

Abstract
Background
Aminoglycoside-containing regimens may be an effective treatment option for infections caused by carbapenem-resistant Klebsiella pneumoniae (CR-Kp), but aminoglycoside-resistance genes are common in these strains. The relationship between the aminoglycoside-resistance genes and aminoglycoside MICs remains poorly defined.
Objectives
To identify genotypic signatures capable of predicting aminoglycoside MICs for CR-Kp.
Methods
Clinical CR-Kp isolates (n = 158) underwent WGS to detect aminoglycoside-resistance genes. MICs of amikacin, gentamicin, plazomicin and tobramycin were determined by broth microdilution (BMD). Principal component analysis was used to initially separate isolates based on genotype. Multiple linear regression was then used to generate models that predict aminoglycoside MICs based on the aminoglycoside-resistance genes. Last, the performance of the predictive models was tested against a validation cohort of 29 CR-Kp isolates.
Results
Among the original 158 CR-Kp isolates, 91.77% (145/158) had at least one clinically relevant aminoglycoside-resistance gene. As a group, 99.37%, 84.81%, 82.28% and 10.76% of the CR-Kp isolates were susceptible to plazomicin, amikacin, gentamicin and tobramycin, respectively. The first two principal components explained 72.23% of the total variance in aminoglycoside MICs and separated isolates into four groups with aac(6′)-Ib, aac(6′)-Ib′, aac(6′)-Ib+aac(6′)-Ib′ or no clinically relevant aminoglycoside-resistance genes. Regression models predicted aminoglycoside MICs with adjusted R2 values of 56%–99%. Within the validation cohort, the categorical agreement when comparing the observed BMD MICs with the predicated MICs was 96.55%, 89.66%, 86.21% and 82.76% for plazomicin, gentamicin, amikacin and tobramycin, respectively.
Conclusions
Susceptibility to each aminoglycoside varies in CR-Kp. Detection of aminoglycoside-resistance genes may be useful to predict aminoglycoside MICs for CR-Kp.
Introduction
Carbapenem-resistant Klebsiella pneumoniae (CR-Kp) pose an urgent public health risk given the limited treatment options and high morbidity and mortality associated with serious infections.1 The plasmids containing carbapenemases, such as the K. pneumoniae carbapenemase (encoded by blaKPC) and/or MBL (e.g. encoded by blaNDM), also frequently co-harbour genes that confer resistance to other classes of antibiotics, including aminoglycosides.2–6 Genes encoding aminoglycoside-resistance determinants, such as aminoglycoside-modifying enzymes (AMEs) and 16S ribosomal RNA methyltransferases (16S-RMTases), are especially prevalent in CR-Kp and have been detected in >95% of clinical isolates.3,6–8
AMEs function by inactivating aminoglycosides through enzymatic modification of specific positions on the antibiotic chemical structure.4,8–12 AMEs include three subclasses of enzymes known as acetyltransferases (AACs), phosphotransferases (APHs) and nucleotidyltransferases (ANTs), which modify specific elements of the 2-deoxystreptamine (DOS) aminoglycoside core structure or its sugar components.3,11 Non-AME 16S-RMTases, while not as common, are also an emergent concern because they can prevent a wide variety of aminoglycosides from binding to the ribosome.7,8 16S-RMTases, such as ArmA and RmtA, confer high-level resistance to aminoglycosides by methylation of the aminoglycoside binding site on the 16S subunit of the 30S ribosome, which can prevent drug binding.7,8,13 Unlike 16S-RMTases, which impede all of the most clinically relevant aminoglycosides (amikacin, gentamicin, plazomicin and tobramycin) from binding to the ribosome, each AME confers resistance to only a subset of the aminoglycosides.14,15
The specificity of each AME for some, but not all, of the aminoglycosides presents a potential therapeutic window of opportunity, whereby an aminoglycoside could be theoretically selected based on the AMEs harboured by a specific CR-Kp isolate. Previous studies have investigated whether the presence of some individual AMEs predict aminoglycoside resistance in Enterobacterales.16–19 However, the relationship between CR-Kp genotypes and aminoglycoside MICs remains poorly defined. Identification of aminoglycoside MICs, not just a determination of susceptibility, is critical in the clinical setting to optimize the pharmacokinetics/pharmacodynamics of an appropriate aminoglycoside for each patient. Therefore, the purpose of this study was to determine the influence of all clinically relevant aminoglycoside-resistance genes in our collection of CR-Kp isolates on aminoglycoside MICs.
Materials and methods
Bacterial isolates and WGS
Susceptibility testing and model generation were performed using clinical K. pneumoniae isolates that were resistant to at least one carbapenem (CR-Kp). To derive the models capable of predicting aminoglycoside MICs, 169 clinical CR-Kp isolates were utilized. WGS and assembly had been performed and described previously (BioProject PRJNA395086).20 Assembled genomes were used to identify clonal relatedness and to detect aminoglycoside-resistance genes. kSNP v3.01 was used to produce a core genome single-nucleotide variant alignment from the genome assemblies.21 The core genome was defined as loci with a base present in at least 95% (161) of the 169 isolates. Pairwise comparisons of the core genomes of each isolate were performed using custom software (https://github.com/egonozer/snp_tools/blob/master/ksnp_matrix_to_diff_matrix.pl) to count the number of nucleotide differences between each pair of isolates. Isolates were considered near-clonal and excluded from the study if they had ≤10 nucleotide differences within the core genome compared with an isolate that was already included.
