

Abstract
Human biology is tightly linked to proteins, yet most measurements do not precisely determine alternatively spliced sequences or posttranslational modifications. Here, we present the primary structures of ~30,000 unique proteoforms, nearly 10 times more than in previous studies, expressed from 1690 human genes across 21 cell types and plasma from human blood and bone marrow. The results, compiled in the Blood Proteoform Atlas (BPA), indicate that proteoforms better describe protein-level biology and are more specific indicators of differentiation than their corresponding proteins, which are more broadly expressed across cell types. We demonstrate the potential for clinical application, by interrogating the BPA in the context of liver transplantation and identifying cell and proteoform signatures that distinguish normal graft function from acute rejection and other causes of graft dysfunction.
Human biology is tightly linked to proteins, and mass spectrometry-based proteomics has established a strong linkage between phenotype and protein-level biology (1, 2). Notable efforts for the compositional mapping of proteins include two drafts of the human proteome in 2014 (3, 4); the Human Protein Atlas, with various tissue- and cell-specific resources available (5, 6); and the recent release of the Human Blood Atlas (HBA), with transcriptomic data from 18 cell types (7). However, these datasets do not capture posttranscriptional and posttranslational processing or how mRNA splicing combines with modifications to create protein-level diversity. Measurement of proteoforms (8) can close these gaps by capturing the complete molecular composition of proteins, refining phenotypic correlations. Furthermore, a reference map of experimentally identified proteoforms would serve as a reference for next-generation technologies, including single-cell proteomics (9, 10).
Protein isoforms vary by cell type (11, 12). With the growth in cell atlas projects, including the Human Biomolecular Atlas Program (HuBMAP) (13), the Human Cell Atlas (5), and others (6, 7), conditions are set for cell-based proteomics. Determination of protein composition in specific cell types using bottom-up proteomics has been accomplished in different studies (3, 14, 15). In this study, we used top-down proteomics (TDP) (16), which avoids the problem of inferring proteins using peptide data from shotgun proteomics analysis (17), to obtain cell- and proteoform-specific information (18) from the major cell types present in the blood and bone marrow. In the past decade, TDP has gained momentum, but limitations in protein separation and coverage of large proteoforms (>30 kDa) are still present (19). In this work, we employed negative or positive cell selection using specific antibodies to cell surface markers and fluorescence-activated cell sorting (FACS) to isolate cells of interest that were then analyzed for their proteoform content (table S1). In characterizing proteoforms across hematopoietic cell ontogeny, we took a three-pronged approach to protein fractionation, depending on cell numbers available (Fig. 1A).
