5α) cells with p-Turbo-GFP-B plasmid. Plate images of positive transformant of E. coli (DH5α) cell harboring p-Turbo-GFP-B plasmid on LB agar plate. GFP expression by the recombinant E. coli (DH5α) cells harboring p-Turbo-GFP-B plasmid was visualized under Axio observer microscope equipped with an ApoTome module (Carl Zeiss) (Objective magnification: 60×; Scale bar: 5 μm).
(B) Experimental setup for assessing interbacterial interaction (C. jejuni vs. E. coli) in co-cultures. Each bacterium was mixed and co-incubated for 10 h, washed, and serially diluted to quantify and image the competing bacteria. The red circle on the culture plate shows a GFP-expressing E. coli colony, while the blue circle represents the C. jejuni colony. The CFU values can be obtained by counting the colonies on the plate. These data can be fitted with a mathematical model to extract important parameters for the interspecies interaction. For instance, we demonstrate a mathematical model fitting prey and predator cell densities (denoted by [Ec] and [Cj], respectively). This helps determine the birth rates of each bacteria (rEc and rCj) and the predation rate (α). To visualize the effect on prey bacteria (E. coli) in co-culture, cells were washed and processed for epifluorescence microscopy and observed under an Axio observer microscope equipped with an ApoTome module (Carl Zeiss) (Objective magnification: 60×; Scale bar: 5 μm). The red arrow indicates E. coli cells, and the white arrow indicates C. jejuni cells. To evaluate and observe the interbacterial interaction under stress (bile salt), cells were incubated upon MH agar plate containing bile salt, washed with PBS, treated with H2DCFDA for 45 min, followed by processed for epifluorescence microscopy and observed under Axio observer microscope equipped with an ApoTome module (Carl Zeiss) (Objective magnification: 60×; Scale bar: 5 μm). The red arrow indicates E. coli cells, and the white arrow indicates C. jejuni cells. Differential expression of competing bacterial genes can be analyzed by semi-quantitative RT-PCR. For example, to see the effect of T6SS functionality of C. jejuni on E. coli, transcriptional profiling of cytokinesis gene (ftsz) was analyzed (Lane 1: GFP-E. coli+ T6SS +ve C. jejuni; Lane 2: GFP-E. coli+ T6SS -ve C. jejuni; Lane 3: GFP-E. coli only).
-
11.
Epifluorescence imaging of GFP expression in E. coli.
To visualize the constitutive expression of GFP in E. coli, epifluorescence microscopy can be performed.-
a.Select the green colony only and resuspend it with 200 μL 1× PBS.
-
b.Centrifuge the resuspended cells at 6,000 × g for 5 min and discard the supernatant.Note: Repeat steps a–b twice.
-
c.Add and resuspend the cell pellet in 100 μL of 4% paraformaldehyde for fixation of the cells and incubate it for 20 min at room temperature (∼25°C).
-
d.Harvest the cells by centrifugation (6,000 × g; 5 min), wash it twice with 100 μL 1× PBS, and finally, resuspend it in 100 μL 1× PBS.
-
e.Take a 6 μL aliquot from the sample and make a smear on a grease-free glass slide.
-
f.Mount the coverslips using 5 μL Vecta-shield mounting media (Vector Laboratories, USA) for imaging.
-
g.To visualize GFP-expressing E. coli (DH5α), use a FITC filter with an excitation wavelength of 400–520 nm and emission wavelength of 480–580 nm (Figure 4A).Note: For our experimental purposes, image acquisition was made under an Axio observer microscope equipped with an ApoTome module (Carl Zeiss) at an objective magnification of 60×. Images were processed in Zen software.
-
a.
-
12.
Interbacterial Competition assay.
During the bacterial competition, one type of bacteria behaves like a predator and tries to eliminate other bacteria (prey) from the ecological niche. This assay can be useful to investigate such inter-bacterial interactions.-
a.From an overnight (∼16 h) grown culture, mix 1 mL GFP-E. coli culture (6×107 CFU/mL) and 1 mL T6SS+ve C. jejuni (6×108 CFU/mL) and add fresh MH broth to a total volume of 5 mL.
