

Abstract
1,4-Dihydropyridines (1,4-DHPs) hold a top-notch position in the pharmaceutical world due to a broader spectrum of applications, whereas the carboxylic moiety has been an integral part of the physiological world, effective food preservatives, and antimicrobial agents. Seeking the enormous potential and applications of these two classes, we worked to combine these to synthesize 2,2′-[3,5-bis(ethoxycarbonyl)-4-phenyl-1,4-dihydropyridine-2,6-diyl]diacetic acid the novel dicarboxylic derivatives of 1,4-DHP (9a–k) achieved via the electro-carboxylation of tetrasubstituted-1,4-dihydropyridines (8a–k) derivatives using Mg–Pt electrodes in an undivided cell. The targeted compounds were established by 1H, 13C NMR, IR, and ESI-MS. Further, the synthesized compounds show excellent resistance against various microbes and the activity increased 2–3 folds after the introduction of acid groups. Compound 9b (against E. coli, S. aureus, B. subtilis, A. niger, and P. glabrum), 9d (against E. coli, K. pneumonia, S. aureus, A. janus, and F. oxysporum), 9f (against E. coli and P. fluorescens), and 9k (against F. oxysporum and P. glabrum) were found to be highly active at 4 μg/mL with reference to standard amoxicillin and fluconazole. Further, the present synthetic protocol would open new gates for other researchers to develop new molecules by bioisosteres of these substrates.
Introduction
1,4-Dihydropyridine (1,4-DHP) scaffold is a significant class of pharmacologically active molecules present in the natural system and biomimetic reducing agents because of their unprecedented and broad-spectrum biological applications. Extensive pharmaceutical research furnished an extended catalog of medicines containing 1,4-DHP as the nucleus, and it is evident by the existence in many commercially available drugs such as amlodipine1 (hypertension), nifedipine2 (treats high blood pressure), isradipine3 (calcium channel blocker), nimodipine4 (decreases brain damage), among other drugs. Pharmaceutical potencies of 1,4-dihydropyridines have also been studied in other significant areas like anticancer,5 antimutagenic,6 neuroprotective,7 growth stimulants,8 antioxidant,9 and radioprotective agents.10
Biological potencies of 1,4-DHP skeleton have been greatly enhanced by the structural modifications and these modifications influence the structure–activity relationship.11 Several electron-withdrawing groups (in circles) have been tested as direct substituents or on methylene groups at C-2 and C-6 positions (Scheme 1, Structure 1–6), and robust bioactivity was recorded.12−16
Scheme 1. 1,4-Dihydropyridine and Its Analogs Exhibiting Different Activities Containing Electron-Withdrawing Groups on C-2 or C-6 Positions.

The carboxylic group is one such ubiquitous moiety with valuable intrinsic chemical abilities that contributes to mammalian physiology and endogenous biochemical processes.17 Carboxylic acid ionizes and gives better solubility at pH 7.4 making it a suitable moiety for drug development as it can be easily metabolized (normal pH of blood 7.35–7.45).18 Other properties such as hydrogen bonding and pKa play a crucial role in the ligand-protein binding depending on inductive effect, neighboring groups, chain length, and so forth.19,20 In literature, more than 450 drugs are enlisted containing carboxylic group on aromatic21 (Diclofenac, ibuprofen, aspirin, and so forth), heterocyclic rings22 (Ciprofloxacin, Amoxicillin, Norfloxacin and Levofloxacin), statins23 (Epanova, Atorvastatin, Fluvastatin, and so forth), β-lactam antibiotics24 (Penicillin, Cephalosporins, Cephamycins, and so forth), fibrates25 (Gemfibrozil), NSAIDs26 (Indomethacin, Naproxen, and so forth), and food preservatives.27
1,4-Dihydropyridines substituted with carboxylic groups, that is, chelidemic acid and its analogs (5 and 6) exhibited several biological activities15,28,29 and are strong ligands to form complexes with several elements of the first transition series.30−33 However, lengthy protocols for the synthesis of chelidemic acid derivatives and substitutional constraints at the C-4 position limit potencies to an extent.
Considering the above facts and in continuation to our previous work11,34 a new route for synthesizing 1,4-DHPs substituted with carboxylic groups at C-2 and C-6 positions was planned to achieve highly efficient molecules against microbes. In the present study, an electrochemical synthetic route is adopted to achieve a carboxylic group in a single step from the halide derivatives of 1,4-DHP.
Results and Discussion
Screening of Reaction Conditions
Electrocarboxylation is a powerful and versatile approach for assembling different heterocyclic structures. The electrochemical synthesis of 9a–k from the dibromo series 8a–k in a constant current of CO2 follows the protocol illustrated in Scheme 2. An extensive literature survey and our previous work helped us establish the optimum conditions (selecting the electrode material, supporting electrolyte, temperature, pressure, and suitable solvent) to set up the experiment.34−37 Several factors play a significant role in product selectivity and better yield, for instance, the reaction was conducted with n-butanol, n-pentanol, and acetonitrile as solvent, as acetonitrile is a stronger proton donor that produced better yield in the process. Tetrapropyl ammonium bromide (TPAB), tetrapropyl ammonium chloride (TPAC), and trabutylammonium tetrafluoroborate (TBABF4) as an ionic solvent were tested, and TPAC in combination with acetonitrile exhibited higher efficiency. The reduction process becomes chemically irreversible when the solution gets saturated at 1 atm with CO2.38,39 Electrode material, a constant current, and an appropriate temperature are other factors that hold a significant value in obtaining the desired compounds. Various combinations of electrodes (Ni, Mg, and Al as a sacrificial anode) were critically investigated against Pt (as a cathode) and it was observed that Mg–Pt came out as the most suitable one for this transformation when tested with different solvents, current densities (10–20 mA/cm2), and temperatures (0–25 °C). At last, after a rigorous investigation the reaction was conducted at a current density of 15 mA/cm2 with Mg as the sacrificial anode having Pt as the cathode, TPAC taken as a supporting electrolyte, and acetonitrile as a solvent with continuous bubbling of CO2 at 1 atm and 20 °C temperature. Complete optimization details are available as Supporting Information.