Antimicrobial resistance genes were identified by aligning the CR-Kp genomes against the NCBI Bacterial Antimicrobial Resistance Reference Gene Database (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA313047) and ResFinder v3.122 using BLAST. In order to test the performance of the models, a validation cohort consisting of 29 different CR-Kp isolates from the CDC & FDA Antibiotic Resistance Isolate Bank that previously underwent WGS was used.23 Antimicrobial resistance genes for isolates in the validation cohort were verified by performing a BLAST search with the accessed genomes for each organism against the ResFinder 3.1 database. Aminoglycoside-resistance genes for all isolates were then manually inspected to identify those with frameshift mutations and to detect alleles of aac(6′)-Ib, such as aac(6′)-Ib′, which were not detected by ResFinder 3.1.15
Antibiotic susceptibility testing
Amikacin (Lot# SLBT0718), gentamicin (Lot# SLBT5354) and tobramycin (Lot# SLBS8814) were obtained from Sigma–Aldrich (St Louis, MO, USA) and a commercial plazomicin solution was used (Achaogen, Inc., San Francisco, CA, USA). Drug stocks were filter-sterilized and used within 24 h of reconstitution. The MIC of each aminoglycoside for all CR-Kp isolates was determined via broth microdilution (BMD) according to CLSI guidelines.24 Interpretative criteria for amikacin, gentamicin and tobramycin were based on CLSI guidelines, whereas FDA breakpoints were used for plazomicin (CLSI susceptibility breakpoints: amikacin, ≤16 mg/L; gentamicin/tobramycin, ≤4 mg/L; and plazomicin, ≤2 mg/L).25 Subsequent analyses were also performed using EUCAST and United States Committee on Antimicrobial Susceptibility Testing (USCAST) aminoglycoside breakpoints. MIC values of >64 and ≤0.125 mg/L were replaced by 128 and 0.125 mg/L, respectively, for analysis.
Principal component analysis (PCA)
To get a general sense of how different aminoglycoside-resistance genes influence aminoglycoside MICs, a PCA was performed using the MIC data of the four tested aminoglycosides (amikacin, gentamicin, plazomicin and tobramycin) for the 158 CR-Kp isolates. The MICs were log2-transformed and CR-Kp isolates clustered according to their positions on the first two eigenvectors, which explained most of the variance in aminoglycoside MICs. The PCA was performed using a correlation matrix using the Pearson correlation coefficient. The two eigenvectors with the highest eigenvalues were used as the x- and y-axes for the correlation circle and final PCA plot. The analysis was performed in XLSTAT (version 2019.3.2).
Aminoglycoside MIC prediction model construction
Multiple linear regression via the backwards stepwise approach was utilized to evaluate the relationship between aminoglycoside-resistance genes and the phenotypic MICs of amikacin, gentamicin, plazomicin and tobramycin. Based on previous studies, all aminoglycoside-resistance genes that were identified during molecular analysis of the 158 CR-Kp isolates from the model derivation cohort and known to affect the activity of amikacin, gentamicin, plazomicin or tobramycin were initially entered into the models.7,15,26,27 The clinically relevant aminoglycoside-resistance genes initially entered into the models were aac(3)-II, aac(3)-IV, aac(6′)-Ib, aac(6′)-Ib′, aac(6′)-Ib-cr, ant(2′′)-I, aph(3′)-I, aph(3′)-II and rmtF. Aminoglycoside-resistance genes that were identified in the CR-Kp isolates, but not considered for model development, since they did not modify amikacin, gentamicin, plazomicin or tobramycin, were ant(3′′)-Ia, aph(4)-Ia, aph(6)-Ia and aph(6)-Id. A stepping criterion of F > 0.10 was used for model removal. A P value of ≤0.05 was considered statistically significant in the final model. Model performance was assessed via the adjusted R2 (aR2) value. Collinearity was assessed via tolerance and variance inflation factor. Outliers, highly influential values and leverage points were not removed from models as they were true observed values. All analyses were performed using SPSS Version 26 (IBM Corporation, Armonk, NY, USA).
The ability for each model to prospectively predict the aminoglycoside MICs was evaluated using the 29 CR-Kp isolates in the validation cohort. Predicted MICs for CR-Kp isolates in the validation cohort were calculated for each aminoglycoside by rounding the result from the regression model to the nearest standard MIC dilution. Using the observed BMD MIC as the reference value, essential agreement (EA) was defined as the predicted (regression) MIC within ±1 log2 dilution of the observed (BMD) MIC. We also evaluated the ability for the model to accurately categorize isolates as aminoglycoside susceptible, intermediate or resistant based on CLSI, EUCAST and USCAST breakpoints.25 Categorical agreement (CA) was defined as having the same susceptible, intermediate or resistant interpretation for the observed and predicted susceptibility category. Minor errors (mEs) were identified when the predicted MIC was defined as intermediate, but the observed MIC was susceptible or resistant, or vice versa. Major errors (MEs) were defined as false resistance (reported resistant by the regression equation when the BMD MIC was susceptible). Very major errors (VMEs) were defined as false susceptibility (reported as susceptible by the regression equation when the BMD MIC was resistant). Finally, sensitivity and specificity were calculated based on Equation 1 and Equation 2, respectively. Adequate performance of the linear regression models was defined as having sensitivity and specificity values ≥80.00%.
