

Abstract
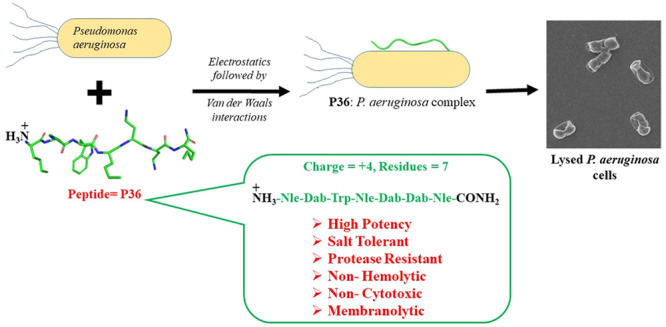
Rapidly growing antimicrobial resistance (AMR) against antibiotics has propelled the development of synthetic antimicrobial peptides (AMPs) as potential antimicrobial agents. An antimicrobial peptide Nle-Dab-Trp-Nle-Dab-Dab-Nle-CONH2 (P36; Nle = norleucine, Dab = diaminobutyric acid, Trp = tryptophan) potent against Pseudomonas aeruginosa (P. aeruginosa) has been developed in the present study. Rational design strategy adopted in this study led to the improvisation of the therapeutic qualities such as activity, salt tolerance, cytotoxicity, and protease resistance of the template peptide P4, which was earlier reported from our group. P36 exhibited salt tolerant antimicrobial potency against P. aeruginosa, along with very low cytotoxicity against mammalian cell lines. P36 was found to be nonhemolytic and resistant toward protease degradation which qualified it as a potent antimicrobial agent. We have investigated the mechanism of action of this molecule in detail using several experimental techniques (spectroscopic, biophysical, and microscopic) and molecular dynamics simulations. P36 was a membrane active AMP with membrane destabilization and deformation abilities, leading to leakage of the intracellular materials and causing eventual cell death. The interaction between P36 and the microbial membrane/membrane mimics was primarily driven by electrostatics. P36 was unstructured in water and upon binding to the microbial membrane mimic SDS, suggesting no influence of secondary structure on its antimicrobial potency. Positive charge, optimum hydrophobic–hydrophilic balance, and chain length remained the most important concerns to be addressed while designing small cationic antimicrobial peptides.
1. Introduction
Growing antimicrobial resistance (AMR) leading to development of multidrug resistant (MDR) microbes1 imposes a great threat to human civilization.2,3 The problem is augmented by the slowdown in the development of new classes of antibiotics.4 Thus, there is an urgent need for development of alternate classes of therapeutic molecules to combat microbial infections. Antimicrobial peptides (AMPs) are a class of small proteins, which have antimicrobial activity and are present in various kingdoms of life. They act as the first line of the innate defense system of the host against several microbes such as bacteria, fungi, yeasts, viruses, and parasites.5 These peptides have diverse sequences and secondary structures. The major advantage of the AMPs over the conventional antibiotics lies in the fact that they develop resistance in microbes much more slowly in comparison to the latter.6 Multiple modes of action of the AMPs, such as membrane disruption, membrane permeabilization leading to severance of intracellular mechanisms and pathways,7−9 and immunomodulatory activities, in contrast to the specific targets of the antibiotics, are responsible for the delayed development of resistance against them in the microbes. Additionally, being constituted of the biocompatible materials, AMPs generate nontoxic metabolites in comparison to the antibiotics. A large class of AMPs are cationic in nature. Electrostatic interaction between the positively charged peptides and the negatively charged membrane surfaces of the microbes lead to membrane binding, depolarization, deformation, membrane leakage, and eventual cell death.10,11
The Gram-negative bacterial infections are more difficult to treat than the Gram-positive bacterial infections.2,4,12 This is owing to the lipopolysaccharide (LPS) layer which forms the outer leaflet of the asymmetrical lipid bilayer in the Gram-negative bacteria.13,14 The AMP encounters the LPS layer in the Gram-negative bacteria before it can assess the inner plasma membrane.15,16 The LPS layer interacts with the antibiotics and the AMPs, rendering them inactive. The resistance of the Gram-negative bacteria against the AMPs is partially attributed to the LPS layer, though there are other known mechanisms of the development of resistance against them such as chemical modification of the lipid membranes, sequestration, proteolytic degradation, release of glycosaminoglycans (GAGs), polysaccharides, and polyanionic scavenging species.17−19
In spite of such huge potential of the AMPs, only a few of them, such as Bacitracin, Daptomycin, Vancomycin, Boceprevir, Telaprevir, etc., are commercially used as antimicrobial therapeutics.20 This is largely owing to the disadvantages that are associated with natural AMPs such as large sequences leading to high cost of synthesis, low serum stability owing to the protease degradation, salt sensitivity of antimicrobial potency,21−24 and toxicity to mammalian cells.25 Cationic antimicrobial peptides have large quantities of lysine (Lys) and arginine (Arg), making them highly susceptible to serine proteases such as trypsin, which specifically hydrolyzes the peptide bond at the C-terminal end of the Lys and Arg. In an effort to overcome the protease degradability of the AMPs, different strategies have been adopted such as (a) insertion of nonstandard ω-amino acids26−28 and side-chain-modified α-amino acids29−31 in the AMP sequence, (b) peptide terminal modifications,29−31 (c) insertion of amino acids of opposite chirality,32−34 (d) cyclization of the AMP sequences,35−37 (e) insertion of peptidomimetic blocks in key positions of the sequence,38−42 (f) pegylation,43−45 (g) lipidation,46,47 and (h) construction of hybrids.48 Of all the strategies employed, modification of the side-chain length of the positively charged amino acid residues, such as Lys and Arg, is the most common one. Many nonstandard amino acids are used as analogs of Lys, such as Ornithine (Orn), Dab (diaminobutyric acid), Dap (diaminopropionic acid), and Arg such as homoarginine 2-amino(3-guanidino)propanoic acid.49−57 In general, the short-side-chain analogs for Lys and the long-side-chain analogs for Arg, improved the activity of the AMPs.
In an earlier study from our group, we had reported a cationic heptapeptide, P4 (LKWLKKLCONH2), with broad spectrum activity against ESKAPE pathogens and fungal strains in vitro.58 Due to the all α-amino acid backbone and high content of Lys, P4 can be anticipated to be susceptible to protease degradation in vivo. In the present study, we wanted to impart protease resistance to P4 by replacing the α-amino acids by their nonstandard analogous amino acids. The designed analogs of P4 (P31–P37, Figure 1) had identical charge but differed in their hydrophobic–hydrophilic balance. Because the hydrophobic–hydrophilic balance plays a crucial role in determining the bioactivities of the AMPs, we investigated the antimicrobial potency, salt tolerance, and cytotoxicity of the designed peptides P31–P37. The mechanism of action of the lead AMP was studied in detail using various biophysical, spectroscopic, microscopic techniques, as well as computational analysis.
Figure 1.

Schematic representation of the heptapeptides (P31–P37) designed from the all α amino acid containing template peptide P4. In peptides P31–P33, polar amino acid Lys of P4 was replaced by unnatural analogs such as Orn, Dab, and Dap; in P34, all of the nonpolar hydrophobic Leu residues in P4 were replaced by Nle; and in P35–37, both the Lys and Leu residues were simultaneously replaced by the unnatural analogs. The side chains of the unnatural amino acid residues are illustrated in the figure.
2. Results and Discussion
2.1. Rational Design of the AMPs
Peptides P31–P37 were designed on the basis of P4, an earlier reported peptide,58 as a template (Figure 1; Supporting Information Tables S3 and S4). In an attempt to improve the activity and protease resistance of P4, Lys and Leu in the sequence were systematically mutated by side-chain-modified analogs of Lys (Orn, Dab, Dap) and Leu (Nle). Mutations were systematically incorporated to change the polar (P31–P33) and hydrophobic (P34) amino acid residues, one type at a time or both simultaneously (P35–P37). The side-chain length and the hydrophobicity of Lys and its analogs diminish in the order of Lys, Orn, Dab, and Dap. Though the side-chain carbon atoms are the same for Nle and Leu, the former is longer, being unbranched in comparison to the later, which is branched. Thus, though all of the P4 analogs (P31–P37) studied here had charge identical to that of P4, their overall hydrophobicities were different due to incorporation of various nonstandard amino acid analogs of Lys and Leu.
2.2. Peptide Synthesis and characterization
All of the designed peptides were synthesized using a solid-phase synthesis protocol as described in the Experimental Section. The purified peptides were characterized using analytical HPLC (Figures S2–S8), ESI-MS (Figures S9–S15), and 1H NMR (Figures S16–S22) spectra.
2.3. Antimicrobial activity of the designed AMPs
Antimicrobial activities of P31–P37 were tested against both Gram-negative (Pseudomonas aeroginosa (P. aeruginosa) and Klebsiella pneumoniae (K. pneumoniae) and Gram-positive (Staphylococcus aureus (S. aureus)) ESKAPE pathogens. The activities of all of the peptides were compared with those of P4 (Table 1; Figure S23). Activities of the peptides were dependent both on the individual sequences and on the bacterial strain. Activity against P. aeruginosa improved upon substitution of Lys with shorter side chains containing amino acids such as Orn and Dab in P31 and P32/P36 respectively. Upon further shortening of the side length of Lys by substituting it with Dap, the activity of P33 diminished against P. aeruginosa. However, upon introducing the second type of substitution of Leu with Nle in P37, in addition to substitution of Lys with Dap, the activity against P. aeruginosa was partially recovered. In the case of P35 and P36 though, substitution of Leu with Nle did not improvise the activities of P31 and P32, respectively. None of the analogs of P4 showed enhanced activity compared to P4 against K. pneumoniae. However, among the various analogs. those containing Dab (P32/P36) mutated in the place of Lys showed the best activity. In the case of Gram-positive bacteria S. aureus, P32 containing Dab residues had improved activity in comparison to P4. The activity of P32 further improved upon additional mutation of the Leu residues to Nle in P36. Activities of P31 containing Lys to Orn substitution was somewhat comparable to that of P4, while the activity was lost upon introducing the double mutation involving both Lys and Leu residues in P35. Peptides P33 and P37 containing Dap mutations were completely inactive against S. aureus. P34, with Leu to Nle mutations, had somewhat diminished activity against P. aeruginosa and completely lost activity against K. pneumoniae and S. aureus in comparison to P4. This differential behavior of the designed AMPs against a specific microbes suggested that the hydrophobic–hydrophilic balance of the AMPs and the length of the side chains of the constituent amino acid residues were very important for their activities. Among the similarly charged cationic AMPs, this might be the factor that modulates activity. Secondly, the differential potency of a particular AMP toward the various microbial strains suggested that the activity depended on the membrane diversity of these strains.