Scheme 2. Synthetic Route for the Novel 1,4-Dihydropyridine Derivatives (9a–k).

Chemistry
Initially, series 7a–k was synthesized via one-pot Hantzsch condensation of ethyl acetoacetate, ammonium acetate, and various aldehydes, which we have reported earlier.40 The primary interpretation was done by the comparison of their melting point (MP) and later the spectrum helped in the proper illustration of the synthesized compounds. IR spectra of compound 7a in series 7 exhibits four major absorptions at 3341, 3064, 2982, and 1686 cm–1 corresponding to the N–H, Ar–H, C–H, and C=O groups, respectively. In 1H NMR spectrum (500 MHz, DMSO-d6), showed a signal at δ 8.80 represented the proton of N-1, a multiplet peak at δ 7.21–7.09 related to aromatic protons, a singlet peak at δ 4.85 corresponding a proton of C-4, a sharp singlet at δ 2.25 related to methyl groups at C-2 and C-6, while a multiplet peak at δ 4.02–3.94 and triplet at δ 1.13–1.11 to the protons of ethyl groups..13C NMR spectrum exhibited C=O, C-2, C-3, C-4, and methyl group on C-2 at δ 169.1, 145.7, 102.3, 42.64, and 17.32 respectively. The data is separately given in the Supporting Information.
In the second step, synthesized compounds were subjected to a slight excess of 2 equiv. moles of NBS using methanol as the solvent at room temperature41 as with a lesser amount of NBS and lower temperature a monosubstituted product is formed.42,43 This step produced diethyl 2,6-bis(bromomethyl)-4-substituted-1,4-dihydropyridine-3,5-dicarboxylate (8a–k) and a mechanism of the transformation is also reported;44 the synthesis was confirmed by two methyl groups disappearing from δ 2.25 and two methylene protons appearing from δ 3.96, also the shift of the singlet peak for a proton of N–H at δ 8.80 ppm, to δ 10.6. Results were verified with existing literature.43 Further, getting [M+2] and [M+4] peaks from the ESI-MS fragmentation confirms the presence of a dibromo compound. These bis(bromomethyl)- derivatives have been used to synthesize different compounds via nucleophilic substitution.45 Taking the same phenomenon with a different approach and in continuation with our previous work,34 the bis(bromomethyl)- derivatives were subjected to electrocarboxylation to obtain a series of novel dicarboxylic-1,4-dihydropyridines (9a–k). The product formation was smooth and without any side-product formation. Adequate spectroscopic techniques confirmed the formation of the desired compounds. In the IR spectra, an additional broad peak at 3517 cm–1 confirms −OH of carboxylic group, in the 1H NMR the strong downfield shift of N–H proton at δ 12.9, the occurrence of −OH peak at δ 11.0 for two symmetrical protons form bis-carboxylic group is a strong evidence of carboxylation. The upfield shift in the signal of methylene protons from δ 5.02 to δ 2.87 is due to the removal of the highly electronegative bromo group with carboxylic group. In 13C NMR two carbonyl signals were at δ 171.7 and 167.2 for carboxylic and ester group, respectively, whereas the other peaks were on their respective positions which confirms the synthesis of the desired compound.
A Plausible Mechanism for Synthesis of Dicarboxylic Derivatives (9a–k)
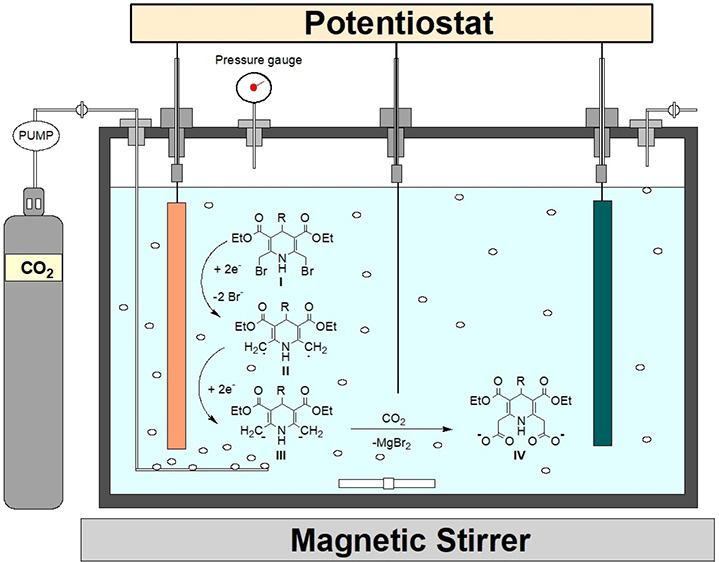
Several reports are available on electrocarboxylation of organic halides, where an undivided cell with magnesium as a stable sacrificial anode and platinum or silver as the cathode gives a much better yield and high carboxylation selectivity.46,47 In the reaction, a reactive radical is formed on reduction and dissociating the bromide ions. Such dissociated bromide ions immediately react with the anode to form MgBr2. Thereafter, the reactive intermediate-II undergoes another reduction to form an intermediate-III anion. This latter nucleophile-III attacks CO2 to yield carboxylate anion-IV (Figure 1). Lastly, the intermediate-IV takes protons from the solution during the workup to yield the final compound.
Figure 1.

Plausible reaction and mechanism for the conversion of dibromo derivatives into dicarboxylic compounds inside the electrochemical cell.