Fig. 1. Workflow and number of identified proteoforms in the Blood Proteoform Atlas.
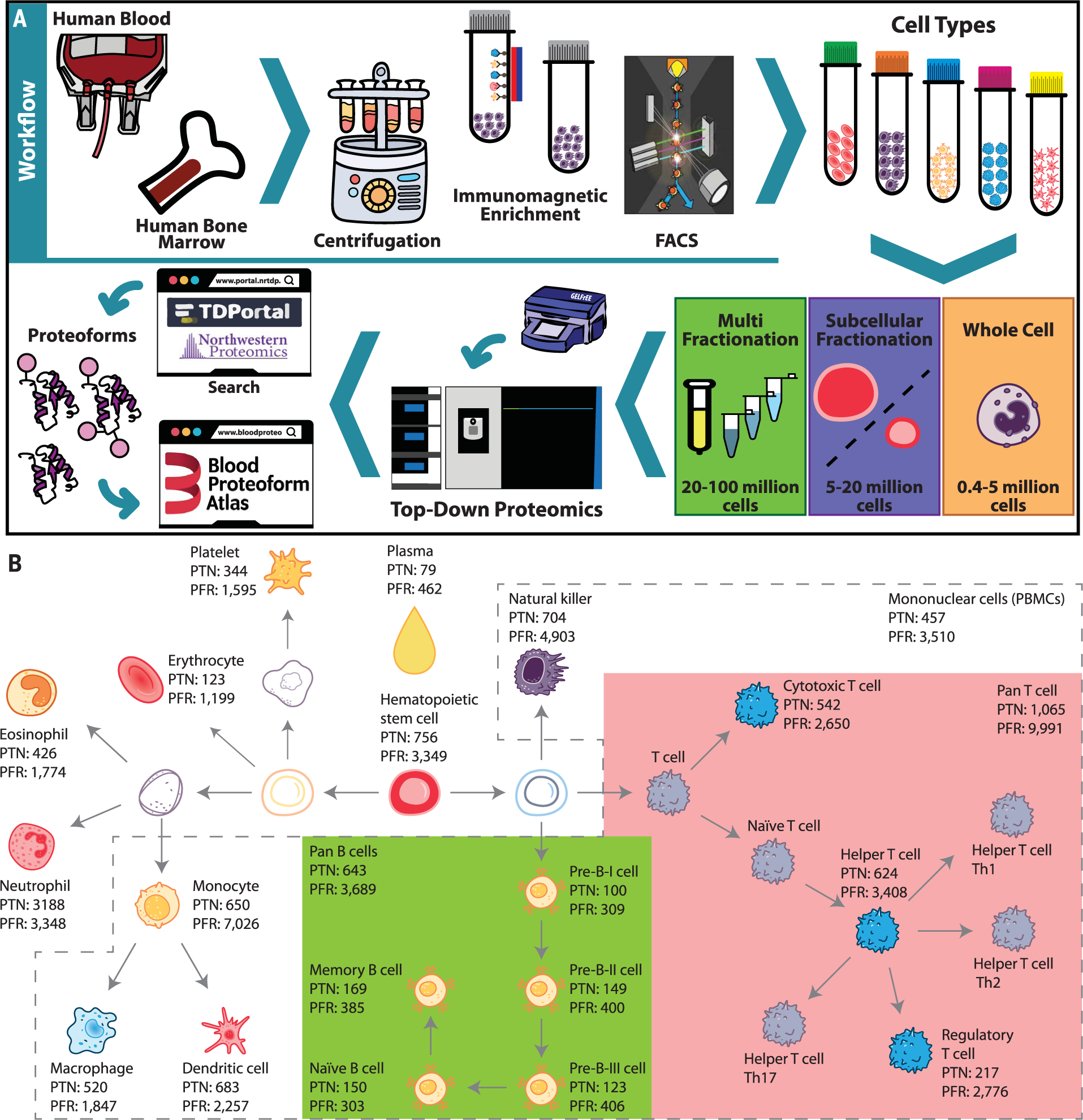
(A) Human blood or bone marrow samples were subjected to centrifugation, immunomagnetic enrichment, and/or FACS. Cell types were submitted to whole-cell, subcellular, and/or protein fractionation on the basis of the obtained cell amounts, followed by systematic proteoform discovery. Proteoforms were identified using a database search against the human proteome and deposited in the Blood Proteoform Atlas (BPA) website. (B) A map of hematopoiesis shows the number of proteoforms identified in each cell type. Certain cell groups (pan B cells, green; pan T cells, pink; and PBMCs, dashed gray lines) were also analyzed in pools. PTN, proteins; PFR, proteoforms.
Proteins and proteoforms
Table 1 captures a total of 29,620 nonredundant proteoforms and 1690 proteins (i.e., specific genes assigned from proteoform spectral matches) across 21 different human hematopoietic cell types and plasma. Many proteoforms discovered have posttranslational modifications; lysine acetylation (32.9%) and C- and N-terminal cleavage (30.6%) are the two most common, with coding polymorphisms (7.6%) or alternative splicing (3.8%) being minority occurrences (fig. S1 and table S2). The number of experimentally determined proteoforms exceeded that of previous reports by ~10-fold (16) and were identified in a dataset comprising 1553 liquid chromatography tandem mass spectrometry (LC-MS/MS) runs performed on Fourier transform (FT) mass spectrometers. In total, 4,042,173 database searches required ~9 days to complete, with a consistent proportion (34%) of them yielding a hit using a conservative 1% global false discovery rate (FDR) at the protein and proteoform levels (20) (see materials and methods section of the supplementary materials). Sample preparation required a total of ~1600 hours for all studies, and acquisition of mass spectra required ~3660 hours. Total proteoforms discovered from each category of cells throughout hematopoiesis are indicated in Fig. 1B and are interactively viewable at http://blood-proteoform-atlas.org. Proteoform identifiers (PFRs) are cross-referenced to gene-specific accessions in UniProtKB/Swiss-Prot and linked to 19,670 transcripts in the HBA (7). For the BPA, the average number of proteoforms arising from each “protein” (i.e., proteoform hits mapped back to their corresponding human gene) was 17.5. Despite the accelerating pace of development for TDP, most identified proteoforms (~93%) are <20 kDa, even analyzing GELFrEE fractions containing proteins up to 50 kDa.
Table 1.