Table 1. MIC99% of P31–P37 against ESKAPE pathogens P. aeruginosa, K. pneumoniae, and S. aureusa.
| MIC99% (μM) |
||||||
|---|---|---|---|---|---|---|
| microbe | Gram
negative bacteria |
Gram
positive bacteria |
||||
|
P. aeruginosa |
K.
pneumoniae |
S. aureus |
||||
| peptide | absence of salts | presence of salts | absence of salts | presence of salts | absence of salts | presence of salts |
| P4 (control) | 30 | ND* | 50 | ND* | 80 | ND* |
| P31 | 20 | ND* | 200 | ND* | 60 | ND* |
| P32 | 20 | 100 | 60 | ND* | 40 | ND* |
| P33 | 200 | ND* | >200 | ND* | 200 | ND* |
| P34 | 60 | ND | 200 | ND | 200 | ND |
| P35 | 60 | ND | 200 | ND | 200 | ND |
| P36 | 20 | 50 | 80 | ND | 20 | ND |
| P37 | 60 | ND | >200 | ND | 200 | ND |
All of the MIC99% are reported in μM. ND = not detectable; ND* = not done.
Because most of the AMPs lose their activity in the presence of the physiological concentration of salt,59 we investigated the salt tolerance of the antimicrobial activity of P31–P37 in the presence of physiological concentrations of NaCl (150 mM), the most abundant salt in the serum. The loss of activity in the presence of salts is generally attributed to the electrostatic reasons that decrease the affinity of the cationic AMPs toward the negatively charged microbial membranes. The presence of salt delays the kinetics of the interaction between the peptide and the membrane surface though, the interaction network in the final peptide: membrane-mimetic complex is salt independent .60 Of all the peptides designed, P36 exhibited moderate NaCl tolerant activity respectively against P. aeruginosa (Figure 2a). P32 and P36 lost their activity completely toward the other strains in the presence of salts. All of the other analogs completely lost their activity in the presence of NaCl against all of the microbial strains tested (Table 1). It is mention-worthy that the template peptide P4 also had a salt sensitive activity toward the microbes. Salt tolerance of the most active peptide P36 against P. aeruginosa was further performed in the presence of some physiologically lesser abundant salts like MgCl2 and CaCl2 (Figure S24). Activity of P36 against P. aeruginosa was completely retained in the presence of CaCl2 (20 μM) while it was partially compromised in the presence of MgCl2 (MIC99% ∼ 60 μM). However, even being compromised in the presence of MgCl2, P36 retains a moderate antimicrobial activity.
Figure 2.

(a) Salt tolerant activity of P32 (black) and P36 (red) against P. aeruginosa. MIC99% was calculated from the microbroth dilution assay performed in the presence of 150 mM NaCl. The control experiment was performed in the presence of 10 μM Polymixin B, and other readings were normalized against it. (b) MTT assay of the peptides P32 (red) and P36 (black) on L132 cell lines. Cell viability was measured by monitoring the absorbance at 570 nm upon addition of increasing concentrations of peptide to the cells. All of the experiments were performed in triplicate.
In summary, from the above discussion, it might be concluded that the activities of the different analogs were dependent on the specific microbial species. This was not surprising, as the membranes of different microbial species were quite unique and most of the cationic AMPs manifested their action through membrane interactions. The designed analogs were most potent against P. aeruginosa. Decrease in the length of the side chain of the charged amino acid residue led to the increase in the activity of the analogs as seen in the Orn and Dab substituted peptides. However, upon decreasing the side-chain length too much as in Dap substituted peptides, there was a loss in activity which could be recovered partially by the second type of substitution of Leu to Nle. Though Leu and Nle have the same hydrophobicities, the length of the side chains in Nle being longer compared to Leu improved the activity. In the cases of peptides P31 or P32, where the charged side chain was longer, there was effective interaction with the microbial membrane already leading to high activity. Thus, in those cases a second type of mutation of Leu to Nle did not improve the activity further. Additionally, in P34, where the hydrophobic–hydrophilic balance was identical to P4 but the length of the hydrophobic chain was varied; the peptide either had diminished or lost activity against the different microbes. This indicated that, in addition to the optimum hydrophobic hydrophilic ratio, the antimicrobial activity depended on the length of the side chains in being able to make the best interaction with the microbial cell membrane.
P32 and P36 were found to be the most active peptides against all of the strains tested, with best activity against P. aeruginosa. Additionally, P36 showed salt tolerant activity against P. aeruginosa. P. aeruginosa is a multidrug resistant opportunist pathogen that causes diseases in plants, animals, and humans. The organism causes serious infection during existing diseases or conditions such as cystic fibrosis or traumatic burns. It is generally problematic to treat infections caused by this organism owing to its intrinsically advanced antibiotic resistance. Though not extremely virulent, this pathogen is capable of extensive colonization and formation of enduring biofilms. Thus, in the later part of the study, we have only focused on the bioactivities, biophysical attributes, and the mode of action in details against P. aeruginosa for the two most active peptides, i.e., P32 and P36.
2.4. Cytotoxicity of the AMPs
Being noncytotoxic to the mammalian cells is one of the basic criteria for being an effective antimicrobial therapeutic. Effects of P32 and P36 on the viability of the human embryonic lungs (L132) cell lines were determined using MTT assay. Upon treatment of the cells for 4 h, with 100 μM (concentration greater than MIC99%) P32 and P36, they exhibited a cell viability of ∼90% (Figure 2b), suggesting a very low cytotoxicity at the biologically relevant concentration.
2.5. Hemolytic activity
Being nonhemolytic is yet another basic criterion for any peptide to be used as a therapeutic molecule, and hence the hemolytic activities of P32 and P36 against human RBCs were studied. Figure S25 represents the digital image of the hemolytic assay performed against human RBCs at different concentrations of P32 and P36 (25, 50, 100, and 200 μM). Incubation of the RBCs with buffer and Triton X-100 were treated as negative and positive controls, respectively. Until 4 h, both P32 and P36 were found to be very weakly hemolytic (hemolysis < 10%) until a concentration of 100 μM, which was much greater than their biologically active concentrations.
2.6. Time Course for Bactericidal Activity of P36 on P. aeruginosa
To determine the time needed by P32 and P36 to express their bactericidal activity against P. aeruginosa at its MIC99%, a time course experiment was performed (Figure 3; Figure S26). Cells were incubated with P32 and P36 at their respective MIC99% for different time spans and then spread onto NB agar plates for CFU counting. After overnight incubation of the agar plate, a number of viable cells were calculated. It was observed that almost complete killing of P. aeruginosa cells was observed within 45 min for P36 (Figure 3) and within 60 min for P32 (Figure S26).
Figure 3.

Time kinetics of the bactericidal activity of P36 at its MIC99% against P. aeruginosa cells. P. aeruginosa cells were treated with P36 at MIC for different time intervals, and cells were spread on NA plate for CFU count after overnight incubation at 37 °C. Bacterial killing percentage was calculated from CFU count of the plate in comparison to the control plate. At 45 min time point, a completely clear plate was observed, suggesting a 100% killing of the microbial cells. (a) Negative control plate (no P36 added), (b) positive control plate (10 μM Polymyxin B treated cells), and (c–e) plates at time points 15, 30, and 45 min, respectively. (f) Time kill kinetics of P32 and P36 against P. aeruginosa, shown in log scale.
2.7. Protease Resistance of P36
The main bottleneck in the application of the AMPs as therapeutic agents arises from their short serum half-life owing to their protease degradability. To test if our lead peptide P36 was immune to protease resistance, we checked its chemical integrity upon incubation with a mixture of various proteases such as trypsin, chymotrypsin, and proteinase K in comparison to the template AMP P4, using analytical HPLC (Figure 4) and ESI-MS (Figures S27–S32). Peptide P36 maintained its retention time when injected into the analytical HPLC, even after incubation with the mixture of proteases until 6 h, suggesting its robustness to the protease degradation (Figure 4a–c). This was corroborated from the ESI-MS analysis of the reaction mixture post-incubation with the enzyme cocktail, which contained only intact P36 (differently ionized, Figures S27–S29) peaks. In contrast, incubation of P4 with proteases resulted in the appearance of several new peaks at different retention times in the HPLC chromatogram (Figure 4d–f). The protease treated P4 reaction mixture was analyzed using ESI-MS, which led to the identification of various degraded species (Figures S30–S32). The above results conclusively proved the resistance of P36 toward enzymatic degradation in comparison to earlier reported all α-amino acid containing P4. Further, to investigate if the activity of P36 was retained upon protease treatment, MIC99% of protease treated P36 was studied against P. aeruginosa. Figure S33 shows that the activity of P36 remained unaltered even upon incubating it with proteases for 6 h, confirming that P36 was indeed protease resistant.
Figure 4.

Protease resistance of P36 and P4. Analytical HPLC traces of the P36 (lLeft panel) and P4 (right panel), after incubation with an enzyme cocktail (trypsin, chymotrypsin, and proteinase K) for different time intervals. Chromatograms of P36 and P4: (a, d) in the absence of enzymes, (b, e) after 1 h. in the presence of enzyme cocktail, and (c, f) after 12 h in the presence of enzyme cocktail.
2.8. Mechanism of Action of P36
In order to study the mechanism of action of P32 and P36 against P. aeruginosa and membrane mimic systems, we performed a series of biophysical experiments as reported below.
2.8.1. Calcein Dye Leakage Assay
In a pursuit to understand the effect of AMPs on the microbial membrane integrity, leakage of calcein dye from POPE/POPG (3:1) large unilamellar vesicles (LUVs) was studied. The composition of the lipids that constitute the LUVs mimic the microbial membrane environment, and thus these LUVs act as microbial cell mimics. DLS studies confirmed the structural stability of the LUVs up to 24 h (Figure 5a, inset). Stability of calcein laden LUVs was also checked by performing a time kinetics of the calcein leakage over 1 h, wherein no detectable calcein fluorescence was observed (Figure 5a). The fluorescence of calcein is not observed when trapped inside the LUVs, while it enhances greatly upon leakage from them. Thus, the enhancement of calcein fluorescence intensity is directly related to the dye leakage from the LUVs and thus membrane disruption. Upon treating the LUVs with 0.5×, 1×, and 2× MIC of P32, about 18, 41, and 81% enhancement of calcein fluorescence intensity was observed, while 0.5×, 1×, and 2× MIC of P36 led to 53, 76, and 85% enhancement of fluorescence signal (Figure 5b). This study clearly indicated a membrane disruptive mode of action for both P32 and P36, with a greater efficiency in the later.