-
b.Incubate it at 37°C under micro-aerophilic conditions for 10 h.
-
c.After 10 h incubation, perform serial ten-fold dilution of the co-culture up to the dilution of 10-5.
-
d.Take a 50 μL aliquot from the last dilution and plate it on an MH agar plate containing X-gal (40 μg/mL) and IPTG (0.1 mM).
-
e.Incubate the plate at 37°C and in micro-aerophilic conditions for 16 h.
-
f.The appearance of the green and white colony on the agar surface is respective to E. coli and C. jejuni. Count the colonies to assess the inter-bacterial interaction. (Figure 4B).Note: We observed a significant reduction in prey (E. coli) cell count (CFU/mL) in the presence of T6SS+ve C. jejuni at 10 h (Figure 4B).
-
a.
-
13.
Cellular imaging of competing bacteria.
During inter-bacterial interaction, one bacteria act as prey while another one predator. Usually, due to predator pressure, prey bacteria get eliminated from the ecological niche. During the inter-bacterial interaction, morphological changes (such as cell blebbing, cell elongation) can be observed in prey bacteria. These morphological changes can be observed using an epifluorescence microscope in the ApoTome module.-
a.Co-culture GFP-E. coli and C. jejuni, as mentioned earlier section (Sub-step 12; a).
-
b.After 10 h, harvest 2 mL of the co-culture by centrifugation at 5,000 × g for 5 min.
-
c.Wash the cell pellet with 200 μL of 1× PBS, followed by centrifugation at 5,000 × g for 5 min to discard the supernatant. Repeat this step another two times.
-
d.Further, resuspend the cell pellet with 100 μL of 4% paraformaldehyde and incubate at room temperature (∼25°C) for 20 min.
-
e.Wash the cell pellet with 200 μL of 1× PBS, followed by centrifugation at 5,000 × g for 5 min to discard the supernatant. Repeat this step another two times.
-
f.Finally, resuspend the washed cells in 500 μL of 1× PBS.
-
g.Next, prepare the slide for epifluorescence microscopy as mentioned earlier (Sub-step 11; e–g).Note: For our experimental purposes, image acquisition was made under an Axio observer microscope equipped with an ApoTome module (Carl Zeiss) at an objective magnification of 60×. Images were processed in Zen software. To measure the bacterial cell length using Zen software, open the software and import the microscopic images of the bacteria. After that, in the ‘Apotome’ section, choose the best-fit option, and from the ‘Graphics’ section select the scale bar to include a scale bar. Next, select the ‘Line Drawing’ tool from the ‘Graphics’ section and draw a line from one end to another end of bacteria to get an approximate bacterial size.(Methods video S5: Zen software user method video to measure cell length of bacteria, related to step 13) (Figure 4B).
-
a.
-
14.
Semi-quantitative RT-PCR to check the gene expression.
Semi-quantitative PCR is a technique used to amplify and simultaneously quantify the gene expression at the transcript level of a targeted DNA. This assay aims to evaluate the gene expression of bacteria after inter-bacterial interaction.-
a.From an overnight grown culture (∼14 h), mix 1 mL GFP-E. coli culture (6 × 107 CFU/mL) with 1 mL T6SS+ve and 1 mL T6SS-ve C. jejuni (6 × 108 CFU/mL) and make the volume 5 mL by adding fresh MH broth. Incubate it at 37°C and in micro-aerophilic condition for 10 h.
-
b.Harvest the bacteria by centrifugation at 5,000 × g for 5 min in a 1.5 mL microcentrifuge tube.
-
c.Resuspend the cell pellet into 300 μL Trizol reagent and incubate it at room temperature (∼25°C) for 20–30 min.Note: After resuspension of bacterial cell pellet into Trizol reagent, the suspension can be stored at −80°C for 7–10 days.CRITICAL: TriZol is a hazardous chemical that can cause severe chemical burns and permanent scarring. This step should be performed in a fume hood with a lab coat and nitrile gloves.
-
d.Add 150 μL of molecular biology grade chloroform and shake it vigorously until the solution turns milky.
-
e.Keep the microcentrifuge tube straight and incubate at room temperature (∼25°C) for 30 min.(Set the centrifuge temperature 4°C).