Equation 1: sensitivity = [isolates with non-susceptible predicted and phenotypic MICs/(isolates with non-susceptible predicted and phenotypic MICs + isolates with non-susceptible predicted MICs and susceptible phenotypic MICs)] × 100
Equation 2: specificity = [isolates with susceptible predicted and phenotypic MICs/(isolates with susceptible predicted and phenotypic MICs + isolates with susceptible predicted MICs and non-susceptible phenotypic MICs)] × 100
Results
Prevalence of antibiotic-resistance genes
Eleven of the 169 CR-Kp isolates were removed from the study, since they were clonally related to another isolate, leaving 158 CR-Kp unique isolates in the model derivation cohort. Carbapenemase genes were detected in >90% of the CR-Kp isolates; 145 isolates had blaKPC-3 (91.77%), 2 isolates had blaKPC-2 and 1 isolate had blaOXA-232. Among them, 91.77% (145/158) had at least one clinically relevant aminoglycoside-resistance gene. The most prevalent aminoglycoside-resistance gene, aac(6′)-Ib, was present in 70.25% (111/158) of isolates. The next most common aminoglycoside-resistance genes were aac(6′)-Ib′ (20.89%; 33/158), aph(3′)-I (17.72%; 28/158), aac(3)-II (6.33%; 10/158), aac(6′)-Ib-cr (4.43%; 7/158) and ant(2′′)-I (3.80%; 6/158). Less than 2% of isolates harboured aac(3)-IV, aph(3′)-II or rmtF, while 8.23% (13/158) of CR-Kp in the derivation cohort had no clinically relevant aminoglycoside-resistance genes. Other clinically relevant aminoglycoside-resistance genes not detected include aac(3)-I, aph(3′)-VI and other 16S-RMTase genes. Similar to the derivation cohort, 72.41% (21/29) of isolates in the validation cohort harboured aac(6′)-Ib. The next most common genes in the validation cohort were aac(6′)-Ib-cr (41.38%; 12/29), aac(3)-II (27.59%; 8/29), aph(3′)-I (24.14%; 7/29) and rmtF (20.69%; 6/29). Less than 7% of isolates harboured aac(3)-IV, aac(6′)-Ib′, ant(2′′)-I or aph(3′)-II. One isolate in the validation cohort had no clinically relevant aminoglycoside-resistance genes.
MIC analysis
Against the 158 non-clonally related clinical CR-Kp isolates in the model derivation cohort, plazomicin (MIC50 = 0.25 mg/L, MIC90 = 0.5 mg/L) was the most active aminoglycoside, where 99.37% of isolates were susceptible at an MIC of ≤2 mg/L (Table 1). There were 84.81% and 82.28% of CR-Kp susceptible to amikacin (MIC50 = 8 mg/L, MIC90 = 32 mg/L) and gentamicin (MIC50 = 0.5 mg/L, MIC90 = 16 mg/L), respectively. Only 10.76% of CR-Kp isolates were susceptible to tobramycin (MIC50 = 16 mg/L, MIC90 = 32 mg/L).
Table 1.
Activity of aminoglycosides against tested CR-Kp isolates (n = 158)
| Aminoglycoside | MIC (mg/L) |
Categorical interpretation (%) |
|||||
|---|---|---|---|---|---|---|---|
| MIC50 | MIC90 | modal MIC | MIC range | susceptible | intermediate | resistant | |
| Amikacin | 8 | 32 | 16 | 0.25 to >64 | 84.81 | 13.92 | 1.27 |
| Gentamicin | 0.5 | 16 | 0.5 | ≤0.125 to >64 | 82.28 | 3.16 | 14.56 |
| Plazomicin | 0.25 | 0.5 | ≤0.125 | ≤0.125 to 64 | 99.37 | 0.00 | 0.63 |
| Tobramycin | 16 | 32 | 8 | ≤0.125 to >64 | 10.76 | 37.97 | 51.27 |
PCA plots
The PCA of the log2-transformed aminoglycoside MICs showed that the first two principal components (PC1 and PC2) explained 72.23% of the total variance in MICs of the four tested compounds for 158 CR-Kp clinical isolates. In the PCA loading plot, the tobramycin MICs had the largest positive loading effect on PC1 (Figure 1). Gentamicin MICs had a relatively equal positive influence on PC1 and PC2, whereas amikacin MICs had a relatively equal negative influence on PC1 and PC2. Plazomicin MICs had the smallest influence on PC1 and PC2. The PCA loading plot also revealed that gentamicin and plazomicin MICs were correlated (small angle between vectors), while amikacin and gentamicin MICs were not correlated (∼90° angle between vectors).
Figure 1.