Proteins and proteoforms identified across 21 human cell types and plasma, aggregated in redundant and nonredundant fashion.
| Cell type | Analysis method | LC-MS/MS runs | Proteins 1% FDR | Unique proteins 1% FDR | Proteoforms 1% FDR (C-score > 30) | Unique proteoforms 1% FDR |
|---|---|---|---|---|---|---|
| Hematopoietic stem cell* | Nuc/Cyt | 57 | 756 | 16 | 3,349 (2,520) | 596 |
| PBMC | WC IEX |
112 | 457 | 27 | 3,510 (2,618) | 1,507 |
| Pan T cell | IEX WC Nuc/Cyt |
367 | 1,065 | 116 | 9,991 (5,839) | 4,163 |
| Cytotoxic T cell | WC Nuc/Cyt |
68 | 542 | 2 | 2,650 (963) | 436 |
| Helper T cell | WC Nuc/Cyt |
89 | 624 | 2 | 3,408 (1,319) | 617 |
| Regulatory T cell | Nuc/Cyt | 58 | 217 | 0 | 2,776 (477) | 899 |
| Pan B cell | WC Nuc/Cyt |
118 | 643 | 8 | 3,689 (1,653) | 745 |
| Pan B cell* | WC | 12 | 334 | 1 | 1,225 (602) | 111 |
| Pre-B-I cell* | WC | 4 | 100 | 0 | 309 (253) | 13 |
| Pre-B-II cell* | WC | 4 | 149 | 0 | 400 (323) | 26 |
| Pre-B-III immature cell* | WC | 4 | 123 | 3 | 406 (334) | 33 |
| Naïve B cell | WC | 5 | 150 | 0 | 303 (176) | 19 |
| Memory B cell | WC | 5 | 169 | 0 | 385 (208) | 20 |
| Natural killer cell | Nuc/Cyt | 98 | 704 | 25 | 4,903 (1,859) | 1,123 |
| Monocyte | Nuc/Cyt | 128 | 650 | 8 | 7,026 (3,029) | 2,391 |
| Immature dendritic cell | Nuc/Cyt | 52 | 683 | 20 | 2,257 (1,343) | 376 |
| Macrophage | Nuc/Cyt | 46 | 520 | 15 | 1,847 (1,082) | 472 |
| Eosinophil | Nuc/Cyt | 52 | 426 | 10 | 1,774 (1,427) | 391 |
| Neutrophil | WC IEX Nuc/Cyt |
104 | 318 | 27 | 3,348 (2,784) | 1,728 |
| Erythrocyte (RBC) | WC PM |
41 | 123 | 6 | 1,199 (555) | 477 |
| Platelet | WC PM |
78 | 344 | 14 | 1,595 (1,122) | 728 |
| Plasma | PM | 50 | 79 | 17 | 462 (326) | 232 |
| Total | – | 1,553 | 9,239 | 317 | 56,813 (30,812) | 17,103 |
| Total (nonredundant) | – | 1,553 | 1,690 | 317 | 29,620 (17,630) | 17,103 |
Analysis methods used: Nuc/Cyt, nuclear and cytosolic fractions were analyzed individually; WC, whole-cell lysate; IEX, ion exchange used before the GELFrEE–LC-MS/MS; PM, ProteoMiner equalized cell lysates.
Cell types isolated from human bone marrow.
Confident assignment of isoforms from RNA splicing
From the HBA, we generated a proteogenomic database with 50,177 protein sequences, corresponding to 95,979 transcripts reconstructed from RNA sequencing (RNA-seq) data for 19 cell types (table S3) (7). Searching 12 cell types shared between the BPA and the HBA, we identified slightly fewer proteins and proteoforms using the HBA database search (801 proteins and 4344 proteoforms; table S4) than with the human UniProtKB/Swiss-Prot database (887 proteins and 4993 proteoforms; table S5). Most proteoforms observed with the HBA database were shared with the UniProtKB/ Swiss-Prot database (82.7%), while 2.2% (114) of proteoforms were only in the HBA database (fig. S2), and of these, 49 (0.96%) represented newly identified proteoforms that are confidently assigned to being derived from transcript isoform or sequence variation (table S6). These results indicate that RNA splicing produces only a handful of new detectable proteoforms <30 kDa, which are expressed from an average of just four introns. However, a few abundant isoforms are missed without cell type–specific RNA splicing information.
Protein-resolved versus proteoform-resolved maps of hematopoietic cell types
Deep TDP of cell populations results in high-dimensional data containing cell type and proteoform identifications (e.g., PFR1033, which maps to the gene-specific accession P62805 in UniProtKB/Swiss-Prot for histone H4). We compared protein- versus proteoform-level data in t-distributed stochastic neighbor embedding (t-SNE) plots (Fig. 2, A and B), accumulation curves (Fig. 2, C and D), and heatmaps after hierarchical clustering (Fig. 2E). Both protein and proteoform data clustered differentiated cell types along primary branches of hematopoiesis at the same level of confidence upon t-SNE analysis. The cluster of antigen-presenting cells (monocytes, macrophages, and dendritic cells) was separated from lymphoid lineage cells [T cells, B cells, and natural killer (NK) cells]. Additionally, the lymphoid lineage clustered together, and the three pre-B cell types from bone marrow formed a distinct cluster (Fig. 2, A and B).