-
f.Next, centrifuge the Trizol-chloroform mix at 10,000 × g for 25 min at 4°C.
-
g.Gently pipette out the upper yellow transparent layer and keep it in another 1.5 mL microcentrifuge tube.
-
h.Add equal volume molecular biology grade isopropanol and keep the solution at −20°C for 12 h.
-
i.After that, centrifuge the vial at 12,000 × g for 30 min at 4°C and discards the supernatant.
-
j.Resuspend the RNA pellet into 100 μL of 70% ethanol and centrifuge the suspension at 12,000 × g for 30 min again at 4°C.
-
k.Resuspend the RNA pellet into 50 μL of nuclease-free water.
-
l.Take the RNA concentration in nanodrop and take 1,000 μg of RNA for further experiment.
-
m.Prepare first-strand cDNA from the RNA sample using Bio-Bharati MuLV Super-transcriptase Kit (BioBharati Life science Pvt. Ltd., India) by following Manufacturers’ protocol (https://www.biobharati.com/product/super-rev-transcriptase-mulv-kit-super-rt-50-rxns-kit/).
-
n.Amplify the E. coli 16S rRNA gene from the cDNA by gene-specific primer set (Table 1) and PCR condition (Table 2) to normalize the cDNA quantity.Note: Use No-RT control (cDNA preparation without reverse transcriptase enzyme) to ensure no genomic DNA contamination in the sample.Note: To validate the PCR amplification of the gene of interest, genomic DNA can be used as a positive control, and a no-template PCR mixture can be used as a negative control. Run equal volume of the PCR product on 1% agarose gel. Quantify the band intensity using ImageLab software. To quantify amplified DNA band intensity during semi-quantitative RT-PCR, first, open the image lab software and import the gel image. Next, click on the ‘Lane and Band’ option. After that, click on the manual option and select the number of required lanes. Next, adjust the box such that it covers the entire band of interest and click on adjust the frame and resize the frame. After that, click on the detect bands, and after detection click on the ‘Report’ option to get the intensity profile of the respective bands. Now use the ‘Total Lane Volume of Intensity’ obtained from the report option to normalize and compare between bands of interest (Methods video S6: ImageLab software user method video to quantify band intensity during semi-quantitative PCR, related to step 14).
-
o.Amplify the ftsZ gene using an equal volume of DNA that has been used to amplify housekeeping gene (16S rRNA) amplification. Run equal volume of the PCR product on 1% agarose gel (Figure 4B).
-
p.Quantify the band intensity using ImageLab software (Methods video S6: ImageLab software user method video to quantify band intensity during semi-quantitative PCR, related to step 14) (Figure 4B).
-
a.
-
15.
Evaluation and observation of interbacterial interaction under stress (bile salt).
To assess the effect of intracellular bile salt accumulation during the interbacterial competition, the production of Reactive Oxygen Species (ROS) is visualized and quantified. For this, C. jejuni and E. coli are co-cultured in the absence or presence of bile salt (mixture of 50% sodium cholate and 50% sodium deoxycholate, w/w).-
a.After co-incubation for 2 h, dilute the culture up to 10-4 dilutions and inoculate 100 μL on an MH agar plate containing bile salt (0.1%, w/v).
-
b.Collect the colonies and resuspend them in 1× PBS to an OD600∼ 0.5.
-
c.Incubate the cells with 2, 7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (20 μM) for 45 min at 37°C.
-
d.Centrifuge the cell suspension at 8,000 × g for 2 min.
-
e.Resuspend the bacteria cell pellet in 1× PBS.
-
f.To measure the fluorescence intensity, take approximately 200 μL of aliquot for analysis using a multi-detection microplate reader with a 485 nm excitation filter and a 535 nm emission filter.Note: We used Spectramax M2e Multi Detection Microplate Readers (Molecular Devices LLC, USA) for our experimental purpose.
-
g.To visualize the ROS production, incubate the cells with H2DCFDA, as mentioned earlier (Sub-step 15; c). Further mount the 4 μL cell suspension onto a glass slide using 2 μL of Vecta-shield mounting media (Vector Laboratories, USA).