Figure 5.

Membrane permeabilization effect of P32 and P36. (a) Structural stability of calcein-entrapped LUVs (3:1 ratio POPE/POPG) in the absence of peptides (inset) monitored over a time period of 3600 s. Unchanged hydrodynamic radius of the LUVs after 24 h (green line) as seen from DLS. Absence of calcein leakage in the untreated LUVs. (b) Calcein dye leakage assay. Bar diagram representing calcein leakage from dye-entrapped LUVs (3:1 ratio of POPE: POPG), a microbial membrane mimic system, upon addition of increasing concentrations of P32 and P36. Triton X-100 (0.1%) was used as the positive control. Time kinetics of (c) NPN and (d) PI uptake after addition of 1× and 2× MIC P36 to P. aeruginosa cells, which indicate the outer and inner membrane permeabilities, respectively. All of the experiments were performed in triplicate.
2.8.2. NPN Uptake Assay (Outer Membrane Permeability)
Outer membrane permeabilization of P. aeruginosa was studied by NPN dye uptake assay. NPN, a hydrophobic dye, is usually excluded by the outer membrane due to permeability barrier from interacting with the membrane lipids. However, disruption of the outer membrane upon treatment with AMPs or other membranolytic agents enables the binding of NPN to the membrane lipids, generating an enhanced fluorescence signal. In the present study, enhancements of 75 and 80% in the fluorescence intensity of NPN were observed upon incubating P. aeruginosa cells with 1× and 2× MIC of P36, respectively (Figure 5c). Similarly, enhancements of 20 and 80% in the fluorescence intensity of NPN were observed upon incubation of P. aeruginosa cells with 1× and 2× MIC of P32, respectively (Figure S34). In the negative control experiment, the untreated cells did not show any increase in the fluorescence of NPN, while, in the positive control experiment, Triton X-100 led to 100% enhancement of the fluorescence signal. It can thus be concluded that P36 causes considerable disruption of the outer membrane of P. aeruginosa and much more efficiently than P32.
2.8.3. PI Uptake Assay (Inner Membrane Permeability)
The inner membrane permeability of P36 for the P. aeruginosa cells was studied using propidium iodide (PI) assay. PI is a fluorophore that generates high fluorescence signal upon intercalating with the bases of DNA. Because PI is impermeable through healthy membrane, and only permeates through compromised membrane, this assay is used to study the inner membrane permeability of the AMPs. Figure 5d shows 20 and 40% increments in intensity of PI upon incubating P. aeruginosa cells with P36 for 0.5 h at its 1× and 2× MIC, respectively. Figure S34 shows ∼15 and 60% increases in the intensity of PI upon using 1× and 2× MIC of P32. In contrast, in the negative control, untreated cells did not show any enhancement in the PI fluorescence. This proved that both P32 and P36 caused inner membrane permeability of the P. aeruginosa cells, with the latter being marginally more efficient at the MIC concentrations
2.8.4. Peptide–Membrane-Mimetic/Peptide–Live-Cell Interactions by Fluorescence Spectroscopy
From the above biophysical studies, P36 has been shown to be highly membrane active. Membrane association is a prerequisite for the membrane activity needed to manifest the antimicrobial activity of AMPs. For understanding the selectivity in the membrane association of P32 and P36, their interactions with the microbial and mammalian membrane mimics and live cells were studied by using intrinsic fluorescence of the aromatic amino acid residue Trp. SDS and DPC micellar systems, containing negative and zwitterionic charges on the surface, respectively, were considered to be microbial and mammalian membrane mimetic systems, respectively.
Upon addition of increasing amounts of SDS micelles to the solutions of P32 and P36, the fluorescence emission of Trp underwent a blue shift of around 22 nm (Figure 6a). This indicated insertion of Trp into the hydrophobic environment of the SDS acyl chains, in turn suggesting an interaction between the peptides and the SDS micelles. Upon similar addition of increasing amounts of DPC micelles to the peptides, no blue shift in the fluorescence emission of Trp was observed, suggesting no interaction between them (Figure 6a). Fluorescence quenching experiments of P36 alone and in membrane-mimetic environments such as SDS and DPC were performed in the presence of a static quencher such as bis(acrylamide) for understanding the solvent exposure (Figure 6b). The extents of quenching of Trp fluorescence of the free peptide and that in the presence of the membrane-mimetic environment were expressed in terms of Stern–Volmer constant (Ksv). It was observed that the Ksv values for the peptide alone and the peptide in DPC were higher than that seen in SDS. This indicated that the peptide was much more solvent exposed in DPC, the mammalian membrane mimetic environment, than in SDS, the microbial membrane mimetic environment. Or, in other words, P36 selectively interacted with the microbial membrane mimetic environment over the mammalian membrane mimetic environment, leading to the lesser exposure of Trp in the former.
Figure 6.

Interactions of P32 and P36 with membrane mimics/live membranes. (a) Change in fluorescence emission maxima (Δλ) of Trp residues of P32 and P36 in the presence of varying ratios of peptide: lipid (SDS and DPC). A blue shift in the emission maxima occurs for both P32 and P36 with increasing concentrations of SDS, a bacterial membrane mimic suggesting an interaction between the two, while there is no change in the fluorescence maxima upon addition of DPC, indicating no interaction between them. (b) Plot of F0/F1 against concentration of peptide alone and in the presence of membrane mimics such as D8PG and DPC. Change in the fluorescence emission maxima of the tryptophan fluorescence of (c) P32 and (d) P36, respectively, in the presence of increasing concentration of live P. aeruginosa cells.
Next, in order to understand the interactions of P32 and P36 with the live P. aeruginosa cell membranes, increasing amounts of cells were added to the peptides while monitoring the Trp fluorescence (Figure 6c,d). Upon increasing the concentration of the cells, an increasing blue shift in the emission maxima was observed for both P32 and P36, which might be attributed to the embedding of Trp in the hydrophobic environment of the microbial membranes. This observation conclusively established the interaction between P32 and P36 with P. aeruginosa cell membranes.
2.9. Determination of the Thermodynamic Factors in the Binding of P32 and P36 with Lipopolysaccharide
P32 and P36 exhibited the best activity toward P. aeruginosa, a Gram-negative bacteria. The outermost leaflet of the asymmetrical bilayer of the outer membrane of the Gram-negative bacteria is composed of lipopolysaccharide (LPS). Hence, any antimicrobial agent that acts on the Gram-negative bacteria first encounters the LPS layer. We wanted to look at the thermodynamic parameters associated with the interaction between the P32/P36 and LPS through ITC (Figure 7). The isotherm obtained for both P32 and P36 indicated a negative enthalpy change or an exothermic reaction with LPS (Figure 7a,b). Gibb’s free energies of the interactions were calculated (Table 2). P36 was found to be marginally preferred over P32 towards LPS binding. The peptide binding to LPS is both entropically and enthalpically favoured.
Figure 7.

Isothermal titration calorimetry (ITC) for P32 and P36 in the presence of LPS (50 μM), a component of the outer leaflet of the bacterial membrane. Exothermic heat of reaction with LPS micelles vs time of interaction for (a) P32 and (b) P36. Plot of enthalpy change per mole of peptide (c) P32 and (d) P36 injection with the peptide:LPS molar ratio.
Table 2. Thermodynamic Parameters in the Interaction of P32 and P36 with SDS Calculated from ITC Experiments Performed at 298 K.
| peptide |
||
|---|---|---|
| thermodynamic parameters | P32 | P36 |
| KA (M–1) | 1.83 × 105 | 4.72 × 105 |
| ΔG (kcal·mol–1) = -RT ln KA | –7.18 | –7.74 |
| N (stoichiometry) | 2.13 | 1.90 |
| ΔH (kcal.mol–1) | –2.26 | –4.08 |
| ΔS (kcal·mol–1·K–1) | 16.6 | 12.4 |
| TΔS(kcal·mol–1) | 4.95 | 3.70- |
| KD (M) | 5.46 × 10–6 | 2.12 × 10–6 |
2.10. Peptide–Live-Cell Interaction Studied through Microbial Surface ζ Potential
Interaction of P36 with the live P. aeruginosa cells was studied by monitoring the surface ζ potential of the cells. Microbial cells are negatively charged and hence have a negative surface ζ potential. Upon binding of the cationic peptides to the negatively charged microbial membranes, the surface ζ potential of the cell surface becomes less negative. Thus, a decreasing negative ζ potential is an indication of the peptide–membrane interaction. Upon addition of P36 to the live cells, the surface ζ potential of the P. aeruginosa cells became less negative and plateaued out beyond 1× MIC to about 0 mV (Figure 8a). This observation conclusively proved binding of P36 to the P. aeruginosa cell membrane.
Figure 8.

(a) Surface ζ potential of live P. aeruginosa cells upon addition of increasing concentration of P36. (b) CD spectra of P36 alone in phosphate buffer and in the presence of live P. aeruginosa cells at different incubation times.
2.11. Structure of AMP upon AMP–Membrane-Mimic/AMP–Live-Cell Interactions Using CD
The secondary structure of the peptides often plays an important role in the peptide–membrane interactions and hence the activity of the peptides. We wanted to study the structure of the two most active peptides P32 and P36 in free state and in the presence of membrane-mimetic systems. Figure S35 shows the change in molar ellipticities of P32 and P36 in water, 50% TFE (a helix promoting solvent), SDS micelles, and DPC micelles. Both peptides adopted a random coil conformation in water and also in the presence of the different membrane-mimetic environments. This suggested that the peptides bound to the membranes mimics as random coils.