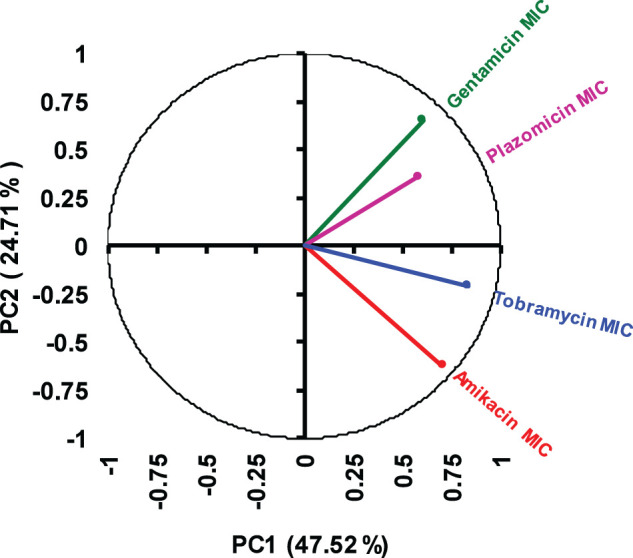
PCA loading plot of the log2-transformed MIC data of the aminoglycosides for 158 CR-Kp clinical isolates. The plot shows how each aminoglycoside MIC influenced the first and second principal components (PC1 and PC2), which together explained 72.23% of the total variance within the dataset. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
The PCA plot generally separated the CR-Kp isolates into four overlapping clusters considering the bacterial genotype and the aminoglycoside MICs (Figure 2). The four clusters corresponded to CR-Kp isolates with (i) aac(6′)-Ib, (ii) aac(6′)-Ib′, (iii) aac(6′)-Ib+aac(6′)-Ib′ or (iv) no clinically relevant aminoglycoside-resistance genes. The lone isolate with rmtF likely marks the approximate location of a fifth cluster for isolates with 16S-RMTases, though this could not be confirmed, since there was only one isolate with an rmtF gene.
Figure 2.

PCA plot based on the MICs of four aminoglycosides for 158 CR-Kp clinical isolates. Each symbol represents an individual CR-Kp isolate. CR-Kp with aac(6′)-Ib are coloured blue, CR-Kp with aac(6′)-Ib′ are coloured red, CR-Kp with aac(6′)-Ib+aac(6′)-Ib′ are coloured yellow and CR-Kp without any of the listed aminoglycoside-resistance genes are coloured green.
Multiple linear regression analysis
Multiple linear regression analysis adequately identified the aminoglycoside-resistance genes that significantly modified the amikacin, gentamicin, plazomicin and tobramycin MICs for CR-Kp isolates (Table 2). The amikacin MICs were significantly modified by aac(3)-IV, aac(6′)-Ib, aac(6′)-Ib′, aph(3′)-I and rmtF. Gentamicin MICs were significantly increased by aac(3)-II, aac(3)-IV, ant(2′′)-I and rmtF. For plazomicin, rmtF was the only gene that altered the MIC. Tobramycin MICs were significantly increased by aac(3)-II, aac(3)-IV, aac(6′)-Ib, ant(2′′)-I and rmtF. The aminoglycoside-resistance gene that increased the aminoglycoside MICs by the greatest magnitude was rmtF, which caused predicted MIC increases of >50 mg/L for all aminoglycosides.
Table 2.
Multiple linear regression equations to predict aminoglycoside MICs based on the aminoglycoside-resistance genes
| Aminoglycoside | Interceptb | Aminoglycoside-resistance genesa |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| aac(3)-II | aac(3)-IV | aac(6′)-Ib | aac(6′)-Ib′ | ant(2′′)-I | aph(3′)-I | rmtF | |||
| Amikacin | βc | 5.8 | –d | 11.1 | 9.0 | −3.7 | – | 3.6 | 122.2 |
| 95% CI | 2.3–9.3 | – | 0.1–22.1 | 5.2–12.7 | −7.9–0.5 | – | −0.4–7.5 | 103.4–141.1 | |
| P | <0.001 | – | 0.048 | <0.001 | 0.084 | – | 0.076 | <0.001 | |
| final equatione | amikacin MIC = 5.8+[11.1×aac(3)-IV]+[9.0×aac(6′)-Ib]+[−3.7×aac(6′)-Ib′]+[3.6×aph(3′)-I]+[122.2×rmtF], aR2 = 0.56 | ||||||||
| Gentamicin | β | 2.1 | 68.1 | 24.9 | – | – | 41.1 | – | 57.8 |
| 95% CI | 0.4–4.6 | 58.0–78.2 | 7.8–41.9 | – | – | 27.8–54.4 | – | 26.9–88.6 | |
| P | 0.092 | <0.001 | 0.005 | – | – | <0.001 | – | <0.001 | |
| final equation | gentamicin MIC = 2.1+[68.1×aac(3)-II]+[24.9×aac(3)-IV]+[41.1×ant(2′′)-I]+[57.8×rmtF], aR2 = 0.64 | ||||||||
| Plazomicin | β | 0.3 | – | – | – | – | – | – | 127.7 |
| 95% CI | 0.3–0.2 | – | – | – | – | – | – | 127.2–128.2 | |
| P | <0.0001 | – | – | – | – | – | – | <0.001 | |
| final equation | plazomicin MIC = 0.3+[127.7×rmtF], aR2 = 0.99 | ||||||||
| Tobramycin | β | 7.1 | 12.4 | 115.0 | 8.8 | – | 49.9 | – | 108.5 |
| 95% CI | 2.9–11.4 | 2.5–22.2 | 98.4–131.7 | 3.7–13.8 | – | 36.9–62.9 | – | 78.3–138.8 | |
| P | 0.001 | 0.014 | <0.001 | 0.001 | – | <0.001 | – | <0.001 | |
| final equation | tobramycin MIC = 7.1+[12.4×aac(3)-II]+[115.0×aac(3)-IV]+[8.8×aac(6′)-Ib]+[49.9×ant(2′′)-I]+[108.5×rmtF], aR2 = 0.66 | ||||||||
aac(6′)-Ib-cr and aph(3′)-II were not retained in any of the multivariate models.