Fig. 2. Display of protein and proteoform analysis for entries in the Blood Proteoform Atlas.
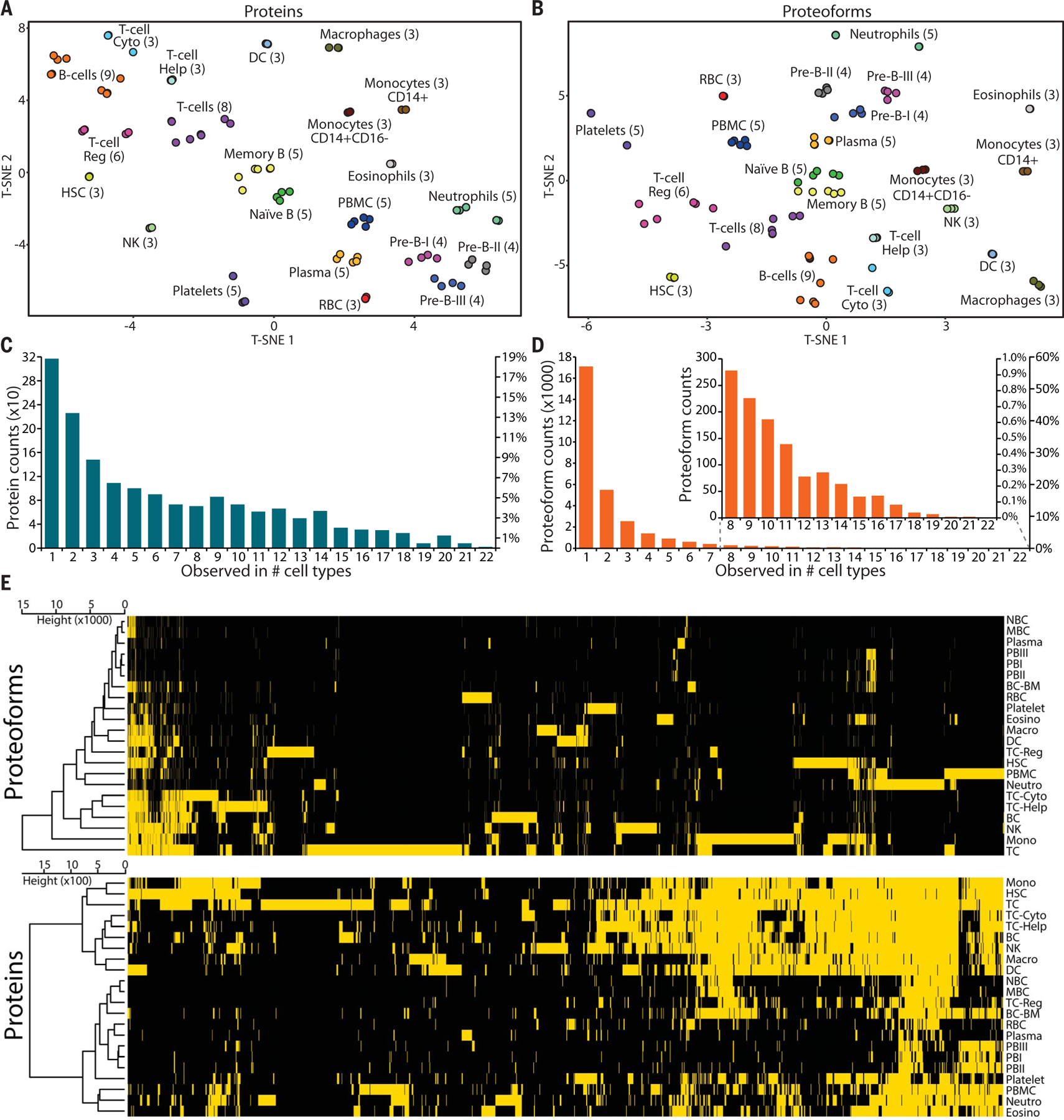
t-SNE plots display cell types grouped by presence or absence of (A) proteins and (B) proteoforms. T-cell Cyto, cytotoxic T cells; T-cell Help, helper T cells; T-cell Reg, regulatory T cells; HSC, hematopoietic stem cells; DC, dendritic cells; RBC, red blood cells. Histograms of (C) proteins and (D) proteoforms shared by different cell types. (E) Heatmaps and cell type hierarchical clustering of identified (yellow) or not-identified (black) proteins (1690) and proteoforms (29,620) at 1% FDR, with proteoforms exhibiting higher specificity for distinct cell types. NBC, naïve B cells; MBC, memory B cells; PBI, pre-B-I cells; BC-BM, B cells from bone marrow; Eosino, eosinophils; Macro, macrophages; Neutro, neutrophils; BC, B cells from blood; Mono, monocytes; TC, T cells.
