

Abstract
Background
Neuroinflammation, which is mainly mediated by excessive microglia activation, plays a major role in ischemic stroke. Overactivated microglia secrete numerous inflammatory cytokines, causing excessive inflammatory responses and ultimately exacerbating ischemic brain injury. Hence, compounds that attenuate neuroinflammation could become promising drug candidates for ischemic stroke. Fraxetin has an anti-inflammatory effect in many inflammatory diseases. However, whether it possesses an anti-inflammatory capacity in microglia-mediated neuroinflammation after ischemic brain injury is unknown. Our study aimed to investigate the suppression effect of fraxetin on neuroinflammation in lipopolysaccharide (LPS)-activated microglia and establish whether fraxetin could alleviate ischemic brain injury in a rodent model of ischemic stroke.
Methods
For the in vitro experiment, primary microglia were obtained from 1-day-old C57/BL6J mice. The cells were activated with LPS and treated with fraxetin at a non-cytotoxic concentration. Real-time PCR, enzyme-linked immunosorbent assays, and immunofluorescence staining were used to evaluate the anti-inflammatory effects of fraxetin. The potential molecular mechanisms were explored and verified through RNA-sequencing analysis, western blotting and real-time PCR. For the in vivo experiment, focal ischemia was induced by middle cerebral artery occlusion (MCAO) in 8-week-old male C57/BL6J mice. Fraxetin (5 mg/kg) or an equal volume of saline was injected into mice intraperitoneally after MCAO, and 2% 2,3,5-triphenyltetrazolium chloride staining was applied to measure infarct volume. Behavioral tests were conducted to measure neurological deficits in the mice. Real-time PCR, western blotting, and immunofluorescence staining were used to examine the expression of inflammatory cytokines and microglia activation in the ischemic penumbra.
Results
Fraxetin effectively inhibited the expression of proinflammatory cytokines including inducible nitric oxide synthase, tumor necrosis factor-α, interleukin-1 beta, and interleukin-6 in LPS-activated microglia. Fraxetin also suppressed the PI3K/Akt/NF-κB signaling pathway in activated microglia, which contributed to its anti-inflammatory effects. Furthermore, the administration of fraxetin attenuated ischemic brain injury and behavioral deficits after stroke. Finally, fraxetin was found to attenuate the activation of microglia both in vitro and in vivo.
Conclusions
Our results suggest that fraxetin has a suppression effect on microglia-mediated neuroinflammation, and this effect is associated with the PI3K/Akt/NF-κB signaling pathway. Fraxetin may therefore have potential neuroprotective properties for ischemic stroke.
Keywords: Stroke, fraxetin, microglia, neuroinflammation
Introduction
Stroke is widely acknowledged as one of the major causes of disability and mortality in the world. The most common type of stroke, ischemic stroke, accounts for approximately 85% of acute cerebral vascular diseases. Currently, there are only two efficacious treatments for ischemic stroke: thrombolysis and endovascular treatment. However, due to their narrow therapeutic window (1) and high risks of intracranial hemorrhage and brain edema (2,3), these treatments are extremely limited in the clinical setting. Therefore, a safer and more effective therapy for ischemic stroke, especially during the early phase, is urgently needed.
Neuroinflammation, which is mainly caused by the overactivation of resident microglia, plays an important role in the pathogenesis of ischemic stroke (4). Although microglia alleviate brain injury at the recovery stage, the inflammatory responses they prompt largely outweigh their beneficial effects at the early stage of ischemic stroke (5,6). Within hours after ischemia onset, activated microglia produce a plethora of proinflammatory mediators [e.g., inducible nitric oxide synthase (iNOS), tumor necrosis factor-α (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6)], chemokines [e.g., monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-2 (MIP-2)], and cytotoxic substances [e.g., reactive oxygen species (ROS) and nitric oxide (NO)] (7,8), which prompts an exaggerated inflammatory response, leading to the eventual destruction of neural cells and exacerbation of brain damage. Therefore, exploring the associated pathological mechanisms and further seeking neuroprotective compounds that can effectively inhibit microglia-mediated neuroinflammation might aid in the discovery of novel therapeutic agents for ischemic stroke.
Fraxetin (7,8-dihydroxy-6-methoxychromen2-one), a coumarin derivative purified from the traditional medicinal plant Fraxinus rhynchophylla, serves as the essential ingredient of many herbal and dietary supplements (Figure 1A) (9-11). Multiple studies have confirmed the many pharmacological activities of fraxetin, including its neuroprotective properties (12), anti-inflammatory activities (13), and ameliorative functions in type 2 diabetes (14) and tumors (15-17). Several studies have evidenced the anti-inflammatory properties of fraxetin in many inflammatory diseases, such as chronic pancreatitis (18), hepatocellular carcinoma (19), and osteoarthritis (20). However, the anti-inflammatory effects of fraxetin on microglia after cerebral ischemia and the mechanisms behind these effects remain elusive.
Figure 1.

Effects of fraxetin on the viability of primary microglia. (A) Chemical structure of fraxetin. (B) Primary microglia were treated with different concentrations of fraxetin (5, 10, 20, 30, 40, or 50 µM). After 24 hours, a CCK-8 assay was performed to detect the viability of the microglia. Control group n=6, other groups n=3. The values represent the mean ± SEM. *P<0.05, **P<0.01 compared with the control group. CCK-8, cell counting kit-8; SEM, standard error of the mean.
Our data showed that fraxetin might have anti-inflammatory effects and alleviate ischemic brain injury by attenuating microglia-mediated neuroinflammation. It was also found that the potential mechanism underlying the anti-inflammatory effect of fraxetin on LPS-activated microglia might be mediated by the PI3K/Akt/NF-κB signaling pathway. To our knowledge, this study is the first comprehensive research to provide novel insights into the therapeutic potential of fraxetin for the regulation of microglia-mediated neuroinflammation after ischemic brain injury. We present the following article in accordance with the ARRIVE reporting checklist (available at https://atm.amegroups.com/article/view/10.21037/atm-21-4636/rc).
Methods
Reagents
Fraxetin (CAS: 574-84-5, purity: ≥98%), purchased from Aladdin Ltd. (Shanghai, China), was dissolved in media containing 0.1% dimethyl sulfoxide (DMSO) for the in vitro experiments. For the in vivo experiments, fraxetin was dissolved in 0.9% saline with 5% DMSO. Lipopolysaccharide (LPS; Escherichia coli 055: B5) was bought from Sigma-Aldrich (St. Louis, MO, USA). The PI3K inhibitor LY294002 was purchased from MCE (Shanghai, China).