Antimicrobial Evaluation of series 7a–k and 9a–k
The synthesized compounds (7a–k and 9a–k) were subjected to various strains to explore the antimicrobial potencies by minimum inhibitory concentration (MIC) following the guidelines set by the Clinical and Laboratory Standards Institute (CLSI). The results obtained were analyzed in contrast with the reference drugs Amoxicillin and Fluconazole in their respective field at 4 μg/mL and 2 μg/mL respectively. Table 1 illustrated that series 7a–k exhibits moderate to good activity against Escherichia coli, Klebselliapneumonia, Pseudomonasaeruginosa, Staphylococcusaureus (Gram-negative), Bacillussubtilis, Pseudomonasfluorescens, Streptococcuspyogenes (Gram-positive), Aspergillusjanus, Aspergillusniger, Aspergillussclerotiorum, Fusariumoxysporum, and Pencilliumglabrum as fungal strain, whereas almost all of the compounds exhibited activity against E. coli, S. aureus, B. subtilis, and A. niger. However, compounds 7b, 7d, and 7f were more active than the others. To be more specific, 7b was active against E. coli, K. pneumonia, S. aureus, B. subtilis, P. fluorescens, A. janus, A. niger, F. oxysporum, and P. glabrum at 8 μg/mL. Compound 7d was more active against E. coli, P. aeruginosa, S. aureus, P. fluorescens, A. niger, A. sclerotiorum, and F. oxysporum at MIC 8 μg/mL, whereas 7f was active against bacterial species K. pneumonia, S. aureus, B. subtilis, and S. pyogenes only.
Table 1. Minimum Inhibitory Concentration (MIC in μg/mL) of Synthesized 7a–k Compounds against the Selected Penal of Microbial Agentsa.
| Gram-negative
bacteria |
Gram-positive
bacteria |
Fungi |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| compound | E. coli | K. pneumonia | P. aeruginosa | S. aureus | B. subtilis | P. fluorescens | S. pyogenes | A. janus | A. niger | A. sclerotiorum | F. oxysporum | P. glabrum |
| 7a | – | – | – | 32 | 32 | – | – | 32 | 16 | – | 32 | – |
| 7b | 8 | 8 | 16 | 8 | 8 | 8 | 16 | 8 | 8 | 16 | 8 | 8 |
| 7c | 16 | – | 16 | – | 16 | 16 | 32 | 32 | 32 | 16 | – | – |
| 7d | 8 | 16 | 8 | 8 | 16 | 8 | 16 | 16 | 8 | 8 | 8 | 16 |
| 7e | 16 | 32 | – | 16 | 16 | – | 16 | 32 | – | – | 32 | 32 |
| 7f | 32 | 8 | 32 | 8 | 8 | 16 | 8 | – | 32 | 16 | 16 | 64 |
| 7g | 64 | – | – | 16 | 8 | 32 | 16 | 32 | 16 | – | – | – |
| 7h | – | – | 32 | – | 64 | – | – | – | 32 | 64 | – | – |
| 7i | 32 | 32 | – | 32 | 64 | 32 | – | – | 16 | 64 | 32 | – |
| 7j | – | – | – | 64 | 32 | – | – | – | 32 | – | – | – |
| 7k | 64 | 32 | – | 32 | 16 | – | – | 32 | 16 | 32 | 16 | 16 |
| amoxicillin | 4 | 4 | 4 | 4 | 4 | 4 | 4 | – | – | – | – | – |
| fluconazole | – | – | – | – | – | – | – | 2 | 2 | 2 | 2 | 2 |
Where “–” indicates no antimicrobial activity.
On the other hand, it was evident from Table 2 that after carboxylation the activity of certain compounds was enhanced by 2–3 folds, possibly by the hydrogen bonding and van der Waals interaction of the compounds (ligand) with the microbes (protein).15 Compound 9b was found to be equipotent with the reference drug at 4 μg/mL against E. coli, S. aureus, B. subtilis, A. niger, and P. glabrum. Compound 9d was active against E. coli, P. aeruginosa, S. aureus, A. janus, and F. oxysporum at MIC 4 μg/mL. Compound 9f was active against E. coli, B. subtilis, and P. fluorescens. The interesting observation was that compound 7k was moderately active against various microbial agents, however after carboxylation the corresponding compound 9k showcased exemplary activity especially against F. oxysporum and P. glabrum at 4 μg/mL.
Table 2. Minimum Inhibitory Concentration (MIC in μg/mL) of Synthesized 9a–k Compounds against the Selected Penal of Microbial Agentsa.
| Gram-negative
bacteria |
Gram-positive
bacteria |
Fungi |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| compound | E. coli | K. pneumonia | P. aeruginosa | S. aureus | B. subtilis | P. fluorescens | S. pyogenes | A. janus | A. niger | A. sclerotiorum | F. oxysporum | P. glabrum |
| 9a | – | – | – | 32 | 32 | – | 32 | 32 | 16 | – | 32 | – |
| 9b | 4 | 8 | 16 | 4 | 4 | 8 | 16 | 8 | 4 | 16 | 8 | 4 |
| 9c | 8 | 16 | 32 | 16 | 16 | 32 | 32 | 16 | 16 | – | – | |
| 9d | 4 | 4 | 8 | 4 | 8 | 16 | 16 | 4 | 8 | 8 | 4 | 8 |
| 9e | 8 | 32 | 32 | 8 | 16 | 16 | 16 | 32 | – | – | 32 | 16 |
| 9f | 4 | 8 | 16 | 8 | 8 | 4 | 8 | 16 | 8 | 16 | 8 | 8 |
| 9g | 16 | 16 | – | 16 | 8 | 32 | 16 | 16 | 16 | – | – | – |
| 9h | – | 32 | 32 | – | 32 | – | – | – | 32 | 32 | – | – |
| 9i | 16 | 8 | – | 16 | 16 | 8 | – | 32 | 16 | 32 | 16 | – |
| 9j | 32 | – | – | 16 | 32 | – | – | – | 8 | – | – | 32 |
| 9k | 16 | 16 | – | 8 | 16 | – | 16 | 8 | 8 | 8 | 4 | 4 |
| Amoxicillin | 4 | 4 | 4 | 4 | 4 | 4 | 4 | – | – | – | – | – |
| Fluconazole | – | – | – | – | – | – | – | 2 | 2 | 2 | 2 | 2 |
Where “–” indicates no antimicrobial activity.