-
h.To observe ROS production in bacterial cells under bile salt-induced stress, epifluorescence microscopy under a FITC filter can be used (with an excitation wavelength of 400–520 nm and emission wavelength of 480–580 nm).Note: For our experimental purposes, image acquisition was made under an Axio observer microscope equipped with an ApoTome module (Carl Zeiss) at an objective magnification of 60×. Images were processed in Zen software (Figure 4B).
-
a.
Host-bacteria interaction
T6SS positive C. jejuni can invade the host cells and can promote pathogenesis (Lertpiriyapong et al., 2012). Thus, the host bacterial interaction between C. jejuni and its host is an important area to study. This detailed protocol can be followed to study the host-bacteria interaction.
-
16.In-vitro cell invasion assay (Gentamicin protection assay).CRITICAL: When giving infection, use an antibiotic-free cell culture medium to avoid bacterial death before even they enter the host cells.Note: To avoid cross-contamination to control cells (cells without C. jejuni infection), separate culture plates should be used.
-
a.Grow monolayer of human INT407 cells in 24-well tissue culture plate (3×105 cells/well).
-
b.Add C. jejuni at MOI of 300:1 when the cells become >70% confluent.
-
c.Incubate for 3 h at 5% CO2 and 37°C in DMEM supplemented with 10% FBS (Do not add any antibiotics at this stage).
-
d.After incubation, remove the culture medium and wash the cells with 300 μL/well of 1× PBS two times.
-
e.Next, treat the cells with 500 μL of gentamicin (150 μg/mL) for an additional 2 h.
-
f.After treatment with gentamicin, wash the cells with 300 μL/well of 1× PBS twice.
-
g.After that, add 200 μL/well of 1% Triton X-100 (Sigma-Aldrich) to lyse cells.
-
h.Transfer the suspension into a 1.5 mL centrifuge tube.
-
i.Serially dilute (10-fold) the suspension up to 10-3.
-
j.Perform a spread plate using 50 μL of the 10-3 diluted suspension on an MH agar plate.
-
k.Further, incubate the plate overnight (∼14 h) at 37°C under micro-aerophilic conditions.
-
l.Count the colonies that appeared on the plate (Figure 5A).
-
a.
-
17.Cellular imaging of invaded bacteria.
-
a.Grow monolayers of human INT407 cells in the presence of coverslips in the 6-well tissue culture plate (1.2×106/well).
-
b.Before infection, incubate C. jejuni cells for 1 h with DAPI (40 μg/mL) in the dark.
-
c.Add C. jejuni at MOI 300:1.
-
d.Incubate for 3 h at 5% CO2 and 37°C in DMEM (Invitrogen) supplemented with 10% FBS (Do not add any antibiotics at this stage).
-
e.Following incubation, remove the culture medium and wash the cells with 500 μL/well of 1× PBS.
-
f.After washing, treat the cells with 150 μg/mL of gentamicin (total volume maximum up to 500 μL/well) for another 2 h.
-
g.Wash the cells three times with 500 μL of 1× PBS.
-
h.Mount the coverslips using 5 μL Vecta-shield mounting media for fixing.
-
i.To visualize the invaded C. jejuni cells, use a DAPI filter with an excitation wavelength of 300–450 nm and an emission wavelength of 380–650 nm.
-
a.
Note: For our experimental purposes, we used the Leica confocal microscope for image acquisition at an objective magnification of 60× (Figure 5B).
-
18.
Semi-quantitative RT-PCR to check the hcp gene expression.
To examine whether the host responses were associated with the expression of the major T6SS gene, the transcriptional profile of hcp can be studied.-
a.Grow monolayer of human INT407 cells in 24-well tissue culture plate (3 × 105 cells/well).
-
b.Add C. jejuni at MOI 300:1 when the cells become 70% confluent.
-
c.Incubate for 5 h at 5% CO2 and 37°C in DMEM and supplemented with 10% FBS.
-
d.Following incubation, remove the culture media and wash the cells with 500 μL of 1× PBS three times.
-
e.Add 300 μL TrizoL reagent.CRITICAL: TriZol is a hazardous chemical that can cause severe chemical burns and permanent scarring. This step should be performed in a fume hood with a lab coat and nitrile gloves.