Cell culture
Primary microglia were obtained from 1-day-old C57/BL6J mice as previously described (21) and cultured in Dulbecco’s Modified Eagle Medium (Invitrogen, Frederick, MD, USA), which was supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA) and 100 U/mL antibiotics, at 37 ℃ in a humid atmosphere containing 5% CO2. After 10 days of culture, the flasks were shaken at 180 rpm for 10 minutes to separate the primary microglia, and the cells were replanted into new plates. The purity of the primary microglia was higher than 95%, as detected by Iba-1 staining (1:500, Wako, Japan).
Assessment of cell viability
Microglial viability was evaluated using cell counting kit-8 (CCK-8) (Dojindo Laboratories, Tokyo, Japan). Primary microglia were seeded into 96-well plates, and different doses of fraxetin were used to treat the cells. After 24 hours, the medium was removed and CCK-8 reagent was added to the 96-well plates at 10 μL per well. After 2-hour incubation, the optical density (OD) was detected at 450 nm using a microplate reader (Bio-Rad, Hercules, CA, USA). Cell survival rates were calculated as the mean percentage of the OD value of fraxetin-treated cells over that of cells without any treatment.
Drug treatment
For the in vitro experiment, the primary microglia were divided into five groups as follows: a control group, an LPS-treated group, a fraxetin-treated group, an LY294002-treated group, and an LY294002 + fraxetin-treated group. LPS (100 ng/mL) was administrated in all groups except the control group. Cells were treated with 10 μM of LY294002 for 30 minutes before the administration of fraxetin (10, 20, or 30 μM). After 2 hours, LPS was added, and the cells were cultured for a further 24 hours. For the in vivo experiment, fraxetin (5 mg/kg) or an equal volume of saline were randomly injected into mice intraperitoneally at 15 minutes, 24 hours, and 48 hours after middle cerebral artery occlusion (MCAO).
Nitrite analysis
After seeding microglia cells in 24-well plates, we pretreated primary microglia with fraxetin for 2 hours and then added LPS (100 ng/mL) for a further 24 hours. The nitrite concentration in the culture supernatant was evaluated using a Griess reaction kit (Beyotime Biotech, China). Absorbance was quantified at 540 nm using a microplate reader.
Middle Cerebral Artery Occlusion (MCAO) model establishment
Eight-week-old male C57/BL6J mice were purchased from the Animal Model Center of Nanjing Medical University (Nanjing, Jiangsu, China). All the mice weighed 20 to 25 grams. The mice were housed under temperature-controlled conditions with free access to standard water and food.
Experiments were performed under a project license (No. 2019AE01073) granted by the Animal Care and Use Committee at Nanjing University, in compliance with the institutional guidelines for the care and use of animals. The mice were randomly divided into a sham operation group and an MCAO model group. The MCAO model group comprised two subgroups: a saline-treated MCAO group and a 5 mg/kg fraxetin-treated MCAO group. During the operation, pentobarbital sodium (45 mg/kg i.p.) was used for anesthetization. The body of the mice temperature was kept at 37.0±0.5 ℃. After the internal carotid artery (ICA) and the middle cerebral artery (MCA) had been surgically exposed, a 6/0 surgical suture was inserted into the MCA through the ICA until the ipsilateral blood flow of the area supplied by the MCA decreased to below 30% of the baseline. The process was monitored using laser Doppler flowmetry (Perimed Corporation, Stockholm, Sweden). After 60 minutes of occlusion, the filament was withdrawn to allow blood reperfusion. The mice in the sham-operated group underwent all these operations except for the nylon insertion. The 3-day post-surgical mortality rate was about 10%.
Infarct size quantification
Three days after the MCAO procedure, the brains of the mice were removed and sliced into 1.0-mm-thick coronal slices. The samples were then immersed in 2% 2,3,5-Triphenyltetrazolium chloride (TTC, Sigma-Aldrich) at room temperature for 15 minutes for staining. The infarct volume of each slice was calculated and analyzed using ImageJ (NIH). The formula used for calculation was as follows: (volume of contralateral hemisphere − volume of ipsilateral normal hemisphere)/(2 × volume of contralateral hemisphere) × 100% (22).
Quantitative real-time PCR
Primary microglia were pretreated with fraxetin for 2 hours and then stimulated with LPS for 24 hours. 3 days after the MCAO procedure, mice were decapitated by cervical dislocation after anesthesia. TTC staining was used to define the area of cerebral infarction and penumbra. Then we dissected the penumbral tissue in cortex by operating apparatus such as smooth forceps and small sharp scissors. For mice in sham group, we dissected the tissue in the same area as that of penumbral tissue of MCAO mice. The total RNA of tissues and cells were isolated using TRIzol (Invitrogen). After the addition of chloroform to the TRIzol, the mixture was agitated for 15 seconds and centrifuged at 13,000 ×g at 4 ℃ for 30 minutes. The supernatant was then extracted and mixed with isopropanol for further centrifugation, after which the supernatant was removed again and 75% ethanol was added. Finally, after centrifugation once more, the supernatant was removed and RNase-free water was added. DNA wiper mix was added for the removal of genomic DNA, and a PrimeScript RT Reagent Kit (Vazyme, Nanjing, China) was used in line with the manufacturer’s instructions. Complementary DNA synthesis was also performed using the PrimeScript RT Reagent Kit according to the manufacturer’s instructions.
Real-time PCR (RT-PCR) was conducted on the Step One Plus PCR system (Applied Biosystems, Foster City, CA, USA) with 20 μL reaction mixture using a SYBR Green Kit (Applied Biosystems). The primer sequences used for the experiment were as follows:
TNF-α: forward, 5'- CAAGGGACAAGGCTGCCCCG-3', reverse, 5'-GCAGGGGCTCTTGACGGCAG-3'; IL-1β: forward, 5'- AAGCCTCGTGCTGTCGGACC-3', reverse, 5'-TGAGGCCCAAGGCCACAGGT-3'; IL-6: forward, 5'- GCTGGTGACAACCACGGCCT-3', reverse, 5'-AGCCTCCGACTTGTGAAGTGGT-3'; iNOS: forward, 5'-GTTTGACCAGGACCCAGA-3', reverse, 5'-GTGAGCTGGTAGGTTCCTGT-3'. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH): forward, 5'-GCCAAGGCTGTGGGCAAGGT-3', reverse, 5'-TCTCCAGGCGGCACGTCAGA-3'.
Enzyme-linked immunosorbent assay
Primary microglia were pretreated with fraxetin for 2 hours and then activated for 24 hours with LPS. The expression levels of the cytokines including TNF-α, IL-6, and IL-1β in the culture supernatant were measured using commercial enzyme-linked immunosorbent assay (ELISA) kits in accordance with the manufacturer’s instructions (Cusabio Biotech, Wuhan, China).