We further wanted to study the P36–P. aeruginosa interactions through circular dichroism (CD) spectroscopy. CD was performed on P36 alone in phosphate buffer and on P36–P. aeruginosa cell mixture at different time points (Figure 8b). Later, fresh P36 was added to the peptide–live-cell mixtures and CD was studied. P36 formed a random coil conformation with a negative peak at 200 nm. The live P. aeruginosa cells in phosphate buffer at pH 7.4 showed a positive peak at 200 nm and a negative peak at around 228 nm. Upon addition of 1× MIC of P36 to the cells, the positive and the negative CD peaks red-shifted to around 205 and 232 nm, respectively. The positive peak intensity was significantly diminished upon addition of P36. This was due to the cancellation of a positive peak from the P. aeruginosa cells by the negative peak of P36. The negative peak of the cells remained almost similar in intensity in the P36:P. aeruginosa complex due to absence of any peaks from P36 in this region. The slight red shift in the peak positions of the final P36–P. aeruginosa system was owing to the interactions between them. The spectra of the P36–P. aeruginosa mixture remained almost unchanged from 15 min to 2.5 h, indicating completion of interaction within the first 15 min. Upon addition of fresh P36 after 2.5 h, a negative peak reappeared at 195 nm, suggesting random coil conformation of the freshly added P36 in addition to the negative Cotton effect peak at ∼230 nm from the P36–P. aeruginosa complex. The positive peak of the P36:P. aeruginosa cell complex was not observed as it was canceled by the negative peak of the free peptide. The peptide added in the second slot was excess in comparison to the cells, which were already completely complexed with the peptide added in the first slot. Hence, the peak due to the free P36 reappeared in this case. Thus, we concluded that the peptide existed as random coil in the free state as well as in the live P. aeruginosa cell bound systems. Adoption of random coil conformation in the presence of membrane mimics and live cells may be attributed to the very small size of the AMPs.
2.12. P36–Membrane-Mimic Interaction through NMR
Next, we wanted to study the interaction of P36 with (a) SDS, the microbial membrane mimic; (b) LPS, constituent of the Gram-negative microbial membrane; and (c) DPC, the mammalian membrane mimic using NMR. Figure 9 shows the stacked plots of the aromatic region of the 1H NMR spectra of P36 (1 mM) in 10% D2O (containing 10 mM phosphate buffer, pH 6.5) and in the presence of membrane-mimetic environments such as SDS, LPS, and DPC. The sharp NMR signals of P36 became broad in the presence of SDS and LPS, while it remained unaffected in the presence of DPC. Broadening of the NMR signals was a clear indication of the interaction between P36 and SDS and LPS. Unchanged line widths of P36 in the presence of the DPC micellar system suggested the absence of interaction between them. These observations conclusively explained the high antimicrobial potency of P36 against the P. aeruginosa in contrast to its noncytotoxicity toward mammalian cells.
Figure 9.

Stacked plots of the NH region of the 1H NMR spectra of P36 (1 mM) alone and in the presence of SDS, LPS, and DPC. Broadening of the line widths and the change in the chemical shifts of the peptide signals in the presence of SDS and LPS indicate interaction between them. Unchanged line widths and the chemical shifts of the peptide in the presence of DPC suggest absence of interaction between them.
2.12.1. Live-Cell NMR
To understand the details of the mechanism of action of P36 on P. aeruginosa cells, we performed a real time NMR experiment with cells added to the peptide (Figure S36). Line width broadening of all of the signals of the peptide was observed upon incubation with the cells, due to the increase in the t2 relaxation time, suggesting an interaction between P36 and the live cells. Additionally, new peaks attributed to the leaked metabolites from the lysed, wounded, and dead cells were observed as early as 30 min. The time kill kinetics of bactericidal activity of P36 against P. aeruginosa cells, which had earlier shown considerable killing within 30 min of incubation, corroborated with the NMR observation. The appearance of the metabolite peaks supported the previous biophysical studies and indicated the membranolytic mode of action for P36.
2.13. Microscopic Visualization of the Membranolytic Mechanism of Action of P36
In order to visualize the effect of P36 on the membrane integrity and the morphology of P. aeruginosa cells, field emission scanning electron microscopy (FESEM) was performed. P. aeruginosa cells were incubated with the P36 at their 1× and 2× MIC concentrations to study the peptide-mediated deformation and cell lysis. Cells not treated with P36 retained their healthy morphology with a smooth outer membrane, while the cells treated with Triton X for 4 h showed complete disruption of the cellular morphology and the presence of cell debris (Figure 10a,b). Upon addition of 1× and 2× MIC concentrations of P36 to the P. aeruginosa cells, cellular morphology was severely deformed accompanied by the disruption of the cell membrane (Figure 10c,d).
Figure 10.

Field emission scanning electron microscopic (FESEM) images of the P. aeruginosa cells in the absence and presence of P36. (a) Negative control and (b) positive control in the presence of 10 μM Polymixin B. Considerable deformation in cellular morphology observed upon incubation of cells with (c) 1× MIC and (d) 2× MIC of P36.
2.14. Atomistic Visualization of P36 Structure and Membrane Interaction: Insight from MD Simulations
MD simulations of the free peptides (P36, P32) showed that the peptides remained as a random coil along the MD trajectory (Table S5), in lines with the CD experiment. SDS micelle is a popular mimic of the bacterial membrane70−72 and was adopted for simulation studies in this work. No secondary structure of the peptide was attained in response to SDS micelle binding (Table S5). Micelle structure (eccentricity, micellar radius) was independent of peptide binding (Table S5). Noticeable structural features observed from the MD structure of the P36–SDS complex were as follows: (1) The peptide laid over the surface of the micelle with noticeable local deformation (∼11 Å width and ∼13 Å deep peptide binding pocket, Figure 11a) of the micelle surface. (2) Trp3 and Nle4 side chains were found to be on the same side relative to peptide backbone. (3) Trp3 was found to be buried in the dry hydrophobic core of the SDS micelle, in lines with the experimentally observed shift in the emission maxima of the tryptophan fluorescence (Figure 6a,c). No water was found within 3.4 Å of the Trp3 side chain. The hydrogen bonding requirement of the polar “-NH” of the indole ring of Trp3 was found to be satisfied by establishing direct interaction with the oxygen of the terminal sulfate of SDS (Figure 11a). (4) Hydrophobic side chains of Nle1 and Nle4 were found to be more buried relative to Nle7 in the hydrophobic core of SDS micelle (Figure 11a,b). (5) High solvent exposure was observed for the positively charged side chains of Dab2, Dab5, and Dab6 (Figure 11b). The interaction network of the positively charged terminals of Dab2, Dab5, and Dab6, were of three kinds: (I) direct interaction with the negatively charged sulfate oxygens of SDS (Figure S39), (II) water-mediated interaction with SDS, and (III) interaction with the bulk water. (6) The peptide backbone satisfied its hydrogen bonding requirement by forming H-bonds with water molecules or sulfates of the SDS micelle or both (Figure 11a; Figure S40). The above structural observations were robust features of our MD simulations (confirmed from six independent MD runs differing in the initial structural model and initial velocities; see Experimental Section). A similar observation was confirmed from P32 simulations in the presence of SDS micelles (Figures S37 and S38).
Figure 11.

(a) Representative snapshot of P36:micelle (SDS) complex (without ions and water) after 50 ns of MD. Peptide P36 is shown as yellow sticks (nitrogen, blue; oxygen, red) and SDS micelle shown as surface representation. Zoomed-in views of the peptide:micelle binding pockets are shown in the black-broken-line boxes: (1) SDS micelle pocket hosting the peptide shown as yellow mesh (dimensions: width, ∼10–12 Å; depth, ∼12–15 Å). (2) Direct peptide:micelle interaction. Trp3 side chain is found to be buried in a dry (no waters found within 3.4 Å of Trp side chain) hydrophobic core of the micelle that satisfied the hydrogen bonding requirement of the indole–NH, forming direct interaction with sulfate of SDS (shown explicitly in the solid box). Hydrophobic side chains (Nle1, Nle4, and Nle7) are also found to be buried in the SDS core. Positively charged side chains (Dab2, Dab5, and Dab6) are exposed to bulk water and establish direct salt bridges with negatively charged SDS surfaces. (3) Peptide water interactions, in which peptide satisfied its backbone hydrogen bonding requirement by involving waters. Water-mediated peptide, in which micelle interaction was observed. SDS sulfates within 3.4 Å of the peptide are shown explicitly in sticks form (sulfur, yellow; oxygen, red). Black broken lines represent interactions (distance between two heavy atoms < 3.4 Å). Hydrogens are not shown for clarity. (b) Side-chain solvent accessibility of P36 estimated by calculating the solvent-accessible surface area (SASA). The SASA was averaged over the last 30 ns of the 50 ns MD trajectory. The solvent exposure (yellow area in the net plot) is shown with broken lines of constant exposure in the net plot.
P32 and P36 binding to the SDS micelles were found to be significantly faster relative to zwitterionic DPC micelles, due to obvious electrostatic reasons (Figure S41). P36 was able to bind to the DPC micelle surface after 38 ns of dynamics, much slower relative to the initial SDS binding event which took place within 1–3 ns (Figure S44). The results indicated that electrostatic interactions were crucial for initial peptide:micelle binding kinetics. Although the kinetics of the initial peptide binding event depended on the nature of the micelles (SDS or DPC), the interaction network between the peptide and micelle in the final equilibrated peptide:micelle complex was more or less similar between peptide:SDS and peptide:DPC micelles (Figures S37–S41).
3. Conclusions
We have successfully designed a lead protease resistant AMP P36, with high salt tolerant potency against Gram-negative bacteria P. aeruginosa. P36 manifested noncytotoxic and nonhemolytic behavior against mammalian cell lines. We established the membranolytic mode of action of P36 using several biophysical, spectroscopic, and microscopic tools. The study illustrated that the antimicrobial activity of AMPs was dependent on the hydrophobic–hydrophilic balance and the length of the side chains of amino acid residues, disruption of which led to the loss in activity or increase in the cytotoxicity. In this work, we have overcome several significant shortcomings of the AMPs, viz., the salt induced inactivation of the bioactivity protease degradability and cytotoxicity which limit their therapeutic potential and applicability. Antimicrobials such as P36 hold immense potential for being commercialized as antimicrobial agents of the future.
4. Experimental Section
Peptide synthesis, purification, and characterization details for the peptides P31–P37 have been described in detail in the Supporting Information (SI).
4.1. Microbroth Dilution Assay
Antimicrobial activity of the peptides was studied using standard microdilution broth assay.58 The mid log-phase cultures of P. aeruginosa, K. pneumoniae, and S. aureus were obtained from overnight-grown cultures of the respective microbes. The cell suspensions were centrifuged at 6000 rpm for 5 min. Cell pellets were washed three times each with 10 mM phosphate buffer (pH 7.4) alone or in the presence of (a) 150 mM sodium chloride (NaCl), (b) 1.25 mM magnesium chloride (MgCl2), and (c) 1 mM calcium chloride (CaCl2). Thereafter, the cell pellets were resuspended in the same buffers to obtain a cell suspension of 105 CFU/mL. The reaction was performed in a 96 well plate, where 50 μL of the cell suspension was incubated with different concentrations of peptide (ranging from 1 to 200 μM), prepared from 1 mM peptide stock in phosphate buffer (pH 7.4). The reaction was incubated at 310 K for 4 h. A negative control containing only cell suspension and a positive control containing 1 μM Polymyxin B with cell suspension were maintained. Next, 150 μL of suitable medium was added to each well and the reaction was incubated overnight with constant shaking at 310 K temperature. Absorbance of the culture was monitored at 630 nm to monitor microbial growth. The positive control Polymyxin B was used to normalize all of the other readings. The peptide concentration at which 99% growth inhibition was observed served as its MIC99%. All experiments were performed in triplicate.