-
f.Do vigorous pipetting to detach the cells. Take the solution in a 1.5 mL micro-centrifuge tube at room temperature (∼25°C) for 20–30 min.
-
g.Isolate the RNA and prepare the cDNA as mentioned earlier (Sub-step 14; c–m).
-
h.Using PCR cycling conditions, amplify the C. jejuni 16S rRNA gene from the cDNA (Table 2).
-
i.Run equal volume of the PCR product on 1% agarose gel. Quantify the band intensity using ImageLab software (Methods video S6: ImageLab software user method video to quantify band intensity during semi-quantitative PCR, related to step 18).
-
j.Amplify hcp gene using equal volume cDNA. Run equal volume of the PCR product on 1% agarose gel.
-
k.Quantify the band intensity using ImageLab software (Methods video S6: ImageLab software user method video to quantify band intensity during semi-quantitative PCR, related to step 18) (Figure 5C).Note: To investigate the role of the major T6SS gene (like hcp) in modulating host invasion, semi-quantitative PCR of hcp gene can be performed. We observed the up-regulation of hcp gene of C. jejuni during semi-quantitative PCR.
-
a.
Figure 5.

A schematic of C. jejuni invasion of the host cell by gentamicin protection assay
The confluent monolayers of human INT407 cells co-incubated with T6SS+ve C. jejuni for 7 h. Next, cells were washed to remove the extracellular bacteria while adhered bacteria were killed by gentamicin treatment (150 μg/mL) for 2 h. The washed cell was either processed for imaging or lysed to count the intracellular bacteria. This module can also be used for analyzing the expression of core genes of functional T6SS, including hcp.
(A) Colonies that appeared on the MH agar plate can be utilized to count the number of invaded bacteria present in human INT407 cells.
(B) Representative images of intracellular C. jejuni (stained with DAPI) in human INT407 cells (Scale bar: 50 μm).
(C) Representative agarose gel image showing the hcp gene expression of T6SS+ve C. jejuni.
Expected outcomes
The protocol described herein is unambiguous and transparent for isolating and characterizing T6SS+ve C. jejuni from the primary host, such as chickens. Although we demonstrated the usefulness of our method by isolating highly virulent and T6SS armed C. jejuni, the protocol is also applicable for isolating C. jejuni from other environmental sources, such as water, soil, meat, milk, animal feeds, etc.
C. jejuni is a Gram-negative, helical-shaped, non-spore-forming, microaerophilic, non-fermenting motile bacterium with a single flagellum at one or both poles, which is also oxidase and catalase-positive and grow optimally at 37°C–42°C (Balaban and Hendrixson, 2011). It forms a milky-white, rough-edged, glistening colony on blood-free Campylobacter selective agar media. As C. jejuni is Gram-negative, it acquired the color of counterstain safranin (pink) and appeared pink under the light microscope during the Gram staining procedure. Motility assay resulted in the outspread of the bacterial spot from the center to the periphery (hallow formation) on the soft-agar surface, confirming the motility phenotype of C. jejuni. We have tested the bacteria for Catalase and Oxidase activity for biochemical characterization. As C. jejuni is catalase-positive, it produces O2 bubbles when added with H2O2. Catalase enzyme causes the breakdown of H2O2 into water and O2. Catalase activity of bacteria helps protect them from oxidative damage by reactive oxygen species (ROS).
The readout for oxidase assay is the formation of blue color in the presence of N, N, N′, N′-tetramethyl-p-phenylenediamine dihydrochloride reagent. Oxidase-positive bacteria possess cytochrome oxidase (in the case of C. jejuni) or indophenol oxidase, which catalyze the transport of electrons between electron donors compounds (NADH) present in the bacteria and a redox dye (electron acceptor), which in turn deduced it to a blue colored compound indophenol blue. Hence the formation of indophenol blue in the oxidase test indicates that C. jejuni is positive for oxidase activity.
C. jejuni can hydrolyze hippurate (a glycine conjugate of benzoic acid) to glycine and benzoate by the hippuricase enzyme. Hippurate hydrolysis assay thus can help distinguish C. jejuni from other Campylobacter spp. since the latter lacks the enzyme. The free amino groups (-NH2) of glycine react with ninhydrin (2, 2-Dihydroxyindane-1, 3-dione) to form the blue product.