Western blotting
Cells and brain tissues from the ischemic penumbra were lysed in lysis buffer (Thermo Fisher Scientific, Rockford, IL, USA) for 30 minutes. Then, the lysates were centrifuged at 13,000 ×g at 4 ℃ for 30 minutes. The protein concentrations were quantified using a BSA (bovine serum albumin) kit (Beyotime) and diluted with a 5× loading buffer solution. After being separated by 10% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis), the samples were transferred to polyvinylidene fluoride membranes. The membranes were blocked with 5% skim milk for 2 hours at room temperature and then immersed in primary antibodies against IL-1β, IL-6, TNF-α, PI3K, p-PI3K, Akt, p-Akt, NF-κB p65, p-p65 (1:1,000, Cell Signaling Technology, Hertfordshire, UK), and β-actin (1:5,000, Bioworld Biotechnology, Minneapolis, MN, USA) at 4 ℃ overnight. Next, the membranes were immersed in secondary antibodies for an additional 2 hours at room temperature and then photographed using a Gel-Pro System (Tanon Technologies, China). Western blot quantification was conducted using ImageJ software (ImageJ 1.5, NIH, USA).
Immunofluorescence staining
Three days after the MCAO procedure, 0.9% saline and 4% paraformaldehyde were applied to perfuse the mice transcardially. Brain tissues were cut into 20-μm-thick sections following dehydration. Then, 0.1% Triton X-100 was used to permeabilize the brain sections or primary microglia for 10 minutes, and a 2% BSA blocking buffer was used to saturate the excess protein-binding sites for 90 minutes. The tissue sections and cell coverslips were incubated overnight at 4 ℃ with primary antibodies against Iba1 (1:500, Cambridge, UK). Subsequently, the brain sections and microglia were incubated with goat anti-rabbit secondary antibodies conjugated to Alexa594 (1:200; Abcam) for 2 hours at 37 ℃. The cell nuclei were stained using DAPI (4',6-diamidino-2-phenylindole, 5 g/mL) for 20 minutes. A fluorescence microscope (Olympus BX51, Japan) was used to acquire images.
Measurement of neurological deficits
Modified neurological severity score (mNSS) tests, comprising motor, sensory, and reflex tests, were conducted to measure neurological impairment at 1 and 3 days after MCAO. The scores ranged from 0 to 18 points, with a higher score representing more serious impairment.
The rotarod test was used to examine motor balance and coordination. Before the MCAO procedure, the mice underwent two trials a day on a rotarod device (RWD Life Science, Shenzhen, China) for 3 days. During trials, the mice were put on an accelerating rotarod (acceleration from 10 to 40 rpm for 5 minutes). At 1 and 3 days after MCAO, the length of time that each mouse was able to stay on the rotating beam was recorded along with the maximum speed.
Forelimb grip strength was evaluated using a grip strength meter (GS3, Bioseb, France) at 1 and 3 days after MCAO. During the test, the tail of the mouse was held, and the forelimbs were placed on a platform. The tail was then pulled back straight until the forelimbs released. The grip force on the screen was noted down to measure the maximum forelimb muscle strength.
RNA-sequencing analysis
Primary microglia were treated with DMSO or 30 μM fraxetin for 2 hours and then stimulated with LPS for 24 hours. Total RNA was extracted from the cells with TRIzol (Invitrogen). RNA-sequencing transcriptome analysis and downstream analysis were performed by Shanghai Majorbio Bio-pharm Technology Co., Ltd. Differentially downregulated genes were processed for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway significant enrichment analysis. Differentially expressed genes (DEGs) with a fold change of ≥1.5 and an adjusted P-value of <0.05 were picked out. Western blot was used to verify the candidate pathway which may be associated with the protective effect of fraxetin.
Statistical analysis
GraphPad Prism 8 was used to perform the statistical analysis. Data were expressed as the mean ± SEM. The Shapiro-Wilk test was used to test the normality assumption of the data. If data were normally distributed, Student’s t test was used to evaluate the statistical significance between two groups, while the Mann-Whitney test was applied for comparison of non-normally distributed variables. For multiple groups, statistical significance was analyzed by one-way analysis of variance (ANOVA) followed by either Bonferroni's post hoc test or the Kruskal-Wallis test followed by Dunn’s multiple comparison test. Differences were considered statistically significant at P<0.05.
Results
Effects of fraxetin on the viability of primary microglia
To examine the safety and feasibility of fraxetin, we tested its cytotoxicity in primary microglia. Primary microglia were exposed to different concentrations of fraxetin (0 to 50 μM) for 24 hours. As shown in Figure 1B, fraxetin at lower concentrations (5 to 30 μM) had no effect on microglial viability, whereas at higher concentrations (40 to 50 μM), it decreased microglial viability significantly (F(6, 17) =4.023; P=0.0109). This finding indicated that fraxetin is safe within the range of 5 to 30 μM, and the concentration of fraxetin was set as ≤30 μM for the subsequent experiments.
Fraxetin reduces proinflammatory cytokine production and attenuates the activation of LPS-stimulated microglia
As specific cytokines released by microglia (such as IL-1β, IL-6, TNF-α, and NO) induce inflammatory responses which exacerbate tissue injury (23), we next evaluated whether fraxetin can affect the LPS-induced release of proinflammatory factors.
First, primary microglia were pretreated with fraxetin for 2 hours and then activated with LPS for a further 24 hours. The results demonstrated that NO production, which was increased with LPS stimulation, was suppressed by fraxetin at specific concentrations (F(4, 16) =1420; P<0.0001) (Figure 2A). RT-PCR was then used to determine whether fraxetin has a suppression effect on iNOS mRNA expression. The mRNA expression of iNOS in microglia increased significantly after LPS stimulation (F(4, 17) =27.79; P<0.0001) (Figure 2B), but fraxetin suppressed this increase in a concentration-dependent manner (10, 20, and 30 μM). Next, we evaluated the messenger RNA (mRNA) levels of TNF-α, IL-1β, and IL-6 in the primary microglia. The results showed that the mRNA expression of TNF-α (F(4, 15) =12.88; P<0.0001), IL-1β (F(4, 10) =7.818; P=0.0040), and IL-6 (F(4, 10) =5.442; P=0.0137) exhibited significant decreases in fraxetin-treated microglia (Figure 2C-2E).
Figure 2.

Fraxetin reduces proinflammatory cytokine production and attenuates activation of LPS-stimulated microglia. (A) The concentration of NO in the supernatant was examined using a Griess assay. n=4–5 per group. (B) iNOS mRNA was quantified using RT-PCR. n=4–5 per group (C-E). TNF-α (C), IL-1β (D), and IL-6 (E) mRNA levels were quantified by RT-PCR. n=3–4 per group (F-H). The protein concentrations of TNF-α (F), IL-1β (G), and IL-6 (H) were measured using an ELISA kit. n=4–7 per group. The values represent the mean ± SEM. **P<0.01, ***P<0.001 and ****P<0.0001 compared with the control group. #P<0.05, ##P<0.01, ###P<0.001, ####P<0.0001 compared with groups only stimulated with LPS. (I) Microglia were treated with 30 µM fraxetin and then stained with DAPI and Iba-1. Scale bars: 30 µm. LPS, lipopolysaccharide; NO, nitric oxide; iNOS, inducible nitric oxide synthase; RT-PCR, real-time polymerase chain reaction; TNF, tumor necrosis factor; IL-1β, interleukin-1 beta; IL-6, interleukin-6; ELISA, enzyme-linked immunosorbent assay; DAPI, 4',6-diamidino-2-phenylindole.