Conclusion
In summary, we have reported the synthesis of novel dicarboxylic derivatives of 1,4-DHP (9a–k) via a highly efficient electrocarboxylation method. Carboxylic moiety substituted on methylene group at C-2 and C-6 position of the 1,4-DHP core activates the molecule by forming hydrogen bonds and assisting the ligand-protein interactions. The same was evident from the antimicrobial evaluation of the precursor compounds (7a–k) and final derivatives (9a–k). Results revealed that the activity of dicarboxylic substituted derivatives (9a–k) escalated by 2–3 folds and compounds 9b, 9d, and 9k at 4 μg/mL were found to be relatively more active than others against bacterial and fungal strains in comparison to reference drug Amoxicillin and Fluconazole, respectively. This active moiety can be further utilized to develop novel and unconventional derivatives via bioisosteres of the carboxylic group either by the replacement of hydroxyl moiety only or carbonyl and hydroxyl both. This synthetic protocol is very attractive and can be applied to construct a wide range of symmetrical molecules; hence, this opens the gates for new areas in research.
Materials and Methods
High-graded chemicals were purchased from Merck and Sigma-Aldrich and were used as such without further purification. Although solvents of HPLC grade were procured from Loba Chemie, acetonitrile was first distilled at 80–82 °C and kept in P2O5 in A4 molecular sieves overnight, followed by a second distillation to get pure and dry CH3CN, which was stored in a closed amber-colored bottle. Direct current for electrocarboxylation was supplied using an electrophoresis power supply (Toshniwal) and was fixed to a voltmeter (range 0–300 V) and an ammeter (0–100 mA). An undivided Pyrex glass cell was used having two different openings for cathode (platinum) and anode (magnesium) and third opening for continuous CO2 bubbling throughout the reaction. Digital melting point apparatus and open-end-capillary method was used for the melting point. PerkinElmer (Spectrum-II) with ATR mode for IR and Bruker Advanced NMR spectrometer for 1H NMR and 13C NMR data were used having DMSO as the solvent and TMS as the internal standard. The MS was recorded on LC-MS Spectrometer Model Q-ToF Micromass.
Experimental Section
Synthesis
Synthesize Diethyl 2,6-dimethyl-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate (7a–k)
Ethyl acetoacetate and benzaldehyde (2:1) were refluxed with the excess of ammonium acetate in the presence of glycerol as an environmentally benign solvent at 90 °C for 1–2 h;40 the reaction was allowed to cool down before working up in ice-cold water. The obtained yellow color solid was further recrystallized with ethanol and activated charcoal to obtain opaque white crystalline compound.
7a
Yield 94%, white crystalline solid, MP 158–159 °C. IR spectrum, υ, cm–1: 3341 (N–H), 3064 (Ar–H), 2982 (C–H) and 1686 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 8.80 (s, 1H, NH), 7.21–7.18 (t, J0 = 7.65 Hz, 2H, H-3′, H-5′), 7.15–7.13 (d, J0 = 8.40 Hz, 2H, H-2′ and H-6′), 7.10–7.09 (t, J0 = 7.15 Hz, 1H, H-4′), 4.85 (s, 1H, H-4), 4.12 (q, 4H, CH2CH3), 2.25 (s, 6H, CH3), 1.13–1.11 (t, 6H, CH2CH3). 13C NMR spectrum, δ, ppm: 166.8 (C=O), 141.9 (C-2 and C-6), 137.8 (C-1′), 129.0 (C-3′ and C-5′), 126.3 (C-2′ and C-6′), 124.5 (C-4′), 102.5 (C-3 and C-5), 61.4 (−CH2CH3), 42.6 (C-4), 16.5 (−CH3), 14.2 (−CH2CH3).
Synthesize Diethyl 2,6-bis(bromomethyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate (8a–k)
To synthesize compound 8a, 5 mmol of precursor 7a was dissolved in 50 mL of methanol and the reaction proceeded in the dark with the addition of 10 mmol of NBS (light-sensitive) in small fractions at ambient temperature, followed by stirring at room temperature for 3 h,41Scheme 3. The reaction was monitored using TLC with chloroform–methanol in 95:05 ratio. Obtained precipitates were processed for further reaction after washing with water. This general protocol was adopted to synthesize 8a–k (Table 3).
Scheme 3. Synthesis of Dibromo-1,4-dihydropyridine derivatives (8a–k) from Simple 1,4-Dihydropyridines (7a–k).

Table 3. Derivatives of Series 8 Synthesized with Various Aldehydes along with Obtained Results.
| entry | producta | R | yieldb (%) | Rf value | melting point (°C) |
|---|---|---|---|---|---|
| 1 | 8a | C6H5 | 67 | 0.76 | 158–15943,48 |
| 2 | 8b | 4-NO2-C6H4 | 74 | 0.53 | 172–173 |
| 3 | 8c | 4-Me-C6H4 | 69 | 0.72 | 148–150 |
| 4 | 8d | 4-OMe-C6H4 | 66 | 0.67 | 156–157 |
| 5 | 8e | 2-OH-C6H4 | 71 | 0.61 | 176–178 |
| 6 | 8f | 3-NO2-C6H4 | 70 | 0.57 | 167–16842 |
| 7 | 8g | 4-OH, 3-OCH3 C6H3 | 70 | 0.48 | 181–182 |
| 8 | 8h | C6H4-CH=CH | 66 | 0.65 | 164–165 |
| 9 | 8i | 2-pyridinyl | 42 | 0.61 | 145–148 |
| 10 | 8j | 2-furyl | 76 | 0.69 | 147–149 |
| 11 | 8k | 2-thiophenyl | 74 | 0.66 | 151–152 |
Products were characterized with standard and reliable spectral techniques.
Yield refers to the total extraction from different crops.