Intercept is the aminoglycoside MIC value when none of the aminoglycoside-resistance genes present in the final model is present in an isolate.
β is the estimated increase in aminoglycoside MIC (mg/L) caused by the presence of the aminoglycoside-resistance gene.
Indicates that this gene was not retained in the multivariate model.
Final equations predict the MIC of each aminoglycoside depending on which aminoglycoside-resistance genes are present; input ‘1’ in place of the aminoglycoside-resistance gene name if an isolate has that gene or input a ‘0’ if it does not have the gene.
The linear regression models were adequately able to predict the aminoglycoside MICs for the 29 CR-Kp isolates in the prospective validation cohort (Table 3). The model predicted BMD MIC values for each isolate in the validation cohort as reported in Table S1 (available as Supplementary data at JAC Online). Using CLSI aminoglycoside breakpoints (FDA breakpoints for plazomicin), the plazomicin model performed the best, with 96.55% EA and CA compared with the reference BMD MICs. The plazomicin model’s sensitivity and specificity were both >95% and it had ME and VME rates of 0% and an mE rate of 3.45%. For the amikacin, gentamicin and tobramycin models, sensitivity and specificity were between 84.21% and 100% (tobramycin specificity could not be calculated). The amikacin model had 72.41% and 86.21% EA and CA, respectively, and, although the mE and VME rates were >10%, there were no MEs. Despite a CA of 89.66%, the gentamicin model only had an EA compared with the BMD MIC of 55.17%. For the 13 isolates that did not have an accurate MIC prediction, the median difference between the predicted MIC range and the actual MIC was 0.875 mg/L. There were 6.90% mEs and 8.33% MEs for the gentamicin model, but none of the isolates displayed a VME. For the tobramycin model, EA and CA were 82.76%. There were no VMEs, but the mE and ME rates were >10%.
Table 3.
Performance of linear regression model for predicting aminoglycoside MICs for 29 CR-Kp isolates in the prospective validation cohort using CLSI breakpoints (FDA breakpoints were used for plazomicin)
| Amikacin | Gentamicin | Plazomicin | Tobramycin | |
|---|---|---|---|---|
| EA | 72.41% | 55.17% | 96.55% | 82.76% |
| CA | 86.21% | 89.66% | 96.55% | 82.76% |
| Sensitivity | 90.00% | 94.44% | 100.00% | 89.66% |
| Specificity | 84.21% | 100.00% | 95.65% | −a |
| mE | 10.34% | 6.90% | 3.45% | 13.79% |
| ME | 0.00% | 8.33% | 0.00% | 33.33% |
| VME | 14.29% | 0.00% | 0.00% | 0.00% |
Could not be calculated, since there were no isolates predicted to be tobramycin susceptible.
The linear regression models also performed well for the 29 CR-Kp isolates in the prospective validation cohort when EUCAST (FDA breakpoints for plazomicin) or USCAST breakpoints were applied (Tables S2 and S3).
Discussion
Aminoglycosides are an important treatment option for CR-Kp, since the majority of isolates are susceptible to one or more of the aminoglycosides. In this study, 96.84% of CR-Kp isolates were susceptible to at least two aminoglycosides. The WGS and PCA findings revealed that AME-encoding genes aac(6′)-Ib and aac(6′)-Ib′ were prevalent and influential regarding aminoglycoside MICs for CR-Kp isolates. Linear regression models were able to predict the BMD aminoglycoside MICs with sensitivity and specificity values ≥84.21% for all models.
Defining the relationship between the bacterial genotype and resistance phenotype may improve antibiotic selection for each patient’s infection. For example, aminoglycoside-resistance genes that are determined to influence aminoglycoside MICs could be integrated into molecular rapid diagnostic panels that can detect their presence in clinical isolates. Rapid diagnostic tests consistently shorten the time it takes to administer active antibiotic therapy and can improve clinical outcomes.28,29 Thus, a molecular-based approach could estimate aminoglycoside MICs 1–2 days before traditional MICs become available. This may also improve aminoglycoside selection for use in combinations against CR-Kp.30 Furthermore, unlike traditional MIC testing, which is typically only performed on a subset of aminoglycosides for an isolate, a molecular testing approach in conjunction with a robust MIC prediction model could rapidly predict MICs of all aminoglycosides. Specific MICs can be predicted based on a molecular test, while clinical MIC testing often does not detect a precise MIC (e.g. MIC >16 mg/L). The specific MIC can be important for selection of an optimal dose.
The gentamicin, plazomicin and tobramycin linear regression models in the present study only retained aminoglycoside-resistance genes previously shown to confer resistance to each of these aminoglycosides, as expected.27 Plazomicin is not vulnerable to modification by the AMEs presently known to be harboured by K. pneumoniae; therefore, its model only retained the 16S-RMTase gene.11 Interestingly, the amikacin model retained aac(3)-IV and indicates that it increases the amikacin MIC by an average of 11.1 mg/L. The AME encoded by aac(3)-IV does not acetylate amikacin and its inclusion in the final model may be an artefact of the small number of isolates that harboured this gene in the derivation cohort (1.90%; 3/158). Two of these aac(3)-IV isolates were amikacin non-susceptible, but they also co-harboured aac(6′)-Ib and aph(3′)-I, which could both account for the increased amikacin MIC. Interestingly, each of the aac(3)-IV genes had a frameshift mutation that led to a premature stop codon near the 3′ end of the gene. Since mutations occurring near the 3′ end of the gene often do not lead to loss of function,31 we coded these genes as functional in our analysis. However, additional studies would be required to confirm that the AAC(3)-IV enzymes in these isolates were active.