To further identify C. jejuni as a T6SS+ve strain, molecular detection of major T6SS genes can be performed using PCR. We have amplified selected T6SS genes (vasC, vasD, vasK, hcp, vasE) to characterize C. jejuni isolate for our study.
Since Hcp is known as a major effector protein secreted by a functional T6SS of several gut pathogens (Zhou et al., 2012), the presence of Hcp in the bacterial culture supernatant can serve as a hallmark for T6SS functionality. We optimized the detection of Hcp in the bacterial culture supernatant probing with an anti-Hcp antibody while the amount of protein present in the medium was quantified by indirect ELISA. To obtain further insights into the hcp gene transcriptional profile upon host cell invasion, we performed RT-PCR to quantitate hcp gene expression (Singh and Mallick, 2019).
Given that functional T6SS targets prey bacteria and the host cell, we have established methods for direct visualization and quantification of interbacterial and host-bacterial interaction (Ringel et al., 2017). Though we used E. coli (DH5α) as a model prey and human intestinal cell line (INT407) as hosts for demonstration, other targets of T6SS can also be explored using this protocol. Under that, we observed T6SS+ve C. jejuni caused morphological changes (elongation) of prey (E. coli) and significantly eliminated the prey from the niche.
Finally, the above methods can also be applied to test how environmental factors (such as bile salt) modulate the T6SS functionality. Nevertheless, using this protocol, we can address open questions and test hypotheses on how environmental factors exacerbate or impair the process of bacterial pathogenesis and come up with effective therapeutic measures for improving gut health.
Limitations
Though our protocol is ideal for probing the predatory and pathogenic ability of natural isolates of T6SS-positive C. jejuni, any phenotypic changes exhibited by T6SS-negative C. jejuni should be compared with an isogenic mutant of T6SS genes. Furthermore, our protocol for observing inter-bacterial interaction crucially depends on the visual separation of the competing bacterial species. We used a GFP-expressing recombinant E. coli to investigate the inter-bacterial interaction; our protocol can be extended to visualize a multi-species interaction using different fluorescence labeling of bacterial species.
Troubleshooting
Problem 1
During the sample processing to isolate Campylobacter spp., many bacterial colonies are often formed after plating the cecum sample on Blood free Campylobacter selective media. This may create a problem in isolation of a single colony for further study (see Isolation of Campylobacter spp.; Sub-Section 1).
Potential solution
-
•
Allow solidifying of the agar plate completely before plating.
-
•
Dilute the fecal sample up to 10-4or more dilutions and inoculate a maximum volume of up to 100 μL.
Problem 2
Smear on bacteria observed under FESEM (see Identification of C. jejuni; sub-section 3-b).
Potential solution
-
•
Take the bacteria from the broth and wash with 1× PBS at least thrice.
Problem 3
During the hippurate hydrolysis test, negative control, as well as test samples, may have false-positive results (see Identification of C. jejuni; Sub-section 5-c).
Potential solution
If an organism possesses a hippuricase enzyme, it can hydrolyze hippuric acid to glycine and benzoic acid (Krieg and Padgett, 2011). In the presence of ninhydrin (oxidizing agent), glycine becomes deaminated (Bottom et al., 1978). Sufficiently wash the bacterial cells with 1× PBS, at least thrice. A trace amount of amino acid can produce false-positive results.
Problem 4
RNA contamination in genomic DNA (see characterization of C. jejuni; sub section 6-a).
Potential solution
-
•
RNA contamination may hinder the PCR reaction. To avoid this, add RNase A to the cell lysis buffer.
Problem 5
No amplification during PCR from genomic DNA (see characterization of C. jejuni section; sub section 6-b).
Potential solution
-
•
Use 300 ng–500 ng genomic DNA as a template.
-
•
RNA contamination may interfere with target gene amplification.
-
•
To amplify the target gene, the initial denaturation step should be at least for 3 min–5 min cycling.
Problem 6
Smear formation in Western Blot while TCA precipitated proteins are used (see functional characterization of T6SS+ve C. jejuni; sub-section 8-d).