The effects of fraxetin on TNF-α (F(4, 15) =321.4; P<0.0001), IL-1β (F(4, 21) =21.22; P<0.0001), and IL-6 (F(4, 15) =46.04; P<0.0001) at the protein level were then analyzed using an ELISA kit. The results consistently revealed a similar tendency in protein expression as that observed for mRNA (Figure 2F-2H).
Next, the effects of fraxetin on the morphological character of microglia were examined. LPS treatment obviously changed the morphology of primary microglia, which showed a larger cell body and shorter processes, whereas pretreatment with 30 μM fraxetin morphologically suppressed microglial activation (Figure 2I). The above findings suggested that fraxetin suppressed TNF-α, IL-1β, and IL-6 levels in LPS-activated primary microglia.
Fraxetin regulates PI3K/Akt/NF-κB signaling in LPS-activated microglia
Next, the potential molecular mechanisms underlying the anti-inflammatory effects of fraxetin on LPS-activated microglia were explored. RNA-sequencing analysis was performed, and the results revealed that 471 genes were upregulated and 307 were downregulated after fraxetin treatment (Figure 3A). KEGG functional enrichment analysis showed that the PI3K-Akt signaling pathway was downregulated after fraxetin treatment (Figure 3B). The PI3K-Akt signaling pathway has been reported to be strongly associated with microglia-mediated inflammation (24,25). Further, this pathway regulates the activation of NF-κB, which is critical in neuroinflammation (26). Therefore, Western blotting was subsequently performed to verify the effects of fraxetin on the PI3K/Akt/NF-κB pathway. Results showed that LPS induced the phosphorylation of Akt (F(4, 10) =19.43; P=0.0001), PI3K (F(4, 10) =6.488; P=0.0077) and NF-κB (F(4, 10) =19.39; P=0.0001) (Figure 3C,3D). Intriguingly, pretreatment with fraxetin suppressed the phosphorylation of these molecules in a dose-dependent manner.
Figure 3.

Fraxetin regulates the PI3K/Akt/NF-κB signaling pathway in LPS-activated microglia. (A) Volcano plot for LPS-activated primary microglia between the control and fraxetin-treated groups. DEGs are listed. n=3 per group. (B) Differentially downregulated genes were processed for functional enrichment analysis of the KEGG pathway. (C) Western blotting was used to detect the protein levels of p-PI3K, p-Akt, and p-p65, n=3 per group. (D) Western blot quantification. The values represent mean ± SEM. (E-G) TNF-α (E), IL-1β (F), and IL-6 (G) mRNAs were quantified by RT-PCR. n=4 per group. **P<0.01 and ****P<0.0001 vs. control group. #P<0.05, ##P<0.01, ###P<0.001, ####P<0.0001 vs. groups only treated with LPS. NS means no significant difference. LPS, lipopolysaccharide; DEG, differentially expressed gene; KEGG, Kyoto Encyclopedia of Genes and Genomes; SEM, standard error of the mean.
To further explore the relationship of the PI3K/Akt/NF-κB pathway with the anti-inflammatory effects of fraxetin, primary microglia were treated with 10 μM of the PI3K inhibitor LY294002. LY294002 treatment decreased the mRNA levels of TNF-α (F(4, 10) =25.42; P<0.0001), IL-1β (F(4, 10) =20.04; P<0.0001), and IL-6 (F(4, 10) =30.18; P<0.0001) in LPS-activated primary microglia (Figure 3E-3G), and cotreatment with LY294002 and fraxetin did not enhance the anti-inflammatory effect. These data suggested that fraxetin attenuated the expression of proinflammatory cytokines via the PI3K/Akt/NF-κB signaling pathway.
To sum up, the above results indicated that fraxetin inhibited the activation of the PI3K/Akt/NF-κB signaling pathway in LPS-activated microglia, which might partly contribute to its anti-inflammatory effect.
Fraxetin alleviates ischemic brain injury in mice
Since fraxetin had exhibited anti-inflammatory effects on LPS-activated microglia, we next investigated whether fraxetin could protect mice against ischemic brain injury. Neurological scores and brain infarct volumes were evaluated 3 days after MCAO. TTC staining showed that fraxetin administration significantly decreased the infarct volume compared with the MCAO saline group (Figure 4A,4B). Additionally, a series of behavioral tests (mNSS, grip strength test, and a rotarod test) were used to assess neurological deficits in MCAO mice. Consistent with previous results, fraxetin treatment significantly ameliorated the behavioral deficit after MCAO, as evidenced by stronger grip strength (t(20) =3.055; P=0.0062), improved motor function (t(20) =2.503; P=0.0211), and lower scores in the mNSS test (t(20) =2.330; P=0.0304) (Figure 4C-4E).
Figure 4.