8a
Yield 67%, pale yellow solid, MP 158–159 °C. IR spectrum, υ, cm–1: 3181 (N–H), 3104 (Ar–H), 2974 (C–H) and 1713 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 10.63 (brs, 1H, NH), 7.31–7.25 (m, 4H, H-3′, H-5′, H-2′ and H-6′), 7.21–7.18 (t, 1H, H-4′), 5.02–4.89 (q, 4H, CH2CH3), 4.58 (s, 1H, H-4), 3.96 (s, 4H, CH2), 1.79 (t, 6H, CH2CH3). 13C NMR spectrum, δ, ppm: 168.4 (C=O), 144.7 (C-2 and C-6), 142.0 (C-1′), 129.2 (C-2′ and C-6′), 128.7 (C-3′ and C-5′), 125.4 (C-4′), 105.7 (C-3 and C-5), 61.6 (-CH2CH3), 41.9 (C-4), 36.2 (−CH2), 14.1 (−CH2CH3). Mass spectrum, m/z(Irel,%): 491 (M+4). Anal. Calcd for C19H21Br2NO4: C, 46.84; H, 4.34; N, 2.88. Found: C, 46.72; H, 4.31; N, 2.87%.
Synthesize 2,2′-[3,5-Bis(ethoxycarbonyl)-4-phenyl-1,4-dihydropyridine-2,6-diyl]diacetic acid (9a–k)
Precursor compound from series 8a–k (1 mmol) was added to 100 mL of acetonitrile containing TPAC (5 mmol) as the supporting electrolyte and an undivided cell having Pt as the inert cathode and Mg as the sacrificial anode to obtain series 9a–k (Table 4). A 15 mA/cm2 constant current density was maintained at 20 °C. A continuous supply of CO2 (bubbling) at 1 atmospheric pressure was maintained throughout the reaction period of 7–8 h, Scheme 4. The excessive solvent was removed using a rota-evaporator and the solid residue was collected upon the completion of the reaction. The desired compound was obtained after recrystallization with ethanol.
Table 4. Derivatives of Series 9 Synthesized with Various Aldehydes along with Obtained Results.
| entry | producta | R | yield (%) | Rf value | melting point (°C) |
|---|---|---|---|---|---|
| 1 | 9a | C6H5 | 91 | 0.45 | 219–220 |
| 2 | 9b | 4-NO2-C6H4 | 94 | 0.69 | 233–235 |
| 3 | 9c | 4-Me-C6H4 | 85 | 0.59 | 194–196 |
| 4 | 9d | 4-OMe-C6H4 | 89 | 0.52 | 209–210 |
| 5 | 9e | 2-OH-C6H4 | 83 | 0.64 | 221–222 |
| 6 | 9f | 3-NO2-C6H4 | 85 | 0.60 | 229–230 |
| 7 | 9g | 4-OH, 3-OCH3 C6H3 | 90 | 0.61 | 247–249 |
| 8 | 9h | C6H4-CH=CH | 81 | 0.53 | 216–217 |
| 9 | 9i | 2-pyridinyl | 64 | 0.66 | 209–210 |
| 10 | 9j | 2-furyl | 92 | 0.72 | 189–191 |
| 11 | 9k | 2-thiophenyl | 90 | 0.68 | 192–193 |
Products were characterized with standard and reliable spectral techniques.
Scheme 4. Synthesis of Dicarboxylic-1,4-dihydropyridine Derivatives (9a–k) from Dibromo-Derivatives (8a–k).

9a
Yield 91%, white solid, MP 218–219 °C. IR spectrum, υ, cm–1: 3517 (OH), 3343 (N–H), 3057 (Ar–H), 2984 (C–H) and 1687 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 12.9 (brs, 1H, NH), 11.0 (s, 2H, OH), 7.27–7.24 (dd, J0 = 7.50 Hz, 2H, H-3′ and H-5′), 7.17–7.14 (t, J0 = 7.50 Hz, 1H, H-4′), 7.10–7.08 (d, J0 = 8.40 Hz, 2H, H-2′, and H-6′), 5.53 (s, 1H), 4.08 (q, 4H, CH2CH3), 2.87 (s, 4H, CH2), 1.06 (t, 6H, CH2CH3). 13C NMR spectrum, δ, ppm: 171.7 (−COOH), 167.2 (C=O), 142.2 (C-2 and C-6), 138.1 (C-1′), 128.2 (C-3′ and C-5′), 126.8 (C-2′ and C-6′), 125.8 (C-4′), 102.3 (C-3 and C-5), 61.7 (-CH2CH3), 42.6 (C-4), 31.2 (−CH2), 14.2 (−CH2CH3). Mass spectrum, m/z (Irel,%): 418 (M+1). Anal. Calcd. for C21H23NO8, C, 60.43; H, 5.55; N, 3.36; found, C, 59.22; H, 5.43; N, 3.27%.
Antimicrobial Evaluation
To analyze the antimicrobial assay of the newly synthesized compounds 3a–k, they were subjected to a list of Gram-positive, Gram-negative, and fungal strains. Bacterial and fungal penal selected for this study is Escherichia coli (MTCC 443), Klebsellia pneumonia (MTCC 3384), Pseudomonas aeruginosa (MTCC 424), Staphylococcus aureus (MTCC 96) as Gram-negative, Bacillus subtilis (MTCC 441), Pseudomonas fluorescens (MTCC 103), Streptococcus pyogenes (MTCC 442) as Gram-positive, Aspergillus janus (MTCC 2751), Aspergillus niger (MTCC 281), Aspergillus sclerotiorum (MTCC 1008), Fusarium oxysporum (MTCC 2480), and Pencillium glabrum (MTCC 4951) as fungal strain. Bacterial samples were incubated at 37 °C for 24 h and the nutrient broth was utilized for their storage. On the other hand, malt extract at 28 °C for 72 h was used to grow the fungal strains before the inoculation. The triplicates of all the synthesized compounds on dissolving in DMSO were tested via a serial dilution method at concentrations of 128, 64, 32, 16, 8, 4, and 2 μg/mL. Amoxicillin (bacterial) and Fluconazole (fungal) at 4 and 2 μg/mL, respectively, were taken as reference drugs to compare the results and effectiveness of the compounds.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group no (RGP- 070). Special thanks go to Chandigarh University, Gharuan and Punjabi University, Patiala for providing basic facilities for research and liberal support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01316.
Optimization of conditions and characterization (1H NMR, 13C NMR, and MS) of synthesized compounds (PDF)
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript
The authors declare no competing financial interest.