Cell relatedness at the protein versus proteoform levels
To probe the specificity of proteins versus proteoforms, we compared their incidence frequency across all cell types studied. The histogram in Fig. 2C shows that most proteins are shared between two or more cell types (81%). In contrast, the majority of proteoforms (58%) were identified in only one cell type (Fig. 2D), which shows that uniqueness is significantly more pronounced with proteoform-level information [χ2(21) = 0.519, P = 4.73 × 10−14]. Figure 2E shows the heatmap for all proteins and proteoforms identified. The high number of unique proteoforms per cell type is evident, and the clustering distance for proteoforms is one order of magnitude higher than for proteins. The average number of cell types in which a proteoform was found was 2.19, whereas for proteins it was 6.51. The mean number of identified nonredundant proteoforms per cell type was 1346, compared with only 76 for proteins (more statistical measures are presented in table S7). These results indicate that proteoforms are better markers of a cell type than monitoring gene expression using just protein-level assignments.
Depth of proteome coverage
The BPA currently provides 8.3% coverage (fig. S3) of the total human proteome (20,395 genes) and 16% of the predicted proteome <30 kDa in UniProtKB/Swiss-Prot. However, the accumulation curve for proteins shows that ~80% of possible protein identifications (fig. S3) were made from the analyzed cell types using current workflows. In contrast, the collector’s curve for proteoforms did not reach a plateau (fig. S3), with just ~37% of projected proteoforms identified. This modeling indicates that saturation of primary hematopoietic cells would uncover >50,000 additional proteoforms in this size regime. We expect that larger proteins that act as hubs of cellular decision-making could have more proteoforms per protein [e.g., tumor suppressor protein p53 (21)]. Further, to estimate the number of human proteoforms, we multiplied the theoretical number of proteins by three times the standard deviation of the mean number of proteoforms observed per protein. From this, we estimated the number of proteoforms to be ~1.1 million in a human cell type, close to a previous estimation (22). Hence, this study likely accounts for at most ~3% of human proteoforms distributed from 1 to 59 kDa (fig. S3), demonstrating a clear need to improve technologies for systematic proteoform discovery (23).
Quantitative TDP of hematopoietic cell types
Quantitative comparison of proteoforms using label-free TDP (24) was applied to compare pools of B cells (CD19+) against T cells (CD3+) from the same donor (fig. S4 and supplementary text in the supplementary materials). We also compared five B cell subtypes from a single donor sorted by FACS, including pre-B-I, pre-B-II, and pre-B-III from bone marrow and memory and naïve B cells from blood (table S1 and Fig. 3). Many proteins or proteoforms are shared by cells from the bone marrow or between their mature forms in the blood, indicating that these cell types are distinct (Fig. 3, A and B). The same pattern is observed in the expression heatmap generated from the standardized intensities scores (Fig. 3C), separating the two cell groups by >66% of the total clustering distance. Three clusters of proteoforms were observed: (i) those up-regulated in naïve and memory B cells, (ii) those up-regulated in pre-B cells, and (iii) those with a random regulation pattern.
Fig. 3. Comparison of B cell subtypes using quantitative top-down proteomics.

Venn diagram analysis of (A) proteins and (B) proteoforms observed in pre-B-I, pre-B-II, and pre-B-III cells; naïve B cells; and memory B cells. (C) Heatmap and hierarchical clustering of quantified proteoforms from B cells subtypes. Numbers 1 to 3 represent the three major clusters of proteoforms found. (D) Volcano plot of up- and down-regulated proteoforms from naïve B cells (NB) relative to memory B cells (MB). (E) Box-and-whisker plots of proteoforms PFR1464 (TMSB4X) and PFR1215 (S100A6) levels show their variance among the five B cell subtypes. Fragmentation maps of (F) PFR1464 and (G) PFR1215. Red box indicates N-terminal acetylation. Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr. (H) Distributions of proteoforms PFR1464 and PFR1215 in blood cell types of the BPA.
A closer look into the differentially regulated proteoforms from the mature B cells showed 10 proteoforms with increased relative abundance in naïve B cells, with 50 elevated in memory B cells (Fig. 3D). The proteoform PFR1464 from thymosin beta-4 (Fig. 3F) was elevated by ~10-fold in naïve B cells relative to memory cells, whereas proteoform PFR1215 from the protein S100A (Fig. 3G) was increased in memory relative to naïve B cells. Exploring the presence of these two proteoforms in the five B cell subtypes showed that PFR1215 is present at low levels in naïve B cells and at high levels in pre-B cells and memory B cells (Fig. 3E). PFR1464 is present mainly in naïve B cells and has low levels in pre-B cells (Fig. 3E). Expanding this comparison to all cell types in the BPA, we observe that PFR1464 is more abundant in naïve B cells, whereas PFR1215 is more widely observed in different cell types (Fig. 3H). Quantitative results were consistent using spectral counting and intact proteoform quantification methods in both cases.