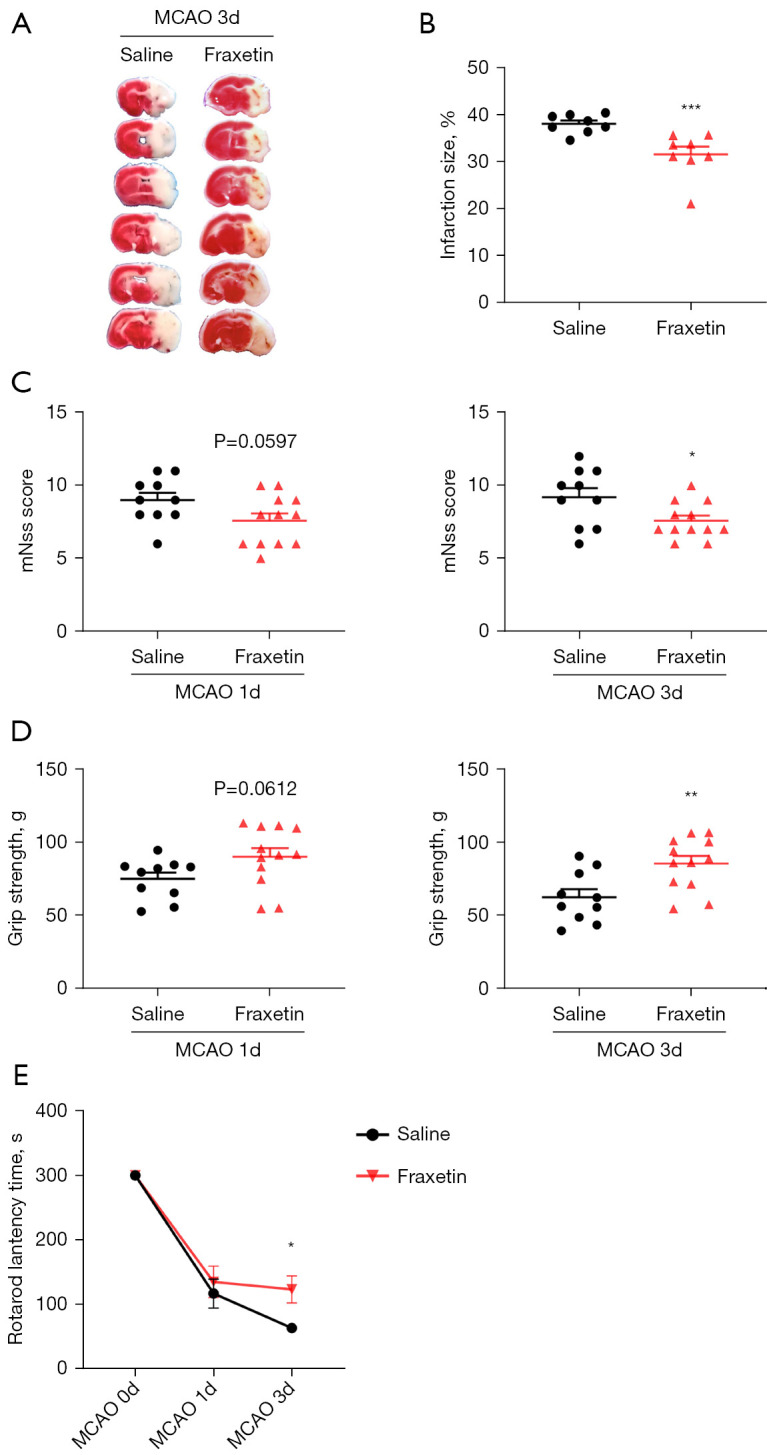
Fraxetin alleviates ischemic brain injury in mice. (A) Representative TTC staining images at day 3 after MCAO; (B) quantification of TTC staining (n=8); (C-E) mNSS score (C), grip strength (D), and a rotarod test (E) were used to detect neurological deficits after MCAO. Saline group n=10, fraxetin group n=12. The values represent mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 vs. MCAO-saline group. TTC, 2,3,5-triphenyltetrazolium chloride; MCAO, middle cerebral artery occlusion; mNSS, modified neurological severity score; SEM, standard error of the mean.
The above results demonstrated that fraxetin attenuated ischemic brain injury and improved neurological deficits of MCAO mice.
Fraxetin attenuates proinflammatory cytokine expression and microglial activation after MCAO
To verify the protective effects of fraxetin on microglia-mediated neuroinflammation after brain ischemia, we evaluated proinflammatory cytokine production in the penumbra tissues of mice after MCAO. At 3 days after the MCAO procedure, increased mRNA levels of proinflammatory cytokines (TNF-α, IL-6, and IL-1β) were observed. However, the administration of fraxetin significantly decreased the expression of MCAO-induced TNF-α (F(2, 9) =6.858; P=0.0155), IL-1β (F(2, 11) =6.666; P=0.0127) and IL-6 (F(2, 6) =10.80; P=0.0103) mRNA expression (Figure 5A-5C). Also, western blotting was used to detect the protein levels of TNF-α (F(2, 6) =21.22; P=0.0019), IL-1β (F(2, 6) =9.224; P=0.0148), and IL-6 (F(2, 6) =5.442; P<0.0001), and the expression patterns were found to be similar to those observed at the mRNA level (Figure 5D,5E).
Figure 5.

Fraxetin attenuated proinflammatory cytokine expression and microglial activation after MCAO. (A-C) TNF-α (A), IL-1β (B), and IL-6 (C) mRNAs of the penumbra tissues were quantified using RT-PCR. n=3–6 per group. (D-E) TNF-α, IL-6, and IL-1β proteins in the penumbra tissues were evaluated using western blotting (D) and quantified (E). n=3 per group. (F) Representative images of tissue sections from the ischemic penumbra were collected 3 days after MCAO, and stained with Iba1 and DAPI. The inset on the right side shows a digitally magnified field of cells from the image. Scale bars: 100 and 40 µm. The values represent mean ± SEM. *P<0.05, **P<0.01 and ****P<0.0001 vs. sham group. #P<0.05, ####P<0.0001 vs. MCAO-saline groups. MCAO, middle cerebral artery occlusion; TNF, tumor necrosis factor; IL-1β, interleukin-1 beta; IL-6, interleukin-6; RT-PCR, real-time polymerase chain reaction; DAPI, 4',6-diamidino-2-phenylindole; SEM, standard error of the mean.
We also used immunofluorescence staining to evaluate the activation state of microglia in the penumbra (Figure 5F). The activation of microglia (Iba1+) induced by ischemic brain injury was reversed by fraxetin treatment. These results indicated that fraxetin inhibited the activation and inflammatory response of microglia in penumbra after ischemic stroke.
Discussion
In the current study, we showed that fraxetin suppressed excessive inflammatory cytokine production induced by LPS in microglia and that its inhibitory effects were dose-dependent, thus demonstrating that this compound has protective and anti-inflammatory effects. We also observed that fraxetin convincingly inhibited PI3K/Akt/NF-κB signaling pathway activation in LPS-activated microglia. Accordingly, in our in vivo experiment, fraxetin significantly alleviated ischemic brain injury and exerted an anti-inflammatory effect by decreasing the production of proinflammatory factors, such as IL-6 and IL-1β, in the early phase of ischemic stroke in mice. Collectively, these results demonstrate that fraxetin can mitigate ischemic brain injury by suppressing microglia-mediated neuroinflammation, which indicates its potential as a therapeutic compound to treat ischemic stroke.
Neuroinflammation caused by microglial overactivation plays an important role in the pathogenetic process following ischemic brain injury (27). Several studies have demonstrated that LPS can bind to the CD14 receptors on microglial membranes, leading to microglial activation, inducing neuroinflammation, producing neurotoxic substances, and ultimately damaging the brain (28,29). LPS is therefore widely used to activate microglia cultured in vitro, which can mimic microglia-mediated neuroinflammation in vivo (30-32). In our study, primary microglia treated with LPS were used as an in vitro model to examine the anti-inflammatory effects of fraxetin and the underlying molecular mechanisms. In accordance with previous studies (33,34), after being stimulated with LPS, proinflammatory microglia were excessively activated, and secreted inflammatory mediators and oxidative metabolites, including iNOS, TNF-α, IL-1β, IL-6, and NO. We found that fraxetin significantly reduced LPS-induced inflammatory cytokine secretion in primary microglia in a concentration-dependent manner (Figure 2). Thus, we can conclude that the neuroprotective role of fraxetin may originate from its anti-inflammatory effect on microglia.