There were also a few aminoglycoside-resistance genes not included in the final models that encode enzymes expected to increase aminoglycoside MICs. The aac(6′)-Ib-cr gene encodes for an AME that acetylates amikacin, tobramycin and some of the gentamicin components (acetylates gentamicin C1a and C2, but not gentamicin C1),32 but was not retained in any of the final models. This was likely in part due to the low prevalence of aac(6′)-Ib-cr (4.43%; 7/158) and wide range of amikacin, gentamicin and tobramycin MICs for the CR-Kp isolates with aac(6′)-Ib-cr. The lower aminoglycoside acetylation rate by AAC(6′)-Ib-cr compared with AAC(6′)-Ib and the inter-isolate enzyme expression variability have previously been shown to affect aminoglycoside MICs for isolates with aac(6′)-Ib-cr33,34 and may also explain why this gene was not retained in the final models, while aac(6′)-Ib was. It is not surprising that the gene for APH(3′)-II was not retained by any model, since this AME has a high Km value for amikacin and does not phosphorylate gentamicin, plazomicin or tobramycin. It has only rarely been shown to confer amikacin resistance in situations where there is high-level gene expression and/or impaired aminoglycoside uptake.35,36 The exclusion of aac(6′)-Ib′ from the gentamicin and tobramycin models was unexpected, since the AME product of this gene has been shown to cause 4 to 7 log2 increases in amikacin and tobramycin MICs.37–39 In the present study, CR-Kp with aac(6′)-Ib′ had gentamicin and tobramycin MICs that were within 1 log2 dilution of isolates without aac(6′)-Ib′. Isolates with aac(6′)-Ib′ were ∼3–5 times less likely to co-harbour another gene predicted to cause gentamicin or tobramycin resistance. These disparate rates of co-harboured aminoglycoside-resistance genes may have increased baseline MICs for the isolates without aac(6′)-Ib′, making it difficult to pinpoint the influence of aac(6′)-Ib′. Future studies may include samples enriched with aminoglycoside-resistance genes that were under-represented in the current study.
Despite the mis-incorporation of some aminoglycoside-resistance genes, the aminoglycoside models adequately predicted the BMD MICs for isolates in the prospective validation cohort. CA was >80% for each model, while sensitivity and specificity were between 84.21% and 100%. However, there was some mis-specification of the models. Only the plazomicin model had EA and CA values ≥90%, which CLSI defines as its cut-off value for a clinically acceptable assay to determine MICs.40 Both plazomicin and gentamicin models had mE rates below the CLSI cut-off of ≤10%, while amikacin and tobramycin fell above this threshold and only the amikacin and plazomicin models displayed ME rates <3%. The gentamicin and tobramycin models each had higher false resistance rates (ME >3%), which indicates that there could be aminoglycoside-resistance genes included in the model that are not equally expressed in all CR-Kp isolates. The gentamicin, plazomicin and tobramycin models met the CLSI criteria for acceptable VME rates of <3%.40 The elevated rate of false susceptibility for the amikacin model (VME = 14.29%), however, indicates that there may be additional variables that need to be incorporated into this model to improve its accuracy, such as the estimated gene copy number. The models generally performed slightly better when using EUCAST or USCAST breakpoints, which may be more accurate.41
To the best of our knowledge, this is the first study to attempt to build models that predict aminoglycoside MICs specifically for CR-Kp. CR-Kp often simultaneously co-harbour multiple aminoglycoside-resistance genes; approximately 1/3 of the isolates in the derivation cohort of the present study had at least two clinically relevant aminoglycoside-resistance genes. The presence of multiple aminoglycoside-resistance genes may have different phenotypic effects on the MICs compared with isolates that harbour the genes individually. Most of the previous studies that have built phenotypic prediction models have attempted to classify isolates categorically as susceptible or resistant to a single aminoglycoside, but not predict specific MICs. For example, several studies with Enterobacterales have shown that gentamicin resistance can be adequately predicted by the presence of specific aminoglycoside-resistance genes.16–19 Long et al.42 used machine learning in a large collection of ESBL-producing K. pneumoniae to develop models that sufficiently predicted which isolates were resistant to amikacin, gentamicin or tobramycin (F1 scores >85). There has been at least one attempt to predict aminoglycoside MICs based on genotype. Nguyen et al.43 used machine learning and targeted resistance gene approaches to predict MICs of 20 antibiotics, including amikacin, gentamicin and tobramycin, for K. pneumoniae that were mostly carbapenem susceptible (>70%). The models performed well and the EA rates for each aminoglycoside were ≥95%; however, there were few amikacin-resistant isolates in the dataset, which led to a VME rate of 29.8% for the amikacin model. Interestingly, they found that the machine-learning approach did not significantly improve the model compared with the method that only incorporated known antibiotic-resistance genes. The present study expands on this growing body of evidence, suggesting that genotypic models may be useful to predict aminoglycoside activity.