BPA and peripheral blood mononuclear cells from liver transplant recipients
With a reference set of proteoforms, the BPA can inform clinical research and care with more precise protein information, for example, the prostate-specific antigen isoform test in prostate cancer (25). One area of unmet need is organ transplantation, in particular liver transplantation (LT), where episodes of acute rejection (AR) limit survival (26). AR arises from an imbalance of immune activation (IA) related to cellular and humoral antidonor responses (e.g., effector CD4+, cytotoxic CD8+ T cells, and donor-specific antibodies) over immune quiescence (IQ) suppressor countermechanisms (e.g., regulatory T cells) (27–30). This key balance of IA versus IQ relating to AR risk is, however, difficult to determine without available objective markers in LT recipients (LTRs). Given this critical clinical scenario, we sought to discover and validate cell-based proteoforms as indicators of LTR immune status for future clinical applications.
We first conducted an untargeted quantitative TDP analysis of peripheral blood mononuclear cells (PBMCs) using the 0 to 30 kDa GELFrEE fractions of whole PBMC lysates from a cohort of 75 LTRs (Fig. 4, A and B). Patients were initially divided into three groups: transplant excellent (TX representing IQ and healthy graft function; n = 25), acute dysfunction no rejection (ADNR, representing nonrejection causes of graft injury; n = 25), and AR (n = 25). The AR and ADNR phenotypes were diagnosed by needle biopsy, and TX by clinical and laboratory criteria, as previously described (31–34). The absence of biopsies in TX was a limitation; however, transplant centers do not generally perform surveillance biopsies in LTR with healthy graft function. Figure 4 shows the results for AR versus non-AR (TX+ADNR) and TX versus non-TX (AR+ADNR), grouped for simplification and alignment with clinical utility. We identified a total of 198 differentially expressed proteoforms (DEPs) from 99 proteins (Fig. 4, C and D), many of which were detected in our small exploratory study (31). Pathway and process analysis was performed on the identified proteins from each group using Metascape (35) (fig. S5 and table S8). Between the groups, we found some commonly enriched pathways involved in T cell activation and graft migration, including RHO GTPase effectors (36). TX-specific pathways include regulated exocytosis, platelet degranulation, and cytoskeletal organization. These pathways could be related to exocytosis of granules from cytotoxic T cells, platelet-mediated thrombosis, and remodeling of the actin cytoskeleton (37).
Fig. 4. Quantitative top-down proteomics analysis of PBMC proteoforms from liver transplant recipients.

(A) Workflow used to compare patients with transplant excellent (TX); with acute dysfunction, no rejection (ADNR); and with acute rejection (AR). (B) The number of patients whose PBMCs were analyzed in an untargeted fashion. Volcano plot showing differentially expressed proteoforms in (C) AR patients relative to non-AR (TX+ADNR) and (D) TX patients relative to non-TX (ADNR+AR). (E) The number of patients whose PBMCs were analyzed for targeted proteoforms. Volcano plot with the relative levels of the 24 targeted proteoforms in (F) AR versus non-AR and (G) TX versus non-TX. (H) Distributions of the normalized spectral (N.S.) counts of proteins (left) and proteoforms (right) observed in BPA cell types.
Next, we performed a targeted validation study using some of the most significant proteoforms from discovery (table S9). A panel with 24 proteoforms from 23 proteins (table S10) was deployed on a new cohort of 59 patient PBMCs (TX = 36, ADNR = 10, and AR = 13) (Fig. 4E) from a multicenter LT study (NIAID CTOT-14; NCT01672164) (32). We note that the number of AR subjects is small but reflects the ~20% prevalence of AR in LTR (26, 38–40). Statistical analysis comparing AR and non-AR or TX and non-TX populations confirmed significantly up-regulated proteoforms from the TX or non-AR groups (Fig. 4, F and G, and table S10). The proteoforms differentially regulated in TX were platelet factor 4 (PF4) N-terminally truncated with five extra amino acids, PFR18631; nonhistone chromosomal protein HMG-17 (HMGN2) canonical sequence, PFR1006; and a C terminus part of cytoplasmic actin 1 (ACTB) from amino acid positions 330 to 375, PFR69028. On the other hand, 15 proteoforms were significantly increased in non-AR (table S10). The three proteoforms with the higher q values were a piece of serum deprivation-response protein (CAVIN2) from amino acid positions 297 to 343, PFR70141; a C-terminal portion of high mobility group protein B1 (HMGB1) from amino acid positions 107 to 214, PFR69103; and the canonical sequence of profilin-1 (PFN1) N-terminally acetylated, PFR1439.