The assay was also performed with P36 incubated with an enzyme cocktail (trypsin, chymotrypsin, and Proteinase K) for 4 h and subsequently quenched, using the same method as explained above.
4.2. Cell Viability Assay
Cell viability effects of the most active peptides were studied using MTT assay. L132 cells were seeded into a 96 well plate at the density of 104 cells/well. After incubating the cells overnight, they were treated with different concentrations (0–300 μM) of P32 and P36 for 24 h. Subsequently, MTT (0.3 mg/mL) reagent in DMEM was added, the reaction was incubated for 3 h, and the formed formazan crystals were solubilized by adding 200 μL of DMSO. Cell viability was calculated by measuring absorbance at 570 nm by using a Tecan plate reader.
4.3. Hemolytic Assay
Fresh human erythrocyte cells (RBC) were collected in EDTA vials. RBCs were pelleted upon centrifugation at 8000g for 10 min at 4 °C, followed by washing with PBS (pH = 7.4) three times. Cells were resuspended in PBS buffer at pH 7.4 to a final concentration of 2 × 108 cells/mL. The cells were incubated with Triton X and various concentrations of peptides (until 100 μM), for 4 h at 37 °C while shaking. The incubated samples were centrifuged at 8000g for 10 min at 4 °C. RBC lysis and heme release were quantitatively estimated by monitoring the optical density of the supernatant solution at 414 nm. Heme released upon treatment with 1% Triton X-100 was taken as 100%, and the data from other experiments were normalized against it. All of the experiments were performed in triplicate. Digital photographs of the centrifuged eppendorfs were taken. The color of the supernatant solution indicated the extent of hemolysis qualitatively.
4.4. Time Course of Bactericidal Activity
Overnight-grown culture of P. aeruginosa was centrifuged at 6000 rpm for 5 min, and the pellet was washed three times with 10 mM phosphate buffer (pH 7.4). Thereafter, the pellet was resuspended to obtain 105 CFU/mL suspension of cells. A 50 μL aliquot of bacterial cell suspension was incubated with MIC of P32 and P36 at 310 K for different time intervals (5–120 min). After each incubation time, a 5 μL aliquot was taken from each reaction volume and diluted with the same amount of buffer. The reaction mixture was then spread onto the NB agar plates. The plates were incubated overnight at 310 K, and CFU counting was done. All experiments were performed in triplicate.61
4.5. Proteolytic Stability Assay
4.5.1. HPLC Experiment
A 10 mM amount of P4 and 10 mM P36 were treated with a 1:1:1 mixture of enzymes trypsin, chymotrypsin, and protease K (0.3 mg/mL) at 37 °C for 0–6 h. After incubation, 50 μL of reaction solution was taken and mixed with 50 μL of acetonitrile with 1% TFA at 4 °C and kept for 15 min to inactivate the enzymes. The mixture was then diluted with HPLC grade water, and 20 μL was injected onto the C18 reverse-phase column for HPLC analysis. Samples were eluted by a linear gradient of 10–100% CH3CN/H2O in 0.1% TFA at a flow rate of 1 mL/min for 25 min. The UV absorbance of the eluted peptides was detected at 214 and 280 nm with a UV monitor.
4.5.2. Mass Spectrometry
In order to determine the chemical integrity of the most active peptides P4 and P36 upon protease degradation, a mass spectrometric analysis of the peptide–enzyme reaction mixture, post-incubation, and the subsequent deactivation of the enzymes were carried out on an Agilent-Q-TOF 6500 instrument equipped with Mass Hunter workstation software in the electrospray ionization positive mode.
-
(a)
In order to see if P36 retained its activity upon incubation with the proteases, MIC of the enzyme incubated P36 against P. aeruginosa was delineated (Figure S33). P36 was shown to retain most of its activity even after protease treatment, proving the protease resistance of the peptides.
4.6. Calcein Leakage Assay
To prepare an anionic bacterial membrane-mimicking model, LUVs were made from 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoetanolamine (POPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) in 3:1 ratio, with a final concentration of 2 mg/mL. All lipid mixtures were dissolved in chloroform. A lipid film was prepared by passing N2 gas over it and lyophilizing overnight. Calcein-entrapped vesicles were prepared, thereafter, by hydrating the lipid films with 70 mM calcein (in 10 mM Tris-HCl buffer, pH 7.4). The reaction mixture was vortexed, followed by five freeze–thaw cycles. During the freeze–thaw cycles, liquid nitrogen and hot water (60 °C) were used for freezing and thawing, respectively. To prepare large unilamellar vesicles, calcein-entrapped vesicles were passed through a mini extruder (Avanti Polar Lipids, Alabaster, AL) using two-stacked polycarbonate membrane filters of 100 nm pore size for 27 times. Subsequently, the sample was passed through a gel-filtration-based hydrated Centrisep-spin column by centrifuging at 3000 rpm for 2 min, to remove the free calcein.
Calcein release from 20 μM dye-entrapped liposomes in 600 μL of extravesicular buffer (10 mM Tris, 100 mM NaCl, pH 7.4) was determined by monitoring the emitted fluorescence at 520 nm from liposome dispersion. A Hitachi F-7000 FL spectrometer with a slit width of 2.5 nm was used to monitor the fluorescence. After the stabilization of calcein fluorescence, peptide was added in increasing concentrations and fluorescence enhancement was measured after 5 min of each addition. An absolute leakage of calcein from the LUVs was obtained by disrupting the liposomes at the end of each experiment by addition of 0.1% per volume of Triton X-100. All measurements were performed in triplicate at 298 K. An independent control, containing only 0.1% per volume of Triton X-100, was added to calcein loaded LUVs. This ascertained the maximum disruption and calcein release possible. Similar results were obtained in both the cases.
Percent leakage was calculated using the equation below:
where F, Fo, and FT were (1) fluorescence intensity after addition of peptide, (2) basal fluorescence intensity, and (3) maximum fluorescence intensity obtained after addition of 0.1% per volume of Triton X-100, respectively.
4.7. Assay of Membrane Permeabilization
4.7.1. Inner Membrane Permeabilization
The mid-logarithmic phase of P. aeruginosa cells was obtained from overnight-grown cultures. The cells were pelleted, and the cell pellet was washed in sodium phosphate buffer (pH 7.4) and resuspended in the same to obtain a concentration of 106 CFU/mL of P. aeruginosa cells. A 10 μM amount of propidium iodide dye was added to 1 mL of the cell suspension and incubated at room temperature for 30 min under shaking conditions. The fluorescence of the dye was measured at 298 K for 1500 s at an excitation wavelength of 535 nm (slit width, 10 nm) and emission wavelength of 617 nm (slit width, 10 nm) using the Hitachi F-7000 FL spectrophotometer. The fluorescence intensity of the free cell suspension was monitored for any fluctuations in the intensity to test the stability of the cells. After a stable intensity was obtained, P36 and P32 were added to the bacterial suspension at different concentrations (1× or 2× MIC). An increase in dye fluorescence suggested the permeabilization of the inner membrane. Peptide induced permeabilization of the inner membrane allowed entry of PI into the cells, which bound with the DNA resulting in an increase of the fluorescence intensity.
4.7.2. Outer Membrane Permeabilization
NPN uptake assay was performed to determine the outer membrane permeabilization ability of P36 and P32. An overnight-grown culture of P. aeruginosa cells was washed and resuspended in 10 mM sodium phosphate buffer (pH 7.4) to a final cell concentration of 106 CFU/mL. The fluorescence of the dye in the presence of cells alone was monitored for 20 min at room temperature to study the stability of the cells. P36 and P32 were added with increasing concentrations (1× and 2× MIC) to a cuvette containing 1 mL of cells and 10 μM NPN. Thereafter increased fluorescence after peptide addition, was monitored for 20 min using the HORIBA JOBIN YVON Fluoromax-4 spectrometer at an excitation wavelength of 350 nm (slit width, 5 nm) and emission wavelength of 410 nm (slit width, 5 nm) to determine the NPN uptake.
4.8. Fluorescence Experiments
4.8.1. Blue Shift Experiment
4.8.1.1. Shift in Presence of Membrane Mimetics
The intrinsic tryptophan fluorescence of P32/P36 was utilized to analyze their interaction with the membrane mimics. SDS and DPC were taken to mimic bacterial and mammalian membranes, respectively. P32/P36 at their respective MIC concentrations (against P. aeruginosa) were titrated against increasing concentration of SDS and DPC at 25 °C. The molar ratio of the peptide:peptide-mimic systems was varied from 1:0.25 to 1:10. The change in the fluorescence emission intensity (fluorescence emission range, 295–520 nm) of Trp against an excitation wavelength of 280 nm was monitored on the HORIBA JOBIN YVON Fluoromax-4 spectrophotometer.61
4.8.1.2. Shift in Presence of Live Cells
Intrinsic tryptophan fluorescence was utilized to analyze the interaction between the peptide and live P. aeruginosa cells. P. aeruginosa cells were collected, washed, and resuspended in 10 mM phosphate buffer (pH 7.4). Cells (5 μL) were added (stock, 108 CFU/mL) in subsequent steps to the P32 and P36 solutions. Trp fluorescence emission was monitored (for an excitation wavelength of 280 nm, excitation and emission slit widths of 2.5 nm) only for P32 and P36 and in the presence of live P. aeruginosa cells.