Supplementary Material
References
- Fares H.; DiNicolantonio J. J.; O’Keefe J. H.; Lavie C. J. Amlodipine in Hypertension: A First-Line Agent with Efficacy for Improving Blood Pressure and Patient Outcomes. Open Hear. 2016, 3 (2), e000473. 10.1136/openhrt-2016-000473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancia G.; Parati G.; Bilo G.; Choi J.; Kilama M. O.; Ruilope L. M. Blood Pressure Control by the Nifedipine GITS-Telmisartan Combination in Patients at High Cardiovascular Risk: The TALENT Study. J. Hypertens. 2011, 29 (3), 600–609. 10.1097/HJH.0b013e328342ef04. [DOI] [PubMed] [Google Scholar]
- Johnson B. A.; Ait-Daoud N.; Wells L. T. Effects of Isradipine, a Dihydropyridine-Class Calcium Channel Antagonist, on D-Methamphetamine-Induced Cognitive and Physiological Changes in Humans. Neuropsychopharmacology 2000, 22 (5), 504–512. 10.1016/S0893-133X(99)00116-5. [DOI] [PubMed] [Google Scholar]
- Marinho M. M. F.; De Bruin V. M. S.; De Sousa F. C. F.; Aguiar L. M. V.; De Pinho R. S. N.; Viana G. S. B. Inhibitory Action of a Calcium Channel Blocker (Nimodipine) on Seizures and Brain Damage Induced by Pilocarpine and Lithium-Pilocarpine in Rats. Neurosci. Lett. 1997, 235 (1–2), 13–16. 10.1016/S0304-3940(97)00687-3. [DOI] [PubMed] [Google Scholar]
- Goto R. N.; Sobral L. M.; Sousa L. O.; Garcia C. B.; Lopes N. P.; Marín-Prida J.; Ochoa-Rodríguez E.; Verdecia-Reyes Y.; Pardo-Andreu G. L.; Curti C.; Leopoldino A. M. Anti-Cancer Activity of a New Dihydropyridine Derivative, VdiE-2N, in Head and Neck Squamous Cell Carcinoma. Eur. J. Pharmacol. 2018, 819, 198–206. 10.1016/j.ejphar.2017.12.009. [DOI] [PubMed] [Google Scholar]
- Ošiņa K.; Rostoka E.; Isajevs S.; Sokolovska J.; Sjakste T.; Sjakste N. Effects of an Antimutagenic 1,4-Dihydropyridine AV-153 on Expression of Nitric Oxide Synthases and DNA Repair-Related Enzymes and Genes in Kidneys of Rats with a Streptozotocin Model of Diabetes Mellitus. Basic Clin. Pharmacol. Toxicol. 2016, 119 (5), 458–463. 10.1111/bcpt.12617. [DOI] [PubMed] [Google Scholar]
- Michalska P.; Mayo P.; Fern C.; Tenti G.; Men J. C.; Le R. Profiles of Novel 1, 4-Dihydropyridine Derivatives for the Treatment of Alzheimer’s Disease. Antioxidants 2020, 9 (8), 650–669. 10.3390/antiox9080650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milkovic L.; Vukovic T.; Zarkovic N.; Tatzber F.; Bisenieks E.; Kalme Z.; Bruvere I.; Ogle Z.; Poikans J.; Velena A.; Duburs G. Antioxidative 1,4-Dihydropyridine Derivatives Modulate Oxidative Stress and Growth of Human Osteoblast-like Cells in Vitro. Antioxidants 2018, 7 (9), 123. 10.3390/antiox7090123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh R.; Maheswari S.; Murugesan S.; Sandhiya R.; Lalitha A. Catalyst-Free One-Pot Synthesis and Antioxidant Evaluation of Highly Functionalized Novel 1,4-Dihydropyridine Derivatives. Res. Chem. Intermed. 2015, 41 (11), 8233–8243. 10.1007/s11164-014-1887-z. [DOI] [Google Scholar]
- Donkor I. O.; Zhou X.; Schmidt J.; Agrawal K. C.; Kishore V. Synthesis and Radioprotective Effects of Adamantyl Substituted 1,4-Dihydropyridine Derivatives. Bioorg. Med. Chem. 1998, 6 (5), 563–568. 10.1016/S0968-0896(98)00017-0. [DOI] [PubMed] [Google Scholar]
- Malhi D. S.; Kaur M.; Sohal H. S. Effect of Substitutions on 1, 4-Dihdropyridines to Achieve Potential Anti-Microbial Drugs: A Review. ChemistrySelect 2019, 4 (38), 11321–11336. 10.1002/slct.201902354. [DOI] [Google Scholar]
- Nordqvist A.; Granberg K. L.. Mineralocorticoid Receptor Antagonists, 1st ed.; Elsevier Inc., 2019; Vol. 109. [DOI] [PubMed] [Google Scholar]
- Muñoz-Torrero D.; Lavilla R.; Pérez-Areales F. J.; Ghashghaei O. Multicomponent Reactions: A Mighty Journey Partner for Infectious Tropical Disease Drug Discovery. Annu. Rep. Med. Chem. 2019, 53, 181–217. 10.1016/bs.armc.2019.05.005. [DOI] [Google Scholar]
- Nasrollahi S. M. H.; Ghasemzadeh M. A.; Zolfaghari M. R. Synthesis and Antibacterial Evaluation of Some New 1,4-Dihydropyridines in the Presence of Fe3O4@silica Sulfonic Acid Nanocomposite as Catalyst. Acta Chim. Slov. 2018, 65 (1), 199–207. 10.17344/acsi.2017.3820. [DOI] [PubMed] [Google Scholar]
- Credille C. V.; Morrison C. N.; Stokes R. W.; Dick B. L.; Feng Y.; Sun J.; Chen Y.; Cohen S. M. SAR Exploration of Tight-Binding Inhibitors of Influenza Virus PA Endonuclease. J. Med. Chem. 2019, 62 (21), 9438–9449. 10.1021/acs.jmedchem.9b00747. [DOI] [PubMed] [Google Scholar]