With a cell-based expression atlas in hand, we could identify the cell types in which these 24 proteoform targets are typically present. Figure 4H shows the heatmaps of proteins (left) and their proteoforms (right). Most proteoforms are found in a narrower range of cell types; for example, PFR70141 from the CAVIN2 gene was only identified in NK cells, naïve B cells, B cells, and T cells, while at the protein level (O95810) it was additionally observed in platelets, PBMCs, and hematopoietic stem cells (HSCs). Moreover, transcriptome data from the HBA point to the CAVIN2 gene as highly expressed in PBMCs, supporting the results. All proteoforms from the panel were identified in T cells, 23 in B cells, and 22 in NK cells–collectively the most abundant PBMC types (41). Five were identified in red blood cells (RBCs), neutrophils, and plasma, suggesting nonspecific cell proteoforms. On the basis of the protein and proteoform hits in Fig. 4H, we performed a cell enrichment test against all BPA identifications (fig. S6). Consistent with a narrower distribution of proteoforms, one proteoform of PF4, PFR18631, was observed in six cell types (platelets, plasma, B cells, T cells, and HSCs). A second proteoform, PFR18628, was identified in 11 cell types, and the corresponding protein (P02776) in 15 different cell types. In this case, platelets showed the highest normalized spectral counts for the protein that is an archetype of the chemokine family essential in platelet aggregation and inflammation (42).
Some of the identified proteoforms derive from proteins with immune functions, including PF4 and PFN1. PF4 expression is reduced in humans and mice with acute liver injury and inhibits ischemia-reperfusion injury in LT mouse models (43, 44). PFN1 is involved in actin organization, which inhibits CD8+ cytotoxicity by reducing migration and degranulation (45). Box-and-whisker plots showing the prominent striation in individual patient responses are presented in fig. S7. Proteoforms related to ACTB, PFN1, and PF4 were statistically significant in TX and non-AR groups, supporting the discovery-stage experiments. Notably, the protein PF4 had two proteoforms in the 24 proteoform panel, and only PFR18631 was 1.9-fold up-regulated in the TX group compared with the non-TX group. This proteo form has four extra amino acids (FASA) on the N terminus compared with PFR18628, which maps to the canonical isoform in UniProtKB/UniprotKB/Swiss-prot (42) and was not differentially expressed. The N-terminal processing of PFR18631 may represent an essential mechanism for modulating PF4 activity similar to the one described to inhibit endothelial cell growth (42). Additional studies are underway to monitor proteoform changes over time and in specific cell types in LTR patients (32).
The results from this small cohort suggest that in the clinical context of liver transplantation (i) leukocyte proteoform levels might have diagnostic value for IA versus IQ, and (ii) clinically relevant immunoproteoforms are present in select blood cell populations. The novelty of direct proteoform measurement versus less specific epitope- or peptide-based methods could advance care by identifying early specific signs of IA versus IQ to personalize LTR immunosuppressive therapy monitoring and modulation.
Summary
By mapping ~57,000 redundant proteoforms present in human blood, bone marrow, plasma, and within main hematopoietic cell types, we have advanced fundamental knowledge of protein components present in the human body. At the transcript level, the field is advancing single-cell RNA-seq from a compositional tool to connect with spatial localization (46). A reference map of human proteoforms can serve a similar function at the protein level as we seek to understand the spatial and temporal dynamics of proteins operative in human tissue (13). Here, both cell- and proteoform-specific information in the context of organ transplantation were provided as a potential clinical application. This cellular and molecular specificity can help advance the future of protein-level diagnostics and broader goals for understanding human biology.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank N. Haverland and the following members of the Robert H. Lurie Comprehensive Cancer Center Flow Cytometry Core Facility for helpful discussions and experimental assistance with FACS methodology: S. Swaminathan, P. Mehl, and C. Ostiguin. We thank P. Oksvold for discussions and assistance with making the HPA Blood Cell Atlas data available. Analysis of these transcriptomic data was performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under project sens2019032. We also thank L. M. Smith for additional computational resources for proteomic analysis with MetaMorpheus.
Funding:
Funding was provided by Paul G. Allen Frontiers Program award 11715 (N.L.K.); HuBMAP grant UH3 CA246635–02 (N.L.K.); the National Institute of General Medical Sciences of the National Institutes of Health under grants P41 GM108569 (N.L.K.), R21LM013097 (P.M.T.), T32 GM105538 (T.K.T.), and R21 AI135827 (J.L.); Knut and Alice Wallenberg Foundation grant 2016.0204 (A.J.C. and E.L.); and Swedish Research Council grant 2017–05327 (E.L.). A portion of this work was performed at the Ion Cyclotron Resonance User Facility at the National High Magnetic Field Laboratory, which is supported by the National Science Foundation Division of Materials Research and Division of Chemistry through DMR-1644779, and the State of Florida.