In conclusion, models using aminoglycoside-resistance genes to predict aminoglycoside MICs were developed and showed reasonable accuracy for CR-Kp. Only rmtF was retained in the final plazomicin model, while the amikacin, gentamicin and tobramycin models each retained at least four aminoglycoside-resistance genes. Prospective validation experiments revealed that the plazomicin model displayed the best performance overall. Although our aminoglycoside MIC prediction models are preliminary, they are an encouraging step toward clinical translation and improve our understanding of the effect of aminoglycoside-resistance genes on each aminoglycoside against CR-Kp.
Data availability
Sequencing reads and assembled genomes have been submitted to GenBank under BioProject PRJNA395086.
Funding
The project was funded by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust (to Z.P.B. and A.R.H.). This project has also been funded in part by the National Institutes of Health (NIH), under Grants R01AI118257, U19AI135964, K24AI04831 and R21AI153953 (all to A.R.H.). Z.P.B. was supported in part by the National Center for Advancing Translational Sciences, NIH, under Grant KL2TR002002.
Transparency declarations
E.W. serves on the speaker’s bureau for Allergan plc, Melinta Therapeutics and Astellas Pharma, and on the advisory board for GenMark Diagnostics and Shionogi. All other authors: none to declare.
Disclaimer
The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Supplementary data
Tables S1 to S3 are available as Supplementary data at JAC Online.
Supplementary Material
References
- 1.Logan LK, Weinstein RA.. The epidemiology of carbapenem-resistant Enterobacteriaceae: the impact and evolution of a global menace. J Infect Dis 2017; 215 Suppl 1: S28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kitchel B, Rasheed JK, Patel JBet al. Molecular epidemiology of KPC-producing Klebsiella pneumoniae isolates in the United States: clonal expansion of multilocus sequence type 258. Antimicrob Agents Chemother 2009; 53: 3365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castanheira M, Deshpande LM, Woosley LNet al. Activity of plazomicin compared with other aminoglycosides against isolates from European and adjacent countries, including Enterobacteriaceae molecularly characterized for aminoglycoside-modifying enzymes and other resistance mechanisms. J Antimicrob Chemother 2018; 73: 3346–54. [DOI] [PubMed] [Google Scholar]
- 4.Eljaaly K, Alharbi A, Alshehri Set al. Plazomicin: a novel aminoglycoside for the treatment of resistant Gram-negative bacterial infections. Drugs 2019; 79: 243–69. [DOI] [PubMed] [Google Scholar]
- 5.Richter SS, Marchaim D.. Screening for carbapenem-resistant Enterobacteriaceae: who, when, and how? Virulence 2017; 8: 417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathers AJ, Stoesser N, Sheppard AEet al. Klebsiella pneumoniae carbapenemase (KPC)-producing K. pneumoniae at a single institution: insights into endemicity from whole-genome sequencing. Antimicrob Agents Chemother 2015; 59: 1656–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw KJ, Rather PN, Hare RSet al. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev 1993; 57: 138–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doi Y, Wachino JI, Arakawa Y.. Aminoglycoside resistance: the emergence of acquired 16S Ribosomal RNA methyltransferases. Infect Dis Clin North Am 2016; 30: 523–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haidar G, Alkroud A, Cheng Set al. Association between the presence of aminoglycoside-modifying enzymes and in vitro activity of gentamicin, tobramycin, amikacin, and plazomicin against Klebsiella pneumoniae carbapenemase- and extended-spectrum-β-lactamase-producing Enterobacter species. Antimicrob Agents Chemother 2016; 60: 5208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bremmer DN, Clancy CJ, Press EGet al. KPC-producing Klebsiella pneumoniae strains that harbor AAC(6′)-Ib exhibit intermediate resistance to amikacin. Antimicrob Agents Chemother 2014; 58: 7597–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krause KM, Serio AW, Kane TRet al. Aminoglycosides: an overview. Cold Spring Harb Perspect Med 2016; 6: a027029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shields RK, Clancy CJ, Press EGet al. Aminoglycosides for treatment of bacteremia due to carbapenem-resistant Klebsiella pneumoniae. Antimicrob Agents Chemother 2016; 60: 3187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zárate SG, De la Cruz Claure ML, Benito-Arenas Ret al. Overcoming aminoglycoside enzymatic resistance: design of novel antibiotics and inhibitors. Molecules 2018; 23: 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miro E, Grunbaum F, Gomez Let al. Characterization of aminoglycoside-modifying enzymes in Enterobacteriaceae clinical strains and characterization of the plasmids implicated in their diffusion. Microb Drug Resist 2013; 19: 94–9. [DOI] [PubMed] [Google Scholar]
- 15.Ramirez MS, Nikolaidis N, Tolmasky ME.. Rise and dissemination of aminoglycoside resistance: the aac(6′)-Ib paradigm. Front Microbiol 2013; 4: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ginn AN, Zong Z, Wiklendt AMet al. Limited diversity in the gene pool allows prediction of third-generation cephalosporin and aminoglycoside resistance in Escherichia coli and Klebsiella pneumoniae. Int J Antimicrob Agents 2013; 42: 19–26. [DOI] [PubMed] [Google Scholar]
- 17.Almaghrabi R, Clancy CJ, Doi Yet al. Carbapenem-resistant Klebsiella pneumoniae strains exhibit diversity in aminoglycoside-modifying enzymes, which exert differing effects on plazomicin and other agents. Antimicrob Agents Chemother 2014; 58: 4443–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoesser N, Batty EM, Eyre DWet al. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J Antimicrob Chemother 2013; 68: 2234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pesesky MW, Hussain T, Wallace Met al. Evaluation of machine learning and rules-based approaches for predicting antimicrobial resistance profiles in Gram-negative bacilli from whole genome sequence data. Front Microbiol 2016; 7: 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bulman ZP, Krapp F, Pincus NBet al. Genomic features associated with the degree of phenotypic resistance to carbapenems in carbapenem-resistant Klebsiella pneumoniae. mSystems 2021; 6: e00194-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gardner SN, Slezak T, Hall BG.. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 2015; 31: 2877–8. [DOI] [PubMed] [Google Scholar]
- 22.Zankari E, Hasman H, Cosentino Set al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 2012; 67: 2640–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li B, Tsui HC, LeClerc JEet al. Molecular analysis of mutS expression and mutation in natural isolates of pathogenic Escherichia coli. Microbiology (Reading) 2003; 149: 1323–31. [DOI] [PubMed] [Google Scholar]
- 24.CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically—Ninth Edition: M07. 2012.