- Marchalín Š.; Valigura D.; Varečka L.; Lakatoš B.; Vaneková M.; Baran P.; Lawson A. M.; Daïch A. Synthesis of Novel Chiral 1,4-Dihydropyridinyl Schiff-Base Ligands, with Characterization and Evaluation of Calcium Channel Blocker Activity. Monatshefte fur Chemie 2014, 145 (5), 835–847. 10.1007/s00706-014-1164-2. [DOI] [Google Scholar]
- Vázquez J. A.; Durán A.; Rodríguez-Amado I.; Prieto M. A.; Rial D.; Murado M. A. Evaluation of Toxic Effects of Several Carboxylic Acids on Bacterial Growth by Toxicodynamic Modelling. Microb. Cell Fact. 2011, 10, 100. 10.1186/1475-2859-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z. J.; Roesner J. M.; Lutz R.; Liang Y.; Baker J.; Smith D. M.; Fan P. W. Carboxylesterase Catalyzed 18O-Labeling of Carboxylic Acid and Its Potential Application in LC-MS/MS Based Quantification of Drug Metabolites. Drug Metab. Pharmacokinet. 2019, 34 (5), 308–316. 10.1016/j.dmpk.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Coelho J. F.; Ferreira P. C.; Alves P.; Cordeiro R.; Fonseca A. C.; Góis J. R.; Gil M. H. Drug Delivery Systems: Advanced Technologies Potentially Applicable in Personalized Treatments 2010, 1, 164–209. 10.1007/s13167-010-0001-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunta M. J. R. It Is Important to Compute Intramolecular Hydrogen Bonding in Drug Design?. Am. J. Model. Optim. 2017, 5 (1), 24–57. 10.12691/ajmo-5-1-3. [DOI] [Google Scholar]
- Lou Y.; Zhu J. Carboxylic Acid Nonsteroidal Anti-Inflammatory Drugs (NSAIDs). Bioact. Carboxylic Compd. Classes Pharm. Agrochem. 2016, 221–236. 10.1002/9783527693931.ch16. [DOI] [Google Scholar]
- Asahina Y.; Araya I.; Iwase K.; Iinuma F.; Hosaka M.; Ishizaki T. Synthesis and Antibacterial Activity of the 4-Quinolone-3-Carboxylic Acid Derivatives Having a Trifluoromethyl Group as a Novel n-1 Substituent. J. Med. Chem. 2005, 48 (9), 3443–3446. 10.1021/jm040204g. [DOI] [PubMed] [Google Scholar]
- Nicholls S. J.; Lincoff A. M.; Bash D.; Ballantyne C. M.; Barter P. J.; Davidson M. H.; Kastelein J. J. P.; Koenig W.; McGuire D. K.; Mozaffarian D.; Pedersen T. R.; Ridker P. M.; Ray K.; Karlson B. W.; Lundström T.; Wolski K.; Nissen S. E. Assessment of Omega-3 Carboxylic Acids in Statin-Treated Patients with High Levels of Triglycerides and Low Levels of High-Density Lipoprotein Cholesterol: Rationale and Design of the STRENGTH Trial. Clin. Cardiol. 2018, 41 (10), 1281–1288. 10.1002/clc.23055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elander R. P. Industrial Production of β-Lactam Antibiotics. Appl. Microbiol. Biotechnol. 2003, 61 (5–6), 385–392. 10.1007/s00253-003-1274-y. [DOI] [PubMed] [Google Scholar]
- Kimoto E.; Li R.; Scialis R. J.; Lai Y.; Varma M. V. S. Hepatic Disposition of Gemfibrozil and Its Major Metabolite Gemfibrozil 1-O-β-Glucuronide. Mol. Pharmaceutics 2015, 12 (11), 3943–3952. 10.1021/acs.molpharmaceut.5b00411. [DOI] [PubMed] [Google Scholar]
- Lucas S. The Pharmacology of Indomethacin. Headache 2016, 56 (2), 436–446. 10.1111/head.12769. [DOI] [PubMed] [Google Scholar]
- Corrales M.; Fernández A.; Han J. H.. Antimicrobial Packaging Systems, 2nd ed.; Academic Press, 2013. [Google Scholar]
- El Bakali J.; Muccioli G. G.; Renault N.; Pradal D.; Body-Malapel M.; Djouina M.; Hamtiaux L.; Andrzejak V.; Desreumaux P.; Chavatte P.; Lambert D. M.; Millet R. 4-Oxo-1,4-Dihydropyridines as Selective CB2 Cannabinoid Receptor Ligands: Structural Insights into the Design of a Novel Inverse Agonist Series. J. Med. Chem. 2010, 53 (22), 7918–7931. 10.1021/jm100286k. [DOI] [PubMed] [Google Scholar]
- Loiseau P. M. Action of DNA-Gyrase Inhibiting Derivatives of 4-Oxo-1, 4-Dihydro-3-Pyridinecarboxylic Acid against Trypanosoma Brucei. Int. J. paracytology 1995, 25 (03), 403–405. 10.1016/0020-7519(94)00106-X. [DOI] [PubMed] [Google Scholar]
- Aghabozorg H.; Firoozi N.; Roshan L.; Eshtiagh-Hosseini H.; Salimi A. R.; Mirzaei M.; Ghanbari M.; Shamsipur M.; Ghadermazi M. Supramolecular Structure of Calcium(II) Based on Chelidamic Acid: An Agreement between Theoretical and Experimental Studies. J. Iran. Chem. Soc. 2011, 8 (4), 992–1005. 10.1007/BF03246555. [DOI] [Google Scholar]
- Mirzaei M.; Eshtiagh-Hosseini H.; Shamsipur M.; Saeedi M.; Ardalani M.; Bauzá A.; Mague J. T.; Frontera A.; Habibi M. Importance of Polarization Assisted/Resonance Assisted Hydrogen Bonding Interactions and Unconventional Interactions in Crystal Formations of Five New Complexes Bearing Chelidamic Acid through a Proton Transfer Mechanism. RSC Adv. 2015, 5 (89), 72923–72936. 10.1039/C5RA09526C. [DOI] [Google Scholar]