Footnotes
Competing interests: N.L.K. is involved in entrepreneurial activities in top-down proteomics and consults for Thermo Fisher Scientific. A.I.K. is an employee of Stem Cell Technologies.
SUPPLEMENTARY MATERIALS
Data and materials availability:
All cell types and cell enrichment kits used are commercially available and listed in the material and methods section of the supplementary materials. All .raw files are available at the Proteomics Identifications Database (PRIDE; www.ebi.ac.uk/pride/) under accession numbers PXD026123 to PXD026178. Proteoform information is available in the Blood Proteoform Atlas (https://blood-proteoform-atlas.org).
REFERENCES AND NOTES
- 1.Picotti P et al. , Nature 494, 266–270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Battle A et al. , Science 347, 664–667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim MS et al. , Nature 509, 575–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilhelm M et al. , Nature 509, 582–587 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Thul PJ et al. , Science 356, eaal3321 (2017).28495876 [Google Scholar]
- 6.Uhlén M et al. , Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Uhlen M et al. , Science 366, eaax9198 (2019).31857451 [Google Scholar]
- 8.Smith LM, Kelleher NL; Consortium for Top Down Proteomics, Nat. Methods 10, 186–187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marx V, Nat. Methods 16, 809–812 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Doerr A, Nat. Methods 16, 20 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Yang X et al. , Cell 164, 805–817 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghadie MA, Lambourne L, Vidal M, Xia Y, PLOS Comput. Biol. 13, e1005717 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.HuBMAP Consortium, Nature 574, 187–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doll S et al. , Nat. Commun. 8, 1469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Palma S et al. , J. Proteome Res. 10, 3814–3819 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Tran JC et al. , Nature 480, 254–258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nesvizhskii AI, Aebersold R, Mol. Cell. Proteomics 4, 1419–1440 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Smith LM, Kelleher NL, Science 359, 1106–1107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown KA, Melby JA, Roberts DS, Ge Y, Expert Rev. Proteomics 17, 719–733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LeDuc RD et al. , Mol. Cell. Proteomics 18, 796–805 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uversky VN, Int. J. Mol. Sci. 17, 1874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aebersold R et al. , Nat. Chem. Biol. 14, 206–214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith LM et al. , Sci. Adv 7, eabk0734 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ntai I et al. , Mol. Cell. Proteomics 15, 45–56 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stovsky M et al. , J. Urol. 201, 1115–1120 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Levitsky J et al. , Clin. Gastroenterol. Hepatol. 15, 584–593.e2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreau A, Varey E, Anegon I, Cuturi MC, Cold Spring Harb. Perspect. Med. 3, a015461 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ronca V, Wootton G, Milani C, Cain O, Front. Immunol. 11, 2155 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pons JA et al. , Transplantation 86, 1370–1378 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G, Front. Immunol. 10, 43 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toby TK et al. , Am. J. Transplant. 17, 2458–2467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levitsky J et al. , Am. J. Transplant. 20, 2173–2183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levitsky J et al. , Transplantation 10.1097/TP.0000000000003895 (2021). [DOI] [Google Scholar]
- 34.Levitsky J et al. , Am. J. Transplant ajt.16835 (2021). [Google Scholar]
- 35.Zhou Y et al. , Nat. Commun. 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.El Masri R, Delon J, Nat. Rev. Immunol. 21, 499–513 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Witke W et al. , EMBO J. 17, 967–976 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramji A et al. , Liver Transpl. 8, 945–951 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Uemura T et al. , Clin. Transplant. 22, 316–323 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Thurairajah PH et al. , Transplantation 95, 955–959 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Kleiveland CR, in The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models, Verhoeckx K et al. , Eds. (Springer, 2015), pp. 161–167. [PubMed] [Google Scholar]
- 42.Gupta SK, Hassel T, Singh JP, Proc. Natl. Acad. Sci. U.S.A 92, 7799–7803 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo H, Wang Y, Zhao Z, Shao X, Scand. J. Immunol. 81, 129–134 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Drescher HK et al. , Front. Physiol. 10, 326 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schoppmeyer R et al. , Eur. J. Immunol. 47, 1562–1572 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Stuart T et al. , Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All cell types and cell enrichment kits used are commercially available and listed in the material and methods section of the supplementary materials. All .raw files are available at the Proteomics Identifications Database (PRIDE; www.ebi.ac.uk/pride/) under accession numbers PXD026123 to PXD026178. Proteoform information is available in the Blood Proteoform Atlas (https://blood-proteoform-atlas.org).