4.8.2. Solvent Sccessibility
Intrinsic fluorescence of tryptophan of P36 in 10 mM phosphate buffer in the absence and presence of D8PG (microbial membrane mimetic) and DPC were recorded by using the HORIBA JOBIN YVON Fluoromax-4 spectrophotometer. The intrinsic Trp fluorescence emission spectra of P36 (10 μM) upon titration with increasing concentration (from 10 to 200 μM) of D8PG and DPC were measured at an excitation wavelength of 280 nm and excitation/emission slit of 2.5 nm, over an emission spectral range of 295–520 nm. All of the fluorescence experiments were performed at 25 °C in a quartz cuvette of 1 cm path length. Fluorescence quenching experiments was used to determine the solvent accessibility of P36 in the vicinity of D8PG. Acrylamide, a static quencher, was added to the peptide–D8PG complex as well as free peptide solution up to a final concentration of 0.2 M. The resultant fluorescence intensity of the peptide was analyzed by fitting to the Stern–Volmer equation.
where F0 denotes the initial fluorescence intensity in the absence of the quencher, F stands for the fluorescence intensity at each quencher concentration, and [Q] denotes concentration in terms of molarity. Ksv represents the Stern–Volmer quenching constant expressed in M–1 calculated from the above equation, both in free and bound states of P36.61
4.9. ITC Experiments
Isothermal calorimetry was performed on MicroCal iTC200 (GE Healthcare) to assess the binding interaction between P32 and P36 with lipopolysaccharide (LPS). Peptides and LPS stock solutions were prepared in 10 mM phosphate buffer (pH 7.2). LPS solutions were vortexed for 10 min and sonicated for 5 min before loading into the cell. LPS solution (50 μM) was taken in the cell, and 1 mM concentrations of P32 and P36 were taken in the syringe. Peptide concentration was 20 times greater than the LPS concentration. The concentration of LPS used was above its critical micelle concentration (CMC) values (CMC ∼ 14 μg/mL or 1.6 μM). The reaction was carried out at 298 K with 20 peptide injections (3 μL each), at an interval of 180 s at 300 rpm stirring speed.62 A single site binding model was used to elucidate the equilibrium association constant (Ka) and the enthalpy change (ΔH). Basic thermodynamic equations, as given below, were used to determine the Gibb’s free energy change (ΔG) and entropy change (ΔS).
respectively.
4.10. Circular Dichroism Spectroscopy
Solution structure of the peptides was studied using circular dichroism spectroscopy. The spectra of P31–P37 were recorded on a Jasco J-815 spectropolarimeter (Tokyo, Japan). (±)-10-Camphorsulfonic acid was used for calibration of optical rotation. The spectra were measured over a wavelength range of 260 to 190 nm, using a 1 mm path length Suprasil quartz cuvette at a scan rate of 100 nm/min, interval of 0.5 nm, and time constant of 1 s. An accumulation of three scans at 298 K was collected. P31–P37 were dissolved in different solvents such as deionized water, 50% TFE, 30 mM SDS, and 10 mM DPC to generate a 50 μM final peptide solution.
4.11. Live-Cells CD Spectroscopy
Overnight-grown P. aeruginosa cells were collected, washed, and resuspended in 10 mM phosphate buffer (pH 7.4) to a final cell count of 5 × 105 cells/mL. Cells (50 μL) were added (stock, 108 CFU/mL) to P36 solution (25 μM, 250 μL), and CD spectra were recorded after different incubation times (until 2.5 h) using a 1 cm quartz cell, 260 to 190 nm measurement range, 100 nm/min scanning speed, 2 nm bandwidth, 4 s response time, and 1.0 nm data pitch up to 2.5 h of incubation at 37 °C. Similarly, the CD spectrum for P. aeruginosa cells only was measured under identical conditions. The peptide concentration was 25 μM in 10 mM phosphate buffer (pH 7.4).63
4.12. Live-Cells ζ Potential
ζ potential was measured with the help of a Zetasizer Nano ZS 90 instrument (Malvern, U.K.), containing helium–neon laser (633 nm) as a source of light, with the detection at 90° scattering angle at room temperature (28 °C). Overnight-grown P. aeruginosa cells were washed three times by DI water and resuspended in the same to obtain a cell suspension of 5 × 105 cells/mL. To minimize the effect of buffer on the ζ potential value, cells were suspended in DI water instead of buffer. P36 (1 mM stock solution, 5 μL) was added to 1 mL of P. aeruginosa cells (5 × 105) at different time intervals, and its ζ potential value was checked after each addition.64,65
4.13. NMR Experiments
4.13.1. NMR in the Presence of Microbial Membrane Mimics (SDS and LPS)
One-dimensional proton NMR for 1 mM P36 in 90% of 10 mM phosphate buffer (pH 6.5) and 10% D2O was recorded on a Bruker Avance III 600 MHz NMR spectrometer. 1H NMR was performed for P36 (1 mM) also in the presence of different membrane-mimetic environments, such as 200 mM perdeuterated SDS (microbial membrane mimetic), 3 μM LPS (Gram-negative bacterial membrane component), and ≥2.5 mM DPC micelles (mammalian membrane mimetic).
4.13.2. Live-Cell NMR Experiments
P. aeruginosa cultures were grown overnight and used to perform the experiment. The cell suspensions were centrifuged at 6000 rpm for 5 min. Cell pellets were washed three times with 10 mM phosphate buffer at pH 6.5 and later resuspended in the same buffer to obtain a cell suspension containing 106 CFU/mL. One-dimensional (1D) proton NMR spectra of 1 mM solution of P36 in a mixture of 90% of 10 mM phosphate buffer (pH 6.5) and 10% D2O was recorded on a Bruker Avance III 600 MHz NMR spectrometer. In another experiment, 500 μg of solid peptide P36 was added to a 500 μL solution of the cells (106 CFU/mL), and the mixture was thoroughly vortexed to achieve a final peptide concentration of 1 mM. A series of 1D 1H NMR spectra were recorded at different time points after addition of P36 to the P. aeruginosa cells. The temperature for the experiment was adjusted as per the standard growing temperature of P. aeruginosa at 310 K.
4.14. Field Emission Scanning Electron Microscopy
P. aeruginosa cells from overnight-grown cultures were pelleted down by centrifugation at 6000 rpm for 5 min, washed thrice, and resuspended in 10 mM sodium phosphate buffer at pH 7.4 to a final number of 105 cells/mL. The cell suspensions were incubated with different concentrations (1× and 2× MIC) of P36 for 4 h at 310 K. Untreated cells were set as the control. Post-incubation, the cells were fixed with 2.5% glutaraldehyde for 1 h at 277 K. Next, the cells were washed twice and resuspended in 20 μL of 10 mM sodium phosphate buffer. A 10 μL aliquot of this cell suspension was spotted on a clean glass slide and was allowed to dry overnight. Thereafter, the slides were washed with 50 and 80% ethanol for 5 min each. The samples were finally air-dried, followed by gold coating and observed under the FESEM (Zeiss Gemini).65
4.15. MD Simulations
4.15.1. Modeling of Peptides (P32, P36) in Water
The most active peptides P32 and P36 (in the presence or absence of micelle) were considered for molecular dynamics simulations. The initial linear models of P32 and P36 were generated using PyMOL version 2.4.1 software.66 N- and C-termini of these peptides were modeled as -NH3+ and -CONH2, respectively. A water box of dimensions 60 × 60 × 60 Å3 was overlaid, keeping the linear peptide model at the center. The overall charge (+4) of the peptide was neutralized by adding four chloride ions. A total of ∼21075 atoms were considered for MD simulations of peptides in water. Topology descriptions of the nonstandard amino acids (Dab and Nle) were retrieved from the Swiss Side Chain database.67
4.15.2. Modeling of Membrane-Mimetic Systems (SDS/DPC Micelles)
CHARMM-GUI Micelle Builder68 was used to build SDS/DPC micelles. SDS micelle was modeled by including a total of 60 molecules of SDS. The experimental value of the aggregation number was reported to be close to 60. Simulations with 60 SDS molecule is a popular choice for computational analysis.69−72 The micelle was placed at the center and solvated by overlaying a water box of dimensions 100 × 100 × 100 Å3. For neutralizing the simulation box, 60 Na+ counterions were added. A total of ∼98230 atoms were considered for MD simulations of SDS in water. DPC micelle was modeled in the same way as SDS. DPC is a neutral zwitterionic molecule; thus, no counterion was added. The final snapshot produced from the molecular dynamics was considered for studying peptide–micelle interactions.
4.15.3. Simulation Setup for Studying Peptide–Micelle Interaction
First, the center of mass of the model linear peptide (P32 or P36) was initially placed ∼55 Å away from the center of mass of the micelle (SDS or DPC; Figure S1a). The water padding was extended in the peptide direction and the solvated water box of dimensions 100 × 100 × 140 Å3 was subjected to MD simulation (Figure S1b). More than 145530 atoms were considered for MD simulations. To check convergence and ensure efficient sampling, we performed various independent MD replicas differing in the initial setup (setup A, setup B, and setup C: orientations of the peptide relative to micelle) (Figure S1a) as well as in their initial velocities (Table S1).
4.15.4. Simulation Parameters
All of the simulations in this study were performed in GROMACS version 2019 package,73 with CHARMM36 (version March 2019)74 as force field parameters. The TIP3P75 water model was used to model waters. Energy minimization was performed using the steepest descent algorithm (step size = 0.1 Å, 50000 steps). After energy minimization, 200 ps of equilibration was performed (restraining only the micelle and peptide) considering NVT ensemble (first 100 ps) followed by NPT ensemble. After equilibration, the production run for 50 ns was performed and the last 30 ns was considered for analysis. The simulation parameters adopted for this work are given in Table S2. Coordinates were saved at every 10 ps from the MD trajectories for analysis.
4.15.5. MD Trajectory Analysis
Secondary structural content of the peptides was obtained by processing the MD trajectories using the gmx dssp tool.76 Structural deformation of micelles in response to peptide binding was characterized by estimating the trajectory averaged radius of gyration (RG) and micelle eccentricity using the gmx gyrate tool.73 SASA (solvent-accessible surface area) values are the geometric estimation of the exposure of molecule surface to the bulk solvent. SASA values were computed by gmx sasa(77,78) using a probe radius of 1.4 Å. The gmx distance(73) tool was used to plot the distance between the center of mass of the micelle and peptide as a function of simulation time. Peptide–micelle distance and SASA of peptide were obtained from the MD trajectories and plotted using OriginPro,79 and images were generated using PyMOL visualization software.66
Acknowledgments
S.C. acknowledges CSIR (Grant 01/(2984)/19/EMR-II) and Department of Biotechnology (Grant BT/PR21251/NNT/28/1067/2016) for funding. G.P., T.S., S.D., and S.R.V. thank IIT Guwahati for funding. FIST is acknowledged for the 500 MHz NMR spectrometer. The computing facility by Biomolecular Simulation Lab (BSL, BSBE Department, IIT Guwahati) and BIF, IIT Guwahati; Param-Ishan supercomputing system, IIT Guwahati; Departmental Instrumental Facility, Department of Chemistry and BSBE, IIT Guwahati; and Central Instrument Facility, IIT Guwahati are gratefully acknowledged. Authors thank Dr. A. P. Bidkar for performing the MTT assay.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01089.