- 25.CLSI. Performance Standards for Antimicrobial Susceptibility Testing—Twenty-Ninth Edition: M100. 2019.
- 26.Shakya T, Wright GD.. Mechanisms of aminoglycoside antibiotic resistance. In: Aminoglycoside Antibiotics: From Chemical Biology to Drug Discovery. John Wiley & Sons, Inc., 2007; 119–40. [Google Scholar]
- 27.Ramirez MS, Tolmasky ME.. Aminoglycoside modifying enzymes. Drug Resist Updat 2010; 13: 151–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eby JC, Richey MM, Platts-Mills JAet al. A healthcare improvement intervention combining nucleic acid microarray testing with direct physician response for management of Staphylococcus aureus bacteremia. Clin Infect Dis 2018; 66: 64–71. [DOI] [PubMed] [Google Scholar]
- 29.Walker T, Dumadag S, Lee CJet al. Clinical impact of laboratory implementation of Verigene BC-GN microarray-based assay for detection of Gram-negative bacteria in positive blood cultures. J Clin Microbiol 2016; 54: 1789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y, Sokolowski K, Rana Aet al. Generating genotype-specific aminoglycoside combinations with ceftazidime/avibactam for KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 2021; 65: e0069221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zomer A, Burghout P, Bootsma HJet al. ESSENTIALS: software for rapid analysis of high throughput transposon insertion sequencing data. PLoS One 2012; 7: e43012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bulman ZP, Cirz R, Hildebrandt Det al. Unraveling the gentamicin drug product complexity reveals variation in microbiological activities and nephrotoxicity. Antimicrob Agents Chemother 2020; 64: e00533-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robicsek A, Strahilevitz J, Jacoby GAet al. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med 2006; 12: 83–8. [DOI] [PubMed] [Google Scholar]
- 34.Ruiz E, Ocampo-Sosa AA, Alcoba FJet al. Changes in ciprofloxacin resistance levels in Enterobacter aerogenes isolates associated with variable expression of the aac(6′)-Ib-cr gene. Antimicrob Agents Chemother 2012; 56: 1097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bongaerts GP, Kaptijn GM.. Aminoglycoside phosphotransferase-II-mediated amikacin resistance in Escherichia coli. Antimicrob Agents Chemother 1981; 20: 344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perlin MH, Lerner SA.. High-level amikacin resistance in Escherichia coli due to phosphorylation and impaired aminoglycoside uptake. Antimicrob Agents Chemother 1986; 29: 216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lambert T, Ploy MC, Courvalin P.. A spontaneous point mutation in the aac(6′)-Ib′ gene results in altered substrate specificity of aminoglycoside 6′-N-acetyltransferase of a Pseudomonas fluorescens strain. FEMS Microbiol Lett 1994; 115: 297–304. [DOI] [PubMed] [Google Scholar]
- 38.Casin I, Hanau-Bercot B, Podglajen Iet al. Salmonella enterica serovar Typhimurium blaPER-1-carrying plasmid pSTI1 encodes an extended-spectrum aminoglycoside 6′-N-acetyltransferase of type Ib. Antimicrob Agents Chemother 2003; 47: 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rather PN, Munayyer H, Mann PAet al. Genetic analysis of bacterial acetyltransferases: identification of amino acids determining the specificities of the aminoglycoside 6′-N-acetyltransferase Ib and IIa proteins. J Bacteriol 1992; 174: 3196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Humphries RM, Ambler J, Mitchell SLet al. CLSI Methods Development and Standardization Working Group best practices for evaluation of antimicrobial susceptibility tests. J Clin Microbiol 2018; 56: e01934-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butler DA, Rana AP, Krapp Fet al. Optimizing aminoglycoside selection for KPC-producing Klebsiella pneumoniae with the aminoglycoside-modifying enzyme (AME) gene aac(6′)-Ib. J Antimicrob Chemother 2021; 76: 671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long SW, Olsen RJ, Eagar TNet al. Population genomic analysis of 1,777 extended-spectrum β-lactamase-producing Klebsiella pneumoniae isolates, Houston, Texas: unexpected abundance of clonal group 307. mBio 2017; 8: e00489-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen M, Brettin T, Long SWet al. Developing an in silico minimum inhibitory concentration panel test for Klebsiella pneumoniae. Sci Rep 2018; 8: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing reads and assembled genomes have been submitted to GenBank under BioProject PRJNA395086.