- Ramić E.; Eichel R. A.; Dinse K. P.; Titz A.; Schmidt B. Complexation of Copper(II)-Chelidamate: A Multifrequency-Pulsed Electron Paramagnetic Resonance and Electron Nuclear Double Resonance Analysis. J. Phys. Chem. B 2006, 110 (41), 20655–20663. 10.1021/jp061940u. [DOI] [PubMed] [Google Scholar]
- Eshtiagh-Hosseini H.; Mirzaei M.; Zarghami S.; Bauzá A.; Frontera A.; Mague J. T.; Habibi M.; Shamsipur M. Crystal Engineering with Coordination Compounds of 2,6-Dicarboxy-4- Hydroxypyridine and 9-Aminoacridine Fragments Driven by Different Nature of the Face-to-Face Π···π Stacking. CrystEngComm 2014, 16 (7), 1359–1377. 10.1039/C3CE41730A. [DOI] [Google Scholar]
- Singh K.; Sohal H. S.; Singh B. Synthesis of α-Hydroxycarboxylic Acids from Various Aldehydes and Ketones by Direct Electrocarboxylation: A Facile, Efficient and Atom Economy Protocol. Asian J. Chem. 2021, 33 (4), 839–845. 10.14233/ajchem.2021.23090. [DOI] [Google Scholar]
- Ran C. K.; Liao L. L.; Gao T. Y.; Gui Y. Y.; Yu D. G. Recent Progress and Challenges in Carboxylation with CO2. Curr. Opin. Green Sustain. Chem. 2021, 32, 100525. 10.1016/j.cogsc.2021.100525. [DOI] [Google Scholar]
- Dell’Amico L.; Bonchio M.; Companyó X. Recent Advances in Electrochemical Carboxylation of Organic Compounds for CO 2 Valorization. CO2 as a Build. Block Org. Synth. 2020, 3, 225–252. 10.1002/9783527821952.ch7. [DOI] [Google Scholar]
- Long Ngo H.; Kumar Mishra D.; Mishra V.; Chien Truong C. Recent Advances in the Synthesis of Heterocycles and Pharmaceuticals from the Photo/Electrochemical Fixation of Carbon Dioxide. Chem. Eng. Sci. 2021, 229, 116142. 10.1016/j.ces.2020.116142. [DOI] [Google Scholar]
- Reche I.; Mena S.; Gallardo I.; Guirado G. Electrocarboxylation of Halobenzonitriles: An Environmentally Friendly Synthesis of Phthalate Derivatives. Electrochim. Acta 2019, 320, 134576. 10.1016/j.electacta.2019.134576. [DOI] [Google Scholar]
- Scialdone O.; Galia A.; Isse A. A.; Gennaro A.; Sabatino M. A.; Leone R.; Filardo G. Electrocarboxylation of Aromatic Ketones: Influence of Operative Parameters on the Competition between Ketyl and Ring Carboxylation. J. Electroanal. Chem. 2007, 609 (1), 8–16. 10.1016/j.jelechem.2007.02.014. [DOI] [Google Scholar]
- Sohal H. S.; Goyal A.; Sharma R.; Khare R. One-pot, Multicomponent Synthesis of Symmetrical Hantzsch 1,4-dihydropyridine Derivatives Using Glycerol as Clean and Green Solvent. Eur. J. Chem. 2014, 5 (1), 171–175. 10.5155/eurjchem.5.1.171-175.943. [DOI] [Google Scholar]
- Petrova M.; Muhamadejev R.; Vigante B.; Duburs G.; Liepinsh E. Intramolecular Hydrogen Bonds in 1,4-Dihydropyridine Derivatives. R. Soc. open Sci. 2018, 5 (6), 180088–180102. 10.1098/rsos.180088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenouz Y. R. M.; Moshtaghi A. Selective Mono Bromination of 1,4-Dihydropryidines. Iran. J. Chem. Chem. Eng. 1997, 16 (1), 29–32. 10.30492/IJCCE.1997.10330. [DOI] [Google Scholar]
- Petrova M.; Muhamadejev R.; Vigante B.; Cekavicus B.; Plotniece A.; Duburs G.; Liepinsh E. Intramolecular C-H···O Hydrogen Bonding in 1,4-Dihydropyridine Derivatives. Molecules 2011, 16 (9), 8041–8052. 10.3390/molecules16098041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova M.; Muhamadejev R.; Cekavicus B.; Vigante B.; Plotniece A.; Sobolev A.; Duburs G.; Liepinsh E. Experimental and Theoretical Studies of Bromination of Diethyl 2, 4, 6-Trimethyl-1, 4-dihydropyridine-3, 5-dicarboxylate. Heteroat. Chem. 2014, 25 (2), 114–126. 10.1002/hc.21145. [DOI] [Google Scholar]
- Kulakov I. V.; Turdybekov D. M. Synthesis and Crystal Structure of 2, 6-Bis (N-Cytisinomethyl)-4-Phenyl-1, 4-Dihydropyridine-3, 5-Dicarboxylic Acid Diethyl Ester. Chem. Heterocycl. Compd. 2010, 46 (7), 839–843. 10.1007/s10593-010-0591-1. [DOI] [Google Scholar]
- Matthessen R.; Fransaer J.; Binnemans K.; De Vos D. E. Electrocarboxylation: Towards Sustainable and Efficient Synthesis of Valuable Carboxylic Acids. Beilstein J. Org. Chem. 2014, 10, 2484–2500. 10.3762/bjoc.10.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S. F.; Wang H.; Lan Y. C.; Liu X.; Lu J. X.; Zhang J. Influences of the Operative Parameters and the Nature of the Substrate on the Electrocarboxylation of Benzophenones. J. Electroanal. Chem. 2012, 664, 105–110. 10.1016/j.jelechem.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziaie M.; Dilmaghani K. A.; Tukmechi A. Synthesis and Biological Evaluation of 1, 2, 4-Triazoles and 1, 3, 4-Oxadiazoles Derivatives Linked to 1, 4-Dihydropyridines Scaffold. Acta Chim. Slov. 2017, 64 (4), 895–901. 10.17344/acsi.2017.3506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.