The PI3K/Akt/NF-κB pathway involved in inflammation mediation has been extensively studied in cerebral ischemia. A growing number of studies have suggested that activated PI3K/Akt phosphorylates various downstream factors, including NF-κB, mTOR, and STAT3, triggering an inflammatory response in ischemia-reperfusion injuries (35-37). A recent study demonstrated that the triggering receptor expressed on myeloid cells 2 (TREM2) attenuates LPS-induced inflammatory responses in BV2 cells by regulating PI3K/Akt signaling (38). Treatments that inhibit this pathway may therefore offer effective prevention of microglial inflammation. In our study, we confirmed that the PI3K/Akt pathway was phosphorylated after LPS stimulation, and that fraxetin attenuated the = PI3K/Akt activation (Figure 3), which suggests that the PI3K/Akt signaling pathway is involved in the anti-inflammatory effects of fraxetin on LPS-activated microglia. Also, previous studies showed that PI3K/Akt mediates microglial inflammation via NF-κB. β-HIVS attenuates the expression of the proinflammatory mediators in LPS-activated BV2 cells by suppressing the PI3K/Akt-dependent NF-κB pathway (39). Trans-isoferulic acid inhibits the same pathway and plays a role in anti-inflammation (37). As the main downstream molecular target of PI3K/Akt, NF-κB is a common nuclear transcription factor and contributes to the production of proinflammatory cytokines (40). LPS stimulation causes the phosphorylation of NF-κB p65, which upregulates the expression of proinflammatory cytokines such as iNOS (41). A study by Zhang et al. showed that NF-κB signaling modulates microglia-mediated neuroinflammation (42). Kaempferol has also been documented to suppress neuroinflammation via the NF-κB signaling pathway (43). In our study, we consistently found that phosphorylation of NF–κB p65 was upregulated in LPS-stimulated microglia, while fraxetin reversed this increase (Figure 3). Also, to understand the possible mechanisms underlying the anti-inflammatory capacity of fraxetin, LY294002, a highly selective inhibitor of PI3K, was used to further verify the effects of fraxetin on the PI3K/Akt/NF-κB pathway during the inflammatory process. Our results showed that the combination of LY294002 and fraxetin had the same anti-inflammatory effects as treatment with LY294002 alone (Figure 3). Collectively, our findings confirmed that pretreatment with fraxetin suppressed inflammatory responses in LPS-activated microglia by modulating the PI3K/Akt/NF-κB pathway.
To date, fraxetin has attracted much attention for its anti-tumor (44), anti-inflammatory (35), antibacterial (45), and radical-scavenging (46) properties in different pathological processes. Despite the protective effects of fraxetin in many diseases, few studies have reported its effects in cerebro-cardiovascular diseases. Thuong et al. reported that fraxetin directly protected against low-density lipoprotein oxidation and inhibited vascular proliferation via Nrf2/ARE activation in atherosclerosis (47). In our study, we found that fraxetin treatment led to reduced infarct volume and attenuated neurological deficits in mice after cerebral ischemia (Figure 4). Notably, microglial activation and proinflammatory cytokine expression were significantly reduced in the ischemic penumbra of mice treated with fraxetin at 3 days after MCAO (Figure 5), indicating that fraxetin could exert a neuroprotective effect by suppressing inflammatory responses after ischemia.
However, to avoid the excessive use of mice in this study, only a limited number of experimental animals were subjected to ischemic brain injury, which may have led to potential experimental errors. The recent studies has indicated that the polarization of microglia may be a therapeutic targets for focal cerebral ischemia (21,48,49). Microglia, serving as resident immune cells in CNS, undergo pro-inflammatory phenotype (M1) or anti-inflammatory phenotype (M2) in response to the microenvironmental changes after cerebral ischemia (50). Although the differentiation of M1 and M2 microglia is certainly oversimplified, changing the activation state of microglia appears to be an intriguing therapeutic strategy for cerebral ischemia. Growing evidence has revealed that microglia predominantly switch to M1 phenotype from 24 to 72 h after MCAO, which produces pro-inflammatory cytokines, such as IL-1b, TNF-α and INF-γ, and amplifies ischemic injury in acute phase after ischemic stroke (6,21). In the present study, we aimed to explore the effect of fraxetin on suppressing neuroinflammation mediated by proinflammatory microglia. Consistent with the previous studies, we detected a significant activation of microglia and production of pro-inflammatory cytokines simultaneously in the ischemic hemisphere 3 d after MCAO (Figure 5). Meanwhile, fraxetin reversed microglia activation and decreased pro-inflammatory cytokines production, which might indicate an attenuated proinflammatory microglia activation (Figure 5). However, whether fraxetin can directly inhibit the overactivation of M1 microglia and switch them to the M2 phenotype for protection will be explored in our further research, which can make our study more meaningful.
Our data provide strong evidence that fraxetin can mitigate inflammatory responses in microglia and alleviate ischemic brain injury. Uncovering the signaling pathways regulated by fraxetin might provide new insights into therapeutic targets for ischemic stroke.
Conclusions
Overall, our results show that fraxetin is a promising compound for attenuating microglia-mediated inflammation in acute ischemic stroke. Furthermore, since fraxetin is a clinically approved traditional Chinese medicine, it may hold great promise for stroke therapeutics.
Supplementary
The article’s supplementary files as
Acknowledgments
The authors sincerely thank Jennifer Reynolds, Lisa-Jane Roberts, Min Sun and Hui-Ya Li for providing language help.
Funding: This research was supported by the National Natural Science Foundation of China (81630028, 81920108017, and 81701168), the Key Research and Development Program of Jiangsu Province of China (BE2020620), and Jiangsu Province Key Medical Discipline (ZDXKA2016020).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Experiments were performed under a project license (No. 2019AE01073) granted by the Animal Care and Use Committee at Nanjing University, in compliance with the institutional guidelines for the care and use of animals.
Footnotes
Reporting Checklist: The authors have completed the ARRIVE reporting checklist. Available at https://atm.amegroups.com/article/view/10.21037/atm-21-4636/rc
Data Sharing Statement: Available at https://atm.amegroups.com/article/view/10.21037/atm-21-4636/dss
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://atm.amegroups.com/article/view/10.21037/atm-21-4636/coif). The authors have no conflicts of interest to declare.