(Figures) MD setup, characterizations by analytical HPLC, HRMS, and 1H NMR, MIC bar diagrams, hemolytic assay, bactericidal time kinetics, live-cell NMR, CD interactions, residue specific solvent exposure, peptide–micelle distance; (tables) peptide physicochemical properties and MD simulations and structural parameters; general procedures for peptide synthesis, purification and characterization, materials, and microbial culture and cell culture details (PDF)
Author Contributions
S.C. designed the project, G.P. performed most of the experiments (synthesis, purification, characterization the peptides, biological assays, and biophysical (CD, fluorescence) experiments), T.S. performed FESEM experiments and some biophysical experiments, V.S.R did the molecular dynamics simulations, and S.D. performed all of the NMR experiments. S.C. and P.S. analyzed the data and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Magiorakos A. P.; Srinivasan A.; Carey R. B.; Carmeli Y.; Falagas M.; Giske C.; Harbarth S.; Hindler J.; Kahlmeter G.; Olsson-Liljequist B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- Delcour A. H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta, Proteins Proteomics 2009, 1794, 808–816. 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duin D.; Paterson D. L. Multidrug-resistant bacteria in the community: trends and lessons learned. Infect. Dis. Clin. North Am. 2016, 30, 377–390. 10.1016/j.idc.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair J.; Webber M. A.; Baylay A. J.; Ogbolu D. O.; Piddock L. J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- Sarkar T.; Chetia M.; Chatterjee S. Antimicrobial peptides and proteins: From nature’s reservoir to the laboratory and beyond. Front. Chem. 2021, 9, 691532. 10.3389/fchem.2021.691532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G.; Baeder D. Y.; Regoes R. R.; Rolff J. Predicting drug resistance evolution: Insights from antimicrobial peptides and antibiotics. Proc. R. Soc. B: Biol. Sci. 2018, 285, 20172687. 10.1098/rspb.2017.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epand R. M.; Vogel H. J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta, Biomembr. 1999, 1462, 11–28. 10.1016/S0005-2736(99)00198-4. [DOI] [PubMed] [Google Scholar]
- Nguyen L. T.; Haney E. F.; Vogel H. J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. 10.1016/j.tibtech.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Nicolas P. Multifunctional host defense peptides: intracellular-targeting antimicrobial peptides. FEBS J. 2009, 276, 6483–6496. 10.1111/j.1742-4658.2009.07359.x. [DOI] [PubMed] [Google Scholar]
- Lohner K.; Sevcsik E.; Pabst G.. Liposome-Based Biomembrane Mimetic Systems: Implications for Lipid-Peptide Interactions. In Advances in Planar Lipid Bilayers and Liposomes, 1st ed.; Vol 6; Leitmannova Liu A., Ed.; Academic Press: Waltham, MA, USA, 2008; pp 103–132. 10.1016/S1554-4516(07)06005-X. [DOI] [Google Scholar]
- Yeaman M. R.; Yount N. Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- Miller S. I. Antibiotic resistance and regulation of the Gram-negative bacterial outer membrane barrier by host innate immune molecules. mBio 2016, 7, e01541-16. 10.1128/mBio.01541-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy T. J.; Kahne D.; Walker S. The bacterial cell envelope. Perspect. Biol. 2010, 2, a000414–a00429. 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder D. S.; McIntosh T. J. The lipopolysaccharide barrier: correlation of antibiotic susceptibility with antibiotic permeability and fluorescent probe binding kinetics. Biochemistry 2000, 39, 11777–11787. 10.1021/bi000810n. [DOI] [PubMed] [Google Scholar]
- Rosenfeld Y.; Barra D.; Simmaco M.; Shai Y.; Mangoni M. L. A synergism between temporins toward gram-negative bacteria overcomes resistance imposed by the lipopolysaccharide protective layer. J. Biol. Chem. 2006, 281, 28565–28574. 10.1074/jbc.M606031200. [DOI] [PubMed] [Google Scholar]
- LaRock C. N.; Nizet V. Cationic antimicrobial peptide resistance mechanisms of streptococcal pathogens. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 3047–3054. 10.1016/j.bbamem.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maria-Neto S.; de Almeida K. C.; Macedo M. L. R.; Franco O. L. Understanding bacterial resistance to antimicrobial peptides: From the surface to deep inside. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 3078–3088. 10.1016/j.bbamem.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Matamouros S.; Miller S. I. S. Typhimurium strategies to resist killing by cationic antimicrobial peptides. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 3021–3025. 10.1016/j.bbamem.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan B. H.; Gaynord J.; Rowe S. M.; Deingruber T.; Spring D. R. The multifaceted nature of antimicrobial peptides: Current synthetic chemistry approaches and future directions. Chem. Soc. Rev. 2021, 50, 7820–7880. 10.1039/D0CS00729C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. H.; Cho Y.; Lehrer R. I. Effects of pH and salinity on the antimicrobial properties of clavanins. Infect. Immun. 1997, 65, 2898–2903. 10.1128/iai.65.7.2898-2903.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H. L.; Yu H. Y.; Yip B. S.; Chih Y. H.; Liang C. W.; Cheng H. T.; Cheng J. W. Boosting salt resistance of short antimicrobial peptides. Antimicrob. Agents Chemother. 2013, 57, 4050–4052. 10.1128/AAC.00252-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman M. J.; Anderson G. M.; Stolzenberg E. D.; Kari U. P.; Zasloff M.; Wilson J. M. Human β-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997, 88, 553–560. 10.1016/S0092-8674(00)81895-4. [DOI] [PubMed] [Google Scholar]
- Rothstein D. M.; Spacciapoli P.; Tran L. T.; Xu T.; Roberts F. D.; Dalla Serra M.; Buxton D. K.; Oppenheim F. G.; Friden P. Anticandida activity is retained in P-113, a 12-amino-acid fragment of histatin 5. Antimicrob. Agents Chemother. 2001, 45, 1367–1373. 10.1128/AAC.45.5.1367-1373.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falagas M. E.; Kasiakou S. K. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit. Care 2006, 10, R27. 10.1186/cc3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raguse T. L.; Porter E. A.; Weisblum B.; Gellman S. H. Structure– activity studies of 14-helical antimicrobial β-peptides: probing the relationship between conformational stability and antimicrobial potency. J. Am. Chem. Soc. 2002, 124, 12774–12785. 10.1021/ja0270423. [DOI] [PubMed] [Google Scholar]
- Ghosh C.; Haldar J. Membrane-active small molecules: designs inspired by antimicrobial peptides. ChemMedChem 2015, 10, 1606–1624. 10.1002/cmdc.201500299. [DOI] [PubMed] [Google Scholar]
- Molchanova N.; Hansen P. R.; Franzyk H. Advances in development of antimicrobial peptidomimetics as potential drugs. Molecules 2017, 22, 1430–1489. 10.3390/molecules22091430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Kizhakkedathu J. N.; Straus S. K. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8, 4. 10.3390/biom8010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q. Y.; Yan Z. B.; Meng Y. M.; Hong X. Y.; Shao G.; Ma J. J.; Cheng X. R.; Liu J.; Kang J.; Fu C. Y. Antimicrobial peptides: Mechanism of action, activity and clinical potential. Mil. Med. Res. 2021, 8, 48. 10.1186/s40779-021-00343-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentilucci L.; De Marco R.; Cerisoli L. Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. 10.2174/138161210793292555. [DOI] [PubMed] [Google Scholar]
- Bottger R.; Hoffmann R.; Knappe D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS One 2017, 12, e0178943–0178957. 10.1371/journal.pone.0178943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold N.; Czihal P.; Fritsche S.; Sauer U.; Schiffer G.; Knappe D.; Alber G.; Hoffmann R. Novel apidaecin 1b analogs with superior serum stabilities for treatment of infections by gram-negative pathogens. Antimicrob. Agents Chemother. 2013, 57, 402–409. 10.1128/AAC.01923-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.; Liu K.; Zhao P.; Zhou J.; Zhang F.; He Y.; Zhang H.; Fareed M. S.; Lu Y.; Xu Y.; et al. The effects of incorporation of the counterparts and mimics of l-lysine on the antimicrobial activity, hemolytic activity, cytotoxicity and tryptic stability of antimicrobial peptide polybia-MPII. Amino Acids 2022, 54, 123–135. 10.1007/s00726-021-03099-0. [DOI] [PubMed] [Google Scholar]
- Nguyen L. T.; Chau J. K.; Perry N. A.; De Boer L.; Zaat S. A.; Vogel H. J. Serum stabilities of short tryptophan-and arginine-rich antimicrobial peptide analogs. PLoS One 2010, 5, e12684–12691. 10.1371/journal.pone.0012684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dathe M.; Nikolenko H.; Klose J.; Bienert M. Cyclization increases the antimicrobial activity and selectivity of arginine-and tryptophan-containing hexapeptides. Biochemistry 2004, 43, 9140–9150. 10.1021/bi035948v. [DOI] [PubMed] [Google Scholar]
- Oh D.; Sun J.; Nasrolahi Shirazi A.; LaPlante K. L.; Rowley D. C.; Parang K. Antibacterial activities of amphiphilic cyclic cell-penetrating peptides against multidrug-resistant pathogens. Mol. Pharmaceutics 2014, 11, 3528–3536. 10.1021/mp5003027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekaran P.; Kim E. Y.; Lee J.; Ryu E. K.; Shin S. Y.; Bang J. K. Synthesis of Fmoc-Triazine amino acids and its application in the synthesis of short antibacterial peptidomimetics. Int. J. Mol. Sci. 2020, 21, 3602–3622. 10.3390/ijms21103602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Kang D.; Choi J.; Huang W.; Wadman M.; Barron A. E.; Seo J. Effect of side chain hydrophobicity and cationic charge on antimicrobial activity and cytotoxicity of helical peptoids. Bioorg. Med. Chem. Lett. 2018, 28, 170–173. 10.1016/j.bmcl.2017.11.034. [DOI] [PubMed] [Google Scholar]