Potential solution
-
•
Acetone treatment is essential after TCA precipitation, excluding residual TCA from the pellet. Residual TCA in the pellet can change the pH of the polyacrylamide gel, and a smear may appear.
-
•
Completely dry the residual acetone present in the pellet after acetone wash.
-
•
After adding 1× Laemmli buffer, boil the sample in a dry bath at 100°C for 3–5 min.
-
•
Use freshly prepared SDS-PAGE running buffer and check the pH.
Problem 7
During Western blot analysis, no corresponding band at the expected size or different size bands were observed (see functional characterization of T6SS+ve C. jejuni; sub-section 8-d).
Potential solution
-
•
A freshly prepared blocking buffer should be used.
-
•
Primary antibody dilution needs to be optimized; for this case, do not use less than 1:2,000 dilution.
Problem 8
Bacterial lawn formation after transformation of the p-turbo-GFP-B plasmid into E. coli (DH5α) cells (see interbacterial interaction; sub-section 10).
Potential solution
-
•
When adding antibiotics, cool down the media and mix properly. Uneven distribution of antibiotics in the molten LB agar may allow untransformed bacterial cells to overgrow the positive transformants due to lack of selection pressure and create a lawn. Insufficient dilution may create bacterial lawn formation. Do the pipetting carefully to make the bacterial dilution to avoid inappropriate mixing.
Problem 9
DNA contamination after RNA isolation (see interbacterial interaction; sub section 14).
Potential solution
-
•
Thaw the RNA sample on ice. After that, mix 2 μL of 10× DNase I reaction buffer, 1 unit of DNase I, and 1–2 μg of RNA. Then, make the final volume 20 μL with RNase-free H2O and incubate the sample for 30 min at 37°C. Now, inactivate the DNase by incubating for 5–10 min at 65°C–75°C.
Problem 10
How to confirm DNA contamination in the RNA sample (see interbacterial interaction; sub-section 14)?
Potential solution
-
•
No-RT (Reverse Transcriptase enzyme) control can be used as a confirmatory control to check possible DNA contamination in RNA samples. In No-RT control, no Reverse Transcriptase enzyme should be added during cDNA preparation. The gene of interest cannot be amplified from the cDNA sample if no DNA contamination is there.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Amirul I. Mallick (amallick@iiserkol.ac.in).
Materials availability
This study did not generate new unique reagents. Please contact Dr. Amirul I. Mallick (amallick@iiserkol.ac.in) to inquire about accessing other materials in this manuscript.
Acknowledgments
S.G., A.K., and K.M. thank UGC, MoE, Govt. of India, and P.B. thanks CSIR, MoST, Govt. of India for their fellowship support. A.I.M. and D.D. acknowledge IISER Kolkata for experimental facilities. We are grateful to Dr. Asish Mukhopadhaya, Principal Scientist, NICED (ICMR), Kolkata; and our former lab member, Dr. Ankita Singh, PhD, India, for their support in establishing C. jejuni culture in our laboratory. We thankfully acknowledge Dr. P.P. Datta for providing pTurbo-GFP-B plasmid and Dr. R. Das of IISER Kolkata for his helpful suggestions.
Author contributions
S.G. performed all experiments. A.K. assisted in phenotypic characterization. P.B. assisted in biochemical and molecular characterization. K.M. assisted in the sequence alignment and phylogenetic tree construction. A.I.M. and D.D. developed the concept and wrote the manuscript. S.S. contributed to the critical revision of the final version of the manuscript. The protocols for experimental works were supervised by A.I.M., while D.D. supervised quantitative works.
Declaration of interests
The authors declare no competing interests.
Institutional permissions
The chicken experimentation protocol was approved by the Institute Animal Ethics Committee (IAEC), Indian Institute of Science Education and Research Kolkata. All procedures were conducted in accordance with the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) guidelines, MoFAH&D, Govt. of India. The permit number of the experimental protocols approved by the IAEC was IISERK/IAEC/OA/2018/001.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101368.
Data and code availability
This study did not generate any new code. All data is available from the corresponding author upon reasonable request.