References
- 1.Berkhemer OA, Fransen PS, Beumer D, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med 2015;372:11-20. 10.1056/NEJMoa1411587 [DOI] [PubMed] [Google Scholar]
- 2.Bai J, Lyden PD. Revisiting cerebral postischemic reperfusion injury: new insights in understanding reperfusion failure, hemorrhage, and edema. Int J Stroke 2015;10:143-52. 10.1111/ijs.12434 [DOI] [PubMed] [Google Scholar]
- 3.Khatri R, McKinney AM, Swenson B, et al. Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology 2012;79:S52-7. 10.1212/WNL.0b013e3182697e70 [DOI] [PubMed] [Google Scholar]
- 4.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 2010;87:779-89. 10.1189/jlb.1109766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics 2010;7:378-91. 10.1016/j.nurt.2010.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu X, Li P, Guo Y, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012;43:3063-70. 10.1161/STROKEAHA.112.659656 [DOI] [PubMed] [Google Scholar]
- 7.Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol 2006;84:49-59. 10.1139/Y05-143 [DOI] [PubMed] [Google Scholar]
- 8.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 2009;4:399-418. 10.1007/s11481-009-9164-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaur M, Kohli S, Sandhu S, et al. Coumarin: a promising scaffold for anticancer agents. Anticancer Agents Med Chem 2015;15:1032-48. 10.2174/1871520615666150101125503 [DOI] [PubMed] [Google Scholar]
- 10.Kundu J, Chae IG, Chun KS. Fraxetin Induces Heme Oxygenase-1 Expression by Activation of Akt/Nrf2 or AMP-activated Protein Kinase α/Nrf2 Pathway in HaCaT Cells. J Cancer Prev 2016;21:135-43. 10.15430/JCP.2016.21.3.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin C, Zhou LQ, Ma XT, et al. Dual Functions of Microglia in Ischemic Stroke. Neurosci Bull 2019;35:921-33. 10.1007/s12264-019-00388-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molina-Jiménez MF, Sánchez-Reus MI, Cascales M, et al. Effect of fraxetin on antioxidant defense and stress proteins in human neuroblastoma cell model of rotenone neurotoxicity. Comparative study with myricetin and N-acetylcysteine. Toxicol Appl Pharmacol 2005;209:214-25. 10.1016/j.taap.2005.04.009 [DOI] [PubMed] [Google Scholar]
- 13.Witaicenis A, Seito LN, da Silveira Chagas A, et al. Antioxidant and intestinal anti-inflammatory effects of plant-derived coumarin derivatives. Phytomedicine 2014;21:240-6. 10.1016/j.phymed.2013.09.001 [DOI] [PubMed] [Google Scholar]
- 14.Yao Y, Zhao X, Xin J, et al. Coumarins improved type 2 diabetes induced by high-fat diet and streptozotocin in mice via antioxidation. Can J Physiol Pharmacol 2018;96:765-71. 10.1139/cjpp-2017-0612 [DOI] [PubMed] [Google Scholar]
- 15.Kimura Y, Sumiyoshi M. Antitumor and antimetastatic actions of dihydroxycoumarins (esculetin or fraxetin) through the inhibition of M2 macrophage differentiation in tumor-associated macrophages and/or G1 arrest in tumor cells. Eur J Pharmacol 2015;746:115-25. 10.1016/j.ejphar.2014.10.048 [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Wang L, Deng Y, et al. Fraxetin Suppresses Proliferation of Non-Small-Cell Lung Cancer Cells via Preventing Activation of Signal Transducer and Activator of Transcription 3. Tohoku J Exp Med 2019;248:3-12. 10.1620/tjem.248.3 [DOI] [PubMed] [Google Scholar]
- 17.Sánchez-Reus MI, Peinado II, Molina-Jiménez MF, et al. Fraxetin prevents rotenone-induced apoptosis by induction of endogenous glutathione in human neuroblastoma cells. Neurosci Res 2005;53:48-56. 10.1016/j.neures.2005.05.009 [DOI] [PubMed] [Google Scholar]
- 18.Balaha M, Ahmed N, Geddawy A, et al. Fraxetin prevented sodium fluoride-induced chronic pancreatitis in rats: Role of anti-inflammatory, antioxidant, antifibrotic and anti-apoptotic activities. Int Immunopharmacol 2021;93:107372. 10.1016/j.intimp.2021.107372 [DOI] [PubMed] [Google Scholar]
- 19.Song J, Ham J, Hong T, et al. Fraxetin Suppresses Cell Proliferation and Induces Apoptosis through Mitochondria Dysfunction in Human Hepatocellular Carcinoma Cell Lines Huh7 and Hep3B. Pharmaceutics 2021;13:112. 10.3390/pharmaceutics13010112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q, Zhuang D, Feng W, et al. Fraxetin inhibits interleukin-1β-induced apoptosis, inflammation, and matrix degradation in chondrocytes and protects rat cartilage in vivo. Saudi Pharm J 2020;28:1499-506. 10.1016/j.jsps.2020.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng HL, Li XX, Chen YT, et al. Neuronal Soluble Fas Ligand Drives M1-Microglia Polarization after Cerebral Ischemia. CNS Neurosci Ther 2016;22:771-81. 10.1111/cns.12575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Guo M, Liu Y, et al. RNPS1 inhibition aggravates ischemic brain injury and promotes neuronal death. Biochem Biophys Res Commun 2020;523:39-45. 10.1016/j.bbrc.2019.11.185 [DOI] [PubMed] [Google Scholar]
- 23.Qin Z, Zhang B, Yang J, et al. The Efflux Mechanism of Fraxetin-O-Glucuronides in UGT1A9-Transfected HeLa Cells: Identification of Multidrug Resistance-Associated Proteins 3 and 4 (MRP3/4) as the Important Contributors. Front Pharmacol 2019;10:496. 10.3389/fphar.2019.00496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong L, Li YZ, An HT, et al. The E3 Ubiquitin Ligase c-Cbl Inhibits Microglia-Mediated CNS Inflammation by Regulating PI3K/Akt/NF-κB Pathway. CNS Neurosci Ther 2016;22:661-9. 10.1111/cns.12557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harikrishnan H, Jantan I, Haque MA, et al. Anti-Inflammatory Effects of Hypophyllanthin and Niranthin Through Downregulation of NF-κB/MAPKs/PI3K-Akt Signaling Pathways. Inflammation 2018;41:984-95. 10.1007/s10753-018-0752-4 [DOI] [PubMed] [Google Scholar]