- Chongsiriwatana N. P.; Patch J. A.; Czyzewski A. M.; Dohm M. T.; Ivankin A.; Gidalevitz D.; Zuckermann R. N.; Barron A. E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2794–2799. 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhee S.; Hu Y.; Niu Y.; Bai G.; Wu H.; Costanza F.; West L.; Harrington L.; Shaw L. N.; Cao C.; et al. Non-hemolytic α-AApeptides as antimicrobial peptidomimetics. Chem. Commun. 2011, 47, 9729–9731. 10.1039/c1cc13684d. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Yin G.; Yan Z.; Sang P.; Wang M.; Brzozowski R.; Eswara P.; Wojtas L.; Zheng Y.; Li X.; et al. Helical sulfono-γ-AApeptides with aggregation-induced emission and circularly polarized luminescence. J. Am. Chem. Soc. 2019, 141, 12697–12706. 10.1021/jacs.9b05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting D. S. J.; Beuerman R. W.; Dua H. S.; Lakshminarayanan R.; Mohammed I. Strategies in translating the therapeutic potentials of host defense peptides. Front. Immunol 2020, 11, 983–998. 10.3389/fimmu.2020.00983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura Y.; Nishida M.; Ogawa Y.; Takakura Y.; Matsuzaki K. Action mechanism of tachyplesin I and effects of PEGylation. Biochim. Biophys. Acta, Biomembr. 2007, 1768, 1160–1169. 10.1016/j.bbamem.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Singh S.; Papareddy P.; Morgelin M.; Schmidtchen A.; Malmsten M. Effects of PEGylation on membrane and lipopolysaccharide interactions of host defense peptides. Biomacromolecules 2014, 15, 1337–1345. 10.1021/bm401884e. [DOI] [PubMed] [Google Scholar]
- Albada H. B.; Prochnow P.; Bobersky S.; Langklotz S.; Schriek P.; Bandow J. E.; Metzler-Nolte N. Tuning the activity of a short Arg-Trp antimicrobial peptide by lipidation of a C-or N-terminal lysine side-chain. ACS Med. Chem. Lett. 2012, 3, 980–984. 10.1021/ml300148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makovitzki A.; Avrahami D.; Shai Y. Ultrashort antibacterial and antifungal lipopeptides. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15997–16002. 10.1073/pnas.0606129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T.; Wu D.; Li W.; Zheng X.; Li W.; Shan A. High specific selectivity and membrane-active mechanism of synthetic cationic hybrid antimicrobial peptides based on the peptide FV7. Int. J. Mol. Sci. 2017, 18, 339–355. 10.3390/ijms18020339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri M.; Hancock R. E.. High-performance liquid chromatography and mass spectrometry-based design of proteolytically stable antimicrobial peptides. Antimicrobial Peptides. Methods in Molecular Biology, Vol. 1548; Hansen P., Ed.; Humana Press: New York, 2017; pp 61–71. 10.1007/978-1-4939-6737-7_5. [DOI] [PubMed] [Google Scholar]
- Bagheri M.; Arasteh S.; Haney E. F.; Hancock R. E. Tryptic stability of synthetic bactenecin derivatives is determined by the side chain length of cationic residues and the peptide conformation. J. Med. Chem. 2016, 59, 3079–3086. 10.1021/acs.jmedchem.5b01740. [DOI] [PubMed] [Google Scholar]
- Karstad R.; Isaksen G.; Wynendaele E.; Guttormsen Y.; De Spiegeleer B.; Brandsdal B. O.; Svendsen J. S.; Svenson J. Targeting the S1 and S3 subsite of trypsin with unnatural cationic amino acids generates antimicrobial peptides with potential for oral administration. J. Med. Chem. 2012, 55, 6294–6305. 10.1021/jm3002058. [DOI] [PubMed] [Google Scholar]
- Henklein P.; Bruckdorfer T. Shorter arginine homologues to stabilize peptides towards tryptic digestion. Chim. Oggi (Chem. Today) 2008, 26, 12–15. [Google Scholar]
- Chen C.; Hu J.; Zeng P.; Chen Y.; Xu H.; Lu J. R. High cell selectivity and low-level antibacterial resistance of designed amphiphilic peptide G (IIKK) 3I-NH2. ACS Appl. Mater. Interfaces 2014, 6, 16529–16536. 10.1021/am504973d. [DOI] [PubMed] [Google Scholar]
- Arias M.; Piga K. B.; Hyndman M. E.; Vogel H. J. Improving the activity of Trp-rich antimicrobial peptides by Arg/Lys substitutions and changing the length of cationic residues. Biomolecules 2018, 8, 19–35. 10.3390/biom8020019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L. T.; de Boer L.; Zaat S. A.; Vogel H. J. Investigating the cationic side chains of the antimicrobial peptide tritrpticin: hydrogen bonding properties govern its membrane-disruptive activities. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 2297–2303. 10.1016/j.bbamem.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Russell A. L.; Williams B. C.; Spuches A.; Klapper D.; Srouji A. H.; Hicks R. P. The effect of the length and flexibility of the side chain of basic amino acids on the binding of antimicrobial peptides to zwitterionic and anionic membrane model systems. Bioorg. Med. Chem. 2012, 20, 1723–1739. 10.1016/j.bmc.2012.01.015. [DOI] [PubMed] [Google Scholar]
- Lu J.; Xu H.; Xia J.; Ma J.; Xu J.; Li Y.; Feng J. D- and unnatural amino acid substituted antimicrobial peptides with improved proteolytic resistance and their proteolytic degradation characteristics. Front. Microbiol 2020, 11, 563030. 10.3389/fmicb.2020.563030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit G.; Ilyas H.; Ghosh S.; Bidkar A. P.; Mohid S. A.; Bhunia A.; Satpati P.; Chatterjee S. Insights into the mechanism of antimicrobial activity of seven-residue peptides. J. Med. Chem. 2018, 61, 7614–7629. 10.1021/acs.jmedchem.8b00353. [DOI] [PubMed] [Google Scholar]
- Aoki W.; Ueda M. Characterization of antimicrobial peptides toward the development of Novel Antibiotics. Pharmaceuticals. 2013, 6, 1055–81. 10.3390/ph6081055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S.; Pandit G.; Debnath S.; Chatterjee S.; Satpati P. Effect of monovalent salt concentration on the secondary structure in peptide-micelle binding. RSC Adv. 2021, 11, 36836–36849. 10.1039/D1RA06772A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit G.; Chowdhury N.; Abdul Mohid S.; Bidkar A. P.; Bhunia A.; Chatterjee S. Effect of secondary structure and side chain length of hydrophobic amino acid residues on the antimicrobial activity and toxicity of 14-residue-long de novo AMPs. ChemMedChem 2021, 16, 355–367. 10.1002/cmdc.202000550. [DOI] [PubMed] [Google Scholar]
- Pandit G.; Biswas K.; Ghosh S.; Debnath S.; Bidkar A. P.; Satpati P.; Bhunia A.; Chatterjee S. Rationally designed antimicrobial peptides: Insight into the mechanism of eleven residue peptides against microbial infections. Biochim. Biophys. Acta, Biomembr. 2020, 1862, 183177–183192. 10.1016/j.bbamem.2020.183177. [DOI] [PubMed] [Google Scholar]
- Romoli O.; Mukherjee S.; Mohid S. A.; Dutta A.; Montali A.; Franzolin E.; Brady D.; Zito F.; Bergantino E.; Rampazzo C.; et al. Enhanced silkworm cecropin B antimicrobial activity against Pseudomonas aeruginosa from single amino acid variation. ACS Infect. Dis. 2019, 5, 1200–1213. 10.1021/acsinfecdis.9b00042. [DOI] [PubMed] [Google Scholar]
- Gaglione R.; Cesaro A.; Dell’Olmo E.; Della Ventura B.; Casillo A.; Di Girolamo R.; Velotta R.; Notomista E.; Veldhuizen E. J.; Corsaro M. M.; et al. Effects of human antimicrobial cryptides identified in apolipoprotein B depend on specific features of bacterial strains. Sci. Rep. 2019, 9, 6728. 10.1038/s41598-019-43063-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder S.; Yadav K. K.; Sarkar R.; Mukherjee S.; Saha P.; Haldar S.; Karmakar S.; Sen T. Alteration of zeta potential and membrane permeability in bacteria: a study with cationic agents. SpringerPlus 2015, 4, 672. 10.1186/s40064-015-1476-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L.The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, 2017.
- Gfeller D.; Michielin O.; Zoete V. SwissSidechain: a molecular and structural database of non-natural sidechains. Nucleic Acids Res. 2012, 41, D327–D332. 10.1093/nar/gks991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Jo S.; Lee H. S.; Klauda J. B.; Im W. CHARMM-GUI micelle builder for pure/mixed micelle and protein/micelle complex systems. J. Chem. Inf. Model. 2013, 53, 2171–2180. 10.1021/ci4002684. [DOI] [PubMed] [Google Scholar]
- Mackerell A. D. Jr Molecular dynamics simulation analysis of a sodium dodecyl sulfate micelle in aqueous solution: decreased fluidity of the micelle hydrocarbon interior. J. Phys. Chem. 1995, 99, 1846–1855. 10.1021/j100007a011. [DOI] [Google Scholar]
- Rakitin A. R.; Pack G. R. Molecular dynamics simulations of ionic interactions with dodecyl sulfate micelles. J. Phys. Chem. B 2004, 108, 2712–2716. 10.1021/jp030914i. [DOI] [Google Scholar]
- Wang Q.; Hong G.; Johnson G. R.; Pachter R.; Cheung M. S. Biophysical properties of membrane-active peptides based on micelle modeling: a case study of cell-penetrating and antimicrobial peptides. J. Phys. Chem. B 2010, 114, 13726–13735. 10.1021/jp1069362. [DOI] [PubMed] [Google Scholar]
- Lebecque S.; Crowet J. M.; Nasir M. N.; Deleu M.; Lins L. Molecular dynamics study of micelles properties according to their size. J. Mol. Graphics Modell. 2017, 72, 6–15. 10.1016/j.jmgm.2016.12.007. [DOI] [PubMed] [Google Scholar]
- Abraham M.; van der Spoel D.; Lindahl E.; Hess B.; G. Development Team . GROMACS User Manual 2019; https://www.gromacs.org
- Best R. B.; Zhu X.; Shim J.; Lopes P. E.; Mittal J.; Feig M.; MacKerell A. D. Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durell S. R.; Brooks B. R.; Ben-Naim A. Solvent-induced forces between two hydrophilic groups. J. Phys. Chem. 1994, 98, 2198–2202. 10.1021/j100059a038. [DOI] [Google Scholar]
- Kabsch W.; Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Eisenhaber F.; Lijnzaad P.; Argos P.; Sander C.; Scharf M. The double cubic lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 1995, 16, 273–284. 10.1002/jcc.540160303. [DOI] [Google Scholar]
- Bondi A. v. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. 10.1021/j100785a001. [DOI] [Google Scholar]
- Origin Pro; OriginLab Corp.: Northampton, MA, USA, 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.