References
- Balaban M., Hendrixson D.R. Polar flagellar biosynthesis and a regulator of flagellar number influence spatial parameters of cell division in Campylobacter jejuni. PLoS Pathog. 2011;7:e1002420. doi: 10.1371/journal.ppat.1002420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottom C.B., Hanna S.S., Siehr D.J. Mechanism of the ninhydrin reaction. Biochem. Educ. 1978;6:4–5. doi: 10.1016/0307-4412(78)90153-X. [DOI] [Google Scholar]
- Bridier A., Sanchez-Vizuete P., Guilbaud M., Piard J.-C., Naïtali M., Briandet R. Biofilm-associated persistence of food-borne pathogens. Food Microbiol. 2015;45:167–178. doi: 10.1016/j.fm.2014.04.015. [DOI] [PubMed] [Google Scholar]
- Chen W.P., Kuo T.T. A simple and rapid method for the preparation of gram-negative bacterial genomic DNA. Nucleic Acids Res. 1993;21:2260. doi: 10.1093/nar/21.9.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coico R. Gram staining. Curr. Protoc. Microbiol. 2005;3:3C. doi: 10.1002/9780471729259.mca03cs00. [DOI] [PubMed] [Google Scholar]
- Gupta S., Ray S., Khan A., China A., Das D., Mallick A.I. The cost of bacterial predation via type VI secretion system leads to predator extinction under environmental stress. iScience. 2021;24:103507. doi: 10.1016/j.isci.2021.103507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg N.R., Padgett P.J. In: Methods in Microbiology, Taxonomy of Prokaryotes. Rainey F., Oren A., editors. Academic Press; 2011. 3 - phenotypic and physiological characterization methods; pp. 15–60. [DOI] [Google Scholar]
- Lertpiriyapong K., Gamazon E.R., Feng Y., Park D.S., Pang J., Botka G., Graffam M.E., Ge Z., Fox J.G. Campylobacter jejuni type VI secretion system: roles in adaptation to deoxycholic acid, host cell adherence, invasion, and in vivo colonization. PLoS ONE. 2012;7:e42842. doi: 10.1371/journal.pone.0042842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani K. Dr. Hans Christian Jaochim Gram: inventor of the Gram stain. Prim. Care Update OB/GYNS. 2003;10:235–237. doi: 10.1016/S1068-607X(03)00055-6. [DOI] [Google Scholar]
- Mah T.-F. Biofilm-specific antibiotic resistance. Future Microbiol. 2012;7:1061–1072. doi: 10.2217/fmb.12.76. [DOI] [PubMed] [Google Scholar]
- Peng Y., Wang X., Shou J., Zong B., Zhang Y., Tan J., Chen J., Hu L., Zhu Y., Chen H., Tan C. Roles of Hcp family proteins in the pathogenesis of the porcine extraintestinal pathogenic Escherichia coli type VI secretion system. Sci. Rep. 2016;6:26816. doi: 10.1038/srep26816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relucenti M., Familiari G., Donfrancesco O., Taurino M., Li X., Chen R., Artini M., Papa R., Selan L. Microscopy methods for biofilm imaging: focus on SEM and VP-SEM pros and cons. Biology. 2021;10:51. doi: 10.3390/biology10010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringel P.D., Hu D., Basler M. The role of type VI secretion system effectors in target cell lysis and subsequent horizontal gene transfer. Cell Rep. 2017;21:3927–3940. doi: 10.1016/j.celrep.2017.12.020. [DOI] [PubMed] [Google Scholar]
- Singh A., Mallick A.I. Role of putative virulence traits of Campylobacter jejuni in regulating differential host immune responses. J. Microbiol. 2019;57:298–309. doi: 10.1007/s12275-019-8165-0. [DOI] [PubMed] [Google Scholar]
- Tamura K., Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Tao J., Yu H., Ni J., Zeng L., Teng Q., Kim K.S., Zhao G.-P., Guo X., Yao Y. Hcp family proteins secreted via the type VI secretion system coordinately regulate Escherichia coli K1 interaction with human brain microvascular endothelial cells. Infect. Immun. 2012;80:1243–1251. doi: 10.1128/IAI.05994-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any new code. All data is available from the corresponding author upon reasonable request.