- 26.Sochocka M, Diniz BS, Leszek J. Inflammatory Response in the CNS: Friend or Foe? Mol Neurobiol 2017;54:8071-89. 10.1007/s12035-016-0297-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol 2007;184:53-68. 10.1016/j.jneuroim.2006.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martins IJ. Overnutrition Determines LPS Regulation of Mycotoxin Induced Neurotoxicity in Neurodegenerative Diseases. Int J Mol Sci 2015;16:29554-73. 10.3390/ijms161226190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qin L, Wu X, Block ML, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007;55:453-62. 10.1002/glia.20467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao X, Jin Y, Zhang H, et al. The Anti-inflammatory Effects of 4-((5-Bromo-3-chloro-2-hydroxybenzyl) amino)-2-hydroxybenzoic Acid in Lipopolysaccharide-Activated Primary Microglial Cells. Inflammation 2018;41:530-40. 10.1007/s10753-017-0709-z [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Deng S, Zhang Z, et al. 6-Gingerol attenuates microglia-mediated neuroinflammation and ischemic brain injuries through Akt-mTOR-STAT3 signaling pathway. Eur J Pharmacol 2020;883:173294. 10.1016/j.ejphar.2020.173294 [DOI] [PubMed] [Google Scholar]
- 32.Lv Z, Liu C, Zhai M, et al. LPS Pretreatment Attenuates Cerebral Ischaemia/Reperfusion Injury by Inhibiting Inflammation and Apoptosis. Cell Physiol Biochem 2018;45:2246-56. 10.1159/000488170 [DOI] [PubMed] [Google Scholar]
- 33.Meng T, Fu S, He D, et al. Evodiamine Inhibits Lipopolysaccharide (LPS)-Induced Inflammation in BV-2 Cells via Regulating AKT/Nrf2-HO-1/NF-κB Signaling Axis. Cell Mol Neurobiol 2021;41:115-27. 10.1007/s10571-020-00839-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han L, Yin K, Zhang S, et al. Dalesconols B inhibits lipopolysaccharide induced inflammation and suppresses NF-κB and p38/JNK activation in microglial cells. Neurochem Int 2013;62:913-21. 10.1016/j.neuint.2013.03.003 [DOI] [PubMed] [Google Scholar]
- 35.Chen G, Liu J, Jiang L, et al. Galangin Reduces the Loss of Dopaminergic Neurons in an LPS-Evoked Model of Parkinson's Disease in Rats. Int J Mol Sci 2017;19:12. 10.3390/ijms19010012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong J, Qiu X, Yu Q, et al. A novel polysaccharide from Acorus tatarinowii protects against LPS-induced neuroinflammation and neurotoxicity by inhibiting TLR4-mediated MyD88/NF-κB and PI3K/Akt signaling pathways. Int J Biol Macromol 2020;163:464-75. 10.1016/j.ijbiomac.2020.06.266 [DOI] [PubMed] [Google Scholar]
- 37.Dilshara MG, Lee KT, Jayasooriya RG, et al. Downregulation of NO and PGE2 in LPS-stimulated BV2 microglial cells by trans-isoferulic acid via suppression of PI3K/Akt-dependent NF-κB and activation of Nrf2-mediated HO-1. Int Immunopharmacol 2014;18:203-11. 10.1016/j.intimp.2013.11.020 [DOI] [PubMed] [Google Scholar]
- 38.Li C, Zhao B, Lin C, et al. TREM2 inhibits inflammatory responses in mouse microglia by suppressing the PI3K/NF-κB signaling. Cell Biol Int 2019;43:360-72. 10.1002/cbin.10975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jayasooriya RG, Lee KT, Lee HJ, et al. Anti-inflammatory effects of β-hydroxyisovalerylshikonin in BV2 microglia are mediated through suppression of the PI3K/Akt/NF-kB pathway and activation of the Nrf2/HO-1 pathway. Food Chem Toxicol 2014;65:82-9. 10.1016/j.fct.2013.12.011 [DOI] [PubMed] [Google Scholar]
- 40.Genovese T, Melani A, Esposito E, et al. The selective adenosine A2A receptor agonist CGS 21680 reduces JNK MAPK activation in oligodendrocytes in injured spinal cord. Shock 2009;32:578-85. 10.1097/SHK.0b013e3181a20792 [DOI] [PubMed] [Google Scholar]
- 41.Wu PS, Ding HY, Yen JH, et al. Anti-inflammatory Activity of 8-Hydroxydaidzein in LPS-Stimulated BV2 Microglial Cells via Activation of Nrf2-Antioxidant and Attenuation of Akt/NF-κB-Inflammatory Signaling Pathways, as Well As Inhibition of COX-2 Activity. J Agric Food Chem 2018;66:5790-801. 10.1021/acs.jafc.8b00437 [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Gao W, Yang K, et al. Salt-Inducible Kinase 1 (SIK1) is Induced by Alcohol and Suppresses Microglia Inflammation via NF-κB Signaling. Cell Physiol Biochem 2018;47:1411-21. 10.1159/000490831 [DOI] [PubMed] [Google Scholar]
- 43.Liu Z, Yao X, Sun B, et al. Pretreatment with kaempferol attenuates microglia-mediate neuroinflammation by inhibiting MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. Free Radic Biol Med 2021;168:142-54. 10.1016/j.freeradbiomed.2021.03.037 [DOI] [PubMed] [Google Scholar]
- 44.Ren S, Xing Y, Wang C, et al. Fraxetin inhibits the growth of colon adenocarcinoma cells via the Janus kinase 2/signal transducer and activator of transcription 3 signalling pathway. Int J Biochem Cell Biol 2020;125:105777. 10.1016/j.biocel.2020.105777 [DOI] [PubMed] [Google Scholar]
- 45.Wang H, Zou D, Xie K, et al. Antibacterial mechanism of fraxetin against Staphylococcus aureus. Mol Med Rep 2014;10:2341-5. 10.3892/mmr.2014.2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, Ying X, Sun W, et al. The therapeutic effect of fraxetin on ethanol-induced hepatic fibrosis by enhancing ethanol metabolism, inhibiting oxidative stress and modulating inflammatory mediators in rats. Int Immunopharmacol 2018;56:98-104. 10.1016/j.intimp.2018.01.027 [DOI] [PubMed] [Google Scholar]
- 47.Thuong PT, Pokharel YR, Lee MY, et al. Dual anti-oxidative effects of fraxetin isolated from Fraxinus rhinchophylla. Biol Pharm Bull 2009;32:1527-32. 10.1248/bpb.32.1527 [DOI] [PubMed] [Google Scholar]
- 48.Meng H, Zhao H, Cao X, et al. Double-negative T cells remarkably promote neuroinflammation after ischemic stroke. Proc Natl Acad Sci U S A 2019;116:5558-63. 10.1073/pnas.1814394116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Won S, Lee JK, Stein DG. Recombinant tissue plasminogen activator promotes, and progesterone attenuates, microglia/macrophage M1 polarization and recruitment of microglia after MCAO stroke in rats. Brain Behav Immun 2015;49:267-79. 10.1016/j.bbi.2015.06.007 [DOI] [PubMed] [Google Scholar]
- 50.Zhou X, Zhang YN, Li FF, et al. Neuronal chemokine-like-factor 1 (CKLF1) up-regulation promotes M1 polarization of microglia in rat brain after stroke. Acta Pharmacol Sin 2021. [Epub ahead of print]. 10.1038/s41401-021-00746-w [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as